

Graphical abstract
Keywords: 1,2,3-Triazole; Click chemistry; Copper-catalysed 1,3-dipolar cycloaddition (CuAAC); Huisgen azide-alkyne cycloaddition; Biological activity; Lead compounds
Abstract
The 1,2,3-triazole ring is a major pharmacophore system among nitrogen-containing heterocycles. These five-membered heterocyclic motifs with three nitrogen heteroatoms can be prepared easily using ‘click’ chemistry with copper- or ruthenium-catalysed azide-alkyne cycloaddition reactions. Recently, the ‘linker’ property of 1,2,3-triazoles was demonstrated, and a novel class of 1,2,3-triazole-containing hybrids and conjugates was synthesised and evaluated as lead compounds for diverse biological targets. These lead compounds have been demonstrated as anticancer, antimicrobial, anti-tubercular, antiviral, antidiabetic, antimalarial, anti-leishmanial, and neuroprotective agents. The present review summarises advances in lead compounds of 1,2,3-triazole-containing hybrids, conjugates, and their related heterocycles in medicinal chemistry published in 2018. This review will be useful to scientists in research fields of organic synthesis, medicinal chemistry, phytochemistry, and pharmacology.
1. Introduction
Heterocyclic organic chemistry is one of the most important and well-studied branches of medicinal chemistry. An important feature of heterocyclic bioactive compounds is their various constituent heteroatoms, including nitrogen,1, 2 sulphur,3, 4, 5, 6 oxygen,7, 8 and others.9 These heteroatoms directly affect the reactivity of the target skeleton, activity (or toxicology) of the compounds, interactions between target drugs and different target inhibitors, as well as able to influence of metabolism and pharmacokinetics.
The most promising heterocyclic compounds are azoles,10, 11, 12, 13, 14 which are five-membered nitrogen heterocycles. The nitrogen-containing azole skeleton offers the advantage of two nitrogen heteroatoms in the ring, which has an important effect on structural modifications and biological interactions. Over the past decade, the chemistry and biology of azole derivatives have emerged as very popular topics. We performed a search of the Scopus database (31 December 2018) using each of the azoles (‘imidazole’, ‘pyrazole’, ‘triazole’, ‘tetrazole’, and ‘pentazole’) as keywords to identify different published reports, and found that triazole motifs were the most frequently studied (Fig. 1 ),15 particularly 1,2,3-triazoles.
Fig. 1.

Number of reports on each azole in the Scopus database for 2008–2018.
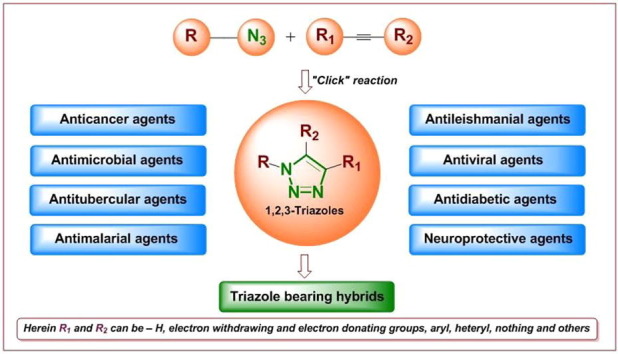
It should be noted here that the advances in click chemistry (the copper-catalysed azide-alkyne cycloaddition [Cu-AAC] reaction), various synthetic methodologies for the 1,2,3-triazole scaffolds (e.g., derivatives, hybrids, and conjugates) have been used in medicinal chemistry (Fig. 2 ).16 In 2018, various biological screenings were conducted17, 18, 19, 20 which led to the identification of anticancer,21, 22, 23 antimicrobial,24, 25, 26, 27, 28 anti-infective,29, 30 and antioxidant31, 32, 33 properties of the studied 1,2,3-triazole-bearing hybrids. Moreover, triazole-linked derivatives were suggested to affect adenosine diphosphate ribosylation biology,34 and were used widely in peptides to mimic a trans-amide bond, despite their hazardous effects on native peptide activity.35
Fig. 2.

Bioactive 1,2,3-triazoles using ‘click’ reaction.
In addition, a 1,2,3-triazole core provides diverse pharmacophore properties and hybrids are most commonly considered ‘lead compounds’ when they contain or are fused by a 1,2,3-triazole ring. Thus, in view of the above factors and the recent focus of researchers on 1,2,3-triazole compounds, we have discussed and highlighted the diverse biological activities of the 1,2,3-triazole hybrids, conjugates, and their related compounds as leads in medicinal chemistry, based on articles published in 2018.
2. Chemistry: synthesis and properties of the 1,2,3-triazoles
The general synthetic route of 1,2,3-triazoles using click chemistry is described in Scheme 1 A. The most popular route to 1,2,3-triazoles is Cu(I)-catalysed Huisgen 1,3-dipolar cycloaddition, which is the interaction between organic azides and alkynes that forms only the 1,4-regioisomeric 1,2,3-triazoles (Scheme 1, B).36 If ruthenium catalysts are used instead of Cu(I) for this reaction, 1,5-regioisomeric 1,2,3-triazoles are the major products,37 and there is a known non-catalytic thermal pathway that produces both regioisomeric 1,2,3-triazoles (Scheme 1B). Several other methods for the synthesis of 1H-1,2,3-triazoles including Cu-AAC; solid-phase synthesis; the three-component reaction of aldehydes, nitromethane, and sodium azide (Scheme 1C); the reaction of enamines with 4-methylbenzenesulfonyl azide (Scheme 1D); and the interaction of β-bromostyrenes with sodium azide in a palladium-catalysed reaction (Scheme 1E), were reviewed by Efimov in 2019.38 In 2015, novel ‘click’ methods for 1,2,3-triazoles using classical reactions were reviewed by Totobenazara and Burke.39
Scheme 1.

Synthetic routes to 1,2,3-triazoles.
Generally, triazoles are stable to hydrolysis under acidic or basic conditions, metabolic degradation, and redox conditions. These compounds can form H-bonds and π-π stacking interactions. Although the aromatic ring in 1,2,3-triazoles is fairly stable, it becomes more reactive when bonded to electronegative groups. For example, 1,2,3-triazole can form 1-CF3-1,2,3-triazole, which decomposes at approximately 170 °C and generates highly reactive intermediates.40 In addition, triazoles are weakly acidic and weakly basic. They are rarely oxidised but may tautomerise to 2H-1,2,3-triazoles. They are more sensitive to reducing agents. Furthermore, several other properties including strong dipole moments of the triazole system, bioisosteric effects,41 and the nature of the heteroatoms all make triazole units an important scaffold in medicinal chemistry.
3. Biology
3.1. Anticancer activity
3.1.1. 1,2,3-Triazole containing derivatives of natural compounds
Natural compounds and combination therapy are used widely in cancer therapies.42, 43 Although the 1,2,3-triazole moiety does not exist in nature, it has attracted interest as a candidate anticancer drug, particularly for the production of ‘1,2,3-triazole-natural compound’ hybrids.44 In this field, Ding et al.45 reported the Cu2O nanoparticle-catalysed ‘click’ reaction that enabled the synthesis of melampomagnolide B-triazole conjugates, and evaluated their anticancer activities. The most potent derivative 1 (Fig. 3 ) (HCT116, IC50 = 0.43 µM) that exhibited almost 11.5-fold potency than Melampomagnolide B (HCT116, IC50 = 4.93 µM), did not affect normal cells (FHC, HPDE6-C7, 3T3), and significantly induced apoptosis as well as inhibition of proliferation and migration of cancer cell lines. The effect of 1 on HCT116 cell migration was measured using the Transwell assay and the number of migrated cells was distinctly lower after exposure to melampomagnolide B-triazole 1 for 48 h. In addition, the expression of epithelial proteins such us snail, zonula occludens-1 (ZO-1), and E-cadherin, as well as mesenchymal marker proteins such as vimentin, zinc finger E-box binding homeobox 1 (ZEB1), and N-cadherin was productively decreased after treatment with derivative 1, which confirmed that triazole compound 1 may inhibit and affect the migration of HCT116 cells. Another melampomagnolide B-triazole conjugate, compound 2 (Fig. 3), was effective and potent against several cancer cell lines including leukaemia (GI50 = 0.10–0.23 µM), colon (GI50 = 0.14–1.17 µM), melanoma (GI50 = 0.15–1.47 µM), renal (GI50 = 0.02–0.70 µM), ovarian (GI50 = 0.15–1.86 µM), prostate (GI50 = 0.72–0.83 µM), and breast (GI50 = 0.17–1.03 µM) cancer cell line subpanels.46 Further investigation indicated that triazole compound 2 was a potent inhibitor of nuclear factor (NF)-kB and cell proliferation in TMD-231 (MDA-MB-231) cell line. Moreover, treatment of TMD-231 cells with derivative 2 decreased the DNA binding activity of NF-kB by inhibiting p65 phosphorylation mediated by inhibitor of NF-kB kinase (IKK)-b and increasing basal IkBα levels via inhibition of substantial NF-kB activation and IkBα turnover.
Fig. 3.

1,2,3-Triazole-bearing natural compounds (1–16) analogues as anticancer leads.
Yu et al.47 synthesised a series of 1,2,3-triazolo-dihydroartemisinin-coumarin hybrids and screened their anticancer potentials in two types of cancer cells. These hybrids displayed modest cytotoxicity towards HT-29 and MDA-MB-231 cell lines, particularly under hypoxic condition. However, hybrid 3 (Fig. 3) was more active on HT-29 than MDA-MB-231 cell (normoxic IC50 = 1.5 µM and under hypoxia IC50 = 0.01 µM). Furthermore, this hybrid arrested HT-29 cells in the G0/G1 phase, inhibited the migration of tumour cells, and caused a large reduce in mitochondrial membrane capability, leading to apoptosis of HT-29 cells.
Diterpenoids with the 1,2,3-triazole moiety in Jiyuan Oridonin A were tested for their antiproliferative properties.48 Derivative 4 (Fig. 3), containing a triazole ring, also displayed better antiproliferative activity with IC50 values of 2.7 µM against the Eca109 cell line and 1.5 µM against the MCF-7 breast cancer cell line than against the other tested cells. Moreover, the IC50 value of 4 was also evaluated in MGC-803 (0.6 µM) and PC-3 (0.6 µM) cancer cells. Cellular mechanistic studies demonstrated that compound 4 arrested the cell cycle at the G1 phase and induced potent apoptosis of the SMMC-7721 cell line; it also inhibited colony migration and formation through the Wnt signalling pathway in these cells. Another two series of ent-kaurene diterpenoid derivatives were screened for their antiproliferative properties in four cancer cell lines (Eca109, EC9706, SMMC7721, and MCF-7 cells).49 Compound 5 (Fig. 3) displayed highly anticancer potential in all tested cell lines (IC50 values of 2.70, 5.04, 4.44, and 4.76 µM, against the Eca109, EC9706, SMMC7721, and MCF-7 cell lines, respectively) than the positive control Oridonin did, confirming that 1,2,3-triazole is a beneficial heterocyclic moiety that affects the cytotoxicity of triazole-containing hybrids. Investigation of the anticancer mechanism showed that 5 increased reactive oxygen species (ROS) levels in cancer cell lines, leading to the decline of mitochondrial membrane potential and the release of cytochrome c into the cytoplasm. Furthermore, this effect was further enhanced and activated by caspase-9 to induce apoptosis.
Wei et al.50 synthesised a series of 1,2,3-triazole-containing albiziabioside A derivatives and evaluated their anticancer potentials in vitro and in vivo. The target lead (6, Fig. 3) showed significant activity on HCT116 cells (IC50 = 5.19 µM). Moreover, this triterpene saponin triazole derivative demonstrated favourable selectivity, and was effective on multidrug resistance (MDR) cancer cells where it induced ferroptosis and apoptosis as a p53 activator via the mitochondrial pathway. In vivo investigations revealed that the introduction of the triazole fragment into the triterpene skeleton considerably inhibited tumorigenesis without causing toxicity against normal cells (for 6). Indeed, this study provided clinical evidence to support the consideration of the albiziabioside A derivative 6 as a potential cancer drug candidate with enhanced activity, and further information on its novel mechanism of action. Another series of 1,2,3-triazole-linked saponin hederacolchiside A1 with aryl/amide portion were synthesised to improve the inhibitory activity, metabolic stability, and comprehensively determine the pharmacological mechanism.51 The antiproliferative activities of the synthesised analogues were assayed on six types of human cancer cell lines, PC3, HT29, HepG2, A549, HL60, and U937 cell lines. The results revealed that most of the synthesised hybrids with the para- and meta-substituents demonstrated strong cytotoxicity towards HL60 and U937 cells. In particular, derivative 7 (Fig. 3) showed strong anticancer potential against all selected cells compare with the positive controls 5-fluorouracil and hederacolchiside A1. Moreover, the results of the cell cycle and apoptosis screening indicated that 7 clearly inhibited the proliferation of HepG2 cells via the induction of apoptosis and cell cycle arrest of the in the G1 and S phases.
Biologically relevant natural, synthetic, and hybrid compounds are a great resource for lead formation and the development of novel pharmaceuticals. Recently, 60 novel 1,2,3-triazole-containing pharmacophores related to the allogibberic acid fragments were synthesised by Wu et al.52 The cytotoxic potential of all hybrid allogibberic triazole hybrids was tested to determine their anticancer activities on the A-549, HL-60, SMMC-7721, MCF-7, and SW480 cell lines. The most potent compound (8, Fig. 3) demonstrated satisfactorily higher cytotoxicity than the positive control drug cisplatin did towards all five tested tumour cell lines, with IC50 values of 0.25–1.70 µM. Mechanistic exploration revealed that hybrid compound 8 induced cell cycle arrest at the S phase and apoptosis of SMMC-7721 cells.
A quinine derivative containing a triazolyl-phenol moiety (9, Fig. 3) was identified as a selective hybrid with activity against HT-29 cells (IC50 = 0.65 μg/mL or 1.21 μM).53 In addition, 9 and other analogues were evaluated in three different solid tumour (HT-29, MCF-7, A549, and DU-145) cell lines and one normal human epithelial (MCF-10A) cell line. The triazole-bearing alkaloid 9 also displayed good antiproliferative properties against the cells listed above; however, it may be a lead compound against HT-29 colon cancer, against which it was eight times more potent than cisplatin.
The acetoxymethyl 1,2,3-triazole compound 10 (Fig. 3) synthesised using Huisgen azide-alkyne cycloaddition, which is a synthetic analogue of natural meiogynin A, was investigated as a pan- B-cell lymphoma-2 (Bcl-2) inhibitor. This triazole hybrid induced apoptosis and was strongly cytotoxic to BL2, RS4;11, and H929 cells at rates of 93, 75.5, and 37.5, and 20.5%, respectively in Remb1 cells.54 Here, it should be noted that the selected target proteins such as Bcl-2-associated X protein (Bcl-xL), myeloid cell leukaemia 1 (Mcl-1), and Bcl-2 and their interaction between new triazole hybrids were screened using fluorescence polarisation, and the results indicated that modulation of the lateral chain dramatically affected the compound by considerably modifying its activity on target proteins. Other triazole-natural compound hybrids, triterpene derivative-linked 1,2,3-triazole moieties were prepared and their anticancer potentials were investigated on the selected cancer cells such as C-32, T47D, and SNB-19 using the WST-1 assay.55 Hybrid derivative 11 (Fig. 3) displayed a significant IC50 value (0.17 µM) toward the human glioblastoma SNB-19 cell line, which was nearly 5-fold higher that the value of the reference drug cisplatin.
Novel 4β-amidotriazole-containing podophyllotoxin hybrids were synthesised using click chemistry method and were screened for DNA topoisomerase-IIα inhibitory activity, as well as anticancer properties.56 Aryl triazolic-amide derivatives containing chloro, fluoro, 3,4-methylenedioxy, 3,4-dimethoxy, and 3,4,5-trimethoxy substituents exhibited satisfactory cytotoxicity. For example, derivatives 12–14 (Fig. 3) showed promising inhibition of selected human cancer cell lines. Moreover, these three experimental compounds displayed IC50 values of 0.99 (12), 0.70 (13), and 089 (14) µM on DU-145 cancer cells, and induced apoptosis of a prostate cancer cell line. The DNA topoisomerase-II inhibition assay showed that catenated DNA in the presence of topoisomerase II incubated with compounds 12–14 inhibited topoisomerase II activity represented by the presence of connected DNA in the wells. However, compounds 12 and 13 demonstrated clear linear DNA synthesis, and were analogous to etoposide in the well-characterised inhibition of topo-II.
Wogonin-based proteolysis-targeting chimeras related to the 1,2,3-triazole moiety have been developed and evaluated to study their mechanism of action on the degradation of CDK9 protein in breast cancer MCF-7 cells.57 The most active triazole hybrid 15 (Fig. 3, IC50 = 17 μM) demonstrated better antiproliferative potential on the MCF-7 cell line than wogonin (IC50 = 30 μM) did at lower concentrations. The western blotting revealed that derivatives containing a triazole linker selectively downregulated the intracellular CDK9 level. Sample 15 was identified as a selective and leading chemical degrader of CDK9 with modest antiproliferative potential in various cancer cell lines and decreased the levels of Mcl-1, which is a prosurvival protein.
Novel derivatives of bavachinin-containing 1,2,3-triazoles were synthesised and evaluated for anticancer effects in four selected cancer cell lines.58 The results of the 1,2,3-triazole analogue 16 (Fig. 3) showed IC50 values of 7.72, 16.08, 7.13, and 11.67 µM against A549, PC-3, HCT-116, and MCF-7 cancer cell lines, respectively. Moreover, derivative 16 induced apoptotic cell death and morphological changes, and inhibited cell migration and colony formation.
3.1.2. Quinazoline-based triazole hybrids
Quinazoline and quinazolinone derivatives linked to 1,2,3‐triazoles form part of a good arsenal of potential anticancer agents.59, 60 4-Anilinoquinazoline-substituted 1,2,3-triazoles were screened for cytotoxic potential against HepG2, KB, and SK-Lu-1 cells by Le-Nhat-Thuy et al.61 The most active compound, 17 (Fig. 4 , IC50, 0.04, 0.14, and 1.03 µM against KB, HepG2, and SK-Lu-1, respectively), displayed up to 100-fold stronger activity than the positive control erlotinib did. Docking simulations of the lead compounds, including 17, into the active ATP-binding site of various epidermal growth factor receptor (EGFR) kinases was conducted and the interaction of H-bonds of the 1,2,3-triazole fragment and the dense hydrophobisation of di-oxygenated portion of quinazolines with the remnants in the ATP pocket, was a major points in EGFR binding.
Fig. 4.

