

Abstract
Current dogma holds that the innate immune system primes the adaptive immune system in response to infection, which in turn amplifies innate responses in a positive loop to effectively control pathogens. Therefore, it is accepted in most cases that T-cell deficient hosts die of acute infection because of the impaired ability of the innate immune system to control pathogens. Recent studies, however, reveal that adaptive immune cells actively dampen initial innate responses. In contrast to current understanding, there is now evidence that an insufficient number of T cells results in loss of control of innate immune responses. This raises new questions regarding the, as of yet underappreciated, role of the adaptive immune system in early infection and inflammation.
The innate and adaptive immune systems collaborate to protect hosts from infection
The mammalian host is armed with the innate and adaptive immune systems. The innate immune system is evolutionarily conserved among multi-cellular organisms and provides rapid defense against invading microbes within hours – long before the adaptive immune system mounts an antigen specific response a few days later. Cells of the innate immune system recognize microbial structures called pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) including Toll-like receptors (TLRs) 1, 2, 3. This interaction mobilizes the innate immune response, leading to upregulation of both major histocompatibility complex (MHC) class I and II and co-stimulatory molecules, in addition to secretion of inflammatory cytokines that more efficiently prime T cells and help to guide the subsequent adaptive response 1, 4. In a positive feed-forward activation loop, activated adaptive immune cells further drive innate cells to amplify anti-pathogen responses. Type 1 helper T (Th1) cells activate macrophages through both cell–cell contact and interferon-gamma (IFN-γ) secretion 5, 6, 7, Th2 cells activate eosinophils through cytokine release 8, 9, 10 and B cells secrete antibodies to activate the cascade of complement proteins, phagocytes, natural killer (NK) cells and mast cells 11, 12, 13, 14, 15. Current dogma, thus, centers on both the sequential activation of the two arms of the immune system and the mutual amplification of responses to effectively and efficiently combat microbial invaders over time.
What is the role of adaptive cells in the early phase of infection: negative or positive regulators?
In response to infection, sufficient immune responses are required to protect hosts from invading pathogens. Excessive activation of immune cells, however, can lead to microcirculatory dysfunction, tissue damage, shock or even death of the host 3, 16, 17, 18, 19. These damaging responses are referred to collectively as immunopathology 3, 16, 17, 18, 19. Hosts utilize various components of the immune system to carefully maintain the delicate balance between allowing the immune response to target the pathogen and preventing widespread over-activation that can lead to immunopathology. Negative regulation of TLR signaling, via soluble decoy TLRs and intracellular negative regulators, has been reported to be crucial for host survival and exists at multiple levels [20]. It is accepted, in most cases, that innate immune cells help prime lymphocytes during acute infection and, in a reciprocal fashion, antigen-specific lymphocytes further activate innate cells in the effector phase to clear the pathogen. It is not clear, however, whether or how lymphocytes that normally take days to activate can regulate innate cells in the early phase of infection. To study the early interactions between adaptive immune cells (such as T cells) and innate cells [21], nude (i.e. T-cell deficient) and wild type mice were inoculated with murine hepatitis virus strain A59 (MHV-A59), a coronavirus that infects mouse liver and brain. MHV-A59 is an RNA virus that can target TLR3. After challenged with a sublethal dose of the virus, nude mice quickly died. Unexpectedly, the cause of death was determined to be because of excessive production of pro-inflammatory cytokines rather than from uncontrolled viral load, which indicated that a lack of T cells in the system might have resulted in a stronger innate immune response. To confirm that the death of immunocompromised mice was caused by a cytokine storm, non-infectious ligands for TLRs instead of infectious viruses were utilized. RNA virus-related polyinosinic:polycytidylic acid (poly I:C) (a non-infectious ligand for TLR3) and lippolysaccharide (LPS) (a bacterial product for TLR4) were used to ensure that both wild type and immunocompromised mice were exposed to similar levels of TLR stimulation over time. In the same way, treatment of nude mice or recombinant activation gene-1 (RAG-1) knockout mice (which also lack B cells) with poly I:C caused rapid death that could be rescued by antibody mediated tumor necrosis factor (TNF) blockade. Therefore, a cytokine storm resulting from insufficient T-cell numbers in the system seemed to directly cause the lethality observed in both nude and RAG-1 knockout mice. In further experiments to test the hypothesis, specific depletion of T cells in wild type mice using both, not single, anti-CD4 and anti-CD8 monoclonal antibodies replicated the cytokine storm seen in nude or RAG-1 Knockout mice. In addition, the introduction of T cells by adoptive transfer of T lymphocytes into RAG-1 deficient mice protected the mice from the cytokine storm [21]. The introduction of either a CD4+ or a CD8+ population sufficiently reduced innate responses to TLRs in vitro, indicating that either subset is sufficient to repress the innate responses. Collectively, these data indicate that T cells are both necessary and sufficient to temper the early innate immune response triggered by viral infection or TLR stimulation.
