

Abstract
The c-Jun N-terminal kinases (JNKs) have been the subject of intense interest since their discovery in the early 1990s. Major research programs have been directed to the screening and/or design of JNK-selective inhibitors and testing their potential as drugs. We begin this review by considering the first commercially-available JNK ATP-competitive inhibitor, SP600125. We focus on recent studies that have evaluated the actions of SP600125 in lung, brain, kidney and liver following exposure to a range of stress insults including ischemia/reperfusion. In many but not all cases, SP600125 administration has proved beneficial. JNK activation can also follow infection, and we next consider recent examples that demonstrate the benefits of SP600125 administration in viral infection. Additional ATP-competitive JNK inhibitors have now been described following high throughput screening of small molecule libraries, but information on their use in biological systems remains limited and thus these inhibitors will require further evaluation. Peptide substrate-competitive ATP-non-competitive inhibitors of JNK have also now been described, and we discuss the recent advances in the use of JNK inhibitory peptides in the treatment of neuronal death, diabetes and viral infection. We conclude by raising a number of questions that should be considered in the quest for JNK-specific inhibitors.
Abbreviations: CVB3, Coxsackievirus B3; HIV, Human immunodeficiency virus; JNKs, c-Jun N-terminal kinases; MAPKs, mitogen-activated protein kinases; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; PARP-1, poly(ADP-ribose) polymerase-1; PDB, Protein DataBase; ROS, Reactive oxygen species; RNAi, RNA interference
Keywords: c-Jun N-terminal Kinase; NS3, Non-structural protein 3; SP600125; Small molecule ATP-competitive inhibitors; JNK inhibitory peptides; Ischemia/reperfusion damage; Viral infection
1. Introduction
The c-Jun N-terminal kinases (JNKs) were initially described in the early 1990s as a family of serine/threonine protein kinases, activated by a range of stress stimuli and able to phosphorylate the N-terminal transactivation domain of the c-Jun transcription factor. This phosphorylation enhances c-Jun-dependent transcriptional events in mammalian cells [1], [2]. Further research has revealed three JNK genes (JNK1, JNK2 and JNK3) and their spliceforms as well as the range of external stimuli that lead to JNK activation. A number of independent approaches have since suggested the importance of JNK-dependent signalling events in both normal development and in disease. This has been highlighted by the striking beneficial phenotypes of JNK gene knockout mice in disease models, including neuroprotection against stroke and improved insulin responsiveness in diabetes (reviewed in [3]).
Inhibitors have been used increasingly to explore the biological functions of JNK in mammalian systems without the need for JNK gene knockout. In this review, we explore recent developments in the discovery of JNK inhibitors and their potential in the treatment of human disease. We first focus on small molecule, ATP-competitive JNK inhibitors as summarised in Fig. 1 . Our initial discussion centres on SP600125 developed by Signal Pharmaceuticals/Celgene. In addition, we provide a brief overview of an increasing number of other small molecule ATP-competitive JNK inhibitors now described in the published literature. We then discuss the recent advances in the use of ATP-non-competitive JNK inhibitory peptides. These inhibitors are also highlighted in Fig. 1.
Fig. 1.
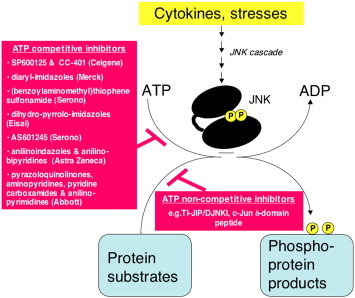
General overview of the reaction catalysed by active JNK, highlighting the different classes of JNK inhibitors described to date. JNK is activated via a specific protein kinase cascade when eukaryotic cells are exposed to cytokines or stress. Active JNK catalyses a phosphorylation reaction in which the γ-phosphoryl residue from ATP is transferred to a range of protein substrates [92]. This review centres on the JNK inhibitors described in the published literature. These inhibitors can be divided into those that directly interfere with ATP-binding and thus termed “ATP-competitive” or JNK inhibitory peptides that compete with the protein substrates and thus termed “ATP-non-competitive”.
Lastly, we consider questions that arise with the development of JNK inhibitors and their possible therapeutic application. These questions centre on the controls needed to establish specificity of actions of JNK inhibitors, whether JNK isoform-selective inhibitors are possible or desirable, whether other compounds have off-target effects to inhibit JNK, and what concerns accompany the chronic use of JNK-specific inhibitors. Further work will be needed to address these issues, however the demonstrated efficacy of the current generation of JNK inhibitors in improving outcomes in disease models suggests that this further effort will be worthwhile.
2. SP600125, an anthrapyrazolone inhibitor of JNK
2.1. Discovery and initial characterisation of SP600125
In late 2001, the small molecule JNK inhibitor, SP600125 (anthra[1,9]pyrazol-6(2H)-one), was reported following the screening of a proprietary library (Celgene/Signal Pharmaceuticals) for inhibitors of JNK2 activity towards the c-Jun transactivation domain [4]. The chemical structure of SP600125 is shown in Table 1 , alongside the structures of other small molecule inhibitors of JNK discussed in subsequent sections of this review.
Table 1.
Small molecule JNK inhibitors identified in high throughput screening of proprietary chemical libraries
| Core structure/Class description in initial publications | Inhibitor Structure (note: Compound number refers to that given in the original report) | Identification | IC50 (and/or Ki when reported) | PDB entry | Reference |
|---|---|---|---|---|---|
| Anthrapyrazolone |
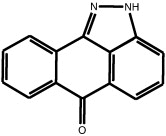 SP600125 SP600125 |
High throughput screening of Celgene compound collection using JNK2 activity assay, followed by cell-based testing | IC50 JNK1 = 40 nM IC50 JNK2 = 40 nM IC50 JNK3 = 90 nM | 1UKI 1PMV | [5] |
| Diaryl-imidazoles |
 Compound 1 Compound 1 |
Reported by scientists at Merck, but discovery approach not described. Note, this compound also interacts with and inhibits p38 with higher potency (IC50 p38 = 0.078 nM) | IC50 JNK3 = 7 nM | 1PMN | [5] |
| (Benzoylaminomethyl) thiophene sulfonamides |
 Compound 50a Compound 50a |
High throughput screening of Serono compound collection using JNK3 activity assay, followed by structure–activity relationship studies and neuronal cell based assays for the inhibition of NGF-deprivation-induced cell death. | IC50 JNK2 = 650 nM IC50 JNK3 = 150 nM | – | [58] |
| Dihydro-pyrrolo imidazoles |
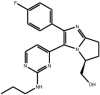 Compound (S)-5 Compound (S)-5 |
Design by Eisai scientists based on structures of known p38 inhibitors with additions to increase JNK3 inhibitory potency and selectivity; testing inhibition of c-Jun phosphorylation and survival following K+ withdrawal from cerebellar granule neurons. Note, this compound inhibits p38 with lower potency (IC50 p38 = 28 nM) | IC50 JNK1 = 2.5 nM | – | [59] |
| (Benzothiazol-2-yl) acetonitrile |
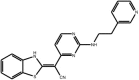 Compound 59 (AS601245) Compound 59 (AS601245) |
High throughput screening of Serono compound collection using JNK3 activity assay, followed by structure–activity relationship studies, cell-based assays and anti-inflammatory effects in vivo. | IC50 JNK1 = 150 nM IC50 JNK2 = 220 nM IC50 JNK3 = 70 nM | – | [60] |
| Anilinoindazoles |
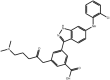 Compound 10 Compound 10 |
High throughput screening of Astra Zeneca compound collection using JNK3 activity assay, followed structure–activity relationship studies. | IC50 JNK1 = 101 nM IC50 JNK3 = 3 nM | 2B1P | [61] |
| Pyrazoloquinolinones |
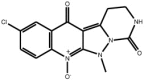 Compound 16 Compound 16 |
High throughput screening of Abbott compound collection using JNK1 activity assay, followed by testing in cell-based assays for inhibition of tumour necrosis-factor stimulated c-Jun phosphorylation | IC50 JNK1 = 290 nM | 2GO1 (structure with a related compound) | [63] |
| Aminopyridines |
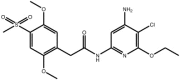 Compound 35 Compound 35 |
High throughput screening of Abbott compound collection using JNK1 activity assay, followed by testing in cell-based assays for inhibition of tumour necrosis-factor stimulated c-Jun phosphorylation | IC50 JNK1 = 36 μM (Ki JNK1 = 3 nM) IC50 JNK2 = 70 μM (Ki JNK2 = 13 nM)(Ki JNK3 = 61 nM) | 2GMX (structure with a related compound) | [64] |
| Pyridine carboxamide |
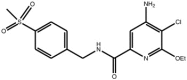 Compound 12 Compound 12 |
High throughput screening of Abbott compound collection using JNK1 activity assay, followed by testing in cell-based assays for inhibition of tumour necrosis-factor stimulated c-Jun phosphorylation. | IC50 JNK1 = 24 nM) IC50 JNK2 = 74 nM) | 2H96 (structure with a related compound) | [65] |
| Anilino-bipyridines |
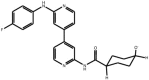 Compound 11 Compound 11 |
High throughput screening of Astra Zeneca compound collection using JNK3 activity assay, followed structure–activity relationship studies. Note, this compound also inhibits p38 with comparable potency (IC50 p38 = 40 nM) | IC50 JNK1 = 88 nM IC50 JNK3 = 15 nM | 2EXC | [62] |
| Anilino-pyrimidines |
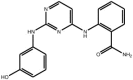 Compound 2b Compound 2b |
High throughput screening of Abbott compound collection using JNK1 activity assay, followed by rational structural design then testing in cell-based assays for inhibition of tumour necrosis-factor stimulated c-Jun phosphorylation. | IC50 JNK3 = 9 nM | 2NO3 | [66] |
The highly planar nature of SP600125 and poor solubility in aqueous solution, both consequences of its anthrapyrazolone core structure, were noted in its initial description [4]. JNK inhibition by SP600125 was further observed to be reversible and ATP-competitive, showing IC50 values for JNK inhibition in the range of 40–90 nM (see Table 1) with > 300-fold selectivity over the related mitogen-activated protein kinases (MAPKs), ERK1 and p38-2 and between 10-fold and 100-fold selectivity over another 14 protein kinases tested [4]. These results suggested high affinity and specific interactions of SP600125 with residues in the JNK ATP-binding site.