1,2,3-Triazole-quinazolines as anticancer leads (17–20).
The phosphoinositide 3-kinase (PI3K)-γ-specific properties of 1,4-substituted 1,2,3-triazolo-quinazolinones has been investigated via in in vitro cytotoxicity test against several cancer cell lines (HL-60, Colo-205, HCT-116, MCF-7, and A549 cells).62 Two compounds (18 and 19, Fig. 4) demonstrated less toxicity against HL-60 cells (IC50 = 1 µM) than the other compounds did. The specificity of compounds 18 and 19 against various isoforms of PI3-Ks was examined using western blotting and they exhibited selectivity against PI3Kγ isoforms, with IC50 values of 1 and 3 µM, respectively.
Kettle et al.63 discovered that the quinazoline-triazole conjugate 20 (Fig. 4) is a potential Pan-KIT mutant inhibitor towards gastrointestinal stromal tumours. Triazole compound 20, containing an –OCH3 group at the C7 position, was expected to have an optimal balance between KDR selectivity and KIT mutant potency, and exhibited potential growth inhibition of KIT mutant cell lines, with a satisfactory KDR margin, and considerable improvements in the absorption, distribution, metabolism, and elimination (ADME) and physico-chemical properties. Evaluation of triazole hybrid 20 in a mouse allograft tumour model of Ba/F3 cells showed that 20 caused the tumour to decrease at a dose of 20 mg/kg. Importantly, the preclinical in vivo potency of 20, which is marked as AZD3229 is encouraging, and may serve in near future treatment of patients with gastrointestinal stromal tumour.
3.1.3. 1,2,3-Triazole-linked peptide type compounds
Peptide-like derivatives are not orally bioavailable; however, protein therapy tends to exhibit specificity against its targets because it interacts with a greater number of substances, but this occurs at the expense of low bio-availability and metabolic instability.64 The introduction of the 1,2,3-triazole ring in these macromolecules also demonstrated interesting effects. Using this connection, Chen et al.65 synthesised a new series of nonpeptide inhibitors bearing two 1,2,3-triazole moieties toward the polo-box domain (PBD) of polo-like kinase 1. Among them, compound 21 (Fig. 5 ) that was related to the 1,4-regioisomer triazole portion showed considerable bioactivity with an IC50 value of 3.37 μM, compared with that reported for phosphate peptide (PLHSpT, IC50 = 0.62 μM), and it showed moderate activity against the PBD of Polo-like kinase 1. However, the authors suggested that the nonpeptide skeleton may confer advantages over other peptide-related inhibitors and further investigations of the novel nonpeptide PBD of Polo-like kinase 1 inhibitors could lead to the development of selective anticancer agents.
Fig. 5.

Structures of the 1,2,3-triazole-linked peptide type compounds (21–25).
Matthiesen et al.66 reported a new series of geranylgeranyl diphosphate synthase (GGDPS) inhibitors based on the α-methylated isoprenoid-triazoles. They focused on a grand design of masking the negatively charged bisphosphonates using pivaloyloxymethyl (POM) groups to use GGDPS inhibitors as anti-myeloma agents. The POM-version of 22 (compound 23, Fig. 5) displayed more augmented cellular activity than the conforming salt did, with a nearly 10-fold improvement in potency.
Cao et al.67 reported that leucine-ureido-triazole conjugates were synthesised as aminopeptidase N inhibitors using the ‘click reaction’. In their study, compound 24 (Fig. 5) exhibited better aminopeptidase N inhibition than other compounds did, with an IC50 of 0.089 µM (approximately 100-fold lower than that of the positive control bestatin). In addition, compared to bestatin, 24 showed favourable in vitro anti-angiogenesis potency in both the HUVEC tubular structure formation bio-assay and rat aortic ring model. Furthermore, in vivo screening of 24 exhibited more promising anti-metastasis in the mouse H22 pulmonary metastasis model with inhibitory rates of 71%, than bestatin, which showed 64% inhibition.
The antiproliferative activity of 1,2,3-triazole-bearing β-glucuronidase-responsive albumin-binding prodrug 25 (Fig. 5) on the A549 cell line after a 3-day treatment showed that it did not affect the cell viability at the elevated dose of 200 nM when incubated alone. However, in combination with β-glucuronidase, the IC50 value of 25 was 9.4 nM.68
3.1.4. Diverse 1,2,3-triazole-containing heterocyclic hybrids
Chen et al.69 reported 1,2,3-triazole-bearing acridine hydroxamic acids as dual Topo and HDAC inhibitors. All the prepared derivatives exhibited potent antiproliferative potentials toward U937 cells. Here, the most active sample, 26 (Fig. 6 ), displayed nanomolar IC50 values on U937 cells, and showed superior HDAC1/6 inhibition potency (IC50 = 3.9 and 2.9 nM, respectively), that was several-fold more potent than the HDAC positive control, SAHA was (IC50 = 14.3 and 6.9 nM, respectively). Moreover, 26 considerably activated cleaved caspases-9, -8, -3, and -7 at 0, 0.1, 0.5, 1, and 2.5 μM for 36 h, indicating that it induced both the death receptor and mitochondrial pathways leading to apoptosis.
Fig. 6.

1,2,3-Triazole leads (26–48) containing various heterocyclic skeletons.
Novel 1,2,3-triazole-containing–isoquinolines were suggested as P-gp inhibitors by Gao et al.70 following cytotoxic and reversed MDR activity bioassays in vitro and in vivo. All synthesised samples demonstrated no or less cytotoxicity (IC50 > 30 µM) in both K562 and K562/A02 cells. However, compound 27 (Fig. 6), with a 4-tert-butylphenyl substituent, reversed MDR in a concentration-dependent manner, with better activity than that of verapamil. Moreover, compound 27 significantly reduced the IC50 of adriamycin (1.22 µM), demonstrating the strongest reversal activity (39-fold), which was close to that of tariquidar (reversal fold = 49.4).
Hybrid compounds containing a thiosemicarbazide moiety and 1,2,3-triazolopyrimide system were evaluated for their antiproliferative potentials against human cancer cell lines such as MGC-803, NCIH1650, and PC-3 cells.71 The results indicated that compound 28 (Fig. 6) had the most potent antiproliferative activity (IC50, 2.37, 5.26, and 7.67 µM in MGC-803, PC-3, and H1650 cells, respectively). Furthermore, mechanistic studies revealed that MGC-803 cells treated with compound 28, showed slight inhibition of colonies (72% inhibition at 2 µM), indicating that the hybrid 28 inhibited the proliferation of cancer cells.
Yamada et al.72 reported the synthesis and antitumor screening of target compounds based on the 4-substituted 1-benzyl-5-diphenylstibano-1,2,3-triazoles using CuAAC of ethynyldiphenylstibane with benzyl azide. The antitumor activities of the novel synthesised triazole samples were evaluated in eight cultured tumour cell lines, including human solid tumour cell lines such as colon, gastric, and breast tumours. Among them, 5-stibanotriazoles (29–33, Fig. 6) showed excellent antitumor activity in all tested tumour cell lines (IC50, 0.25–1.38 µM. The cytotoxicity of 29–33 was tested in normal cells and all 5-stibanotriazoles exhibited higher cytotoxicity than their 5-unsubstituted 1,2,3-triazoles analogues did. In particular, compounds with 4-methylphenyl (30) and 1-cyclohexenyl (33) fragments exhibited satisfactory low cytotoxicity toward the normal cells.
The synthesis, characterisation, and cytotoxicity analysis of 1,2,3-triazoles against human breast cancer cells using the CuAAC reaction were performed by Gilandoust et al.73 Compound 1-(2′-ethoxy-4′-fluoro-[1,1′-biphenyl]-4-yl)-4-phenyl-1H-1,2,3-triazole (34, Fig. 6) demonstrated the greatest cytotoxicity against selected breast cancer cell lines (MCF7, BT474, MDA-MB-231, and Ishikawa cells, with IC50 values of 1.69, 4.08, 4.81, and 1.97 µM, respectively). Thus, 34 was suggested as a lead cytotoxic agent against MCF7 cells and was not toxic to BEAS-2B (normal lung epithelial) cells. In addition, 34 downregulated the expression of VEGFR1 in MCF-7 cells.
Hybrid compounds containing the pyrazolo[3,4-d]pyrimidinone and 1,2,3-triazole portions were evaluated against glioma cells such as human U87 and rat-C6.74 5-((1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (35, Fig. 6) inhibited the proliferation of U87 cells by 47.69% and, owing to its good solubility in DMSO, was chosen for further evaluation. The flow cytometry assay showed that 35 arrested cells in the S phase of the cell cycle, indicating that the cells were unable to duplicate DNA, consequently, leading to a reduction in the M-phase cells. Moreover, the number of dead cells increased with increasing concentration of 35. Furthermore, western blotting showed that p53 was upregulated with increasing concentrations of 35, which confirmed its involvement in apoptosis.
The anticancer activity of the 1,2,3-triazole-tethered dasatinib derivatives were studied in HL60, K562 (leukaemia) and KG1a (leukaemia progenitor) cell lines.75 Among the triazole-containing lead compounds, 36 (Fig. 6) showed superior inhibitory activity (IC50 = 0.14 µM) in the KG1a cell line, following evaluation in MCF-7 and B16BL6 cells using the colony forming and cell wound scratch bioassays, respectively. The dasatinib-triazole hybrid 36 was significantly more potent than dasatinib was. The survival rate of mice dosed with 36 (200 mg/kg) once daily for 2 weeks was 100%, and after increasing the dose, mice treated with 36 also survived, indicating the low toxicity of 36.
Compound 37 (Fig. 6) was discovered as a c-Met-targeting and apoptosis-inducing agent among the synthesised thiopyrimidine-triazole conjugates.76 This lead compound demonstrated 3.7–5.4-fold greater activity than the positive control drug, foretinib, did toward the MCF-7, HepG2, and A549 cell lines with the IC50 values of 1.1, 0.5, and 0.9 µM, respectively. Furthermore, enzyme-based experiments showed that 37 selectively inhibited c-Met, with an IC50 of 16 nM, which was similar to that of foretinib (14 nM). Moreover, 37 induced late apoptosis of HepG2 cells in a concentration-dependent manner.
Newly synthesised imidazopyridine-triazole conjugates were assayed for their cytotoxicity against cancer cell lines such as A549, HCT-116, MDA-MB 231, and DU-145 cells.77 The most active compound (38, Fig. 6) displayed significant antitumor potential in A549 lung cancer cells with IC50 of 0.51 µM, while the value was 1.04 µM in the prostate cancer cell line (DU-145). The effect of 38 on the cell cycle of A549 cells was measured using flow cytometry, and 48 h treatment with 1 µM caused cell cycle arrest in the G2/M phase, suggesting the inhibition of tubulin polymerisation.
The antiproliferative properties of benzimidazole-tethered 1,4-disubstituted 1,2,3-triazoles against selected human tumour cell lines (A549, CFPAC-1, HeLa, and SW620 cells) were evaluated. Compounds 39 and 40 (Fig. 6) exhibited strong and selective antiproliferative potentials in A549 cells with IC50 values of 0.05 and 0.07 µM, respectively, and were further selected to elucidate their mechanism of action.78 Compound 39 markedly decreased the viable cell population by 70.59%, and increased early and late apoptotic/primary necrotic cell populations by 27.81% and 40%, respectively. Furthermore, compound 40 also increased early and late apoptotic/primary necrotic cell populations by 26.97% and 16.37%, respectively, however, the viable cell population decreased to 49.77%. The enzyme p38 mitogen-activated protein kinase (MAPK) was identified as the common target of the triazole leads 39 and 40 that inhibited the enzyme with slightly different activity.
It was suggested that hybrid derivative containing purine and two 1,2,3-triazole portions (41) (Fig. 6), inhibited the virulence of Candida albicans and exhibited in vitro antiproliferative potency and an in vivo nontoxicity. This derivative could be a lead compound for further development by averting cancer-associated biofilm Candida infections.79 Moreover, lead compound 41 displayed high activity against peripheral blood T lymphoblasts (CCRF-CEM, EC50 = 6.5 µM) and modest antineoplastic potential against the MCF-7 cell line (EC50 = 80 µM). In addition, lead compound 41 was selective against C. albicans (lg R ≥ 3 at 20 µM), but not the mammalian normal cell line Vero in vitro (IC50 ≥ 280 µM) and Galleria mellonella in vivo.
Another hybrid, 1,2,3-triazole-related naphthoquinone, inhibited cancer cells such as Caco-2, Calu-3, and MDA-MB231 cells, as well as normal Vero cells.80 Among them, compound 42 (Fig. 6) with a cell viability of 23.92% on Caco-2 cells, showed 6-fold higher selectivity on Caco-2 than Vero cells (selectivity index [SI] = 6.25), confirming the potential of this derivative as a drug-like candidate. Furthermore, hemocompatibility studies revealed that 42 did not affect platelet aggregation caused by arachidonic acid or activated thromboplastin time, indicating that the activation of blood coagulation by the intrinsic and extrinsic pathway was preserved. In addition, 42 demonstrated no significant haemolytic profile after incubation for 3 h, confirming its good safety profile.
1,2,3-Triazole-linked isoxazole-benzothiazole-benzoxazole hybrids were prepared and their anticancer potency was evaluated on cancer A549 and HeLa cells, as well as normal HEK-293 cells.81 The lead compound in the present study was compound 43 (Fig. 6, IC50 = 1.768 [HeLa] and 2.594 [A549] µM). Moreover, the cell cycle of HeLa and A549 cell lines was analysed. Triazole derivative 43 displayed superior potency on A549 cells and in the cell cycle assay, the cell death percentage in the subG1 phase was 49.43%. In HeLa cells, compound 43 (42.58%) exhibited significant and enhanced apoptotic potency in the subG1 phase compared with the reference drugs TAK-165 and GW-610.
Kumar et al.82 studied several 1,2,3-triazoles containing isatin-ospemifene system for their antiproliferative activity against triple-negative MDA-MB-231 (ER-) and MCF-7 (ER+) cell lines. The evaluation studies revealed that compound 44 (Fig. 6) demonstrated high potency with an IC50 of 1.56 µM in the MCF-7 cell line. The structure–activity relationship (SARs) analysis showed that selected triazoles with Br-substituents at the C-5 and C-7 positions of the isatin portion with an ethyl/propyl group as a spacer were more active. For example, the best lead compound exhibited a 30-fold higher potency than tamoxifen in MCF-7 cells.
Using a ‘hybrid conjugation of bioactive ligands’ approach, 1,2,3-triazole-linked indole-3-glyoxamide fragment derivatives have been investigated as potential dual inhibitors of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a cancer chemotherapy.83 Here, lead compound 45 (Fig. 6) not only exhibited dual inhibition of COX-2 and 5-LOX, but also displayed high antiproliferative potential on the prostate cancer cell line DU145, an excellent COX-2SI, as well as satisfactory in vivo anti-inflammatory activity with no ulcerogenic effects. In addition, the effect on tubulin polymerisation revealed that compound 45 interfered with the microtubule dynamics to act as a microtubule-destabilising agent.
Schlapbach et al.84 reported a novel and highly selective MALT1 protease inhibitor based on the N-aryl-piperidine-4-carboxamide derivative related to triazole ring. In the Jurkat T and lymphoma OCI-Ly3 cell lines, hybrids demonstrated activity in a mechanistic T cell activation assay (IL-2 RGA), which suggests the possibility of using MALT1 inhibitors in the treatment of diseases such as autoimmune disorders and B-cell lymphomas with a dysregulated NF-κB pathway. It should be noted that para-substituent tethered to a 1,2,3-triazole moiety was detected as the most beneficial group among other studied substituents (compound 46, Fig. 6). In addition, rat pharmacokinetics showed high in vivo clearance for 46.
The pyrimidine-triazole hybrids have been identified as strong and effective ATP-binding cassette subfamily B member 1 (ABCB1)-dependent MDR modulators.85 The evaluation of their effect on MDR reversal potency in SW620/AD300 cells (paclitaxel resistance) showed that compound 47 (Fig. 6) exhibited a high reversal potential, that was approximately 7-fold higher than that of verapamil (positive control). Compound 47 was more effective on ABCB1-mediated Rh123 accumulation and efflux than the positive control, and 2 µM increased the stockpiling and decreased the efflux of Rh123 in SW620/AD300 cells more than the verapamil did (p < 0.05).
Among 1,2,3-triazole-bearing 2,4-diarylaminopyrimidines, derivative 48 (Fig. 6) was identified as a promising anaplastic lymphoma kinase dual inhibitor (IC50 = 1.4 nM; ROS proto-oncogene 1 [ROS1] IC50 = 1.1 nM).86 Moreover, 48 demonstrated significant inhibitory activities on the ROS1-positive HCC78 (IC50 = 40 nM), anaplastic lymphoma kinase-dependent H2228 (IC50 = 95 nM), and KARPAS299 (IC50 = 21 nM) cell lines. In addition, the acridine orange/ethidium bromide and western blot assays suggested that 48 induced cell apoptosis and highly inhibited cellular anaplastic lymphoma kinase and ROS1 activities.
3.2. Antimicrobial activity
Azoles, particularly triazoles, have been the source of several drugs, such as antibacterial and antifungal agents. In contrast, the recently discovered fused (with different heterocycles) triazoles exhibit novel antimicrobial properties. For example, 1,2,3-triazoles fused with a pyridine ring were investigated as novel lead compounds for the development of potential antimicrobial agents.87 Two selected potential antibacterial agents (49 and 50, Fig. 7 ), which were regioselectively synthesised using the Buchwald’s strategy with the N-heteroarene system. Compound 49 displayed a zone of inhibition of 13 and 10 against Bacillus subtilis and Escherichia coli, compared with 16 and 13 mm, respectively for streptomycin. The hybrid compound 50 displayed a zone of inhibition of 12 and 11 mm towards B. subtilis and E. coli, respectively and these triazolopyridines exhibited minimum inhibitory concentrations (MICs) similar to those of streptomycin on the B. subtilis strain.
Fig. 7.