The complex role of CD4+CD25+ regulatory T cells in innate cell suppression
There is an increasing body of evidence indicating that regulatory T (Treg) cells suppress not only effector T cell function but also cells of the innate immune system 22, 23, 24. Earlier studies of this issue focused on whether suppression of innate immune responses by Treg cells was aimed at limiting late stages of pathogen-specific immunity as a means of minimizing associated tissue damage. CD4+CD25+ Treg cells suppress innate immune pathology through various mechanisms. One study has shown that Treg cells use the immunosuppressive cytokine transforming growth factor beta (TGFβ) to directly inhibit NK cell-mediated cytotoxicity in vitro and in vivo to effectively suppress NK-cell rejection of tumors [23]. Other studies have demonstrated both an IL-10 and TGFβ dependency for the suppression of innate immune responses 22, 24. More recently, it was proposed that Treg cells facilitate early protective responses to local viral infection by allowing a timely entry of immune cells into infected tissue and only later suppress the response [25]. In this study, augmented cytokine production was detected in the draining lymph nodes in Treg-cell-depleted mice, although it was profoundly reduced at the infection site and associated with a delayed migration of innate cells to the site of infection.
It is intriguing that both Treg cells and conventional T cells can efficiently suppress the cytokine surge by innate immune cells in vitro [21]. With the vast number of innate immune cells both inside and outside lymphoid tissues, it is conceivable that a large number of T cells are needed to render efficient suppression at all times. Therefore, both naïve and Treg cells might be required to maintain the inhibition of innate immune responses. Furthermore, the suppression of the innate immune response is mediated by cell–cell contact between T cells and innate cells and is dependent upon MHC, yet independent of antigen specificity [21]. It remains unclear, however, whether these two T-cell populations utilize similar mechanisms to suppress innate immune cells. Identifying the molecules and underlying mechanisms that account for such intercellular suppression might lead to the discovery of new inhibitory networks or reveal novel roles for existing molecules. Several co-inhibitory molecules, such as cytotoxic T lymphocyte antigen (CTLA-4), programmed cell death 1 (PD-1) and B and T lymphocyte attenuator (BTLA), have been found to be imperative in maintaining the balanced immune response and homeostasis of immune cells [26]. The interaction of BTLA with its ligand herpesvirus entry mediator (HVEM) results in the repression of antigen-derived T-cell proliferation and cytokine production 27, 28. It will be interesting to study whether these co-inhibitory molecules have a role in controlling innate responses.
Cells of the innate immune system, including macrophages, dendritic cells (DC) and NK cells, initiate the development of both innate and adaptive immune responses through the release of cytokines 29, 30. In addition, crosstalk among cells of the innate immune system is also an important mechanism to amplify the innate response 31, 32, 33. For example, poly I:C can directly stimulate cytokine production from CD11b+ cells (i.e. macrophages) and to a lesser degree CD11c+ cells (i.e. DC) but not NK or T cells [21]. The addition of NK cells to macrophages in vitro, however, can indirectly activate NK cells to enhance cytokine production in response to poly I:C stimulation. In vivo, it is possible that pathogens can directly stimulate macrophages or DCs through TLR engagement and, at a later phase, these innate cells can then activate NK cells to amplify TNF production. It is likely that the T cell–antigen presenting cell (APC) interaction that dampens the inflammatory innate response via MHC and/or BTLA-HVEM works in a similar way, in which a direct T cell–NK cell interaction during the acute phase of infection is not required (Figure 1 ). In fact, decreased inflammatory cytokines and increased survival rates after NK cell depletion in immunodeficient mice [21] demonstrate that NK cells are responsible for amplifying the cytokine storm.