These interactions of SP600125 with JNK have been further explored following the co-crystallisation of SP600125 with JNK3 [5]. The resulting structure (Protein DataBase (PDB): 1PMV) is shown in Fig. 2 , where the JNK3 residues not conserved in the related MAPK, p38-2, have been highlighted (specifically I70, V79, V196, L206 and Q155). These residues produce a narrow ATP-binding pocket in JNK that accommodated the planar SP600125 molecule and were predicted to contribute to the specificity of SP600125 towards JNK over the p38 MAPKs [5]. Although this prediction was investigated in mutagenesis studies targeting the JNK ATP-binding site residues, single mutations such as I70V or V196A did not significantly alter SP600125 binding to JNK [6]. Further work is needed to evaluate whether the mutation of residues in combination might produce more striking effects. Direct evidence for the JNK3 residues that interact with SP600125 should drive further structural refinements to increase inhibitor affinities and/or specificities.
Fig. 2.
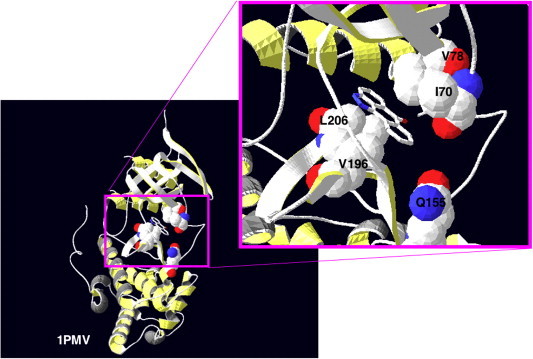
Structure of the JNK complex with the ATP-competitive inhibitor SP600125. SP600125 has been co-crystallised with JNK3, and the resulting structure has been recorded in the Protein DataBase (PDB) as 1PMV [5]. Here, the residues in JNK3 not conserved in p38-2, namely I70, V79, V196, L206 and Q155, are highlighted. Although these residues were predicted to most likely contribute to the specificity of SP600125 towards JNK1/2/3 over the p38 MAPKs [5], subsequent mutagenesis studies further work is required to evaluate which residues make major contributions to binding [6].
2.2. Use of SP600125 to dissect signalling mechanisms and JNK-dependent cell events
In initial testing for biological efficacy of SP600125 in stimulated Jurkat T cell cultures, c-Jun phosphorylation was inhibited with an IC50 of 5 to 10 μM [4]. The concentrations required for intracellular effects were therefore significantly higher than the in vitro IC50 values estimated with the purified JNK proteins (40 to 90 nM, see Table 1). These differences were attributed to the ATP concentrations competing with SP600125 in these different assays; the in vitro biochemical assays were performed at ATP concentrations lower than would be normally found in vivo. Thus, the intracellular IC50 values were higher than those observed in vitro [4].
The use of SP600125 to evaluate JNK-dependent events in cells has grown rapidly since 2001. As > 850 publications have now reported the use of SP600125 in cells or in vivo, we have restricted our discussion here to two broad areas highlighting different areas for possible therapeutic applications of SP600125 and other JNK inhibitors.
We begin by considering the effects of SP600125 to enhance recovery following ischemia/reperfusion damage or other insults in a variety of tissue types. An underlying theme emerges in the actions of SP600125 to prevent cell death. As we will describe, SP600125 can inhibit a number of pro-apoptotic events such as the activation of pro-apoptotic Bcl2 family members, the release of mitochondrial cytochrome c into the cell cytosol, or the activation of pro-apoptotic caspase family of proteases. The reader is referred to recent excellent reviews on apoptosis for further details on the hallmarks of this cell death process [7], [8]. In some cases, it also appears that SP600125 can modulate immune cell responses, and thus provide beneficial effects. We then consider the possible therapeutic applications of SP600125 in the treatment of infectious disease, most notably in its actions to alter the outcomes of viral disease. Taken together, these studies suggest that SP600125 administration will be beneficial in a range of therapeutic applications.
2.3. Efficacy of SP600125 in the treatment of ischemia, ischemia/reperfusion damage and other insults
JNK activation follows insults such as ischemia/reperfusion in many tissues including lung, kidney, liver, brain, and heart [9], [10], [11], [12], [13]. For the lung, a challenge facing its transplantation remains primary graft failure following ischemia/reperfusion injury during the initial removal and subsequent transplantation surgery. The inclusion of SP600125 in the preservation and reperfusion solutions reduced lung injury as visualised directly by histological examination of lung tissue and the assessment of apoptotic cell numbers [14]. These benefits have been accompanied by improved biochemical markers such as decreased release of total protein, lactate dehydrogenase, and tumour necrosis factor-α into the bronchoalveolar lavage fluid, indicating maintenance of tissue integrity despite the ischemia/reperfusion insult [14].
In other forms of lung insult, SP600125 administration has also been beneficial. The administration of SP600125 1 h after smoke inhalation decreased airway cell apoptosis, decreased mucous plugging, lowered the influx of inflammatory cells, decreased the release of cytokines and improved animal survival [15]. These in vivo data suggest a critical role for JNK in smoke-induced lung injury, highlighting the beneficial effects of SP600125. Similarly, the administration of SP600125 has implicated JNK in the regulation of the expression of the acute phase protein, pentraxin 3, in the lung in response to the proinflammatory cytokine, tumour necrosis factor [16]. As higher levels of pentraxin 3 exacerbate lung injury, JNK inhibition is expected to be an attractive therapeutic approach to protect the lung from the increased tumour necrosis factor levels that accompany many inflammatory and other insults.
Ischemia/reperfusion insult can also accompany renal transplantation and surgery, renal failure and trauma. Direct protective effects of SP600125 during kidney ischemia/reperfusion have been observed and have been attributed to JNK inhibition suppressing apoptotic cell death events in a Fas ligand-initiated extrinsic pathway [17]. The involvement of macrophages in renal tissue injury in vivo has also been suggested, with macrophage accumulation being a prominent feature in most forms of human glomerulonephritis and correlating with renal dysfunction. The exposure of bone marrow-derived macrophages to SP600125 prior to transfer into a sheep model of glomerulonephritis caused a 75% reduction in proteinuria, thus highlighting a critical role for the JNK signaling pathway in macrophage-mediated renal injury [18]. The benefits of JNK inhibition in ischemia/reperfusion may therefore include altered inflammatory cell responses that initiate damage.
Possible benefits of SP600125 for the liver following insult have also been demonstrated. Marked protective effects of SP600125 was observed for acetaminophen-induced toxicity both in vitro and in vivo, through the actions of SP600125 were observed to block apoptotic cell death [19]. This has been extended recently to the study of acute hepatic failure following paracetamol poisoning in which SP600125 administration in vivo markedly reduced mortality and hepatic tumour necrosis factor production [20]. Similarly, the subcutaneous injection of SP600125 prior (6 and 2 h) and after (2 h) insult reduced hepatocyte apoptosis, suppressed lethality, and decreased the elevation of serum markers of liver damage in an experimental model of fulminant hepatic failure [21]. In contrast, SP600125 administration was not protective against carbon tetrachloride or concanavalin A toxicity [19]. This highlighted that JNK inhibition will not be beneficial for all forms of hepatic injury, and instead suggests that the targeting of other stress-initiated events should be tested as alternative therapeutic approaches.
Similar, or potentially more extreme, problems also face those striving to improve the survival of neurons following insults to the brain. SP600125 treatment has prevented cell death following ischemia or ischemia/reperfusion of the brain [22], [23], [24], [25]. As one example, SP600125 decreased neuronal apoptosis induced by global ischemia/reperfusion in the hippocampal CA1 subregion. Specifically, SP600125 suppressed the expression of Fas ligand that initiates the extrinsic death pathway, the translocation of the proapoptotic protein Bax to mitochondria, the release of cytochrome c to the cytosol, and the activation of proapoptotic caspases [25]. Similarly, in models of early brain injury after subarachnoid hemorrhage, SP600125 administered intraperitoneally 1 h before and 6 h after haemorrhage demonstrated benefits such as the suppression of caspase activation and concomitant neuronal injury, improved blood–brain barrier preservation, reduced brain swelling, and improved neurological function [26]. SP600125 also prevented apoptosis of dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of Parkinson's Disease [27] as well as neurons in the acute injury accompanying spinal cord trauma [28]. Taken together, these results support the further development of JNK inhibitors as neuroprotective agents and their use in a range of brain insults.
In contrast to the positive findings supporting the benefits of SP600125 administration as described in the preceding paragraphs, detrimental effects of SP600125 have been reported in ischemia/reperfusion injury in other tissues and cell types. For example, when SP600125 was administered both at the onset of partial hepatic ischemia and during the subsequent reperfusion events, a number of markers of liver damage such as serum alanine aminotransferase levels were increased [29]. This was accompanied by deterioration of liver histology and increased neutrophil infiltration that augmented oxidative stress in the reperfused liver tissue [29]. Thus, detrimental effects to the liver appeared to be mediated, at least partially, via circulating immune cells. SP600125 exacerbated these detrimental effects.
There have also been detrimental effects of SP600125 noted for the cells of the heart. SP600125 significantly enhanced the activation of the proapoptotic protease, caspase-3, and increased the numbers of apoptotic cardiac myocytes in culture in response to their energy depletion following exposure to potassium cyanide and 2-deoxy-d-glucose [30]. Similarly, chronic SP600125 treatment in vivo in the cardiomyopathic hamster model of heart failure SP600125 increased the number of apoptotic (TUNEL-positive) myocytes and the area of interstitial fibrosis [31]. This was accompanied by enhanced left ventricular chamber dilation and dysfunction indicating the negative effects on cardiac structure and function [31]. Whilst these results suggest a role for JNK in cardiac myocyte survival, they contradict the observations that SP600125 protected cardiac myocytes from cell death following β-adrenergic stimulation [32]. Again, this has emphasized that the cardiac effects of SP600125 must be evaluated in a range of different insults and pathological conditions.
Additional studies are now needed to explore how SP600125 alters the balance between death and survival in different cell types. At a biochemical level, the cell context-dependent differences, as noted in the preceding paragraphs, may reflect the differences in the expression and/or localisation of JNK substrates within the various cell types. In addition, it is also becoming clearer that defining the impact of JNK signalling on immune cell function will be critical to understanding those diseases in which there is a significant immunological response. The differences observed may also reflect the various administration and insult protocols employed in these studies, or the concentrations of SP600125 achieved in vivo. The availability of additional JNK inhibitors should allow these issues to be addressed directly.
2.4. Efficacy of SP600125 in the treatment of viral infection
Increasingly, it has been shown that viral infection can lead to JNK activation. Examples include infection by Epstein–Barr Virus [33], Herpes Simplex Virus [34], Reovirus [35], Kaposi's Sarcoma Virus [36], or Varicella–Zoster virus [37], [38]. Whilst the exact mechanisms leading to JNK activation remain to be evaluated in many of these cases, it is of interest that Kaposi's Sarcoma Virus encodes the viral kinase ORF36 that interacts with JNK as well as the upstream JNK pathway kinases MKK4 and MKK7. ORF36 expression can lead to the phosphorylation and activation of MKK4/7 and, thus, to JNK activation [36]. Further interventional studies, primarily in cultured cells in vitro, have supported a role for JNK activation in viral infection processes and/or subsequent cellular events. In the following paragraphs, we discuss the results of recent studies evaluating the effects of SP600125 in models of viral infection that suggest that JNK inhibitors may provide new therapeutic interventions.