Antimicrobial 1,2,3-triazole-linked hybrids (49–70).
Szałaj et al.88 investigated the antibacterial properties (targeted towards the inhibition of EcLepB [E. coli SPase I]) of the macrocyclic lipopeptides that contain P2–P1′ boronic ester fragment and 1,2,3-triazole ring. Peptide 51 (Fig. 7) demonstrated activity against all selected gram-negative bacterial strains. However, macrocycle 51 exhibited a 2-fold lower inhibition of EcLepB (IC50 = 175 nM) than other synthesised triazole-peptides whereas, in contrast, macrocycle 51 exhibited a 2-fold stronger antibacterial potency. In addition, peptides were also screened against HepG2 cells to evaluate the cytotoxicity and haemolytic potential in human blood. The results indicated that for compound 51, the cytotoxicity was decreased (8 µM) while the haemolysis was halved (10.6%). The 1,2,3-triazole ring has become one of the major antimicrobial pharmacophore and the effect of the 1,2,3-triazole unit against wild bacterial photogenes is clear. In this regard, a set of 1,2,3-triazole-linked dehydroacetic acid-chalcone derivatives were synthesised for antimicrobial evaluation.89 The hybrid compounds containing a substituted benzene portion exhibited more promising activity than dehydroacetic acid and dehydroacetic acid-chalcone alkyne. Among them, derivative 52 (Fig. 7) was identified as a lead compound, which displayed strong activity against E. coli, with an MIC of 0.0030 µM/mL.
1,2,3-Triazole-bearing benzo[d]-imidazo[2,1-b]thiazoles were also investigated for potential antimicrobial properties.90 Compound 53 (Fig. 7) demonstrated positive anti-E. coli, anti-B. subtilis, anti-Salmonella typhi, and anti-Pseudomonas aeruginosa potency, showing low MIC values (2–8 mg/mL) that were noticeably higher than those of the positive control drugs norfloxacin, chloromycin, and fluconazole. In addition, the interaction of derivative 53 with calf thymus DNA indicated that hybrid 53 might append to DNA, forming a 53-DNA complex that could block DNA replication to impart considerable antimicrobial activities.
Another triazole compound 54 (Fig. 7) showed stronger antifungal activities towards fungal strains, such as Aspergillus niger and C. albicans than the other synthesised hybrids did.91 The zone of inhibition and MIC of 54 were 16 mm for C. albicans and 15 and ˃32 µg/mL for A. niger, respectively. Moreover, the bio-safety analysis of compound 54 against sheep blood cells also indicated it was not cytotoxic at 1 mg/mL.
A set of hybrid compounds tethered to different fragments, which were 1,2,3-triazole, biphenyl, 2-alkoxy-3-phosphorylpropanoate, and an amine linker were synthesised and tested for bacterial transglycosylase inhibitory potency against Acinetobacter baumannii.92 The transglycosylase inhibitory activities of the synthesised samples were lower at 500 µM than those of compounds 55–57 (Fig. 7), which showed much higher transglycosylase inhibitory activity (99% for each sample) at 200 µM. It should be noted that among the prepared 2-alkoxy-3-phosphorylpropanoate triazoles, the biphenyl fragment and methylene bridge affected the activity. In addition, compounds 55 and 57 also exhibited a good antibacterial potency on Staphylococcus aureus (MIC = 6.3 µM)
Another 1,2,3-triazole derivative was studied for antibacterial effects against several selected microorganisms including gram-positive strains, S. aureus, B. subtilis, S. aureus PR and gram-negative pathogens, Klebsiella pneumoniae and E. coli.93 4-(3-((2-Ethylhexyl)oxy)phenyl)-1H-1,2,3-triazole (58) (Fig. 7) was identified as a lead compound (2, 2, 4, and 4 µg/mL) against S. aureus, B. subtilis, S. aureus PR, and K. pneumoniae, respectively. Collectively, this report indicated the potential progress the 1,2,3-triazole moiety as a terminal amide-mimetic element, which maintained and modulated amide-tethered bioactivity.
N-coumaroyltyramines linked to a triazole ring were investigated as potential lead compounds, which positively controlled bacterial biofilms control.94 Three gram-negative strains Pseudoalteromonas ulvae (TC14), Pseudoalteromonas lipolytica (TC8), and Paracoccus sp. (4 M6) species were selected for their ability to form biofilms. The –OCH3 substituent on the tyramine fragment affected the potency and the 4–OCH3 derivative 59 (Fig. 7) displayed the best anti-biofilm activity, which was more effective than ampicillin was against TC14 species (11.3 and 9.3 µM for ampicillin).
Quinazolinones linked by the 1,2,3-triazole moiety showed superior antibacterial potential against MDR S. aureus.95 Compound 60 (Fig. 7) possessed specific and strong potency against S. aureus (MIC = 0.5 µg/mL) and was inactive against gram-negative pathogens A. baumannii and E. coli (MIC ≥ 64 µg/mL). In addition, the hybrid compound 60 was surmounted the MDR mechanism, exhibiting highly potent inhibitory effects on MDR S. aureus including strains resistant to vancomycin (MIC values 0.5–32 µg/mL).
The target compound (61) (Fig. 7), which contained the 4-F-benzyl-, 1,2,3-triazole, –OCH3-naphthalen, and 4-NO2-propen-2 fragments demonstrated a unique activity against bacterial strains such as B. subtilis, Staphylococcus epidermidis, P. aeruginosa, and E. coli, as well as the fungal species, C albicans and Aspergillus niger, with MIC values of 0.0032–0.0063 µmol/mL.96 The 1,2,3-triazole moiety and chalcone fragment clearly synergised the biological activity and the conjugation of these pharmacophoric units, indicating their synergistic properties. In addition, the docking simulation of 61 into E. coli topoisomerase II DNA Gyrase B showed that these triazole-chalcone conjugates may feasibly inhibit DNA topoisomerase.
The indole-triazole hybrid 62 (Fig. 7), named 4-(1-heptyl-1H-1,2,3-triazol-4-yl)-N-(2-phenyl-1H-indol-3-yl)butanamide, was found to have promising quorum sensing inhibitory activity against P. aeruginosa MH602 of 60.82% at 250 μM among the synthesised 2-phenylindole-amide-triazoles.97 SAR analysis showed that an ethyl group at position four of the aryl portion positively affected the activity, whereas other groups, such as the bromo (-Br) derivative, exhibited moderate activity.
A set of 1,4-disubstituted 1,2,3-triazoles tethered to a benzhydrylpiperazine fragment were assayed for their antibacterial potentials against S. aureus and E. coli using the agar well diffusion method.98 The triazole derivatives 63 and 64 (Fig. 7) were found to be more active than ciprofloxacin was (zones of inhibition: 16.33 and 16.45 mm against S. aureus; 15.63 and 16.15 mm against E.coli respectively). The combination of a 1,2,3-triazole ring with the benzhydrylpiperazine moiety increased the biological activity, demonstrating that further modifications of benzhydrylpiperazine scaffolds containing a 1,2,3-triazole portion could be a feasible strategy for developing antibacterial agents.
Several microbes (bacterial and fungal strains) including S. epidermidis, B. subtilis, P. aeruginosa, E. coli, C. albicans, and A. niger were selected to analyse the inhibitory activity of 1,4-disubstituted 1,2,3-triazole scaffolds.99 The synthesised compound 65 (Fig. 7) displayed equal antimicrobial activity on all selected strains (MIC = 0.0153 µmol/mL) except for A. niger, while another lead derivative 66 (Fig. 7) showed comparable antimicrobial potential against all the microbial species (MIC = 0.0138 µmol/mL), except for A. niger and B. subtilis. The introduction of halogen substituents in the thiophenyl/benzyl fragment was found to increase the inhibitory potency.
Moghimi et al.100 were synthesised a triazole-linked aryl urea hybrids as urease inhibitors toward on the ureolytic bacterial infections. The di-chloro-containing triazole compound 67 (Fig. 7) demonstrated superior activity with an IC50 of 22.81 μM in the Berthelot colorimetric assay, indicating it was a urease inhibitor. This compound bearing the 4-OCH3 substituent had acceptable ADME properties and was suggested to be an attractive candidate for further design and modification in the search for novel urease inhibitors.
1,2,3-Triazole-related quinolone conjugates were also assayed in antibacterial screenings against the following selected bacterial strains: Streptococcus pyogenes, S. aureus, E. coli, P. aeruginosa, and Salmonella typhi.101 Compounds 68a-c, 69a-c, and 70a-c (Fig. 7), which bear aryl fragments, demonstrated high antibacterial potency against the selected S. aureus (for 68a-c, 69a-c, and 70a-c, 0.12–0.24 µg/mL), as well as on Salmonella typhi (68a,c; 69c, and 70a-c, 0.12–0.24 µg/mL). Moreover, the statistical two-dimensional quantitative SAR (2D-QSAR) models validated the remarkable ADMET activity compound 70a among all the prepared samples.
3.3. Anti-tubercular activity
Triazoles have been infrequently evaluated for their anti-tubercular properties. In contrast, 1,2,3-triazoles conjugated to diverse heterocyclic fragments were reported to demonstrate potential anti-tubercular properties in 2018. For example, 1,2,3-triazoles containing 1,3,5-triazaspiro[5.5]undeca-2,4-diene and the coumarin moiety were assayed for anti-mycobacterial activity and mycobacterial dihydrofolate reductase (Mtb DHFR) inhibition.102 Among them, compounds 71 and 72 (Fig. 8 ) displayed strong anti-mycobacterial potency (MIC50: 0.01–0.2 µM) on Mycobacterium bovis BCG than other synthesised analogues did. Both compounds also demonstrated anti-tubercular activity with MIC50 values of 0.01 and 0.025 µM against Mycobacterium tuberculosis H37Rv. The cytotoxicity of these two samples was examined in the HepG2 cell line, whereas low haemolytic activity was confirmed using red blood cells (up to 100 µM). Further studies showed that 72 exhibited higher binding affinity to Mtb DHFR than to human DHFR. This investigation was in agreement with the selectivity observation in the enzyme inhibition assay.
Fig. 8.

Structures of the 1,2,3-triazole hybrids (71–81) with anti-tubercular activity.
Spain et al.103 investigated Cu and Zn complexes of the triazole-cyclam bearing ligands with activity against virulent and drug-resistant M. tuberculosis strains. SAR analyses of the cyclam compounds with a cyclic cyclam fragment tethered to two equal naphthalimide pendants through triazole linkers were studied. The 1,2,3-triazole-linked compound 73 (Fig. 8) exhibited good water solubility (10.7 µM) and reduced bacterial load in vivo. It should be noted that zebrafish embryos were used to investigate the in vivo toxicity of a range of doses of 73 complex using the embryo survival assay.
Recently, the alkoxy-triazolequinolones 74 and 75 (Fig. 8, MIC = 6.9 and 6.6 µM, respectively) were found to be the most active compounds against M. tuberculosis H37Rv.104 Moreover, they exhibited IC50 values of 27–28 µM compared to that of ciprofloxacin (10.6 µM) when screened against M. tuberculosis DNA gyrase using a DNA supercoiling activity bio-assay. Compared with clinical strains including MDRs, compound 74 showed similar potency to that exhibited against ciprofloxacin-resistant strains. However, in this case ciprofloxacin may have higher in vitro inhibitory activity against M. tuberculosis DNA gyrase than derivative 74.
A series of propylene-1,2,3-triazoles containing isatin-moxifloxacin hybrids were synthesised an evaluation for their anti-tuberculosis properties.105 In vitro anti-mycobacterial potentials on M. tuberculosis H37Rv and multidrug-resistant tuberculosis (MDR-TB) strains were determined by the rapid direct susceptibility test method. All derivatives (MIC: 0.05–2.0 µg/mL) demonstrated promising MIC values against M. tuberculosis H37Rv and MDR-TB strains, however several of them were notably less active than the positive control moxifloxacin (MIC = 0.10 and 0.12 µg/mL). The most active hybrid 76 (Fig. 8) displayed equal MIC values (0.05 µg/mL) with the reference drug isoniazid, and at the same time was exhibited 2–8-fold more potency than moxifloxacin and rifampicin (MIC = 0.39 µg/mL) against M. tuberculosis H37Rv. Regarding on the MDR-TB, 76 had from 2- to >2048-fold stronger activity compared with reference drugs moxifloxacin, rifampicin, and isoniazid (MIC = 0.12, 32 and >128 µg/mL, respectively).
Another triazole derivative 77 (Fig. 8) (N 1-(β-d-ribofuranosyl)-C4-(4-methylcoumarin-7-oxymethyl)-1,2,3-triazole) synthesised using CuAAC was also found to be a promising anti-M. tuberculosis agent against the H37Rv strain (MIC = 5.1 µM) and MDR clinical isolate (MIC = 10.3 µM).106 It should be noted that the authors envisioned that coumarin and triazole fragments conjugated to a –OCH2- bridge would possess greater pliable as a molecule to bind to the needed or expected target. Furthermore, the minimum bactericidal concentration (MBC) of the triazole conjugates was also determined, and the results revealed that conjugate 77 containing the –OH group demonstrated low potency (MBC = 6.4 µM against the positive control isoniazid [11.5 µM]) and MDR clinical isolate. Moreover, assays on the activity of the test samples on DNA gyrase and bacterial InhA showed that 77 significantly inhibited the enzyme activity.
The in vitro anti-tubercular properties of the triazole-indole hybrid derivatives were evaluated against M. tuberculosis H37Ra in the active and dormant state.107 The most active compound (78, Fig. 8) showed superior anti-tubercular potency against M. tuberculosis H37Ra dormant, with IC50 and MIC values of 1 and 3 µg/mL, respectively. Based on SARs, it was observed that the presence of a Br substituent on indole portion in derivative 78 and other Br-related indole-triazole conjugates conferred significantly higher potency than that of the unsubstituted bis-indole-triazoles.
The anti-mycobacterial evaluation of the 1,2,3-triazole-fused spirochromene scaffolds was conducted using the microplate Alamar Blue assay and the results revealed that most 1,2,3-triazoles condensed with spirochromenes showed modest to good potency.108 Compounds 79–81 (Fig. 8) demonstrated excellent activity: 4.74, 4.34, and 4.11 µM respectively, against M. tuberculosis H37Rv strain.
3.4. Antiparasitic activity
The dipeptidyl nitriles bearing 1,2,3-triazole moiety were evaluated from the structure-diversity design point of view to search for selective trypanocidal rhodesain inhibitors.109 Among them, compound 82 (Fig. 9 ) demonstrated interesting activity by inhibiting the cell growth of Trypanosoma brucei rhodesiense following incubation for 48 h (IC50 = 0.064 μM). In addition, the authors also studied the pharmacokinetic properties of the synthesised compounds to further evaluate their mechanisms of action, and compare their brain penetration (by MDR1 on LLCPK1 cells) with that of other macrocyclic series. Interestingly, the MDR1 efflux ratio of 82 was improved to 6 and 20 in human and mouse MDR1, compared to 26 and 53, respectively exhibited by its lead macrocycle series.
Fig. 9.