Figure 1.
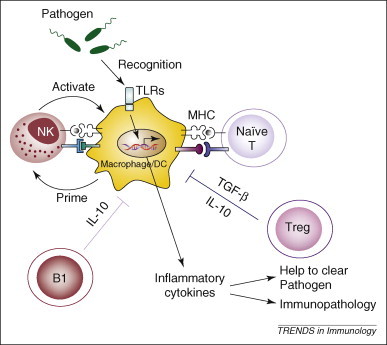
Negative regulation of early innate immune responses by conventional T cells and Treg cells. Pathogens directly stimulate macrophages and DCs through TLR engagement. At the later phase of immune activation, these DC and macrophages prime other innate cells, such as NK cells, to amplify cytokine production. NK cells in turn further activate macrophages to produce more TNFα and other inflammatory cytokines with the potential to cause immunopathology. T cell–APC (i.e. macrophage or DC) interaction via MHC and/or other membrane ligands or receptors dampens the inflammatory innate response during pathogen clearance. Naïve T cells, Treg cells and B1 cells contribute to the inhibition of innate cells by means of, as yet, poorly defined mechanisms but probably involving both cytokines (TGF-β and IL-10) cell–cell interactions (e.g. BTLA–HVEM).
Implications for diseases in immunocompromised hosts
Perhaps, as a reflection of the earlier establishment of the innate immune system in immune phylogeny, the embryonic development of the innate immune system precedes that of the adaptive system. Therefore, an interesting thought arising from the current hypothesis would be to ask whether neonatal innate responses are over-reactive and poorly regulated after infection. Indeed, neonates suffer high morbidity and mortality to infection, but this is often attributed to both weaker innate and adaptive immune systems 34, 35. In accordance with this, it was recently shown that neonatal mice are hyposensitive to TLR stimulation because of the increased IL-10 production by the B1 subset of B cells [36]. Furthermore, neonatal B cells were shown to effectively control the production of proinflammatory cytokines by neonatal dendritic cells that are present in higher percentage in neonatal mice than in adult mice. In the absence of this B1 cell subset, neonatal mice developed stronger inflammatory responses than adult mice and became lethally susceptible to various TLR challenges, indicating that B1 cells might have a unique regulatory role in dampening the neonatal inflammatory response through regulation of dendritic cells. However, in light of the recent evidence indicating that adaptive immunity might be required for controlling innate responses, we questioned whether neonates are truly hypo-responsive. Surprisingly, we discovered that neonatal mice are, in fact, hypersensitive to various forms of TLR stimulation [37]. This discrepancy with earlier studies can be reconciled following a closer investigation of the particular type of TLR stimulation used. In the study reporting neonatal hyposensitivity, a low dose of LPS plus D-galactosamine (D-GalN) was used to challenge the mice [36]; stimulation with high dose LPS alone, however, results in neonatal hyper-sensitivity [37]. D-GalN is widely used with LPS to study LPS-induced liver injury mediated by macrophages 36, 38, 39. After careful comparison of the inflammatory response and liver damage caused by D-GalN in neonatal or adult mice, we discovered that it is D-GalN that causes neonatal resistance to TLR stimulation [37]. Without D-GalN, neonatal mice demonstrate robust inflammatory responses with higher TNFα and IL-6 production than adult mice in response to LPS stimulation. Analogous to immunodeficient adult mice, this uncontrolled proinflammatory innate response is caused by insufficient numbers of neonatal T cells. Infusion of T cells renders newborns resistant to TLR agonists or viral infection. Although the definitive role of B cells in innate suppression is limited by the caveat of adding D-GalN to the stimulation model, these studies indicate that both B and T cells can mediate innate cell suppression under distinct conditions and the number of the lymphocytes determines the caliber of suppression.