In a number of situations following exposure to virus or viral proteins, SP600125 treatment has prevented viral-induced cell death. This is consistent with the studies discussed in the preceding sections that highlighted that SP600125 could prevent cell death in many tissues following a range of different stresses. Specifically, SP600125 treatment prevented apoptotic death following the exposure of human monocytic cells to the Human Immunodeficiency Virus (HIV) accessory protein viral protein Vpr [39]. Similar positive effects to protect cells from death have been observed when SP600125 treatment either rescued influenza epitope-specific human cytolytic T lymphocytes from activation-induced cell death [40] or prevented the death of cultured hippocampal cells exposed to Herpes Simplex Type 1 Virus [41].
Conversely, SP600125 inhibited the proliferation of primary erythroleukemic cells isolated from Friend spleen focus-forming virus-infected mice [42]. Furthermore, in cell lines established from these animals, SP600125 caused significant apoptosis as well as an increase in the fraction of cells in the G2/M phases of the cell cycle and undergoing endoreduplication [42]. These latter data suggest that JNK plays an important role in cell proliferation and/or the survival of erythroleukemia cells, and thus that SP600125 administration could provide a novel approach in the treatment of viral-induced erythroleukemia [42].
In other examples of viral infection, the use of SP600125 has altered viral replication or cellular persistence. For example, rotavirus is a double-stranded RNA virus that affects the gastrointestinal system resulting in vomiting and diarrhoea. The use of SP600125 in combination with p38 MAPK inhibitors has suggested that maximal rotavirus-induced interleukin-8 and c-jun transcription required JNK and p38 activity. Significantly, both p38 and JNK were required for rotavirus replication but not viral structural antigen expression [43]. Similarly, SP600125 used together with inhibitors of phosphatidylinositol 3-kinase inhibited the establishment of persistent SARS-CoV infection in Vero E6 cells [44]. Clearly, there are now many opportunities to evaluate how SP600125 acts in concert with other inhibitors of intracellular signaling pathways to modulate aspects of viral biology. The most appropriate therapeutic strategy may ultimately require combination therapies of signal transduction modulators.
Despite these successes, there have also been some situations when SP600125 treatment has not been beneficial. These have emphasized the need for caution. For example, the use of SP600125 did not significantly change disease progression following infection with Coxsackievirus B3 (CVB3), an enterovirus in the Picornavirus family that is the most common human pathogen associated with myocarditis and idiopathic dilated cardiomyopathy [45]. SP600125 reduced CVB3-induced phosphorylation of activating transcription factor 2 (ATF2), but did not alter CVB3 viral protein synthesis, viral progeny release, cell death, or caspase-3 activation in infected cells. In contrast, p38 MAPK inhibitors altered progeny release [45]. Thus, it remains critical to test the effects of SP600125 on a range of different virus types and cellular effects.
SP600125 treatment may also alter gene expression changes that have significant effects for virus structure and/or life cycle. For Hepatitis C Virus non-structural protein 3 (NS3) protein-expressing cells, exposure to SP600125 abolished a number of transcription factor activities, notably AP1 and ATF2, inhibited c-jun expression, and inhibited NS3-induced cell growth [46]. Similarly, SP600125 blocked Cytomegalovirus IE1-mediated induction of AP-1 and relB promoter activity in NIH 3T3 and cultured smooth muscle cells [47]. Furthermore, nuclear localisation of the viral encoded proteins may be regulated by JNK as seen for the human Papillomavirus E1 DNA helicase [48]. Thus, these newly recognized roles for JNK may open new anti-viral strategies with the use of JNK inhibitors such as SP600125.
2.5. What might the future of SP600125 hold?
Despite the obvious successes of SP600125, and its repeated use in both in vitro and in vivo systems, some scepticism surrounds its continued use, particularly when its specificity for JNK inhibition is more closely evaluated. Despite the initial claims of the selectivity of SP600125, with little or no inhibition shown for 17 tested protein kinases or 18 inflammatory enzymes [4], [49], its subsequent testing has shown inhibition of 13 of 30 tested protein kinases [50]. Notably, serum and glucocorticoid-regulated kinase, p70 ribosomal S6 kinase, AMP-dependent protein kinase, cyclin dependent kinase 3, casein kinase 1δ and dual-specificity tyrosine–regulated kinase 1A were all inhibited by 10 μM SP600125 to a greater extent than the inhibition observed for JNK [50]. Additional data showing SP600125 binding to a range of kinases (39 of the 119 tested protein kinases) in phage interaction screening assays [51], suggests there may be many additional kinase targets of SP600125.
Despite these concerns raised on the specificity of SP600125, its value as a therapeutic agent will be confirmed with its continued usefulness in vivo with minimal toxicity or few undesirable side-effects. Some caution should be exercised when the core anthrapyrazole structure of SP600125 is considered. Anthrapyrazoles have been used as anticancer agents due to their toxic effects associated with reactive oxygen species (ROS) production, topoisomerase inhibition and DNA-interactions [52], [53]. Thus, SP600125 administration in vivo may be associated with similar toxicity that would be undesirable when an aim is to prevent cell death. This will be of greater concern when the effects of long-term dosing are evaluated. Thus, the continued development of SP600125 as a new therapeutic or therapeutic lead will require further evaluation if it shows toxic effects via JNK-independent actions.
A second generation ATP-competitive anthrapyrazolone JNK inhibitor, CC-401, has also been developed by Celgene based on the chemistry of SP600125. Despite limited publicly available details of the compound and its use, Celgene has stated that CC-401 completed a Phase I trial in healthy volunteers. Celgene is also evaluating CC-401 in a phase II clinical trial for acute myelogenous leukemia. Given the anticancer activity of some anthrapyrazoles, further evidence to support the actions of CC-401 via JNK inhibition will be required.
CC-401 has shown efficacy in an experimental model of immune-induced renal injury [54]. Specifically, CC-401 treatment of a rat anti-glomerular basement membrane disease model reduced proteinuria in the first 24 h. The rapid transient neutrophil influx was not affected, but the continued treatment with CC-401 suppressed glomerular and tubulointerstitial damage usually seen at 14 days. As CC-401 had no effect upon glomerular macrophage infiltration at day 14, it was proposed that this protection was due to modulation of macrophage activation. Thus, JNK signalling would appear to promote renal injury in acute and progressive rat anti-glomerular basement membrane disease, so that JNK inhibitors may be a novel therapeutic approach for the treatment of human glomerulonephritis. Similarly, in kidney obstruction, CC-401 significantly reduced tubular apoptosis and inhibited renal fibrosis as shown by interstitial myofibroblast accumulation and collagen IV deposition. This latter effect was attributed to suppression of gene transcription for the profibrotic factors, tumour growth factor-β1 and connective tissue growth factor [55].
CC-401 or related compounds have also been used in models of liver injury. Thus, the inclusion of JNK inhibitory compounds (CC-401, CC0209766, or CC0223105) in a hepatic warm ischemia/reperfusion injury model significantly improved survival rates from < 40% to 60–100% [56]. This decreased mortality was correlated with improved hepatic histology as these compounds significantly inhibited pericentral necrosis, neutrophil infiltration and apoptosis of both hepatocytes and sinusoidal endothelial cells, with decreased caspase-3 activation and cytochrome c release from mitochondria, and lowered levels of lipid peroxidation [56]. As similar beneficial effects were noted following cold ischemic storage of liver tissue followed by its warm reperfusion, benefits would be expected upon the inclusion of these JNK inhibitory compounds in storage and transport solutions used during liver transplantation surgery [57].
To confirm that JNK inhibition is critical for the benefits associated with SP600125 or CC-401 treatment, additional interventions directed towards JNK activity in vivo are needed. In the following paragraphs, new classes of ATP-competitive JNK inhibitors are described that will allow the benefits of JNK inhibition as a new therapeutic approach to be further explored.
3. Emerging new classes of small molecule JNK inhibitors
To date, the other small molecule JNK inhibitors recently disclosed in the publicly available scientific literature have not received the same attention as that directed towards SP600125. In this section, ten additional JNK inhibitors are briefly overviewed. A summary of these inhibitors, together with SP600125 and their chemical structures, is provided in Table 1. This summary is listed chronologically by the first published report of each inhibitor. We also present structures for those inhibitors co-crystallised with JNK proteins (Fig. 3 ). These structures suggest the ATP-competitive nature of these inhibitors.
Fig. 3.
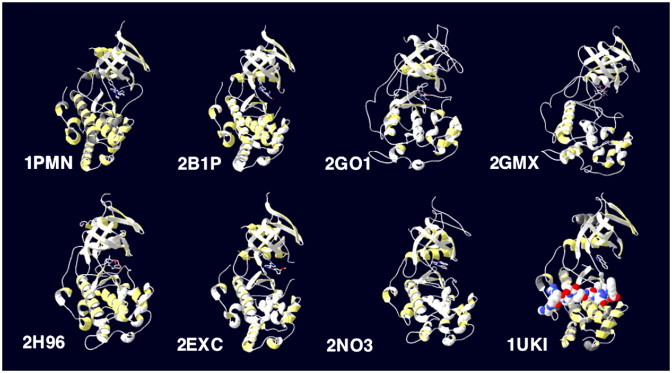
Structures of complexes of JNK with ATP-competitive inhibitors or the JNK-inhibitory peptide derived from JIP. Crystal structures have been recorded in the PDB for JNK in the presence of a number of different ATP-competitive inhibitors. These include 1PMN: diaryl-imidazoles (Merck [5]), 2B1P: anilinoindazoles (Astra Zeneca [61]), 2GO1: pyrazoloquinolinones, 2GMX: aminopyridines and 2H96: pyridine carboxamides (Abbott [64], [65], [67]), 2EXC: anilino-bipyridines (Astra Zeneca [62]) and ZNO3: anilino-pyrimidines (Abbott [66]). The complex of JNK1 with SP600125 and the JNK inhibitory peptide derived from JIP1 is also indicated in 1UKI.