1,2,3-Triazole-bearing antiparasitic leads (82–85).
The imidazole-triazole hybrids bearing NO2-substituent along with a styryl fragment exhibited anti-amoebic potency. The target compound 83 (Fig. 9), which contains the fragments described above, was found to be the most active (0.0084 µM) against Entamoeba histolytica among the metronidazole-triazole-styryl hybrids.110
An analogue of natural lignans named 3-(3,4-dimethoxyphenyl)-5-((4-(4-pentylphenyl)-1H-1,2,3-triazol-1-yl)methyl)isoxazole (84, Fig. 9) was also identified as a lead compound with antiparasitic activity.111 This lead hybrid and other prepared samples inhibited leukaemia THP-1 cells infected with Trypanosoma cruzi amastigotes in vitro. The assays revealed that the potency of derivative 84 was equal to that of the positive control benznidazole (GI50 = 10.2 μM, SI > 49.1). It was observed that the introduction of the hydrophobic 4-pentyl- fragment to the compound 84 increase the potency, and because of the pliable property of the pentyl-moiety, it may interact with the hydrophobic side of the molecular target. In addition, all synthesised compounds, including triazole 84, were assayed against amastigotes of Leishmania amazonensis but no significant results were obtained even at 100 μM.
The anti-Toxoplasma activity of the synthesised arctigenin scaffold bearing a 1,2,3-triazole ring was evaluated in vitro and in vivo.112 The lead compound (85, Fig. 9) displayed strong in vitro anti-Toxoplasma gondii activity and less cytotoxicity (IC50 in T. gondii and HeLa cell line = 17.1 and 600.0 µM, respectively; SI = 35.09), indicating more promising results than those of arctigenin and spiramycin. In addition, pharmacological bioassays suggested that 85 also lead to partial tachyzoite malformation (P < 0.05), which suggested that arctigenin-derived compounds are potential antiparasitic drug-like units that warrant further in-depth mechanistic development.
3.4.1. Antimalarial activity
Malaria still remains one of the most deadly diseases in the world, and natural and synthetic 1,2,3-triazoles have been successfully used against malaria in medicinal chemistry.113, 114 Brandão et al.115 synthesised naphthoquinonolyl-tethered 1,2,3-triazole hybrids using CuAAC, based on a lapachol modification and their antiplasmodial potentials against malaria were reported. Hybrid products were assayed against chloroquine-resistant Plasmodium falciparum (W2) and a HepG2 cell line. Among them, several derivatives, in particular, compound 86 (Fig. 10 , IC50 = 5.2 µM, SI = 197.7) exhibited stronger antimalarial potency and selectivity than lapachol did (IC50 = 123.5 µM, SI ≥ 33.4). Other 1,2,3-triazole hybrids bearing pyrimidine-chloroquinolines were screened for their antiplasmodial potential against chloroquine-sensitive NF54 strains of P. falciparum.116 The lead compound (87, Fig. 10) with IC50 of 0.048 µM demonstrated excellent activity, and a superior SI (317.50; IC50 in Vero cells = 15.24 µM). It should be noted that the SAR analysis revealed that the nature of the substituent on the triazole-linked pyrimidine core (in the case of 87, the p-nitro group) obviously enhanced the antiplasmodial properties.
Fig. 10.

1,2,3-Triazole-bearing antimalarial leads (86–89).
The antiplasmodial activity of the novel coumarin-triazole hybrid 88 (Fig. 10) was evaluated on a chloroquine-sensitive 3D7 strain of P. falciparum, and exhibited higher activity (IC50 = 0.763 mg/mL.117 This lead compound contained two electron-donating –OCH3 substituents in the benzene portion, which apparently affected the antimalarial activity. In addition, the effect of 88 on the supercoiling potency of DNA gyrase relaxed the plasmid DNA and inhibited the supercoiling activity, which could be attributable to the prevention of ATP hydrolysis needed for the inhibition of the gyrase DNA binding and on enzymatic cycle.
Triazole-linked glycohybrids of the isatin hydrazones were synthesised to develop compounds with the antimalarial potential and antiplasmodial properties.118 Derivative 89 (Fig. 10) and its diacetonide galactose analogue (at the sugar portion), which exhibited equal activity to that of the lead compound 88 with a –OCH3 substituent in the benzene ring of hydrazone part, displayed good potency against the sensitive strain 3D7, with IC50 values of 1.27 and 1.64 µM, respectively. In addition, hybrid 89 and its diacetonide galactose analogue with IC50 values <2 µM against Pf3D7 strain, showed strong CC50 values of 62.52 and 105.25 µM, respectively. These results clearly show that these active derivatives had promising CC50 values and their safety was also reflected by their high SI values of 49.22 and 64.17 µM, respectively, for the Pf3D7 strain.
3.4.2. Anti-leishmanial activity
Leishmaniasis is one of the public health problem and its treatment is quite challenging and there are concern for the emergence of resistant strains.119 To address this issue, novel hybrid compounds bearing 1,2,3-triazole and thiosemicarbazone moieties were identified by Temraz et al. [1 2 0] and evaluated for anti-leishmanial potential.120 Notably, hybrid 90 (Fig. 11 ) demonstrated nanomolar activity against Leishmania major promastigotes (IC50 = 140.3 nM), while against axenic amastigotes, the IC50 value was 1 µM, indicating an 8-fold higher activity than that of miltefosine. In vivo acute toxicity testing revealed no toxicity after treatment with the lead compound 90, and the liver, lung, spleen, and kidney isolated from mice administered 75 mg/kg parenterally or 125 mg/kg orally demonstrated normal texture.
Fig. 11.

1,2,3-Triazole-related anti-leishmanial leads (90–93).
Teixeira et al.121 investigated the synthetic pathway and antiparasitic activity of eugenol (a natural compound)-based compounds tethered to 1,2,3-triazole rings, considering the leishmanicidal effect of eugenol compounds and studies that have shown the anti-leishmanial activity of 1,2,3-triazole-containing compounds. A 4-(3-(4-allyl-2-methoxyphenoxy)propyl)-1-(4-methylbenzyl)-1H-1,2,3-triazole (91, Fig. 11) was found to be more active (IC50 = 7.4 µmol L−1) than the other triazole-linked eugenol derivatives were, which was also assayed in Leishmania parasites peritoneal macrophages (IC50 = 1.6 µmol L−1) and showed effects on cell viability. The cytotoxic potential of 91 on macrophage cells was evidenced by an IC50 of 211.9 µmol L−1, while the SI was 132.5. In addition, the triazole derivative 91 demonstrated higher effects than those of pentamidine and glucantime (drugs used clinically).
In anti-leishmanial research, other 1,2,3-triazole-linked heterocycles containing quinolone skeleton were synthesized.122 All triazole hybrids synthesised were evaluated against intracellular amastigotes and extracellular promastigotes of luciferase expressing Leishmania donovani. (1-Benzyl-1H-1,2,3-triazol-4-yl)methyl 6-chloro-2-methyl-4-phenylquinoline-3-carboxylate (92, Fig. 11) exhibited promising anti-amastigote potency with an IC50 of 7 µM. Derivative 92 and some of the other synthesised derivatives were prepared for further in vivo assay. The effects of the compounds in a golden hamster model were evaluated at 50 mg kg− 1 administered intraperitoneally for 5 consecutive days, and triazole 92 exhibited continuous activity (∼46% parasite inhibition) after treatment day 28.
It was suggested that the triazole-based dipeptide containing leucine moiety (93, Fig. 11) has promising anti-leishmanial activity.123 The IC50 value calculated based on normal growth inhibition and IC50 value of 93 on Leishmania major promastigotes was 11 μg/mL. The most relevant factors were the cytotoxicity, lipophilicity and antiparasitic activity of the lead compounds a substantial points for the design of a new anti-leishmanial agents. Furthermore, the dipeptide 93 was more lipophilic than other prepared dipeptides were in this investigation.
3.5. Antiviral activity
Notwithstanding the existence of various types of antiviral drugs in clinical treatment, human immunodeficiency virus (HIV) and acquired immune deficiency syndrome (AIDS) still remain an increasing challenge to public health.124 Therefore, the 1,2,3-triazole hybrids were identified as lead candidates, indicating superior effects on diverse viral types. Anti-hepatitis B virus (HBV) properties of a novel hybrid with 4-monosubstituted 2′-deoxy-2′-β-fluoro-4′-azido-β-d-arabinofuranosyl-1,2,3-triazole scaffolds were studied by Liu et al.125 It was observed that triazole compound 94 (Fig. 12 ) had promising antiviral potentials and impressive activity against the lamivudine-resistant HBV mutants, which were also screened against HBV-infected duck models. The results showed that both the serum (67.4%) and liver duck-HBV DNA levels (53.3%) clearly decreased after treatment with 94.
Fig. 12.

Structures of the 1,2,3-triazole hybrids (94–102) with antiviral activity.
A series of fused 1,2,3-triazole heterocycles and their antiviral properties have been explored by Karypidou et al.126 All synthesised hybrids were tested against several types of viruses such as HIV (types 1 and 2), herpes simplex viruses (types 1, 1 TK-, and 2), adeno virus-2, coronavirus, and vaccinia virus and values were compared with the appropriate positive control drugs (zidovudine, brivudine, cidofovir, ganciclovir, acyclovir, alovudine, and Urtica dioica agglutinin, and zalcitabine). However, antiviral assays indicated that the majority of compounds had slight or no activity against selected viruses. Among them only derivative 95 (Fig. 12) with an EC50 value of 8.95 µM was an averagely active compound against the human coronavirus (229E), but displayed nearly 50-fold lower inhibitory potency than Urtica dioica agglutinin (EC50 = 0.2 µM).
The pentacyclic iminosugar compounds were constructed by fusing triazolo[5,1-c][1,4]oxazepine scaffolds and were investigated for their HIV reverse-transcriptase inhibitory potentials.127 All synthesised samples demonstrated inhibitory activity against reverse-transcriptase. In particular, derivative 96 (Fig. 12), which displayed an IC50 value of 0.69 µM against reverse-transcriptase was identified as a lead compound. Another triazole compound 97 (Fig. 12), which was synthesised by replacing the pyridazine moiety with a 1,2,3-triazole ring, was suggested to have potential non-nucleoside inhibitory activity against hepatitis C virus (HVC) NS5B.128 Compound 97 showed an EC50 of 1.163 nM and a CC50 > 200 nM in a cell-based HCV replicon system experiments, with respect to potency and pharmacokinetics.
Hybrid molecules containing triazole and dihydropyrimidinone rings synthesised via Huisgen azide-alkyne cycloaddition were evaluated against human varicella-zoster virus activity.129 Here, hybrid 98 (Fig. 12) demonstrated a strong antiviral potential, with an EC50 of 3.6 µM on TK+ varicella zoster virus (VZV) strain, which decreased to 7.8 µM against the TK- strain. Additionally, it should be noted that the replacement of the benzyl fragment by 4-NO2-benzyl (in the case of 98) enhanced the antiviral potencies against TK+ VZV strains and considerably reduced the cell growth inhibition.
Antiviral activity investigation of the novel 1,2,3-triazole-phenylalanine derivatives obtained by CuAAC against HIV-1 CA protein inhibitors showed that derivative 99 (Fig. 12) showed superior anti-HIV-1 potency (EC50 = 4.33 µM, SI > 13.33), which was equal to that of the HIV-1 capsid inhibitor 2-methyl-N-[(1S)-2-(methylphenylamino)-2-oxo-1-(phenylmethyl)ethyl]-1H-indole-3-acetamide (EC50 = 5.95 µM, SI > 11.85).130 Moreover, 99 interacted strongly with recombinant HIV-1 CA and exhibited antiviral potential in the early and late stages of HIV-1 replication.
New 1,4-disubstituted 1,2,3-triazole-bearing diarylnicotinamides were examined for anti-HIV potentials against wild-type (WT, IIIB and ROD) and multiple mutant strains by Tian et al.131 Three of the most potent lead derivatives identified were 100 (EC50(IIIB) = 0.020 µM, EC50(E138K) = 0.015 µM, CC50 = 40.15 µM), 101 (EC50(IIIB) = 0.020 µM, EC50(E138K) = 0.014 µM, CC50 = 58.09 µM), and 102 (EC50(IIIB) = 0.020 µM, EC50(E138K) = 0.027 µM, CC50 = 180.90 µM; Fig. 12), which showed equal promising potency against the E138K mutant strain. However, it also demonstrated lower cytotoxicity than the other derivatives. Compounds 101 and 102 were also evaluated for possible inhibitory activity against the reverse transcriptase of HIV-1, and they displayed average inhibitory potency of with IC50 of 2.70 and 1.57 µM, respectively, which are approximately 1–3-fold less potent than that of the reference drug etravirine (0.75 µM). Nevertheless, 101 and 102 were suggested as leading compounds against HIV-1 non-nucleoside reverse transcriptase inhibitors.
3.6. Antidiabetic activity
Quinazolinone-1,2,3-triazole possessing a 4-Br-benzyl fragment in the triazole portion (103, Fig. 13 ) was identified as a superior lead compound with antidiabetic properties and it inhibited α-glucosidase in a competitive manner, with a Ki value of 117 µM. Docking studies of this lead compound confirmed it was well-docked in the active site of α-glucosidase where critical interactions occur.132 α-Glucosidase inhibition is an efficacious method for controlling post-prandial hyperglycaemia in diabetes patients. The recently synthesised lead hybrid, (R)-1-(2-((3,4′-dimethoxy-[1,1′-biphenyl]-4-yl)oxy)propyl)-4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazole (104, Fig. 13), exhibited activity against the target enzyme, which was the best among all the synthesised triazole derivatives, with IC50 value of 14.2 µM in inhibition of the α-glucosidase enzyme.133 Moreover, molecular modelling analysis revealed that 104 displayed a preferential binding mode by interacting with three active site residues Arg312, Glu304, and Phe158, which suggested that further developments of 104 may provide new lead compounds as antidiabetic drugs.
Fig. 13.

Structures of 1,2,3-triazole leads (103 and 104) with antidiabetic activity.
3.7. 1,2,3-Triazoles as neuroprotective agents
Neuroprotection is one of the most challenging aspects of current medical research for the development of treatment of neurological disorders, including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Furthermore, the design, synthesis, and modification of neuroprotective agents based on the 1,2,3-triazole-linked hybrids has remained an important research direction in recent years.134 1,2,3-Triazoles bearing longanlactone analogues were synthesised for potential enhanced neurotrophic activity.135 The in vitro cell cytotoxic potentials of the synthesised natural product derivatives were examined against mouse neuroblastoma cells (Neuro-2a). Among the 1,2,3-triazole-containing derivatives, 105 (Fig. 14 ) demonstrated strong neurotrophic activity in Neuro-2a cells, which was confirmed by a battery of cell-based and neurite outgrowth assays. Derivative 105 distinctly increased neurite length compared to dimethyl sulfoxide (DMSO).
Fig. 14.