Unlike neonatal mice, human newborns have a considerable number of T cells in the periphery at birth [40]. However, the proportion of T cells is lower in peripheral blood in human newborns, especially in infants who are small for gestational age 40, 41, 42. Clinical evidence has shown that overwhelming levels of cytokines can be detected in some human newborn diseases linked to mild infections 43, 44, 45, 46, 47. Therefore, the maturity of the immune system includes the ability not only to produce an effective immune response to clear pathogens but also to regulate the response accordingly. Neonates probably rely heavily on the innate system; although this bias might improve the chances of clearing an infection, it also increases the risk of excessive activation and consequent immunopathology. This might explain why neonatal patients demonstrate high morbidity and mortality in response to infection.
New insights into the evolution and relationship of innate and adaptive immunity
Innate immunity is a phylogenetically ancient defense system, providing efficient and sufficient immunity in invertebrates [48]. For example, Drosophila can survive infections without the need for functional adaptive immune cells. Why then, has the adaptive immune system evolved in vertebrates? One possible explanation is that specific and long-lasting immune memory is required for vertebrates to prevent repeated infection in their, usually, much longer life spans [4]. Both in invertebrates and vertebrates, innate immune responses are tightly regulated by a series of negative regulators at multiple levels to prevent over-reaction and maintain immunological balance 20, 26, 49. It is plausible that the innate immune system in vertebrates is either less potent in its inflammatory response or it is tightly controlled at various checkpoints of the Toll and IMD (immune deficiency) signaling pathways [49]. We speculate that the innate and adaptive arms of the immune system became more specialized during evolution and the auto-regulatory function of innate immunity might have been insufficient (quantitatively and/or qualitatively) to meet all the regulatory requirements for controlling inflammation in vertebrates. Therefore, the self-regulatory mechanism might have required the development of a feedback loop from the adaptive to the innate immune system. Whether the auto-regulation of innate immunity is more sophisticated in invertebrates than vertebrates and how these complicated and difficult biological issues are resolved remains to be determined. It is of interest to note that invertebrates might possess an alternative adaptive immunity [50]. The increasing experimental accessibility of non-mammalian jawed vertebrates, jawless vertebrates, protochordates and invertebrates has provided intriguing new information regarding the likely patterns of emergence of the evolution of alternative mechanisms for receptor diversification. Whether the adaptive immunity-like molecules in invertebrates could also regulate innate immunity is a question of great interest.
It is accepted, in most cases, that the innate and adaptive immune systems respond to infection at different speeds, with the innate response having a dominant, even exclusive, role in the early phase of infection, whereas adaptive immunity comes into full force only after a few days and has the dominant role in the later phase of infection. We now propose that the innate and adaptive immune systems are one integrated defense network that rely on each other not only for amplification but also regulation. The innate immune system has its own auto-regulatory mechanisms including soluble decoy receptors, intracellular negative regulators, transmembrane protein regulators, reduction of TLR expression and regulation by apoptosis [20]. The Treg cell is a well-known population in the adaptive immune system that can regulate both innate and adaptive immune responses by secreting cytokines (e.g. IL-10 and TGFβ) and/or by cell–cell contact 22, 23. Our recent study has reported that conventional T cells of the adaptive immune system could unexpectedly regulate innate response in a cell–cell contact manner [21]. Rather than standing by in the early phase of infection, the adaptive immune system can now be thought to actively serve as an indispensable part of the innate immune response. As with most complex biological systems, a stimulatory signal is accompanied by a protective antagonistic response. Although it has long been assumed that the lack of a proinflammatory response by adaptive cells during early infection equates to inactivity, our data instead indicate that adaptive immune cells are full participants, albeit not in the expected manner. It seems that the ‘black and white’ division of innate and adaptive immunity should be reconsidered. The extent to which innate and adaptive immunity collaborate – efficiently executing a balanced attack against invaders – is likely to exceed our present expectations.