The small molecule JNK inhibitors include examples from the diaryl-imidazoles (as reported by scientists at Merck [5]), (benzoylaminomethyl)thiophene sulfonamides (Serono [58]), dihydro-pyrrolo-imidazoles (Eisai [59]), (benzothiazol-2-yl)acetonitrile (also known as AS601245; Serono [60]), anilinoindazoles and anilino-bipyridines (Astra Zeneca [61], [62]), as well as pyrazoloquinolinones, aminopyridines, pyridine carboxamides and anilino-pyrimidines (Abbott [63], [64], [65], [66]). These compounds have largely been discovered by high throughput screening of compound libraries, typically by testing actions in in vitro kinase assays against purified JNK. Subsequent structure–activity studies and testing in cell culture models has allowed the refinement of these inhibitors. A notable, different approach has also shown the refinement of p38 inhibitors to increase potency towards JNK activity rather than continuing to re-screen libraries directly for JNK inhibitors [59]. Some of the inhibitors have also been reported to show some selectivity towards JNK1 [67], or JNK3 [58], [60], [61], [62], but maximal differences were only approximately 35-fold as seen for the anilinoindazoles with higher affinity for JNK3 [61].
It remains critical to evaluate the biological actions of these new JNK inhibitors. The limited reports that have tested these JNK inhibitors in perfused organ systems or in vivo have shown mixed results. The therapeutic potential for JNK inhibitors is supported by the findings in models of rheumatoid arthritis [60], as well as cerebral and cardiac ischemia [68], [69], and the undisclosed claims for benefits in models of inflammation and diabetes [64]. In contrast, the haemodynamic effects reported for the 4-aminopyridine carboxamide-based JNK inhibitors suggests that further caution may be warranted [70]. Whether undesired side effects arise from JNK-dependent or -independent inhibitor actions must be addressed. Ideally, the effects of many structurally unrelated JNK inhibitory compounds can be compared to determine JNK-independent actions.
4. Natural product inhibitors of JNKs
The success of chemical library screening in identifying JNK inhibitory molecules raises the possibility that additional JNK inhibitors can be found in other sources. A screen of 100,000 natural extracts revealed an extract from the New Guinea vine, Gnetum latifolium, as an in vitro JNK3 inhibitor [71]. Further purification revealed the JNK inhibitory components to be latifolians A and B (Table 2 ). These compounds form part of the 8-benyl-berberine alkaloid structure class distributed across many plant families. The energy-minimised three dimensional structures of these latifolians were determined, together with their IC50 values towards JNK3 (Table 2) [71]. Further studies including kinetic and structural analyses, should address whether the latifolians are ATP-competitive JNK inhibitors, whether all JNK isoforms are targeted equally, and how these molecules interact with the JNK proteins. This information could then direct the development of new classes of JNK inhibitors that exploit the essential structural features of these latifolians without their complex structure.
Table 2.
Latifolians A and B as a new class of natural product JNK inhibitors
| Inhibitor | Inhibitor Structure | Identification | IC50 | PDB entry | Reference |
|---|---|---|---|---|---|
| Latifolian A | 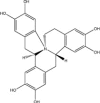 |
High throughput screening of 100,000 natural extracts for inhibitors of JNK3 activity in vitro. | IC50 JNK3 = 13 μM | – | [71] |
| Latifolian B | 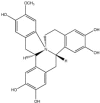 |
High throughput screening of 100,000 natural extracts for inhibitors of JNK3 activity in vitro. | IC50 JNK3 = 10 μM | – | [71] |
5. Peptide inhibitors of JNKs
5.1. A JNK inhibitory peptide derived from the JNK substrate, c-Jun
As recently reviewed, peptide inhibitors of protein kinases have been derived from direct interacting partners of protein kinases, such as their substrates [72]. A cell-permeable peptide JNK inhibitor has been derived from the δ-domain of the JNK substrate, c-Jun [73]. The sequence of this peptide is shown in Table 3 . As the c-Jun δ-domain interacts directly with JNK, this peptide would compete directly with c-Jun substrate binding. This peptide has been used to highlight the complexity of JNK–c-Jun-mediated gene regulation in the response to interleukin-1 [73]. Of interest, differences were observed when the effects of the JNK inhibitory peptide and the ATP-competitive inhibitor, SP600125, were compared [73]. For example, of the interleukin-induced genes, 20 genes were down regulated in the presence of either the c-Jun peptide or SP600125. Of these 20 genes, only 4 were down regulated by both c-Jun peptide and SP600125, 6 genes were affected by c-Jun peptide only and 10 genes were affected by SP600125 only [73]. Whether these differences reflect off-target effects of SP600125, or other differences between these inhibitors such as the mode of action, compartmentalisation or stability of the inhibitors remains to be addressed.
Table 3.
Peptide inhibitors of JNK
| Protein from which inhibitor derived | Peptide name | Amino acid sequence (residue numbers) | Identification | IC50 (and/or Ki when reported) | PDB entry | References |
|---|---|---|---|---|---|---|
| c-Jun | TAT-cJun peptidea | Y-G-R-K-K-R-R-Q-R-R-R-G-I-L-K-Q-S-M-T-L-N-L-A-D-P-V-G-S-L-K-P-H-L-R-A-K-N(33–57) | δ-domain of c-Jun | Not determined, but used in cells at 100 μM concentration | – | [73] |
| JIP1 | TI-JIP | R-P-K-R-P-T-T-L-N-L-F(153–163) | JNK interaction domain of JIP1 | Ki0.39 +/− 0.08 μMin vitro | 1UKH/1UKI | [75], [79] |
| TAT-TIJIPa | G-R-K-K-R-R-Q-R-R-R-P-P-R-P-K-R-P-T-T-L-N-L-F (153–163) | JNK interaction domain of JIP1 | Not determined, but used in cells at 10 μM concentration | – | [78] | |
| JIP1 peptide | R-P-K-R-P-T-T-L-N-L-F-P-Q-V-P-R-S-Q-D(153–171) | JNK interaction domain of JIP1 | Not determined | – | [76] | |
| LJNKi (XG101)a | G-R-K-K-R-R-Q-R-R-R-P-P-R-P-K-R-P-T-T-L-N-L-F-P-Q-V-P-R-S-Q-D (153–171) | IC50 ∼ 1 μM in vitro | – | [76], [77] | ||
| DJNKi (XG102)a,b | t-d-q-s-r-p-v-q-p-f-l-n-l-t-t-p-r-k-p-r-p-p-r-r-r-q-r-r-k-k-r-g (171–153) | Not determined, but 15–20-fold less potent than L-JNKI | – | [76,77] |
Cell-permeable vector sequence is underlined.
Amino acids denoted without capitals are the D-amino acid forms.
5.2. Use of JNK inhibitory peptides derived from the JNK pathway scaffold protein, JIP1
Scaffold proteins, known as JNK-interacting proteins or JIPs, form an additional important feature of the JNK pathway. Of note, JIP1 was first described to inhibit JNK by preventing JNK nuclear translocation, but a short conserved sequence was also identified as critical for the JIP1–JNK interaction [74]. Short JIP-derived peptides have been subsequently shown to inhibit JNK activity in vitro [75]. These peptides, in their cell-permeable form through their conjugation to the Tat peptide [76], [77], [78]), have been used to investigate effects of JNK inhibition in cells and in vivo.
The sequences of the commonly used cell-permeable JNK inhibitory peptides derived from JIP are shown in Table 3. These include the conventional l-amino acid containing peptides TI-JIP, TAT-TIJIP and L-JNKI, as well as the d-amino acid-containing retroinverso peptide, D-JNKI. These JIP-derived peptides inhibitors have been shown kinetically to act in a protein substrate-competitive manner [79], and by co-crystallisation and mutagenesis studies to bind directly to the putative protein substrate docking domain of JNK [80], [81]. More recently, these peptides have been used to evaluate the kinetic mechanism of JNK2 [82]. The results have provided important insights into the biochemistry of JNK including that protein substrate binding is primarily due to the distal contacts in the JNK2 docking groove, that there is minimal allosteric communication between the protein–substrate docking site and the ATP binding site in the active JNK2 catalytic centre, and that phosphorylation proceeds via a random sequential mechanism [82].
A recent review evaluated the studies using the cell-permeable forms of these JNK inhibitory JIP-based peptides [83]. This highlighted the success of these peptides in blocking pancreatic β-cell death [76], cerebral ischemia/stroke [77], [84], and hearing loss induced by aminoglycosides and acoustic trauma [85]. The latter has been extended in recent studies [86], [87]. Here we restrict our attention to reports on the efficacy of JNK inhibitory peptides appearing in the last 2 years since that review and we begin with recent studies on the effects in neuronal cells.
Neuropathic pain often accompanies nerve damage, but there are few options currently available for its effective treatment. In searching for possible targets for therapeutic intervention in the treatment of pain, it had been noted that spinal nerve ligation resulted in a slow but persistent activation of JNK in spinal cord astrocytes [88]. Intrathecal infusion of D-JNKI to spinal fluid did not alter the basal mechanical threshold prior to injury but prevented mechanical allodynia for more than 10 days [88]. It should be noted that the pain returned when the 14-day infusion protocol ended [88]. Thus, D-JNKI treatment provided only temporary pain relief and additional strategies are needed to identify targets for long-term pain relief.
Consistent with the observed benefits of SP600125 or D-JNKI in ischemia and reperfusion, particularly in the brain, TAT-TIJIP also prevented both apoptotic death and necrotic death of neurons in culture [89], [90]. For apoptosis, inhibition of both nuclear and non-nuclear pathways is important [89], [90]. For necrosis, the exact JNK-mediated events remain to be defined, but a number of key findings should direct future studies. Specifically, TAT-TIJIP when applied prior to the transient exposure to glutamate that mimics the excitotoxicity that accompanies stroke, prevented mitochondrial ROS generation, increased cytosolic calcium concentration, and maintained mitochondrial membrane potential [90]. The basic principles of this model involving JNK activation in necrosis are illustrated in Fig. 4 . More recently, the use of SP600125 or JNK knockout cells has shown that JNK mediates necrotic death via its sustained activation of poly(ADP-ribose) polymerase-1 (PARP-1) following exposure to ROS [91]. The direct in vitro phosphorylation assays [91] suggested that PARP-1 can be added to growing list of JNK substrates [92]. It will be of interest to test whether JNK inhibitory peptides can inhibit the actions of JNK on PARP-1 or whether other modified peptide antagonists are required.
Fig. 4.
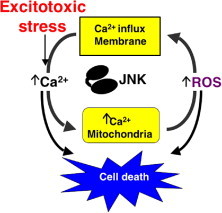
Calcium and reactive oxygen species (ROS) as primary drivers of JNK-dependent necrotic cell death. In this model, calcium and reactive oxygen species (ROS) are involved in an amplification cycle leading to cell death. Calcium overload in the mitochondria causes ROS generation, ROS can in turn increase intracellular calcium by directly activating calcium channels or increasing membrane permeability by lipid peroxidation and inactivating calcium removal transporters, JNK inhibitors break this cycle to attenuate the increase in both ROS and calcium.