Neuroprotective and Alzheimer's disease agents based on 1,2,3-triazole units (105–115).
Lou Gehrig’s disease (also known as amyotrophic lateral sclerosis [ALS]) is a neurological malady of the central nervous system (CNS). The pathology includes a progressive loss of motor neurons in the CNS, which control motor functions that normally leads to death within 3–5 years after diagnosis. Using a set of triazole-riluzole-hybrids and a novel motor neuron screening approach, Sweeney et al.136 identified several novel triazoles as neuroprotective agents. The derivatives with substituents that tethered a pyridyl fragment, such as compounds 106 and 107 (Fig. 14), demonstrated higher neuroprotective potency than riluzole against two independent in vitro bioassays in primary neurons.
Gamma-aminobutyric acid (GABA) is an important inhibitor of neurotransmitters in the CNS. In this regard, Giraudo et al.137 investigated derivatives bearing hydroxy (OH) substituents in the 1,2,3-triazole moiety as promising biomimetics of GABA inhibitors. The authors studied all synthesised triazoles for receptor binding research to determine the binding affinities of the derivatives to native GABA receptor, using rat brain membrane preparations. Pharmacological assays revealed that the diphenylpropyl-substituted lead compound 108 (Fig. 14) showed a higher Ki value (1.6 µM) and molecular modelling analysis showed that 108 adopted a binding pose where the triazole portion was orientated at a 180° flip. In addition, the authors suggested that alkylation of the triazole molecule is a potential strategy for the further development of this heterocyclic system for activity on GABAA receptors.
Novel fused triazole 6-methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridine derivatives were synthesised as P2X7 receptor antagonists by Chrovian et al.138 The 1,2,3-triazole-fused lead 109 (Fig. 14) increased the plasma free fraction that exhibited potential P2X7 receptor occupancy in the hippocampus of rats, with a low ED50 value of 0.07 mg/kg, and unbound plasma EC50 value of 12 ng/mL. Moreover, 109 suppressed brain interleukin (IL)-1β release in vivo in freely moving rats challenged with the P2X7 agonist Bz-ATP. In addition, the hybrid derivative 109 had notable solubility and showed good tolerability in preclinical studies, resulting in an acceptable cardiovascular safety profile in vivo. In addition, the lead compound 109 was selected as a candidate for investigation in clinical trials (phase I) to evaluate the safety and tolerability in healthy humans towards development for the treatment of mood disorders.
Luo et al.139 synthesised several F-18-linked oxadiazole-triazole hybrids with activity on sphingosine-1-phosphate receptor 1 for effects on neuroinflammatory diseases of CNS. The radiosynthesised sample 110 (Fig. 14) was subjected to primary screening to test its permeability across the blood–brain barrier in rodents in vivo, because of its high sphingosine-1-phosphate receptor 1 binding potency (IC50 = 9.7 nM). It was observed that 110 had satisfactory radiochemical yield (∼14.1%), high (>98%) radiochemical purity, nearly 54.1 GBq/µmol specific activity, and showed no defluorination in vivo.
Volinanserin (also known as M100907) is a strong 5-HT2AR receptor antagonist for the treatment of diseases such as sleep disorders and schizophrenia. Gilbertson et al.140 synthesised active, selective (+)-volinanserin enantiomer along with a series of derivatives containing a 1,2,3-triazole ring, which were evaluated to elucidate and develop their activity on 5-HT2AR in the CNS. However, in this investigation, the prepared samples did not exert the antagonistic ability of the bivalent ligand. However, although compound 111 (Fig. 14, pIC50 = 7.44; IC50 = 36.5 nM) exhibited less activity than that of the parent compound (+)-volinanserin (pIC50 = 8.32; IC50 = 4.8 nM), this compound exhibited comparable activity to that of a recently synthesised series of 5-HT2AR bivalent ligands, which bear several ethylene glycol linkers.
3.7.1. 1,2,3-Triazoles as Alzheimer's disease agents
A novel series of 1,2,3-triazol-coumarin-lipoic acid conjugates were investigated for neuroprotective and anti-acetylcholinesterase (anti-AChE) activities as multi-target-directed ligands for the treatment of Alzheimer's disease.141 Specifically, compound 112 (Fig. 14) identified as a potential AChE inhibitor (16.4 µM), displayed satisfactory inhibition of Aβ peptide aggregation (51.2%). In addition, derivative 112 showed selective bio-metal chelation, protecting against intracellular ROS formation, as well as neuroprotection against H2O2- and Aβ1-42-induced cytotoxicity.
To evaluate the neuroprotective effects using hybridisation approach, another benzo[f]coumarin, 113 (Fig. 14), was synthesised and identified as the most active derivative with IC50 values of 7.3 µM for AChE and 68.6 µM for butyrylcholinesterase (BuChE).142 The steady-state inhibition assay showed that high concentrations of derivative 113 showed increase inhibitory potency.
Novel sarsasapogenin-triazolyl hybrids were prepared and reported as promising anti-Alzheimer's agents.143 These hybrids were screened for their Aβ1–42 aggregation inhibitory potentials and several derivatives, including 114 (Fig. 14, 84.74%; positive control curcumin = 5.87%) showed high activity in inhibiting the formation of Aβ1–42 fibrils, and moderate (25 µM) neuroprotective activity against hydrogen peroxide (H2O2)-induced neurotoxicity in SH-SY5Y cells.
To target Alzheimer’s disease, the inhibitory effects of a series of novel tacrine-1,2,3-triazole compounds were investigated on Electrophorus electricus AChE and horse serum BChE.144 Among the synthesised derivatives, hybrid 115 (Fig. 14) strongly inhibited AChE (75%) and BChE (90%) with IC50 values of 4.89 and 3.61 µM at a concentration of 100 µM, respectively. It should be noted that, although the inhibition of cholinesterase by compound 115 was slightly lower than that by the positive control drug tacrine, their unique binding mode suggested by the appropriate analysis made them lead compounds for the development of novel dual inhibitors of AChE and BChE.
3.8. 1,2,3-Triazoles as anti-inflammatory agents
Novel thioquinazolinone-1,2,3-triazole compounds were synthesised using Huisgen azide-alkyne cycloaddition to identify new anti-inflammatory agents targeting COX-2 and LOX inhibition.145 Among them, 116–118 (Fig. 15 ) inhibited COX-2 with IC50 values 0.19, 0.11, and 0.16 µM, respectively while the 15-LOX inhibition analysis showed IC50 values of 4.33, 7.62, and 5.21 µM, respectively. The COX-2 inhibition values of these three compounds were compared to those of celecoxib, diclofenac, and indomethacin, while 15-LOX inhibition rates were compared with zileuton and meclofenamate sodium as reference drugs. In addition, the thioquinazolinone-1,2,3-triazole 118 inhibited monocyte to macrophage differentiation efficiently (IC50 = 5.63 µM, IC50 = 4.86 µM for diclofenac sodium) after challenged by PMA-induced THP-1 differentiation assays.
Fig. 15.

1,2,3-Triazole-bearing lead compounds (116–120) with anti-inflammatory activity.
The 1,2,3-triazole-coumarin hybrids tethered to HO-substituents and phenyl-sulfonate fragment at the position 7, were synthesised and their anti-inflammatory properties, were studied focusing on the assay of proinflammatory cytokines such as tumour necrosis factor (TNF)-α, in the lipopolysaccharide (LPS)-stimulated U937 cell line.146 The most potent compound, 119 (Fig. 15), potently suppressed TNF-α at 10 µM (62%), and the IC50 value in LPS-stimulated U937 cells was 8.01 µM. Moreover, 119 was evaluated for inhibitory activity on TNFSF11-induced osteoclastogenesis in RAW 264.7 cells. The mechanism of action studies showed that the hybrid derivative 119 displayed dose-dependent inhibition of TNFSF11-induced osteoclastogenesis by suppressing the NF-kB pathway. TNFSF11 stimulation considerably enhanced the phosphorylation of NF-kB (ser 536), IkBα (ser 32), and IKKα/β (ser 176/180), while the lead compound, 119, dose-dependently (2.5, 5, and 10 µM) decreased their phosphorylation.
Yu et al.147 reported the activity of a modified 1,2,3-triazole-linked P2Y14R antagonist for the reduction of inflammation. It was observed that compound 112 exhibited the most potent antagonists properties (Fig. 15) in the fluorescent assay, and 112 has three CH2 chains between two amines tethered to the thiophene ring via an amide linker.
3.9. 1,2,3-Triazoles as carbonic anhydrase agents
The 1,2,3-triazole-linked [18F]-PET tracer (121, Fig. 16 ) was identified as a potential carbonic anhydrase IX (CA-IX) inhibitor in both in vitro/in vivo bioassays and mechanism of action was reported by More et al.148 The authors began their in vivo experiments by injecting the lead compound 121 into CA-IX expressing 4T1 and HT-29 skin cells to evaluate the effect of PET tracer on the absorption in cell lines. In particular, evaluation of the activity of compound 121 as a CA-IX inhibitor showed that it was distributed in the bodies of the mice, and after harvesting various body organs, a biodistribution assay also was carried out. The results revealed that sample 121 localised in the tumour in trace amounts but was mostly accumulated in the intestine in both tumour types. The pharmacokinetic of 121 was evaluated by assessing its distribution coefficient, in vivo stability in Balb/C mice, and in vitro stability in human serum.
Fig. 16.

Structures of 1,2,3-triazole hybrids (121–123) with carbonic anhydrase activity.
Benzenesulfonamide derivatives containing the 1,2,3-triazole moiety were evaluated for potential carbonic anhydrase properties on the isoforms CA-I–IX, and the inhibition rates of the target compounds were compared with that of acetazolamide, which is a clinically used positive control drug.149 All the newly synthesised derivatives exhibited superior Ki values (86.8 nM) on the CA I, while their effects against other tumour-associated isoforms were weaker than the reference drug or moderate. Derivative 122 (Fig. 16) demonstrated excellent inhibitory potency against glaucoma-associated CA IV (Ki = 52.4 nM) compared with acetazolamide. It is generally understood that sulphonamide bearing scaffolds are the most important CA inhibitors. Therefore, this strategy was used to develop other novel benzenesulfonamides bearing 1,5-diaryl-1,2,3-triazoles moieties, which were screened for their CA-I–IX inhibition.150 In the stopped flow carbon dioxide hydration assay, all synthesised samples highly inhibited the cytosolic isoform CA-I, displaying Ki values of 53.2 nM to 7.616 µM. Regarding the investigation of the tumour-associated trans-membrane isoform CA-IX, derivative 123 (Fig. 16) demonstrated better inhibition than acetazolamide did (Ki = 14.3 nM).
3.10. 1,2,3-Triazoles towards on treatment of renal anaemia
Wu et al.151 suggested that the triazole compound 124 (Fig. 17 ) is a highly potent candidate for the treatment of renal anaemia. They confirmed the activity of the HIF-PHD inhibitor 124 (IC50 = 62.23 nM), which was 10-fold higher than that of roxadustat (IC50 = 591.4 nM). Furthermore, the in vivo biological assays revealed its upregulation of haemoglobin in cisplatin-induced anaemic mice (120 g/L) to normal levels (160 g/L) with no obvious toxicity.
Fig. 17.

Structure of lead 1,2,3-triazole 124.
3.11. 1,2,3-Triazoles as anti-adipogenic agents
The indole-triazole hybrids were synthesised using the click method and studied for anti-adipogenic properties.152 In this study, a N-((1-(2,3-difluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-4-methyl-N-((1-tosyl-1H-indol-3-yl)methyl)benzenesulfonamide (125, Fig. 18 ) was identified as a promising anti-adipogenic agent with an IC50 of 1.67 µM, which was more potent than that of its parent compound. Further evaluation revealed that the anti-adipogenic activity of this lead compound was evident in the early stage of adipogenesis, which in the mitotic clonal expansion it was controlled the G1 to S phase cell cycle arrest. The mechanistic investigations revealed that the hybrid derivative 125 exhibited anti-adipogenic properties by activating the Wnt/β-catenin pathway, and adipogenic genes such as PPARγ and C/EBPα. Moreover, 125 showed correlating decreased of PPARγ and increased expression of β-catenin in epididymal white adipose tissue in vivo.
Fig. 18.
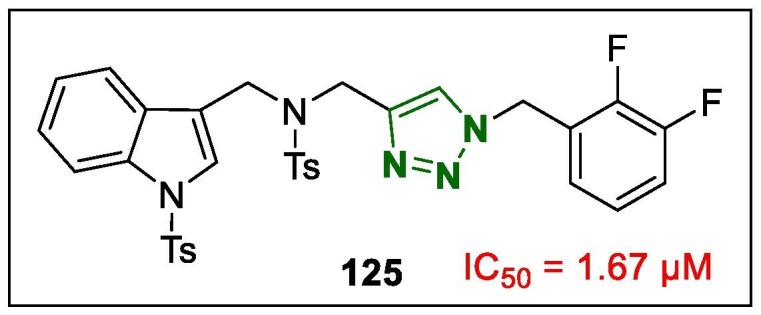
Structure of lead 1,2,3-triazole 125.
3.12. Antioxidant activity
A series of newly functionalised 1,2,3-triazole-pyrimido[4,5-c]isoquinolines were synthesised for antioxidant screening.153 In vitro antioxidant activity analysis (by 2,2-diphenyl-1-picrylhydrazyl (DPPH) method) revealed that compound 126 (Fig. 19 ) exhibited stronger antioxidant potency with an IC50 value 6.02 µM than the positive control drug Trolox did (1.94-fold higher activity than the positive control drug Trolox). It should be noted that the most common derivatives with tethered electron-withdrawing substituents (such as F, Cl and NO2) in the triazole moiety and pyrimidoisoquinoline system showed average to high antioxidant properties.
Fig. 19.
Structure of lead 1,2,3-triazole 126.
3.13. Multi-target investigation of the various activities of 1,2,3-triazoles
Several 1,2,3-triazole-containing coumarino-pyrazole compounds were synthesised and evaluated for AChE, anti-tyrosinase, anti-5–5-LOX, and anticancer potentials.154 The 1,4-substituted triazole regioisomers 127 and 128 (Fig. 20 ) demonstrated satisfactory activities in all target bioassays. For example, the anti-BChE activity results showed that tri-chlorine-related compounds 127 (29.0 µM) and 128 (18.0 µM) were more active than the other synthesised cycloadducts were, while the anti-5-LOX activity assay showed the same observation, i.e., 127 and 128 were displayed strong activity with IC50 values of 24.0 and 21.0 μM, respectively. Anti-tyrosinase activity assays of these two cycloadducts showed they were 58 and 72%, less active than the other samples were. However, compound 128 (IC50 = 6.0 μM) showed the highest cytotoxicity against the HCT-116 cells, among all evaluated 1,2,3-triazole tethered coumarinopyrazole conjugates.
Fig. 20.

Structures of lead 1,2,3-triazoles 127 and 128.
3.14. Miscellaneous activities
1,2,3-Triazole-bearing disaccharides were investigated as RNase A inhibitors for the first time by Kayet et al.155 It was suggested that except a linker type action of the triazole moiety to carbohydrates, these azoles could direct the sugar fragments to the binding sites of enzymes. The bio-physical assays revealed that compound 129 (Fig. 21 ) decreased the ribonucleolytic potency most efficiently, whereas derivative 130 (Fig 21) also exhibited higher inhibitory activity in the agarose gel electrophoresis than other prepared compounds with a missing –COOH substituent did. Kinetics studies showed that the triazole hybrid 130 had weaker RNase A activity (Ki = 65 µM) than its homolog, 129 (Ki = 50 µM), did, which confirm that introducing a carboxylic functional group at C-3 positions of furanose rings decreased the Ki values.
Fig. 21.

Structures of lead 1,2,3-triazoles 129–135.
Kozarski et al.156 reported a set of 7-methylguanosine phosphate substances bearing a 1,2,3-triazole ring at the position 5 as promising inhibitors of human cNIIIB. Among the four prepared analogues, compound 131 (Fig. 21) inhibited cNIIIB potency with an IC50 value of 87.8 µM), while the other three substances were inactive. Moreover, the effect of 131 was studied on the human decapping scavenger enzyme (hDcpS). The results revealed that despite the slight inhibitory potency against hDcpS (5.8-fold less than that of 7-methylguanosine phosphate), 131 was identified as selective inhibitor in the HscNIIIB simple biochemical assay.
Peterson et al.157 prepared symmetrical thiodigalactoside derivatives carrying aryltriazolyl portions to optimise the binding near R144 of galectin-3. Introduction of additional fluorine on the phenyltriazole ring produced the lead 3,4,5-F-phenyltriazole hybrid (132, Fig. 21). This thiodigalactoside slightly increased the galectin-3 affinity and improved the galectin-3 selectivity 50-fold more than that of galectin-1.
The benzamide scaffolds linked by a 1,2,3-triazole ring were discovered to be promising human dihydroorotate dehydrogenase (hDHODH) inhibitors by Lu et al.158 The hDHODH inhibitors play an important role in the rate-limiting step of pyrimidine biosynthesis, while pyrimidine plays a critical role in the DNA and RNA biosynthesis. In this study, the most potent compound was 133 (Fig. 21), which displayed superior suppression of hDHODH with an IC50 of 2.1 µM. The immunosuppression assay revealed that 133 also potently inhibited proliferation of activated peripheral blood mononuclear cells.
Syeda et al.159 described a new ouabagenin triazole analogue in the development of safe male contraceptives, that is a selective and promising inhibitor of Na,K-ATPase α4, and sperm function. Results showed that compound 134 (Fig. 21) specifically targets Na,K-ATPase α4. The activity of the lead derivative 134 was determined in rat sperm Vm, pH, and [Ca2+]i. The prepared cell samples were treated in the presence and absence of 10−8 M 134 for 1 h, and sperm Vm, pH, and [Ca2+]i were measured using various fluorophores and derivative 134 caused sperm plasma membrane depolarisation, escalating Vm by almost 40%, causing intracellular sperm acidification. Overall, except for the decreased ability of sperm to swim and reach the egg, 134 demonstrated decreased hyperactivity of sperm movability. All the above potential activities of 134 could serve in the future development of drug candidates for male contraception.
Several inhibitors of autotaxin that were used to determine the binding mode of the known inhibitor PF-8380 as an example, were developed by Thomson et al.160 The modification results showed that replacing the benzoxazolone fragment with a triazole zinc-binding motif decreased the crystallinity and increased solubility. Among these agents, compound 135 (Fig. 21) was selected for its high affinity as an orally bioavailable inhibitor of autotaxin. The lead 135 concentration-dependently inhibited autotaxin and the formation of LPA in vivo when used at 0.3 mg/kg in Sprague Dawley rats. A robust decrease in all four LPA levels was observed, with the peak reduction of approximately 70–80%, which was in agreement with the Cmax of 135.
4. Conclusion
In conclusion, lead compounds containing the 1,2,3-triazole moiety will serve as effective drug candidates in their current forms or following appropriate chemical and biological modifications in future. Indeed, the developments in Cu-AAC have generated 1,2,3-triazole-hybrids with a broad profile of activities. The present review shows that most synthesised triazoles have favoured activities in which these motifs not only acted as a linker, showing CH-π interaction between the 1,2,3-triazole and enzymes, but also directed the appropriate side-rings (the fused ring in the triazole hybrid system) to the binding sites of enzymes or amino acids. These properties of triazoles in the hybrid molecules make them a unique motif that is an important factor in drug design and delivery. Although the selectivity and solubility of the most common triazole-hybrids have been well improved, the in vivo assay results indicate that further developments are needed. There should also be a focus on fused 1,2,3-triazole heterocyclic compounds, because triazole linkers have been well studied and their mechanism of action is clear. For example, we propose that if a 1,2,3-triazole ring was inserted instead of the imidazole portion in the purine skeleton, this type of fused pyrimidines may exhibit interesting properties for medicinal chemistry. In addition, the present review indicated that a proportion of studies have been dedicated to coumarin modification using Cu-AAC method. In the future, we believe there will be further elucidation of 1,2,3-triazole chemistry and biology, which would facilitate the development of 1,2,3-triazole-based lead compounds with lower toxicity and higher selectivity.
Acknowledgements
This work was funded by grants from the Chinese Academy of Sciences President’s International Fellowship Initiative (K. Bozorov, Grant No. 2019PT054) and the Central Asian Drug Discovery and Development Center of Chinese Academy of Sciences (CAM201803).
Biographies

Khurshed Bozorov was born on March 6, 1984, in Samarkand, Uzbekistan. He obtained a MS degree from Samarkand State University in 2007. He pursued his Ph.D. degree in organic synthesis at the Institute of the Chemistry of Plant Substances working with Professor Khusnutdin M. Shakhidoyatov. He worked on the synthesis, chemical transformation and biological evaluation of thienopyrimidinones. He obtained his Ph.D. degree in 2011. Following his PhD degree he did his postdoctoral research with Professor Haji A. Aisa at the Xinjiang Technical Institute of Physics and Chemistry, CAS between 2013 and 2015. In 2016, he has awarded a National Natural Science Foundation of China, and was appointed to be a Principal Investigator at the Xinjiang Technical Institute of Physics and Chemistry during 2015-2018. In 2019, he has awarded a grant from the Chinese Academy of Sciences President’s International Fellowship Initiative for holding research on Bioactive Purines and its analogues. His main research area belong is design, synthesis and medicinal chemistry of the nitrogen-, sulfur- and oxygen-containing pyrimidine scaffolds, diverse bioactive small molecules, heterocycles and natural compounds.