Acknowledgements
We thank Lishan Su and Mendy Miller for discussion and editing. This work was supported, in part, by National Institutes of Health Grants (to Y-X.F), National Natural Science Foundation of China Grants and Ministry of Science and Technology Grants (to H.T.).
Contributor Information
Hong Tang, Email: tanghong@moon.ibp.ac.cn.
Yang-Xin Fu, Email: yfu@uchicago.edu.
References
- 1.Akira S. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 2.Janeway C.A., Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 3.Poltorak A. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R., Janeway C.A., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 5.Duffield J.S. The inflammatory macrophage: a story of Jekyll and Hyde. Clin. Sci. (Lond.) 2003;104:27–38. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 6.Monney L. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 7.Stout R.D., Bottomly K. Antigen-specific activation of effector macrophages by IFN-gamma producing (TH1) T cell clones. Failure of IL-4-producing (TH2) T cell clones to activate effector function in macrophages. J. Immunol. 1989;142:760–765. [PubMed] [Google Scholar]
- 8.Foster P.S. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwamoto I. Interferon gamma regulates antigen-induced eosinophil recruitment into the mouse airways by inhibiting the infiltration of CD4+ T cells. J. Exp. Med. 1993;177:573–576. doi: 10.1084/jem.177.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohnishi T. IL-5 is the predominant eosinophil-active cytokine in the antigen-induced pulmonary late-phase reaction. Am. Rev. Respir. Dis. 1993;147:901–907. doi: 10.1164/ajrccm/147.4.901. [DOI] [PubMed] [Google Scholar]
- 11.Beaven M.A., Metzger H. Signal transduction by Fc receptors: the Fc epsilon RI case. Immunol. Today. 1993;14:222–226. doi: 10.1016/0167-5699(93)90167-j. [DOI] [PubMed] [Google Scholar]
- 12.Cooper N.R. The classical complement pathway: activation and regulation of the first complement component. Adv. Immunol. 1985;37:151–216. doi: 10.1016/s0065-2776(08)60340-5. [DOI] [PubMed] [Google Scholar]
- 13.Kalesnikoff J. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 2001;14:801–811. doi: 10.1016/s1074-7613(01)00159-5. [DOI] [PubMed] [Google Scholar]
- 14.Leibson P.J. Signal transduction during natural killer cell activation: inside the mind of a killer. Immunity. 1997;6:655–661. doi: 10.1016/s1074-7613(00)80441-0. [DOI] [PubMed] [Google Scholar]
- 15.Takai T. Multiple loss of effector cell functions in FcR gamma-deficient mice. Int. Rev. Immunol. 1996;13:369–381. doi: 10.3109/08830189609061759. [DOI] [PubMed] [Google Scholar]
- 16.Beutler B. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 17.Bjorkbacka H. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 2004;10:416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 18.Danner R.L. Endotoxemia in human septic shock. Chest. 1991;99:169–175. doi: 10.1378/chest.99.1.169. [DOI] [PubMed] [Google Scholar]
- 19.Zipris D. TLR activation synergizes with Kilham rat virus infection to induce diabetes in BBDR rats. J. Immunol. 2005;174:131–142. doi: 10.4049/jimmunol.174.1.131. [DOI] [PubMed] [Google Scholar]
- 20.Liew F.Y. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 21.Kim K.D. Adaptive immune cells temper initial innate responses. Nat. Med. 2007;13:1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maloy K.J. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 2003;197:111–119. doi: 10.1084/jem.20021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smyth M.J. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J. Immunol. 2006;176:1582–1587. doi: 10.4049/jimmunol.176.3.1582. [DOI] [PubMed] [Google Scholar]
- 24.Tiemessen M.M. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lund J.M. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 27.Krieg C. Functional analysis of B and T lymphocyte attenuator engagement on CD4+ and CD8+ T cells. J. Immunol. 2005;175:6420–6427. doi: 10.4049/jimmunol.175.10.6420. [DOI] [PubMed] [Google Scholar]