Continuing these strategies to reduce neuronal cell death, a recent study has shown that D-JNKI is effective in the treatment of Reovirus-induced encephalitis [93]. Infection was achieved by direct injection of high doses of virus into the brain tissue of neonatal rats, with subsequent evaluation of brain pathology and survival. Despite the positive findings with D-JNKI delivered intraperitoneally before or after the viral infection, a number of interesting observations should be further considered. Most notably, the symptoms of myocarditis were not blocked by D-JNKI. Thus, reoviral infection remained lethal due to these cardiac effects. It should be addressed whether JNK activation also underlies this cardiac pathology and whether D-JNKI inhibits JNK activity in the heart. The positive effects of D-JNKI in the heart to reduce ischemia/reperfusion injury and infarct size in vivo have been recently reported, but only when delivered prior to the onset of ischemia [94]. In this latter study, D-JNKI when delivered at the time of reperfusion prevented apoptosis and thus limited the cardiac infarct size but, intriguingly, it did not improve functional recovery [94]. The reasons underlying this discrepancy between cardiac cell death in the infarct zone and functional performance of the heart requires further evaluation.
The JNK inhibitory peptides should also allow greater evaluation of the roles of JNK in infection by other viruses. JNK inhibition by L-JNKI resulted in a 2-fold increase in Varicella–Zoster Virus replication in melanoma cells whereas a strong decrease in virus replication was observed after inhibition of p38 MAPK [37]. It should be noted however that a more recent study has shown SP600125 to cause a dose-dependent reduction in Varicella–Zoster virus yield in primary fibroblasts [38]. The reasons for this discrepancy will require further evaluation, but may include the differences in the cell types evaluated as well as the differences in the mechanism of action of ATP-competitive versus ATP-noncompetitive inhibitors [38]. The recent study showing that alterations in the immune response following JNK2 knockout can influence malarial infection [95] suggests that JNK inhibitors may have far greater use in the treatment of a range of infectious diseases.
The benefits of lowering JNK-dependent signalling in diabetes were first seen in JNK gene knockout studies [96], [97]. This has been extended with observation that the intraperitoneal administration of JNK inhibitory peptides improved insulin resistance and glucose tolerance in diabetic mice [98]. JNK inhibitory peptides have also now been tested for their effects on pancreatic islet β-cells. In transplantation, during the isolation process and subsequent clinical transplantation, islets are subjected to severe adverse conditions that impair survival and ultimately contribute to graft failure. Intraportal injection of JNK inhibitory peptides at islet transplantation reduced JNK activity in insulin target organs, prevented islet graft loss immediately after transplantation, and improved islet transplant outcome thus showing the value of JNK inhibition during these procedures [99]. This has been supported by the independent observation that D-JNKI conferred protection against apoptosis induced during the islet preparation and subsequent exposure to IL-1β [100].
Some controversy remains in this area of islet preservation. A recent report suggested that L-JNKI, but not D-JNKI, would provide protection [101]. The toxicity of D-amino acid-containing peptides, with the paradoxical activation of JNK and p38 MAPKs following exposure of islet β-cells to D-JNKI, was suggested to underlie the observed detrimental effects [101]. Further work is needed to characterize these detrimental effects and to define when D-amino acid-containing peptides may be toxic. However, extending the half-life of the JNK inhibitory peptide may not always be necessary for the desired therapeutic effect. For example, L-JNKI limited lung ischemia/reperfusion damage, and so D-amino acid-containing peptides were not necessary in this system [14]. When rapid, acute treatment is desirable, the prolonged in vivo half-life offered by D-amino acid-containing peptides may not be required.
Lastly, in considering how these peptide inhibitors may advance to clinical trials, Xigen has reported its Phase I trial of XG-102 (D-JNKI). In addition to demonstrating efficacy of the JNK inhibitory peptides, it will be important to optimise in vivo cell-permeable delivery strategies particularly as cytotoxic effects of cell-permeable peptides have been noted [102].
6. Conclusions and perspectives
Despite important advances in recent years in the development of both JNK ATP-competitive and ATP-non-competitive inhibitors, many questions have also arisen. These centre on the controls needed to establish JNK inhibitor specificity, whether JNK isoform-selective inhibitors are possible or desirable, whether other compounds may have off-target effects to inhibit JNK, and what concerns may accompany the chronic use of JNK inhibitors. In concluding this review, we briefly consider these questions that may direct further research efforts to discover and improve JNK inhibitors.
6.1. What controls are needed to establish JNK inhibitor specificity?
The testing of small molecule inhibitors against panels of protein kinases in activity assays in protein-interaction studies has emphasized that off-target effects should always be considered, particularly during the earliest stages of inhibitor/drug development. Although simple concordance between the effects observed with putative JNK inhibitors and the phenotypes of the JNK gene knockout animals may initially support the specificity of inhibitor actions, the use and interpretation of JNK knockout animals can be complicated both by the need to target the different JNK genes and by functional redundancies between the isoforms. A more robust approach has combined genetic and pharmacological approaches to evaluate protein kinase specificity. Modification of the ATP-binding pocket of the protein kinase of interest at the so-called “gate-keeper” residue allows interaction with bulky ATP analogues that may act as either substrates or inhibitors. This approach has aided JNK substrate identification [103], an has been more recently used to inhibit JNK to define JNK2 actions [104] and to determine how JNK activation time courses affect its downstream signalling consequences [105].
6.2. Are JNK isoform inhibitors possible or desirable?
From the phenotypes of JNK1−/−, JNK2−/− or JNK3−/− mice, JNK isoform-selective targeting appears beneficial (see review [3]). Although, high sequence and structure similarity, suggests that this may be difficult to achieve with small molecule inhibitors, in vivo RNA interference (RNAi) remains an option that has been recently used to evaluate the specific role for JNK1 in insulin resistance in a mouse model of diet-induced diabetes [106]. Adenoviral delivery of the RNAi resulted in almost complete knockdown of hepatic JNK1 levels, without affecting JNK1 in other tissues examined. Whilst this was accompanied by reduced circulating glucose levels and enhanced insulin signalling in vitro, plasma triglyceride levels were elevated [106]. This appeared to be the result of the altered expression of several clusters of genes involved in glycolysis and the triglyceride synthesis pathways. Why earlier studies using JNK inhibitors, the overexpression of dominant-negative JNK mutants, or gene knockout studies (e.g. [98], [107], [108]) have not observed similar changes remains to be established. The striking differences when comparing small molecule inhibition or genetic ablation approaches have been recently highlighted [109]. Specifically, for JNK, this has been attributed to compensation in the absence of JNK2 leading to enhanced JNK1 signalling [104].
6.3. Might other compounds currently being evaluated against other targets also inhibit JNK?
Inhibitors initially directed towards other targets in the cell may also interfere with JNK actions. A recent example shows the discovery of an anti-hepatitis C virus compound, 4-[2-(5-bromo-2-fluoro-phenyl)pteridin-4-ylamino]-N-3-(2-oxopyrrolidin-1-yl) propyl] nicotinamide, that inhibits vascular endothelial growth factor receptor kinase as well as JNK activities [110]. Similarly, indirubin-3′-oxime, a known inhibitor of cyclin-dependent kinases and glycogen synthase kinase 3-β, also inhibits JNK [111]. This raises questions on the whether actions of indirubin-3′-oxime to inhibit apoptosis are due to its actions on JNK, cyclin-dependent kinases, or glycogen synthase kinase 3-β, alone or in combination. Similarly, the neuroprotectant 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (PAN-811), may exert its actions to protect against glutamate toxicity via inhibition of both JNK and p38, or leflunomide may protect again acetaminophen-induced liver necrotic injury through its JNK inhibition [112], [113].
6.4. What concerns may accompany chronic use of JNK inhibitors?
The embryonic lethality of the JNK1−/−JNK2−/− mice (see review [3]) has suggested critical roles for JNK in development and homeostasis. JNK has been implicated as critical regulators of neurite formation [114], neuronal axon formation [115], and recently it has been suggested that JNK regulates events associated with both health and degeneration or motoneurons [116]. Furthermore, JNK may play protective roles as demonstrated in thrombin-induced ischemic tolerance in the brain [117], and JNK may aid in regulating circadian rhythms [118]. These roles suggest that chronic JNK inhibition may not be desirable. It will therefore remain a challenge, at least in the short term, to define the range of JNK actions in the cell, as these are likely to be many and diverse. Short-term use of JNK inhibitors remains an attractive option in a number of diseases, and the increasing availability of JNK inhibitors will allow rapid progress in determining inhibitor efficacy.