Jiangyu Zhao was born on May 6, 1983, in Ruicheng, Shanxi, China. She obtained her Master Degree in Organic Chemistry at the Nankai University in 2007. In 2011, she got PhD in Organic Chemistry under the supervision of Prof. Haji A. Aisa and continuing her scientific career at the Xinjiang Technical Institute of Physics and Chemistry, CAS from 2011 until now. Her PhD work was focused on the synthesis and chemical modification of natural products with anti-influenza activities. She worked as a visiting scholar at Paris Diderot University, France in 2011. Her main research interests are the drug design, synthesis and biological screening of active compound from unique medicinal plant resources in Xinjiang. She has hosted one National Key Scientific-Technical Program of China, one National Natural Science Foundation of China, one natural science funds of Xinjiang Uygur Autonomous Region. She has published more than 20 scientific articles in domestic and foreign academic journals, 10 patents were licensed.

Haji A. Aisa was born on September 1, 1965, in Urumqi, Xinjiang, China. He obtained his PhD Degree in Organic chemistry under the supervision of Prof. Cai Junchao at the Shanghai Institute of Materia Medica, CAS in 1999. He was promoted to be a professor in 2000. As a senior visiting scholar, he successively visited Queen’s University, Canada, Paris Diderot University, France and Baylor College of Medicine, USA in 2004, 2012 and 2015. His current research interests are: a) development of bio-resources and indigenous medicinal plants in arid zone and Central Asia; b) the synthesis and drug design in the phytochemistry and organic synthesis; c) investigation and modernization of traditional Chinese medicine. He has been supported by National Science Fund for Distinguished Young Scholars by National Natural Science Foundation of China in 2009. He has published more than 400 scientific articles in domestic and foreign academic journals in the fields of natural product chemistry and organic chemistry, 88 patents were licensed and 26 were put in practice. Based on the fruitful research results, he has won 2 first prizes and 3 second prizes of Xinjiang Science and Technology Progress Awards.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2019.07.005.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Ahmad S., Alam O., Naim M.J., Shaquiquzzaman M., Alam M.M., Iqbal M. Pyrrole: an insight into recent pharmacological advances with structure activity relationship. Eur J Med Chem. 2018;157:527–561. doi: 10.1016/j.ejmech.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Parker W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. 2009;109:2880–2893. doi: 10.1021/cr900028p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bozorov K., Nie L.F., Zhao J., Aisa H.A. 2-Aminothiophene scaffolds: diverse biological and pharmacological attributes in medicinal chemistry. Eur J Med Chem. 2017;140:465–493. doi: 10.1016/j.ejmech.2017.09.039. [DOI] [PubMed] [Google Scholar]
- 4.Bozorov K., Zhao J.-Y., Elmuradov B., Pataer A., Aisa H.A. Recent developments regarding the use of thieno[2,3-d]pyrimidin-4-one derivatives in medicinal chemistry, with a focus on their synthesis and anticancer properties. Eur J Med Chem. 2015;102:552–573. doi: 10.1016/j.ejmech.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Bozorov K., Ma H.-R., Zhao J.-Y., et al. Discovery of diethyl 2,5-diaminothiophene-3,4-dicarboxylate derivatives as potent anticancer and antimicrobial agents and screening of anti-diabetic activity: synthesis and in vitro biological evaluation. Part 1. Eur J Med Chem. 2014;84:739–745. doi: 10.1016/j.ejmech.2014.07.065. [DOI] [PubMed] [Google Scholar]
- 6.Bozorov K., Jy Zhao, Nie L.F., et al. Synthesis and in vitro biological evaluation of novel diaminothiophene scaffolds as antitumor and anti-influenza virus agents. Part 2. RSC Adv. 2017;7:31417–31427. [Google Scholar]
- 7.Khanam H., Shamsuzzaman Bioactive Benzofuran derivatives: a review. Eur J Med Chem. 2015;97:483–504. doi: 10.1016/j.ejmech.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 8.Naik R., Harmalkar D.S., Xu X., Jang K., Lee K. Bioactive benzofuran derivatives: moracins A-Z in medicinal chemistry. Eur J Med Chem. 2015;90:379–393. doi: 10.1016/j.ejmech.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 9.Majumdar P., Pati A., Patra M., Behera R.K., Behera A.K. Acid hydrazides, potent reagents for synthesis of oxygen-, nitrogen-, and/or sulfur-containing heterocyclic rings. Chem Rev. 2014;114:2942–2977. doi: 10.1021/cr300122t. [DOI] [PubMed] [Google Scholar]
- 10.Ayati A., Emami S., Foroumadi A. The importance of triazole scaffold in the development of anticonvulsant agents. Eur J Med Chem. 2016;109:380–392. doi: 10.1016/j.ejmech.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Briguglio I., Piras S., Corona P., et al. Benzotriazole: an overview on its versatile biological behavior. Eur J Med Chem. 2015;97:612–648. doi: 10.1016/j.ejmech.2014.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdellatif K.R.A., Bakr R.B. New advances in synthesis and clinical aspects of pyrazolo[3,4-d]pyrimidine scaffolds. Bioorg Chem. 2018;78:341–357. doi: 10.1016/j.bioorg.2018.03.032. [DOI] [PubMed] [Google Scholar]
- 13.Herr R.J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg Med Chem. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 14.Dymińska L. Imidazopyridines as a source of biological activity and their pharmacological potentials—infrared and Raman spectroscopic evidence of their content in pharmaceuticals and plant materials. Bioorg Med Chem. 2015;23:6087–6099. doi: 10.1016/j.bmc.2015.07.045. [DOI] [PubMed] [Google Scholar]
- 15.Reports regarding the use of triazoles in the Scopus database in: www.scopus.com, 2018.
- 16.Dheer D., Singh V., Shankar R. Medicinal attributes of 1,2,3-triazoles: current developments. Bioorg Chem. 2017;71:30–54. doi: 10.1016/j.bioorg.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Carmona A.T., Carrión-Jiménez S., Pingitore V., Moreno-Clavijo E., Robina I., Moreno-Vargas A.J. Harnessing pyrrolidine iminosugars into dimeric structures for the rapid discovery of divalent glycosidase inhibitors. Eur J Med Chem. 2018;151:765–776. doi: 10.1016/j.ejmech.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Guo T., Dätwyler P., Demina E., et al. Selective inhibitors of human neuraminidase 3. J Med Chem. 2018;61:1990–2008. doi: 10.1021/acs.jmedchem.7b01574. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez C., Sanchez A., Collins J., et al. The 4-N-acyl and 4-N-alkyl gemcitabine analogues with silicon-fluoride-acceptor: application to 18F-radiolabeling. Eur J Med Chem. 2018;148:314–324. doi: 10.1016/j.ejmech.2018.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fjellaksel R., Sundset R., Riss P.J., Hansen J.H. Copper-mediated late-stage iodination and 123 I-labelling of triazole-benzimidazole bioactives. Synlett. 2018;29:1491–1495. [Google Scholar]
- 21.Kommidi H., Guo H., Nurili F., et al. 18F-positron emitting/trimethine cyanine-fluorescent contrast for image-guided prostate cancer management. J Med Chem. 2018;61:4256–4262. doi: 10.1021/acs.jmedchem.8b00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang S., Han Y., Chen M., et al. Radiosynthesis and biological evaluation of 18F-labeled 4-anilinoquinazoline derivative (18F-FEA-Erlotinib) as a potential EGFR PET agent. Bioorg Med Chem Lett. 2018;28:1143–1148. doi: 10.1016/j.bmcl.2017.08.066. [DOI] [PubMed] [Google Scholar]
- 23.Valdomir G., Fernández M.D.L.A., Lagunes I., et al. Oxa/thiazole-tetrahydropyran triazole-linked hybrids with selective antiproliferative activity against human tumour cells. New J Chem. 2018;42:13784–13789. [Google Scholar]
- 24.Zhou J., Stapleton P., Haider S., Healy J. Boronic acid inhibitors of the class A β-lactamase KPC-2. Bioorg Med Chem. 2018;26:2921–2927. doi: 10.1016/j.bmc.2018.04.055. [DOI] [PubMed] [Google Scholar]
- 25.Lopes S.M.M., Novais J.S., Costa D.C.S., et al. Hetero-Diels-Alder reactions of novel 3-triazolyl-nitrosoalkenes as an approach to functionalized 1,2,3-triazoles with antibacterial profile. Eur J Med Chem. 2018;143:1010–1020. doi: 10.1016/j.ejmech.2017.11.052. [DOI] [PubMed] [Google Scholar]
- 26.Fletcher J.T., Sobczyk J.M., Gwazdacz S.C., Blanck A.J. Antimicrobial 1,3,4-trisubstituted-1,2,3-triazolium salts. Bioorg Med Chem Lett. 2018;28:3320–3323. doi: 10.1016/j.bmcl.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezaei-Matehkolaei A., Khodavaisy S., Alshahni M.M., et al. In vitro antifungal activity of novel triazole efinaconazole and five comparators against dermatophyte isolates. Antimicrob Agents Chemother. 2018;62 doi: 10.1128/AAC.02423-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savanur H.M., Naik K.N., Ganapathi S.M., Kim K.M., Kalkhambkar R.G. Click chemistry inspired design, synthesis and molecular docking studies of coumarin, quinolinone linked 1,2,3-triazoles as promising anti-microbial agents. ChemistrySelect. 2018;3:5296–5303. [Google Scholar]
- 29.Tarawneh A.H., Al-Momani L.A., León F., et al. Evaluation of triazole and isoxazole derivatives as potential anti-infective agents. Med Chem Res. 2018;27:1269–1275. doi: 10.1007/s00044-018-2146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batra N., Rajendran V., Agarwal D., et al. Synthesis and antimalarial evaluation of [1,2,3]-triazole-tethered sulfonamide-berberine hybrids. ChemistrySelect. 2018;3:9790–9793. [Google Scholar]
- 31.Rajavelu K., Subaraja M., Rajakumar P. Synthesis, optical properties, and antioxidant and anticancer activity of benzoheterazole dendrimers with triazole bridging unit. New J Chem. 2018;42:3282–3292. [Google Scholar]
- 32.Kumar K.A., Kalluraya B., Kumar S.M. Synthesis and in-vitro antioxidant activities of some coumarin derivatives containing 1,2,3-triazole ring. Phosphorus, Sulfur Silicon Relat Elem. 2018;193:294–299. [Google Scholar]
- 33.Santosh R., Selvam M.K., Kanekar S.U., Nagaraja G.K. Synthesis, characterization, antibacterial and antioxidant studies of some heterocyclic compounds from triazole-linked chalcone derivatives. ChemistrySelect. 2018;3:6338–6343. [Google Scholar]
- 34.Liu Q., Kistemaker H.A.V., Bhogaraju S., et al. A general approach towards triazole-linked adenosine diphosphate ribosylated peptides and proteins. Angew Chem Int Ed. 2018;57:1659–1662. doi: 10.1002/anie.201710527. [DOI] [PubMed] [Google Scholar]
- 35.Ben Haj Salah K., Das S., Ruiz N., et al. How are 1,2,3-triazoles accommodated in helical secondary structures? Org Biomol Chem. 2018;16:3576–3583. doi: 10.1039/c8ob00686e. [DOI] [PubMed] [Google Scholar]
- 36.Kacprzak K., Skiera I., Piasecka M., Paryzek Z. Alkaloids and isoprenoids modification by copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (click chemistry): toward new functions and molecular architectures. Chem Rev. 2016;116:5689–5743. doi: 10.1021/acs.chemrev.5b00302. [DOI] [PubMed] [Google Scholar]
- 37.Johansson J.R., Beke-Somfai T., Said Stålsmeden A., Kann N. Ruthenium-catalyzed azide alkyne cycloaddition reaction: scope, mechanism, and applications. Chem Rev. 2016;116:14726–14768. doi: 10.1021/acs.chemrev.6b00466. [DOI] [PubMed] [Google Scholar]
- 38.Efimov I.V. Recent methods for the synthesis of NH-1,2,3-triazoles (microreview) Chem Heterocycl Compd. 2019;55:28–30. [Google Scholar]
- 39.Totobenazara J., Burke A.J. New click-chemistry methods for 1,2,3-triazoles synthesis: recent advances and applications. Tetrahedron Lett. 2015;56:2853–2859. [Google Scholar]
- 40.Usachev B.I. Chemistry of fluoroalkyl-substituted 1,2,3-triazoles. J Fluorine Chem. 2018;210:6–45. [Google Scholar]
- 41.Bonandi E., Christodoulou M.S., Fumagalli G., Perdicchia D., Rastelli G., Passarella D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov Today. 2017;22:1572–1581. doi: 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Rejhová A., Opattová A., Čumová A., Slíva D., Vodička P. Natural compounds and combination therapy in colorectal cancer treatment. Eur J Med Chem. 2018;144:582–594. doi: 10.1016/j.ejmech.2017.12.039. [DOI] [PubMed] [Google Scholar]
- 43.Prasad S., Tyagi A.K., Aggarwal B.B. In: Bharti A.C., Aggarwal B.B., editors. Vol 2. Academic Press; 2018. Chapter 5 – chemosensitization by ursolic acid: a new avenue for cancer therapy; pp. 99–109. (Role of Nutraceuticals in Cancer Chemosensitization). [Google Scholar]
- 44.Lal K., Yadav P. Recent advancements in 1,4-disubstituted 1H-1,2,3-triazoles as potential anticancer agents. Anti-Cancer Agents Med Chem. 2018;18:21–37. doi: 10.2174/1871520616666160811113531. [DOI] [PubMed] [Google Scholar]
- 45.Ding Y., Guo H., Ge W., et al. Copper(I) oxide nanoparticles catalyzed click chemistry based synthesis of melampomagnolide B-triazole conjugates and their anti-cancer activities. Eur J Med Chem. 2018;156:216–229. doi: 10.1016/j.ejmech.2018.06.058. [DOI] [PubMed] [Google Scholar]
- 46.Janganati V., Ponder J., Balasubramaniam M., et al. MMB triazole analogs are potent NF-κB inhibitors and anti-cancer agents against both hematological and solid tumor cells. Eur J Med Chem. 2018;157:562–581. doi: 10.1016/j.ejmech.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu H., Hou Z., Tian Y., Mou Y., Guo C. Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents. Eur J Med Chem. 2018;151:434–449. doi: 10.1016/j.ejmech.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Ke Y., Liang J.-J., Hou R.-J., et al. Synthesis and biological evaluation of novel Jiyuan Oridonin A-1,2,3-triazole-azole derivatives as antiproliferative agents. Eur J Med Chem. 2018;157:1249–1263. doi: 10.1016/j.ejmech.2018.08.056. [DOI] [PubMed] [Google Scholar]
- 49.Ke Y., Wang W., Zhao L.-F., et al. Design, synthesis and biological mechanisms research on 1,2,3-triazole derivatives of Jiyuan Oridonin A. Bioorg Med Chem. 2018;26:4761–4773. doi: 10.1016/j.bmc.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 50.Wei G., Sun J., Hou Z., et al. Novel antitumor compound optimized from natural saponin Albiziabioside A induced caspase-dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur J Med Chem. 2018;157:759–772. doi: 10.1016/j.ejmech.2018.08.036. [DOI] [PubMed] [Google Scholar]