- 28.Sedy J.R. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 29.Biron C.A. Initial and innate responses to viral infections–pattern setting in immunity or disease. Curr. Opin. Microbiol. 1999;2:374–381. doi: 10.1016/s1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- 30.French A.R., Yokoyama W.M. Natural killer cells and viral infections. Curr. Opin. Immunol. 2003;15:45–51. doi: 10.1016/s095279150200002x. [DOI] [PubMed] [Google Scholar]
- 31.Andoniou C.E. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 2005;6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- 32.Andrews D.M. Functional interactions between dendritic cells and NK cells during viral infection. Nat. Immunol. 2003;4:175–181. doi: 10.1038/ni880. [DOI] [PubMed] [Google Scholar]
- 33.Munz C. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J. Exp. Med. 2005;202:203–207. doi: 10.1084/jem.20050810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adkins B. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 35.Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 2007;7:379–390. doi: 10.1038/nri2075. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X. Type I interferons protect neonates from acute inflammation through interleukin 10-producing B cells. J. Exp. Med. 2007;204:1107–1118. doi: 10.1084/jem.20062013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7528–7533. doi: 10.1073/pnas.0800152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freudenberg M.A. Requirement for lipopolysaccharide-responsive macrophages in galactosamine-induced sensitization to endotoxin. Infect. Immun. 1986;51:891–895. doi: 10.1128/iai.51.3.891-895.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexopoulou L. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 40.Thomas R.M., Linch D.C. Identification of lymphocyte subsets in the newborn using a variety of monoclonal antibodies. Arch. Dis. Child. 1983;58:34–38. doi: 10.1136/adc.58.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heldrup J. Blood T and B lymphocyte subpopulations in healthy infants and children. Acta Paediatr. 1992;81:125–132. doi: 10.1111/j.1651-2227.1992.tb12187.x. [DOI] [PubMed] [Google Scholar]
- 42.Kumar A. Quantitation of T cells in venous blood of healthy neonates. Indian J. Pediatr. 1994;61:711–714. doi: 10.1007/BF02751986. [DOI] [PubMed] [Google Scholar]
- 43.Atici A. Serum tumor necrosis factor-alpha in neonatal sepsis. Am. J. Perinatol. 1997;14:401–404. doi: 10.1055/s-2007-994168. [DOI] [PubMed] [Google Scholar]
- 44.Blackwell C.C. Cytokine responses and sudden infant death syndrome: genetic, developmental, and environmental risk factors. J. Leukoc. Biol. 2005;78:1242–1254. doi: 10.1189/jlb.0505253. [DOI] [PubMed] [Google Scholar]
- 45.Ozdemir A. Neonatal tumor necrosis factor, interleukin-1 alpha, interleukin-1 beta, and interleukin-6 response to infection. Am. J. Perinatol. 1994;11:282–285. doi: 10.1055/s-2007-994592. [DOI] [PubMed] [Google Scholar]
- 46.Vege A. Are elevated cerebrospinal fluid levels of IL-6 in sudden unexplained deaths, infectious deaths and deaths due to heart/lung disease in infants and children due to hypoxia? Acta Paediatr. 1998;87:819–824. doi: 10.1080/080352598750013563. [DOI] [PubMed] [Google Scholar]
- 47.Vege A. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr. 1995;84:193–196. doi: 10.1111/j.1651-2227.1995.tb13608.x. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann J.A. Innate immunity of insects. Curr. Opin. Immunol. 1995;7:4–10. doi: 10.1016/0952-7915(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 49.Aggarwal K., Silverman N. Positive and negative regulation of the Drosophila immune response. BMB Rep. 2008;41:267–277. doi: 10.5483/bmbrep.2008.41.4.267. [DOI] [PubMed] [Google Scholar]
- 50.Litman G.W. Reconstructing immune phylogeny: new perspectives. Nat. Rev. Immunol. 2005;5:866–879. doi: 10.1038/nri1712. [DOI] [PMC free article] [PubMed] [Google Scholar]