References
- 1.Dérijard B., Hibi M., Wu I.-H., Barrett T., Su B., Deng T., Karin M., Davis R.J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 2.Pulverer B.J., Kyriakis J.M., Avruch J., Nikolakaki E., Woodgett J.R. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–673. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 3.Bogoyevitch M.A. The isoform-specific functions of the c-Jun N-terminal kinases (JNKs)—differences revealed by gene targeting. BioEssays. 2006;28:923–934. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- 4.Bennett B.L., Sasaki D.T., Murray B.W., O'Leary E.C., Sakata S.T., Xu W., Leisten J.C., Motiwala A., Pierce S., Satoh Y., Bhagwat S.S., Manning A.M., Anderson D.W. SP600125, and anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scapin G., Patel S.B., Lisnock J., Becker J.W., LoGrasso P.V. The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem. Biol. 2003;10:705–712. doi: 10.1016/s1074-5521(03)00159-5. [DOI] [PubMed] [Google Scholar]
- 6.Fricker M., Lograsso P., Ellis S., Wilkie N., Hunt P., Pollack S.J. Substituting c-Jun N-terminal kinase-3 (JNK3) ATP-binding site amino acid residues with their p38 counterparts affects binding of JNK- and p38-selective inhibitors. Arch. Biochem. Biophys. 2005;438:195–205. doi: 10.1016/j.abb.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Danial N.N., Korsmeyer S. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 8.Jiang X., Wang X. Cytochrome c-mediated apoptosis. Ann. Rev. Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 9.Bogoyevitch M.A., Gillespie-Brown J., Ketterman A.J., Fuller S.J., Ben-Levy R., Ashworth A., Marshall C.J., Sugden P.H. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal protein kinases are activated by ischemia/reperfusion. Circ. Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 10.Bendinelli P., Piccoletti R., Maroni P., Bernelli-Zazzera A. The MAP kinase cascades are activated during post-ischemic liver reperfusion. FEBS Lett. 1996;398:193–197. doi: 10.1016/s0014-5793(96)01228-8. [DOI] [PubMed] [Google Scholar]
- 11.Yin T., Sandhu G., Wolfgang C.D., Burrier A., Webb R.L., Rigel D.F., Hai T., Whelan J. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J. Biol. Chem. 1997;272:19943–19950. doi: 10.1074/jbc.272.32.19943. [DOI] [PubMed] [Google Scholar]
- 12.Herdegen T., Claret F.X., Kallunki T., Martin-Villalba A., Winter C., Hunter T., Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J. Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X., Bedard E.L., Potter R., Zhong R., Alam J., Choi A.M., Lee P.J. Mitogen-activated protein kinases regulate HO-1 gene transcription after ischemia–reperfusion lung injury. Am. J. Physiol. 2002;283:L815–L829. doi: 10.1152/ajplung.00485.2001. [DOI] [PubMed] [Google Scholar]
- 14.Ishii M., Suzuki Y., Takeshita K., Miyao N., Kudo H., Hiraoka R., Nishio K., Sato N., Naoki K., Aoki T., Yamaguchi K. Inhibition of c-Jun NH2-terminal kinase activity improves ischemia/reperfusion injury in rat lungs. J. Immunol. 2004;172:2569–2577. doi: 10.4049/jimmunol.172.4.2569. [DOI] [PubMed] [Google Scholar]
- 15.Syrkina O.L., Quinn D.A., Jung W., Ouyang B., Hales C.A. Inhibition of JNK activation prolongs survival after smoke inhalation from fires. Am. J. Physiol. 2007;292:L984–L991. doi: 10.1152/ajplung.00248.2006. [DOI] [PubMed] [Google Scholar]
- 16.Han B., Mura M., Andrade C.F., Okutani D., Lodyga M., dos Santos C.C., Keshavjee S., Matthay M., Liu M. TNFα-induced long pentraxin PTX3 expression in human lung epithelial cells via JNK. J. Immunol. 2005;175:8303–8311. doi: 10.4049/jimmunol.175.12.8303. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y., Ji H.X., Xing S.H., Pei D.S., Guan Q.H. SP600125, a selective JNK inhibitor, protects ischemic renal injury via suppressing the extrinsic pathways of apoptosis. Life Sci. 2007;80:2067–2075. doi: 10.1016/j.lfs.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Ikezumi Y., Hurst L., Atkins R.C., Nikolic-Paterson D.J. Macrophage-mediated renal injury is dependent on signaling via the JNK pathway. J. Am. Soc. Nephrol. 2004;15:1775–1784. doi: 10.1097/01.asn.0000131272.06958.de. [DOI] [PubMed] [Google Scholar]
- 19.Gunawan B.K., Liu Z.X., Han D., Hanawa N., Gaarde W.A., Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 20.Henderson N.C., Pollock K.J., Frew J., Mackinnon A.C., Flavell R.A., Davis R.J., Sethi T., Simpson K.J. Critical role of c-jun NH2 terminal kinase in paracetamol-induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takamura M., Matsuda Y., Yamagiwa S., Tamura Y., Honda Y., Suzuki K., Ichida T., Aoyagi Y. An inhibitor of c-Jun NH2-terminal kinase, SP600125, protects mice from d-galactosamine/lipopolysaccharide-induced hepatic failure by modulating BH3-only proteins. Life Sci. 2007;80:1335–1344. doi: 10.1016/j.lfs.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 22.Okuno S., Saito A., Hayashi T., Chan P.H. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J. Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guan Q.H., Pei D.S., Zhang Q.G., Hao Z.B., Xu T.L., Zhang G.Y. The neuroprotective action of SP600125, a new inhibitor of JNK, on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampal CA1 via nuclear and non-nuclear pathways. Brain Res. 2005;1035:51–59. doi: 10.1016/j.brainres.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 24.Gao Y., Signore A.P., Yin W., Cao G., Yin X.M., Sun F., Luo Y., Graham S.H., Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J. Cereb. Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 25.Guan Q.H., Pei D.S., Liu X.M., Wang X.T., Xu T.L., Zhang G.Y. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006;1092:36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- 26.Yatsushige H., Ostrowski R.P., Tsubokawa T., Colohan A., Zhang J.H. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J. Neurosci. Res. 2007;85:1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]
- 27.Wang W., Shi L., Xie Y., Ma C., Li W., Su X., Huang S., Chen R., Zhu Z., Mao Z., Han Y., Li M. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson's disease. Neurosci. Res. 2004;48:195–202. doi: 10.1016/j.neures.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Yin K.J., Kim G.M., Lee J.M., He Y.Y., Xu J., Hsu C.Y. JNK activation contributes to DP5 induction and apoptosis following traumatic spinal cord injury. Neurobiol. Dis. 2005;20:881–889. doi: 10.1016/j.nbd.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 29.Lee K.H., Kim S.E., Lee Y.S. SP600125, a selective JNK inhibitor, aggravates hepatic ischemia–reperfusion injury. Exp. Mol. Med. 2006;38:408–416. doi: 10.1038/emm.2006.48. [DOI] [PubMed] [Google Scholar]
- 30.Engelbrecht A.M., Niesler C., Page C., Lochner A. p38 and JNK have distinct regulatory functions on the development of apoptosis during simulated ischaemia and reperfusion in neonatal cardiomyocytes. Basic Res. Cardiol. 2004;99:338–350. doi: 10.1007/s00395-004-0478-3. [DOI] [PubMed] [Google Scholar]
- 31.Kyoi S., Otani H., Matsuhisa S., Akita Y., Tatsumi K., Enoki C., Fujiwara H., Imamura H., Kamihata H., Iwasaka T. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res. 2006;69:888–898. doi: 10.1016/j.cardiores.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Remondino A., Kwon S.H., Communal C., Pimentel D.R., Sawyer D.B., Singh K., Colucci W.S. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ. Res. 2003;92:136–138. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 33.Eliopoulos A.G., Blake S.M., Floettmann J.E., Rowe M., Young L.S. Epstein–Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J. Virol. 1999;73:1023–1035. doi: 10.1128/jvi.73.2.1023-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLean T.I., Bachenheimer S.L. Activation of c-Jun N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 1999;73:8415–8426. doi: 10.1128/jvi.73.10.8415-8426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke P., Meintzer S.M., Widmann C., Johnson G.L., Tyler K.L. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 2001;75:11275–11283. doi: 10.1128/JVI.75.23.11275-11283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamza M.S., Reyes R.A., Izumiya Y., Wisdom R., Kung H.J., Luciw P.A. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 2004;279:38325–38330. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- 37.Rahaus M., Desloges N., Wolff M.H. Replication of varicella–zoster virus is influenced by the levels of JNK/SAPK and p38/MAPK activation. J. Gen. Virol. 2004;85:3529–3540. doi: 10.1099/vir.0.80347-0. [DOI] [PubMed] [Google Scholar]
- 38.Zapata H.J., Nakatsugawa M., Moffat J.F. Varicella–zoster virus infection of human fibroblast cells activates the c-Jun N-terminal kinase pathway. J. Virol. 2007;81:977–990. doi: 10.1128/JVI.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mishra S., Mishra J., Kumar A. Activation of JNK-dependent pathway is required for HIV viral protein R-induced apoptosis in human monocytic cells: involvement of antiapoptotic BCL2 and c-IAP1 genes. J. Biol. Chem. 2007;282:4288–4300. doi: 10.1074/jbc.M608307200. [DOI] [PubMed] [Google Scholar]
- 40.Mehrotra S., Chhabra A., Hegde U., Chakraborty N.G., Mukherji B. Inhibition of c-Jun N-terminal kinase rescues influenza epitope-specific human cytolytic T lymphocytes from activation-induced cell death. J. Leukoc. Biol. 2007;81:539–547. doi: 10.1189/jlb.0706479. [DOI] [PubMed] [Google Scholar]
- 41.Perkins D., Gyure K.A., Pereira E.F., Aurelian L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J. Neurovirology. 2003;9:101–111. doi: 10.1080/13550280390173427. [DOI] [PubMed] [Google Scholar]