- 51.Li H-n, Wang H., Wang Z-p, et al. Synthesis, antitumor activity evaluation and mechanistic study of novel hederacolchiside A1 derivatives bearing an aryl triazole moiety. Biorg Med Chem. 2018;26:4025–4033. doi: 10.1016/j.bmc.2018.06.026. [DOI] [PubMed] [Google Scholar]
- 52.Wu M.-J., Wu D.-M., Chen J.-B., et al. Synthesis and anti-proliferative activity of allogibberic acid derivatives containing 1,2,3-triazole pharmacophore. Bioorg Med Chem Lett. 2018;28:2543–2549. doi: 10.1016/j.bmcl.2018.05.038. [DOI] [PubMed] [Google Scholar]
- 53.Boratyński P.J., Gałęzowska J., Turkowiak K., Anisiewicz A., Kowalczyk R., Wietrzyk J. Triazole biheterocycles from cinchona alkaloids: coordination and antiproliferative properties. ChemistrySelect. 2018;3:9368–9373. [Google Scholar]
- 54.Abou Samra A., Robert A., Gov C., et al. Dual inhibitors of the pro-survival proteins Bcl-2 and Mcl-1 derived from natural compound meiogynin A. Eur J Med Chem. 2018;148:26–38. doi: 10.1016/j.ejmech.2018.01.100. [DOI] [PubMed] [Google Scholar]
- 55.Bębenek E., Kadela-Tomanek M., Chrobak E., Latocha M., Boryczka S. Novel triazoles of 3-acetylbetulin and betulone as anticancer agents. Med Chem Res. 2018;27:2051–2061. doi: 10.1007/s00044-018-2213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reddy V.G., Bonam S.R., Reddy T.S., et al. 4β-amidotriazole linked podophyllotoxin congeners: DNA topoisomerase-IIα inhibition and potential anticancer agents for prostate cancer. Eur J Med Chem. 2018;144:595–611. doi: 10.1016/j.ejmech.2017.12.050. [DOI] [PubMed] [Google Scholar]
- 57.Bian J., Ren J., Li Y., et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81:373–381. doi: 10.1016/j.bioorg.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 58.Gupta N., Qayum A., Raina A., et al. Synthesis and biological evaluation of novel bavachinin analogs as anticancer agents. Eur J Med Chem. 2018;145:511–523. doi: 10.1016/j.ejmech.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Safavi M., Ashtari A., Khalili F., et al. Novel quinazolin-4(3H)-one linked to 1,2,3-triazoles: synthesis and anticancer activity. Chem Biol Drug Des. 2018;92:1373–1381. doi: 10.1111/cbdd.13203. [DOI] [PubMed] [Google Scholar]
- 60.Vasu K.K., Ingawale H.D., Sagar S.R., Sharma J.A., Pandya D.H., Agarwal M. 2-((1H-1,2,3-Triazol-1-yl)methyl)-3-phenylquinazolin-4(3H)-ones: design, synthesis and evaluation as anti-cancer agents. Curr Bioact Compd. 2018;14:254–263. [Google Scholar]
- 61.Le-Nhat-Thuy G., Dinh T.V., Pham-The H., et al. Design, synthesis and evaluation of novel hybrids between 4-anilinoquinazolines and substituted triazoles as potent cytotoxic agents. Bioorg Med Chem Lett. 2018;28:3741–3747. doi: 10.1016/j.bmcl.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 62.Srinivas M., Singh Pathania A., Mahajan P., et al. Design and synthesis of 1,4-substituted 1H–1,2,3-triazolo-quinazolin-4(3H)-ones by Huisgen 1,3-dipolar cycloaddition with PI3Kγ isoform selective activity. Bioorg Med Chem Lett. 2018;28:1005–1010. doi: 10.1016/j.bmcl.2018.02.032. [DOI] [PubMed] [Google Scholar]
- 63.Kettle J.G., Anjum R., Barry E., et al. Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy)quinazolin-4-yl]amino}phenyl)-2-[4-(propan-2-yl)-1 H-1,2,3-triazol-1-yl]acetamide (AZD3229), a Potent Pan-KIT Mutant Inhibitor for the Treatment of Gastrointestinal Stromal Tumors. J Med Chem. 2018;61:8797–8810. doi: 10.1021/acs.jmedchem.8b00938. [DOI] [PubMed] [Google Scholar]
- 64.Craik D.J., Fairlie D.P., Liras S., Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 65.Chen Y., Li Z., Liu Y., et al. Identification of novel and selective non-peptide inhibitors targeting the polo-box domain of polo-like kinase 1. Bioorg Chem. 2018;81:278–288. doi: 10.1016/j.bioorg.2018.08.030. [DOI] [PubMed] [Google Scholar]
- 66.Matthiesen R.A., Varney M.L., Xu P.C., Rier A.S., Wiemer D.F., Holstein S.A. α-Methylation enhances the potency of isoprenoid triazole bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg Med Chem. 2018;26:376–385. doi: 10.1016/j.bmc.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cao J., Ma C., Zang J., et al. Novel leucine ureido derivatives as aminopeptidase N inhibitors using click chemistry. Bioorg Med Chem. 2018;26:3145–3157. doi: 10.1016/j.bmc.2018.04.041. [DOI] [PubMed] [Google Scholar]
- 68.Compain G., Oumata N., Clarhaut J., et al. A β-glucuronidase-responsive albumin-binding prodrug for potential selective kinase inhibitor-based cancer chemotherapy. Eur J Med Chem. 2018;158:1–6. doi: 10.1016/j.ejmech.2018.08.100. [DOI] [PubMed] [Google Scholar]
- 69.Chen J., Li D., Li W., et al. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg Med Chem. 2018;26:3958–3966. doi: 10.1016/j.bmc.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 70.Gao Y., Shi W., Cui J., et al. Design, synthesis and biological evaluation of novel tetrahydroisoquinoline derivatives as P-glycoprotein-mediated multidrug resistance inhibitors. Bioorg Med Chem. 2018;26:2420–2427. doi: 10.1016/j.bmc.2018.03.045. [DOI] [PubMed] [Google Scholar]
- 71.Geng P.-F., Liu X.-Q., Zhao T.-Q., et al. Design, synthesis and in vitro biological evaluation of novel [1,2,3]triazolo[4,5-d]pyrimidine derivatives containing a thiosemicarbazide moiety. Eur J Med Chem. 2018;146:147–156. doi: 10.1016/j.ejmech.2018.01.031. [DOI] [PubMed] [Google Scholar]
- 72.Yamada M., Takahashi T., Hasegawa M., et al. Synthesis, antitumor activity, and cytotoxicity of 4-substituted 1-benzyl-5-diphenylstibano-1H-1,2,3-triazoles. Bioorg Med Chem Lett. 2018;28:152–154. doi: 10.1016/j.bmcl.2017.11.038. [DOI] [PubMed] [Google Scholar]
- 73.Gilandoust M., Harsha K.B., Mohan C.D., et al. Synthesis, characterization and cytotoxicity studies of 1,2,3-triazoles and 1,2,4-triazolo [1,5-a] pyrimidines in human breast cancer cells. Bioorg Med Chem Lett. 2018;28:2314–2319. doi: 10.1016/j.bmcl.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 74.Allam M., Bhavani A.K.D., Mudiraj A., Ranjan N., Thippana M., Babu P.P. Synthesis of pyrazolo[3,4-d]pyrimidin-4(5H)-ones tethered to 1,2,3-triazoles and their evaluation as potential anticancer agents. Eur J Med Chem. 2018;156:43–52. doi: 10.1016/j.ejmech.2018.06.055. [DOI] [PubMed] [Google Scholar]
- 75.Li H-y, He D.-D., Zhao X.-J., et al. Design and synthesis of novel dasatinib derivatives as inhibitors of leukemia stem cells. Bioorg Med Chem Lett. 2018;28:700–706. doi: 10.1016/j.bmcl.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 76.Wang L., Xu S., Liu X., et al. Discovery of thinopyrimidine-triazole conjugates as c-Met targeting and apoptosis inducing agents. Bioorg Chem. 2018;77:370–380. doi: 10.1016/j.bioorg.2018.01.037. [DOI] [PubMed] [Google Scholar]
- 77.Sayeed I.B., Vishnuvardhan M.V.P.S., Nagarajan A., Kantevari S., Kamal A. Imidazopyridine linked triazoles as tubulin inhibitors, effectively triggering apoptosis in lung cancer cell line. Bioorg Chem. 2018;80:714–720. doi: 10.1016/j.bioorg.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 78.Bistrović A., Krstulović L., Harej A., et al. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur J Med Chem. 2018;143:1616–1634. doi: 10.1016/j.ejmech.2017.10.061. [DOI] [PubMed] [Google Scholar]
- 79.Borowiecki P., Wińska P., Bretner M., Gizińska M., Koronkiewicz M., Staniszewska M. Synthesis of novel proxyphylline derivatives with dual Anti-Candida albicans and anticancer activity. Eur J Med Chem. 2018;150:307–333. doi: 10.1016/j.ejmech.2018.02.077. [DOI] [PubMed] [Google Scholar]
- 80.Costa D.C.S., de Almeida G.S., Rabelo V.W.-H., et al. Synthesis and evaluation of the cytotoxic activity of Furanaphthoquinones tethered to 1H-1,2,3-triazoles in Caco-2, Calu-3, MDA-MB231 cells. Eur J Med Chem. 2018;156:524–533. doi: 10.1016/j.ejmech.2018.07.018. [DOI] [PubMed] [Google Scholar]
- 81.Dadmal T.L., Appalanaidu K., Kumbhare R.M., Mondal T., Ramaiah M.J., Bhadra M.P. Synthesis and biological evaluation of triazole and isoxazole-tagged benzothiazole/benzoxazole derivatives as potent cytotoxic agents. New J Chem. 2018;42:15546–15551. [Google Scholar]
- 82.Kumar S., Gu L., Palma G., et al. Design, synthesis, anti-proliferative evaluation and docking studies of 1: H-1,2,3-triazole tethered ospemifene-isatin conjugates as selective estrogen receptor modulators. New J Chem. 2018;42:3703–3713. [Google Scholar]
- 83.Naaz F., Preeti Pallavi M.C., Shafi S., Mulakayala N., Shahar Yar M., Sampath Kumar H.M. 1,2,3-Triazole tethered Indole-3-glyoxamide derivatives as multiple inhibitors of 5-LOX, COX-2 & tubulin: their anti-proliferative & anti-inflammatory activity. Bioorg Chem. 2018;81:1–20. doi: 10.1016/j.bioorg.2018.07.029. [DOI] [PubMed] [Google Scholar]
- 84.Schlapbach A., Revesz L., Pissot Soldermann C., et al. N-aryl-piperidine-4-carboxamides as a novel class of potent inhibitors of MALT1 proteolytic activity. Bioorg Med Chem Lett. 2018;28:2153–2158. doi: 10.1016/j.bmcl.2018.05.017. [DOI] [PubMed] [Google Scholar]
- 85.Wang B., Zhao B., Chen Z.-S., et al. Exploration of 1,2,3-triazole-pyrimidine hybrids as potent reversal agents against ABCB1-mediated multidrug resistance. Eur J Med Chem. 2018;143:1535–1542. doi: 10.1016/j.ejmech.2017.10.041. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y., Chen S., Hu G., et al. Discovery of novel 2,4-diarylaminopyrimidine analogues as ALK and ROS1 dual inhibitors to overcome crizotinib-resistant mutants including G1202R. Eur J Med Chem. 2018;143:123–136. doi: 10.1016/j.ejmech.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 87.Marepu N., Yeturu S., Pal M. 1,2,3-Triazole fused with pyridine/pyrimidine as new template for antimicrobial agents: regioselective synthesis and identification of potent N-heteroarenes. Bioorg Med Chem Lett. 2018;28:3302–3306. doi: 10.1016/j.bmcl.2018.09.021. [DOI] [PubMed] [Google Scholar]
- 88.Szałaj N., Lu L., Benediktsdottir A., et al. Boronic ester-linked macrocyclic lipopeptides as serine protease inhibitors targeting Escherichia coli type I signal peptidase. Eur J Med Chem. 2018;157:1346–1360. doi: 10.1016/j.ejmech.2018.08.086. [DOI] [PubMed] [Google Scholar]
- 89.Lal K., Yadav P., Kumar A., Kumar A., Paul A.K. Design, synthesis, characterization, antimicrobial evaluation and molecular modeling studies of some dehydroacetic acid-chalcone-1,2,3-triazole hybrids. Bioorg Chem. 2018;77:236–244. doi: 10.1016/j.bioorg.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 90.Maddili S.K., Katla R., Kannekanti V.K., et al. Molecular interaction of novel benzothiazolyl triazolium analogues with calf thymus DNA and HSA-their biological investigation as potent antimicrobial agents. Eur J Med Chem. 2018;150:228–247. doi: 10.1016/j.ejmech.2018.02.056. [DOI] [PubMed] [Google Scholar]
- 91.Mishra S., Kaur M., Chander S., et al. Rational modification of a lead molecule: improving the antifungal activity of indole – triazole – amino acid conjugates. Eur J Med Chem. 2018;155:658–669. doi: 10.1016/j.ejmech.2018.06.039. [DOI] [PubMed] [Google Scholar]
- 92.Yu J.-Y., Cheng H.-J., Wu H.-R., et al. Structure-based design of bacterial transglycosylase inhibitors incorporating biphenyl, amine linker and 2-alkoxy-3-phosphorylpropanoate moieties. Eur J Med Chem. 2018;150:729–741. doi: 10.1016/j.ejmech.2018.03.034. [DOI] [PubMed] [Google Scholar]
- 93.Bi F., Ji S., Venter H., Liu J., Semple S.J., Ma S. Substitution of terminal amide with 1H-1,2,3-triazole: identification of unexpected class of potent antibacterial agents. Bioorg Med Chem Lett. 2018;28:884–891. doi: 10.1016/j.bmcl.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 94.Sall C., Ayé M., Bottzeck O., Praud A., Blache Y. Towards smart biocide-free anti-biofilm strategies: click-based synthesis of cinnamide analogues as anti-biofilm compounds against marine bacteria. Bioorg Med Chem Lett. 2018;28:155–159. doi: 10.1016/j.bmcl.2017.11.039. [DOI] [PubMed] [Google Scholar]
- 95.Gatadi S., Gour J., Shukla M., et al. Synthesis of 1,2,3-triazole linked 4(3H)-Quinazolinones as potent antibacterial agents against multidrug-resistant Staphylococcus aureus. Eur J Med Chem. 2018;157:1056–1067. doi: 10.1016/j.ejmech.2018.08.070. [DOI] [PubMed] [Google Scholar]
- 96.Yadav P., Lal K., Kumar L., et al. Synthesis, crystal structure and antimicrobial potential of some fluorinated chalcone-1,2,3-triazole conjugates. Eur J Med Chem. 2018;155:263–274. doi: 10.1016/j.ejmech.2018.05.055. [DOI] [PubMed] [Google Scholar]
- 97.Srinivasarao S., Nizalapur S., Yu T.T., et al. Design, synthesis and biological evaluation of triazole-containing 2-phenylindole and salicylic acid as quorum sensing inhibitors against Pseudomonas aeruginosa. ChemistrySelect. 2018;3:9170–9180. [Google Scholar]
- 98.Govindaiah S., Sreenivasa S., Ramakrishna R.A., Rao T.M.C., Nagabhushana H. Regioselective synthesis, antibacterial, molecular docking and fingerprint applications of 1-benzhydrylpiperazine derivatized 1,4-disubstituted 1,2,3-triazoles. ChemistrySelect. 2018;3:8111–8117. [Google Scholar]
- 99.Kaushik C.P., Pahwa A. Convenient synthesis, antimalarial and antimicrobial potential of thioethereal 1,4-disubstituted 1,2,3-triazoles with ester functionality. Med Chem Res. 2018;27:458–469. [Google Scholar]
- 100.Moghimi S., Goli-Garmroodi F., Allahyari-Devin M., et al. Synthesis, evaluation, and molecular docking studies of aryl urea-triazole-based derivatives as anti-urease agents. Arch Pharm. 2018;351 doi: 10.1002/ardp.201800005. [DOI] [PubMed] [Google Scholar]
- 101.Faidallah H.M., Girgis A.S., Tiwari A.D., et al. Synthesis, antibacterial properties and 2D-QSAR studies of quinolone-triazole conjugates. Eur J Med Chem. 2018;143:1524–1534. doi: 10.1016/j.ejmech.2017.10.042. [DOI] [PubMed] [Google Scholar]
- 102.Yang X., Wedajo W., Yamada Y., et al. 1,3,5-Triazaspiro[5.5]undeca-2,4-dienes as selective Mycobacterium tuberculosis dihydrofolate reductase inhibitors with potent whole cell activity. Eur J Med Chem. 2018;144:262–276. doi: 10.1016/j.ejmech.2017.12.017. [DOI] [PubMed] [Google Scholar]
- 103.Spain M., Wong J.K.H., Nagalingam G., et al. Antitubercular bis-substituted cyclam derivatives: structure-activity relationships and in vivo studies. J Med Chem. 2018;61:3595–3608. doi: 10.1021/acs.jmedchem.7b01569. [DOI] [PubMed] [Google Scholar]