- 42.Nishigaki K., Hanson C., Thompson D., Yugawa T., Ruscetti S. Activation of the Jun N-terminal kinase pathway by friend spleen focus-forming virus and its role in the growth and survival of friend virus-induced erythroleukemia cells. J. Virol. 2005;79:12752–12762. doi: 10.1128/JVI.79.20.12752-12762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holloway G., Coulson B.S. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J. Virol. 2006;80:10624–10633. doi: 10.1128/JVI.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Biophys. Acta. 2005;1741:4–10. doi: 10.1016/j.bbadis.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Si X., Luo H., Morgan A., Zhang J., Wong J., Yuan J., Esfandiarei M., Gao G., Cheung C., McManus B.M. Stress-activated protein kinases are involved in coxsackievirus B3 viral progeny release. J. Virol. 2005;79:13875–13881. doi: 10.1128/JVI.79.22.13875-13881.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassan M., Ghozlan H., Abdel-Kader O. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural protein 3 (NS3)-mediated cell growth. Virology. 2005;333:324–364. doi: 10.1016/j.virol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 47.Wang X., Sonenshein G.E. Induction of the RelB NF-kappaB subunit by the cytomegalovirus IE1 protein is mediated via Jun kinase and c-Jun/Fra-2 AP-1 complexes. J. Virol. 2005;79:95–105. doi: 10.1128/JVI.79.1.95-105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu J.H., Lin B.Y., Deng W., Broker T.R., Chow L.T. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J. Virol. 2007;81:5066–5866. doi: 10.1128/JVI.02480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han Z., Boyle D.L., Chang L., Bennett B., Karin M., Yang L., Manning A.M., Firestein G.S. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bain J., McLauchlan H., Elliott M., Cohen P. The specificities of protein kinase inhibitors: an update. Biochem. J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fabian M.A., Biggs W.H., Treiber D.K., Atteridge C.E., Azimioara M.D., Benedetti M.G., Carter T.A., Ciceri P., Edeen P.T., Floyd M., Ford J.M., Galvin M., Gerlach J.L., Grotzfeld R.M., Herrgard S., Insko D.E., Insko M.A., Lai A.G., Lelias J.M., Mehta S.A., Milanov Z.V., Velasco A.M., Wodicka L.M., Patel H.K., Zarrinkar P.P., Lockhart D.J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–369. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 52.Supino R., Polizzi D., Pavesi R., Pratesi G., Guano F., Capranico G., Palumbo M., Sissi C., Richter S., Beggiolin G., Menta E., Pezzoni G., Spinelli S., Torriani D., Carenini N., Dal Bo L., Facchinetti F., Tortoreto M., Zunino F. A novel 9-aza-anthrapyrazole effective against human prostatic carcinoma xenografts. Oncology. 2001;61:234–242. doi: 10.1159/000055380. [DOI] [PubMed] [Google Scholar]
- 53.Begleiter A., Lin D., Larson K.K., Lang J., Wu X., Cabral T., Taylor H., Guziec L.J., Kerr P.D., Hasinoff B.B., Guziec F.S. Structure–activity studies with cytotoxic anthrapyrazoles. Oncol. Rep. 2006;15:1575–1580. [PubMed] [Google Scholar]
- 54.Flanc R.S., Ma F.Y., Tesch G.H., Han Y., Atkins R.C., Bennett B.L., Friedman G.C., Fan J.H., Nikolic-Paterson D.J. A pathogenic role for JNK signaling in experimental anti-GBM glomerulonephritis. Kidney Int. 2007;72:698–708. doi: 10.1038/sj.ki.5002404. [DOI] [PubMed] [Google Scholar]
- 55.Ma F.Y., Flanc R.S., Tesch G.H., Han Y., Atkins R.C., Bennett B.L., Friedman G.C., Fan J.H., Nikolic-Paterson D.J. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 2007;18:472–484. doi: 10.1681/ASN.2006060604. [DOI] [PubMed] [Google Scholar]
- 56.Uehara T., Bennett B., Sakata S.T., Satoh Y., Bilter G.K., Westwick J.K., Brenner D.A. JNK mediates hepatic ischemia reperfusion injury. J. Hepatol. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 57.Uehara T., Xi Peng X., Bennett B., Satoh Y., Friedman G., Currin R., Brenner D.A., Lemasters J. c-Jun N-terminal kinase mediates hepatic injury after rat liver transplantation. Transplantation. 2004;78:324–332. doi: 10.1097/01.tp.0000128859.42696.28. [DOI] [PubMed] [Google Scholar]
- 58.Ruckle T., Biamonte M., Grippi-Vallotton T., Arkinstall S., Cambet Y., Camp M., Chabert C., Church D.J., Halazy S., Jiang X., Martinou I., Nichols A., Sauer W., Gotteland J.P. Design, synthesis, and biological activity of novel, potent, and selective (benzoylaminomethyl)thiophene sulfonamide inhibitors of c-Jun–N-terminal kinase. J. Med. Chem. 2004;47:6921–6934. doi: 10.1021/jm031112e. [DOI] [PubMed] [Google Scholar]
- 59.Graczyk P.P., Khan A., Bhatia G.S., Palmer V., Medland D., Numata H., Oinuma H., Catchick J., Dunne A., Ellis M., Smales C., Whitfield J., Neame S.J., Shah B., Wilton D., Morgan L., Patel T., Chung R., Desmond H., Staddon J.M., Sato N., Inoue A. The neuroprotective action of JNK3 inhibitors based on the 6,7-dihydro-5H-pyrrolo[1,2-a]imidazole scaffold. Bioorg. Med. Chem. Lett. 2005;15:4666–4670. doi: 10.1016/j.bmcl.2005.07.076. [DOI] [PubMed] [Google Scholar]
- 60.Gaillard P., Jeanclaude-Etter I., Ardissone V., Arkinstall S., Cambet Y., Camps M., Chabert C., Church D., Cirillo R., Gretener D., Halazy S., Nichols A., Szyndralewiez C., Vitte P.A., Gotteland J.P. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J. Med. Chem. 2005;48:4596–4607. doi: 10.1021/jm0310986. [DOI] [PubMed] [Google Scholar]
- 61.Swahn B.M., Huerta F., Kallin E., Malmstrom J., Weigelt T., Viklund J., Womack P., Xue Y., Ohberg L. Design and synthesis of 6-anilinoindazoles as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg. Med. Chem. Lett. 2005;15:5095–5099. doi: 10.1016/j.bmcl.2005.06.083. [DOI] [PubMed] [Google Scholar]
- 62.Swahn B.M., Xue Y., Arzel E., Kallin E., Magnus A., Plobeck N., Viklund J. Design and synthesis of 2'-anilino-4,4'-bipyridines as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg. Med. Chem. Lett. 2006;16:1397–1401. doi: 10.1016/j.bmcl.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 63.Liu M., Xin Z., Clampit J.E., Wang S., Gum R.J., Haasch D.L., Trevillyan J.M., Abad-Zapatero C., Fry E.H., Sham H.L., Liu G. Synthesis and SAR of 1,9-dihydro-9-hydroxypyrazolo[3,4-b]quinolin-4-ones as novel, selective c-Jun N-terminal kinase inhibitors. Bioorg. Med. Chem. Lett. 2006;16:2590–2594. doi: 10.1016/j.bmcl.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 64.Szczepankiewicz B.G., Kosogof C., Nelson L.T., Liu G., Liu B., Zhao H., Serby M.D., Xin Z., Liu M., Gum R.J., Haasch D.L., Wang S., Clampit J.E., Johnson E.F., Lubben T.H., Stashko M.A., Olejniczak E.T., Sun C., Dorwin S.A., Haskins K., Abad-Zapatero C., Fry E.H., Hutchins C.W., Sham H.L., Rondinone C.M., Trevillyan J.M. Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J. Med. Chem. 2006;49:3563–3580. doi: 10.1021/jm060199b. [DOI] [PubMed] [Google Scholar]
- 65.Zhao H., Serby M.D., Xin Z., Szczepankiewicz B.G., Liu M., Kosogof C., Liu B., Nelson L.T., Johnson E.F., Wang S., Pederson T., Gum R.J., Clampit J.E., Haasch D.L., Abad-Zapatero C., Fry E.H., Rondinone C., Trevillyan J.M., Sham H.L., Liu G. Discovery of potent, highly selective, and orally bioavailable pyridine carboxamide c-Jun NH2-terminal kinase inhibitors. J. Med. Chem. 2006;49:4455–4458. doi: 10.1021/jm060465l. [DOI] [PubMed] [Google Scholar]
- 66.Liu M., Wang S., Clampit J.E., Gum R.J., Haasch D.L., Rondinone C.M., Trevillyan J.M., Abad-Zapatero C., Fry E.H., Sham H.L., Liu G. Discovery of a new class of 4-anilinopyrimidines as potent c-Jun N-terminal kinase inhibitors: synthesis and SAR studies. Bioorg. Med. Chem. Lett. 2007;17:668–672. doi: 10.1016/j.bmcl.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 67.Liu G., Zhao H., Liu B., Xin Z., Liu M., Kosogof C., Szczepankiewicz B.G., Wang S., Clampit J.E., Gum R.J., Haasch D.L., Trevillyan J.M., Sham H.L. Aminopyridine carboxamides as c-Jun N-terminal kinase inhibitors: targeting the gatekeeper residue and beyond. Bioorg. Med. Chem. Lett. 2006;16:5723–5730. doi: 10.1016/j.bmcl.2006.08.097. [DOI] [PubMed] [Google Scholar]
- 68.Carboni S., Hiver A., Szyndralewiez C., Gaillard P., Gottel J.P., Vitte P.A. AS601245 (1,3-benzothiazol-2-yl (2-[[2-(3-pyridinyl) ethyl] amino]-4 pyrimidinyl) acetonitrile): a c-Jun NH2-terminal protein kinase inhibitor with neuroprotective properties. J. Pharmacol. Exp. Ther. 2004;310:25–32. doi: 10.1124/jpet.103.064246. [DOI] [PubMed] [Google Scholar]
- 69.Ferrandi C., Ballerio R., Gaillard P., Giachetti C., Carboni S., Vitte P.A., Gottel J.P., Cirillo R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004;142:953–960. doi: 10.1038/sj.bjp.0705873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu G., Zhao H., Liu B., Xin Z., Liu M., Serby M.D., Lubbers N.L., Widomski D.L., Polakowski J.S., Beno D.W., Trevillyan J.M., Sham H.L. Hemodynamic effects of potent and selective JNK inhibitors in anesthetized rats: Implication for targeting protein kinases in metabolic diseases. Bioorg. Med. Chem. Lett. 2007;17:495–500. doi: 10.1016/j.bmcl.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 71.Rochfort S.J., Towerzey L., Carroll A., King G., Michael A., Pierens G., Rali T., Redburn J., Whitmore J., Quinn R.J. Latifolians A and B, novel JNK3 kinase inhibitors from the Papua New Guinean plant Gnetum latifolium. J. Nat. Prod. 2005;68:1080–1082. doi: 10.1021/np049616i. [DOI] [PubMed] [Google Scholar]
- 72.Bogoyevitch M.A., Barr R.K., Ketterman A.J. Peptide inhibitors of protein kinases — discovery, characterisation and use. Biochim. Biophys. Acta. 2005;1754:79–99. doi: 10.1016/j.bbapap.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 73.Holzberg D., Knight C.G., Dittrich-Breiholz O., Schneider H., Dorrie A., Hoffmann E., Resch K., Kracht M. Disruption of the c-JUN–JNK complex by a cell-permeable peptide containing the c-JUN domain induces apoptosis and affects a distinct set of IL-1-induced inflammatory genes. J. Biol. Chem. 2003;278:40213–40223. doi: 10.1074/jbc.M304058200. [DOI] [PubMed] [Google Scholar]
- 74.Dickens M., Rogers J.S., Cavanagh J., Raitano A., Xia Z., Halpern J.R., Greenberg M.E., Sawyers C.L., Davis R.J. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 75.Barr R.K., Kendrick T.S., Bogoyevitch M.A. Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 76.Bonny C., Oberson A., Negri S., Sauser C., Schorderet D.F. Cell-permeable inhibitors of JNK: novel blockers of b-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 77.Borsello T., Clarke P.G., Hirt L., Vercelli A., Repici M., Schorderet D.F., Bogousslavsky J., Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 78.Kendrick T.S., Lipscombe R.J., Rausch O., Nicholson S.E., Layton J.E., Goldie-Cregan L.C., Bogoyevitch M.A. Contribution of the membrane-distal tyrosine in intracellular signaling by the granulocyte colony-stimulating factor receptor. J. Biol. Chem. 2004;279:326–340. doi: 10.1074/jbc.M310144200. [DOI] [PubMed] [Google Scholar]
- 79.Barr R.K., Boehm I., Attwood P.V., Watt P.M., Bogoyevitch M.A. The critical features and the mechanism of inhibition of a kinase interaction motif-based peptide inhibitor of JNK. J. Biol. Chem. 2004;279:36327–38338. doi: 10.1074/jbc.M402181200. [DOI] [PubMed] [Google Scholar]
- 80.Barr R.K., Hopkins R.M., Watt P.M., Bogoyevitch M.A. Reverse two-hybrid screening identifies residues of JNK required for interaction with the kinase interaction motif of JNK-interacting Protein-1. J. Biol. Chem. 2004;279:43178–43189. doi: 10.1074/jbc.M405900200. [DOI] [PubMed] [Google Scholar]
- 81.Heo Y.S., Kim S.K., Seo C.I., Kim Y.K., Sung B.J., Lee H.S., Lee J.I., Park S.Y., Kim J.H., Hwang K.Y., Hyun Y.L., Jeon Y.H., Ro S., Cho J.M., Lee T.G., Yang C.H. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Niu L., Chang K.C., Wilson S., Tran P., Zuo F., Swinney D.C. Kinetic characterization of human JNK2alpha2 reaction mechanism using substrate competitive inhibitors. Biochemistry. 2007;46:4775–4784. doi: 10.1021/bi602423e. [DOI] [PubMed] [Google Scholar]
- 83.Bogoyevitch M.A. Therapeutic promise of JNK ATP-noncompetitive inhibitors. Trends Mol. Med. 2005;11:232–239. doi: 10.1016/j.molmed.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Hirt L., Badaut J., Thevenet J., Granziera C., Regli L., Maurer F., Bonny C., Bogousslavsky J. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke. 2004;35:1738–1743. doi: 10.1161/01.STR.0000131480.03994.b1. [DOI] [PubMed] [Google Scholar]
- 85.Wang J., Van De Water T.R., Bonny C., de Ribaupierre F., Puel J.L., Zine A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J. Neurosci. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eshraghi A.A., Wang J., Adil E., He J., Zine A., Bublik M., Bonny C., Puel J.L., Balkany T.J., Van De Water T.R. Blocking c-Jun-N-terminal kinase signaling can prevent hearing loss induced by both electrode insertion trauma and neomycin ototoxicity. Hear. Res. 2007;226:168–177. doi: 10.1016/j.heares.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 87.Wang J., Ruel J., Ladrech S., Bonny C., van de Water T.R., Puel J.L. Inhibition of the c-Jun N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound-exposed animals. Mol. Pharmacol. 2007;71:654–666. doi: 10.1124/mol.106.028936. [DOI] [PubMed] [Google Scholar]
- 88.Zhuang Z.Y., Wen Y.R., Zhang D.R., Borsello T., Bonny C., Strichartz G.R., Decosterd I., Ji R.R. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J. Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guan Q.H., Pei D.S., Zong Y.Y., Xu T.L., Zhang G.Y. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006;139:609–627. doi: 10.1016/j.neuroscience.2005.11.067. [DOI] [PubMed] [Google Scholar]
- 90.Arthur P.G., Matich G.P., Pang W.W., Yu D.Y., Bogoyevitch M.A. Necrotic death of neurons following an excitotoxic insult is prevented by a peptide inhibitor of c-jun N-terminal kinase. J. Neurochem. 2007;102:65–76. doi: 10.1111/j.1471-4159.2007.04618.x. [DOI] [PubMed] [Google Scholar]
- 91.Zhang S., Lin Y., Kim Y.S., Hande M.P., Liu Z.G., Shen H.M. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ. 2007;14:1001–1010. doi: 10.1038/sj.cdd.4402088. [DOI] [PubMed] [Google Scholar]
- 92.Bogoyevitch M.A., Kobe B. Uses for JNK—the many and varied substrates of c-Jun N-terminal kinases. Micro. Mol. Biol. Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beckham J.D., Goody R.J., Clarke P., Bonny C., Tyler K.L. Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J. Virol. 2007;81:6984–6992. doi: 10.1128/JVI.00467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Milano G., Morel S., Bonny C., Samaja M., von Segesser L.K., Nicod P., Vassalli G. A peptide inhibitor of c-Jun NH2-terminal kinase (JNK) reduces myocardial ischemia/reperfusion injury and infarct size in vivo. Am. J. Physiol. 2007;292:H1828–H1835. doi: 10.1152/ajpheart.01117.2006. [DOI] [PubMed] [Google Scholar]
- 95.Lu Z., Serghides L., Patel S.N., Degousee N., Rubin B.B., Krishnegowda G., Gowda D.C., Karin M., Kain K.C. Disruption of JNK2 decreases the cytokine response to Plasmodium falciparum glycosylphosphatidylinositol in vitro and confers protection in a cerebral malaria model. J. Immunol. 2006;177:6344–6352. doi: 10.4049/jimmunol.177.9.6344. [DOI] [PubMed] [Google Scholar]
- 96.Hirosumi J., Tuncman G., Chang L., Gorgun C.Z., Uysal K.T., Maeda K., Karin M., Hotamisligil G.S. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 97.Jaeschke A., Rincon M., Doran B., Reilly J., Neuberg D., Greiner D.L., Shultz L.D., Rossini A.A., Flavell R.A., Davis R.J. Disruption of the Jnk2 (Mapk9) gene reduces destructive insulitis and diabetes in a mouse model of type I diabetes. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6931–6935. doi: 10.1073/pnas.0502143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kaneto H., Nakatani Y., Miyatsuka T., Kawamori D., Matsuoka T.A., Matsuhisa M., Kajimoto Y., Ichijo H., Yamasaki Y., Hori M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat. Med. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 99.Noguchi H., Nakai Y., Ueda M., Masui Y., Futaki S., Kobayashi N., Hayashi S., Matsumoto S. Activation of c-Jun NH2-terminal kinase (JNK) pathway during islet transplantation and prevention of islet graft loss by intraportal injection of JNK inhibitor. Diabetologia. 2007;50:612–619. doi: 10.1007/s00125-006-0563-2. [DOI] [PubMed] [Google Scholar]
- 100.Abdelli S., Abderrahmani A., Hering B.J., Beckmann J.S., Bonny C. The c-Jun N-terminal kinase JNK participates in cytokine- and isolation stress-induced rat pancreatic islet apoptosis. Diabetologia. 2007;50:1660–1669. doi: 10.1007/s00125-007-0704-2. [DOI] [PubMed] [Google Scholar]
- 101.Fornoni A., Cobianchi L., Sanabria N.Y., Pileggi A., Molano R.D., Ichii H., Rosero S., Inverardi L., Ricordi C., Pastori R.L. The L-isoform but not D-isoforms of a JNK inhibitory peptide protects pancreatic b-cells. Biochem. Biophys. Res. Commun. 2007;354:227–233. doi: 10.1016/j.bbrc.2006.12.186. [DOI] [PubMed] [Google Scholar]
- 102.Cardozo A.K., Buchillier V., Mathieu M., Chen J., Ortis F., Ladriere L., Allaman-Pillet N., Poirot O., Kellenberger S., Beckmann J.S., Eizirik D.L., Bonny C., Maurer F. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. Biochim. Biophys. Acta. 2007;1768:2222–2234. doi: 10.1016/j.bbamem.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 103.Habelhah H., Shah K., Huang L., Burlingame A.L., Shokat K.M., Ronai Z. Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J. Biol. Chem. 2001;276:18090–18095. doi: 10.1074/jbc.M011396200. [DOI] [PubMed] [Google Scholar]
- 104.Jaeschke A., Karasarides M., Ventura J.J., Ehrhardt A., Zhang C., Flavell R.A., Shokat K.M., Davis R.J. JNK2 is a positive regulator of the cJun transcription factor. Mol. Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 105.Ventura J.J., Hubner A., Zhang C., Flavell R.A., Shokat K.M., Davis R.J. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell. 2006;21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 106.Yang R., Wilcox D.M., Haasch D.L., Jung P.M., Nguyen P.T., Voorbach M.J., Doktor S., Brodjian S., Bush E.N., Lin E., Jacobson P.B., Collins C.A., Landschulz K.T., Trevillyan J.M., Rondinone C.M., Surowy T.K. Liver-specific knockdown of JNK1 upregulates PGC-1beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced diabetic mice. J. Biol. Chem. 2007;282:22765–22774. doi: 10.1074/jbc.M700790200. [DOI] [PubMed] [Google Scholar]
- 107.Nakatani Y., Kaneto H., Kawamori D., Hatazaki M., Miyatsuka T., Matsuoka T.A., Kajimoto Y., Matsuhisa M., Yamasaki Y., Hori M. Modulation of the JNK pathway in liver affects insulin resistance status. J. Biol. Chem. 2004;279:45803–45809. doi: 10.1074/jbc.M406963200. [DOI] [PubMed] [Google Scholar]
- 108.Tuncman G., Hirosumi J., Solinas G., Chang L., Karin M., Hotamisligil G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 2006;28:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Knight Z.A., Shokat K.M. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 110.Raboisson P., Lenz O., Lin T.I., Surleraux D., Chakravarty S., Scholliers A., Vermeiren K., Delouvroy F., Verbinnen T., Simmen K. Evaluation of the anti-hepatitis C virus effect of novel potent, selective, and orally bioavailable JNK and VEGFR kinase inhibitors. Bioorg. Med. Chem. Lett. 2007;17:1843–1849. doi: 10.1016/j.bmcl.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 111.Xie Y., Liu Y., Ma C., Yuan Z., Wang W., Zhu Z., Gao G., Liu X., Yuan H., Chen R., Huang S., Wang X., Zhu X., Wang X., Mao Z., Li M. Indirubin-3'-oxime inhibits c-Jun NH2-terminal kinase: anti-apoptotic effect in cerebellar granule neurons. Neurosci. Lett. 2004;367:355–359. doi: 10.1016/j.neulet.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 112.Chen R.W., Lu X.C., Yao C., Liao Z., Jiang Z.G., Wei H., Ghanbari H.A., Tortella F.C., Dave J.R. PAN-811 provides neuroprotection against glutamate toxicity by suppressing activation of JNK and p38 MAPK. Neurosci. Lett. 2007;422:64–67. doi: 10.1016/j.neulet.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 113.Latchoumycandane C., Goh C.W., Ong M.M., Boelsterli U.A. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–421. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- 114.Xiao J., Pradhan A., Liu Y. Functional role of JNK in neuritogenesis of PC12-N1 cells. Neurosci. Lett. 2006;392:231–234. doi: 10.1016/j.neulet.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 115.Oliva A.A., Atkins C.M., Copenagle L., Banker G.A. Activated c-Jun N-terminal kinase is required for axon formation. J. Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Newbern J., Taylor A., Robinson M., Lively M.O., Milligan C.E. c-Jun N-terminal kinase signaling regulates events associated with both health and degeneration in motoneurons. Neuroscience. 2007;147:680–692. doi: 10.1016/j.neuroscience.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 117.Granziera C., Thevenet J., Price M., Wiegler K., Magistretti P.J., Badaut J., Hirt L. Thrombin-induced ischemic tolerance is prevented by inhibiting c-jun N-terminal kinase. Brain Res. 2007;1148:217–225. doi: 10.1016/j.brainres.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 118.Chansard M., Molyneux P., Nomura K., Harrington M.E., Fukuhara C. c-Jun N-terminal kinase inhibitor SP600125 modulates the period of mammalian circadian rhythms. Neuroscience. 2007;145:812–823. doi: 10.1016/j.neuroscience.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]