- 104.Carta A., Bua A., Corona P., et al. Design synthesis and antitubercular activity of 4-alkoxy-triazoloquinolones able to inhibit the M. tuberculosis DNA gyrase. Eur J Med Chem. 2019;161:399–415. doi: 10.1016/j.ejmech.2018.10.031. [DOI] [PubMed] [Google Scholar]
- 105.Yan X., Lv Z., Wen J., Zhao S., Xu Z. Synthesis and in vitro evaluation of novel substituted isatin-propylene-1H-1,2,3-triazole-4-methylene-moxifloxacin hybrids for their anti-mycobacterial activities. Eur J Med Chem. 2018;143:899–904. doi: 10.1016/j.ejmech.2017.11.090. [DOI] [PubMed] [Google Scholar]
- 106.Srivastava S., Bimal D., Bohra K., et al. Synthesis and antimycobacterial activity of 1-(β-d-Ribofuranosyl)-4-coumarinyloxymethyl- / -coumarinyl-1,2,3-triazole. Eur J Med Chem. 2018;150:268–281. doi: 10.1016/j.ejmech.2018.02.067. [DOI] [PubMed] [Google Scholar]
- 107.Danne A.B., Choudhari A.S., Chakraborty S., Sarkar D., Khedkar V.M., Shingate B.B. Triazole-diindolylmethane conjugates as new antitubercular agents: synthesis, bioevaluation, and molecular docking. MedChemComm. 2018;9:1114–1130. doi: 10.1039/c8md00055g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ashok D., Chiranjeevi P., Kumar A.V., et al. 1,2,3-Triazole-fused spirochromenes as potential anti-tubercular agents: synthesis and biological evaluation. RSC Adv. 2018;8:16997–17007. doi: 10.1039/c8ra03197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giroud M., Kuhn B., Saint-Auret S., et al. 2H-1,2,3-triazole-based dipeptidyl nitriles: potent, selective, and trypanocidal rhodesain inhibitors by structure-based design. J Med Chem. 2018;61:3370–3388. doi: 10.1021/acs.jmedchem.7b01870. [DOI] [PubMed] [Google Scholar]
- 110.Negi B., Poonan P., Ansari M.F., et al. Synthesis, antiamoebic activity and docking studies of metronidazole-triazole-styryl hybrids. Eur J Med Chem. 2018;150:633–641. doi: 10.1016/j.ejmech.2018.03.033. [DOI] [PubMed] [Google Scholar]
- 111.Zimmermann L.A., de Moraes M.H., da Rosa R., et al. Synthesis and SAR of new isoxazole-triazole bis-heterocyclic compounds as analogues of natural lignans with antiparasitic activity. Bioorg Med Chem. 2018;26:4850–4862. doi: 10.1016/j.bmc.2018.08.025. [DOI] [PubMed] [Google Scholar]
- 112.Zhang H-b, Shen Q.-K., Wang H., Jin C., Jin C.-M., Quan Z.-S. Synthesis and evaluation of novel arctigenin derivatives as potential anti-Toxoplasma gondii agents. Eur J Med Chem. 2018;158:414–427. doi: 10.1016/j.ejmech.2018.08.087. [DOI] [PubMed] [Google Scholar]
- 113.Hu X.-L., Gao C., Xu Z., Liu M.-L., Feng L.-S., Zhang G.-D. Recent development of coumarin derivatives as potential antiplasmodial and antimalarial agents. Curr Top Med Chem. 2018;18:114–123. doi: 10.2174/1568026618666171215101158. [DOI] [PubMed] [Google Scholar]
- 114.Jarrahpour A., Aye M., Rad J.A., et al. Design, synthesis, activity evaluation and QSAR studies of novel antimalarial 1,2,3-triazolo-β-lactam derivatives. J Iran Chem Soc. 2018;15:1311–1326. [Google Scholar]
- 115.Brandão G.C., Rocha Missias F.C., Arantes L.M., et al. Antimalarial naphthoquinones. Synthesis via click chemistry, in vitro activity, docking to PfDHODH and SAR of lapachol-based compounds. Eur J Med Chem. 2018;145:191–205. doi: 10.1016/j.ejmech.2017.12.051. [DOI] [PubMed] [Google Scholar]
- 116.Chopra R., Chibale K., Singh K. Pyrimidine-chloroquinoline hybrids: synthesis and antiplasmodial activity. Eur J Med Chem. 2018;148:39–53. doi: 10.1016/j.ejmech.2018.02.021. [DOI] [PubMed] [Google Scholar]
- 117.Yadav N., Agarwal D., Kumar S., Dixit A.K., Gupta R.D., Awasthi S.K. In vitro antiplasmodial efficacy of synthetic coumarin-triazole analogs. Eur J Med Chem. 2018;145:735–745. doi: 10.1016/j.ejmech.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 118.Thakur R.K., Joshi P., Baranwal P., et al. Synthesis and antiplasmodial activity of glyco-conjugate hybrids of phenylhydrazono-indolinones and glycosylated 1,2,3-triazolyl-methyl-indoline-2,3-diones. Eur J Med Chem. 2018;155:764–771. doi: 10.1016/j.ejmech.2018.06.042. [DOI] [PubMed] [Google Scholar]
- 119.Oliveira V.G., dos Santos Faiões V., Gonçalves G.B.R., et al. Design, synthesis and antileishmanial activity of naphthotriazolyl-4-oxoquinolines. Curr Top Med Chem. 2018;18:1454–1464. doi: 10.2174/1568026618666181002110116. [DOI] [PubMed] [Google Scholar]
- 120.Temraz M.G., Elzahhar P.A., Bekhit El-Din A., Bekhit A.A., Labib H.F., Belal A.S.F. Anti-leishmanial click modifiable thiosemicarbazones: design, synthesis, biological evaluation and in silico studies. Eur J Med Chem. 2018;151:585–600. doi: 10.1016/j.ejmech.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 121.Teixeira R.R., Gazolla P.A.R., da Silva A.M., et al. Synthesis and leishmanicidal activity of eugenol derivatives bearing 1,2,3-triazole functionalities. Eur J Med Chem. 2018;146:274–286. doi: 10.1016/j.ejmech.2018.01.046. [DOI] [PubMed] [Google Scholar]
- 122.Upadhyay A., Kushwaha P., Gupta S., et al. Synthesis and evaluation of novel triazolyl quinoline derivatives as potential antileishmanial agents. Eur J Med Chem. 2018;154:172–181. doi: 10.1016/j.ejmech.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 123.Maji K., Abbasi M., Podder D., Datta R., Haldar D. Potential antileishmanial activity of a triazole-based hybrid peptide against leishmania major. ChemistrySelect. 2018;3:10220–10225. [Google Scholar]
- 124.Gonzaga D.T.G., Souza T.M.L., Andrade V.M.M., Ferreira V.F., de C. da Silva F. Identification of 1-aryl-1H-1,2,3-triazoles as potential new antiretroviral agents. Med Chem. 2018;14:242–248. doi: 10.2174/1573406413666170906121318. [DOI] [PubMed] [Google Scholar]
- 125.Liu Y., Peng Y., Lu J., et al. Design, synthesis, and biological evaluation of new 1,2,3-triazolo-2′-deoxy-2′-fluoro- 4′-azido nucleoside derivatives as potent anti-HBV agents. Eur J Med Chem. 2018;143:137–149. doi: 10.1016/j.ejmech.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 126.Karypidou K., Ribone S.R., Quevedo M.A., et al. Synthesis, biological evaluation and molecular modeling of a novel series of fused 1,2,3-triazoles as potential anti-coronavirus agents. Bioorg Med Chem Lett. 2018;28:3472–3476. doi: 10.1016/j.bmcl.2018.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yan L., Yin Z., Niu L., Shao J., Chen H., Li X. Synthesis of pentacyclic iminosugars with constrained butterfly-like conformation and their HIV-RT inhibitory activity. Bioorg Med Chem Lett. 2018;28:425–428. doi: 10.1016/j.bmcl.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 128.Liu M., Xu Q., Guo S., et al. Design, synthesis, and structure-activity relationships of novel imidazo[4,5-c]pyridine derivatives as potent non-nucleoside inhibitors of hepatitis C virus NS5B. Bioorg Med Chem. 2018;26:2621–2631. doi: 10.1016/j.bmc.2018.04.029. [DOI] [PubMed] [Google Scholar]
- 129.Kaoukabi H., Kabri Y., Curti C., et al. Dihydropyrimidinone/1,2,3-triazole hybrid molecules: synthesis and anti-varicella-zoster virus (VZV) evaluation. Eur J Med Chem. 2018;155:772–781. doi: 10.1016/j.ejmech.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 130.Wu G., Zalloum W.A., Meuser M.E., et al. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur J Med Chem. 2018;158:478–492. doi: 10.1016/j.ejmech.2018.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tian Y., Liu Z., Liu J., et al. Targeting the entrance channel of NNIBP: discovery of diarylnicotinamide 1,4-disubstituted 1,2,3-triazoles as novel HIV-1 NNRTIs with high potency against wild-type and E138K mutant virus. Eur J Med Chem. 2018;151:339–350. doi: 10.1016/j.ejmech.2018.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Saeedi M., Mohammadi-Khanaposhtani M., Pourrabia P., et al. Design and synthesis of novel quinazolinone-1,2,3-triazole hybrids as new anti-diabetic agents: in vitro α-glucosidase inhibition, kinetic, and docking study. Bioorg Chem. 2019;83:161–169. doi: 10.1016/j.bioorg.2018.10.023. [DOI] [PubMed] [Google Scholar]
- 133.Avula S.K., Khan A., Rehman N.U., et al. Synthesis of 1H–1,2,3-triazole derivatives as new α-glucosidase inhibitors and their molecular docking studies. Bioorg Chem. 2018;81:98–106. doi: 10.1016/j.bioorg.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 134.Kumari M.A., Rao C.V., Triloknadh S., et al. Synthesis, docking and ADME prediction of novel 1,2,3-triazole-tethered coumarin derivatives as potential neuroprotective agents. Res Chem Intermed. 2018;44:1989–2008. [Google Scholar]
- 135.Reddy C.R., Tukaram A.G., Mohammed S.Z., et al. Synthesis and biological evaluation of longanlactone analogues as neurotrophic agents. Bioorg Med Chem Lett. 2018;28:673–676. doi: 10.1016/j.bmcl.2018.01.020. [DOI] [PubMed] [Google Scholar]
- 136.Sweeney J.B., Rattray M., Pugh V., Powell L.A. Riluzole-triazole hybrids as novel chemical probes for neuroprotection in amyotrophic lateral sclerosis. ACS Med Chem Lett. 2018;9:552–556. doi: 10.1021/acsmedchemlett.8b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Giraudo A., Krall J., Nielsen B., et al. 4-Hydroxy-1,2,3-triazole moiety as bioisostere of the carboxylic acid function: a novel scaffold to probe the orthosteric γ-aminobutyric acid receptor binding site. Eur J Med Chem. 2018;158:311–321. doi: 10.1016/j.ejmech.2018.08.094. [DOI] [PubMed] [Google Scholar]
- 138.Chrovian C.C., Soyode-Johnson A., Peterson A.A., et al. A Dipolar cycloaddition reaction to access 6-methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridines enables the discovery synthesis and preclinical profiling of a P2X7 antagonist clinical candidate. J Med Chem. 2018;61:207–223. doi: 10.1021/acs.jmedchem.7b01279. [DOI] [PubMed] [Google Scholar]
- 139.Luo Z., Rosenberg A.J., Liu H., Han J., Tu Z. Syntheses and in vitro evaluation of new S1PR1 compounds and initial evaluation of a lead F-18 radiotracer in rodents. Eur J Med Chem. 2018;150:796–808. doi: 10.1016/j.ejmech.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gilbertson S.R., Chen Y.-C., Soto C.A., et al. Synthesis and activity of functionalizable derivatives of the serotonin (5-HT) 5-HT2A receptor (5-HT2AR) antagonist M100907. Bioorg Med Chem Lett. 2018;28:1381–1385. doi: 10.1016/j.bmcl.2018.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jalili-Baleh L., Forootanfar H., Küçükkılınç T.T., et al. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer's disease based on coumarin and lipoic acid scaffolds. Eur J Med Chem. 2018;152:600–614. doi: 10.1016/j.ejmech.2018.04.058. [DOI] [PubMed] [Google Scholar]
- 142.Jalili-Baleh L., Nadri H., Forootanfar H., et al. Novel 3-phenylcoumarin–lipoic acid conjugates as multi-functional agents for potential treatment of Alzheimer's disease. Bioorg Chem. 2018;79:223–234. doi: 10.1016/j.bioorg.2018.04.030. [DOI] [PubMed] [Google Scholar]
- 143.Wang W., Wang W., Yao G., et al. Novel sarsasapogenin-triazolyl hybrids as potential anti-Alzheimer's agents: design, synthesis and biological evaluation. Eur J Med Chem. 2018;151:351–362. doi: 10.1016/j.ejmech.2018.03.082. [DOI] [PubMed] [Google Scholar]
- 144.Wu G., Gao Y., Kang D., et al. Design, synthesis and biological evaluation of tacrine-1,2,3-triazole derivatives as potent cholinesterase inhibitors. MedChemComm. 2018;9:149–159. doi: 10.1039/c7md00457e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Moussa G., Alaaeddine R., Alaeddine L.M., et al. Novel click modifiable thioquinazolinones as anti-inflammatory agents: design, synthesis, biological evaluation and docking study. Eur J Med Chem. 2018;144:635–650. doi: 10.1016/j.ejmech.2017.12.065. [DOI] [PubMed] [Google Scholar]
- 146.Rama Krishna B., Thummuri D., Naidu V.G.M., Ramakrishna S., Venkata Mallavadhani U. Synthesis of some novel orcinol based coumarin triazole hybrids with capabilities to inhibit RANKL-induced osteoclastogenesis through NF-κB signaling pathway. Bioorg Chem. 2018;78:94–102. doi: 10.1016/j.bioorg.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 147.Yu J., Ciancetta A., Dudas S., Duca S., Lottermoser J., Jacobson K.A. Structure-guided modification of heterocyclic antagonists of the P2Y14 receptor. J Med Chem. 2018;61:4860–4882. doi: 10.1021/acs.jmedchem.8b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.More K.N., Lee J.Y., Kim D.-Y., et al. Acetazolamide-based [18F]-PET tracer: in vivo validation of carbonic anhydrase IX as a sole target for imaging of CA-IX expressing hypoxic solid tumors. Bioorg Med Chem Lett. 2018;28:915–921. doi: 10.1016/j.bmcl.2018.01.060. [DOI] [PubMed] [Google Scholar]
- 149.Kumar R., Vats L., Bua S., Supuran C.T., Sharma P.K. Design and synthesis of novel benzenesulfonamide containing 1,2,3-triazoles as potent human carbonic anhydrase isoforms I, II, IV and IX inhibitors. Eur J Med Chem. 2018;155:545–551. doi: 10.1016/j.ejmech.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 150.Vats L., Sharma V., Angeli A., Kumar R., Supuran C.T., Sharma P.K. Synthesis of novel 4-functionalized 1,5-diaryl-1,2,3-triazoles containing benzenesulfonamide moiety as carbonic anhydrase I, II, IV and IX inhibitors. Eur J Med Chem. 2018;150:678–686. doi: 10.1016/j.ejmech.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 151.Wu Y., Jiang Z., Li Z., Gu J., You Q., Zhang X. Click chemistry-based discovery of [3-Hydroxy-5-(1 H-1,2,3-triazol-4-yl)picolinoyl]glycines as orally active hypoxia-inducing factor prolyl hydroxylase inhibitors with favorable safety profiles for the treatment of anemia. J Med Chem. 2018;61:5332–5349. doi: 10.1021/acs.jmedchem.8b00549. [DOI] [PubMed] [Google Scholar]
- 152.Rajan S., Puri S., Kumar D., et al. Novel indole and triazole based hybrid molecules exhibit potent anti-adipogenic and antidyslipidemic activity by activating Wnt3a/β-catenin pathway. Eur J Med Chem. 2018;143:1345–1360. doi: 10.1016/j.ejmech.2017.10.034. [DOI] [PubMed] [Google Scholar]
- 153.Narsimha S., Battula K.S., Nagavelli V.R. One-pot synthesis of novel 1,2,3-triazole-pyrimido[4,5-c]isoquinoline hybrids and evaluation of their antioxidant activity. Synth Commun. 2018;48:1220–1226. [Google Scholar]
- 154.Chekir S., Debbabi M., Regazzetti A., et al. Design, synthesis and biological evaluation of novel 1,2,3-triazole linked coumarinopyrazole conjugates as potent anticholinesterase, anti-5-lipoxygenase, anti-tyrosinase and anti-cancer agents. Bioorg Chem. 2018;80:189–194. doi: 10.1016/j.bioorg.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 155.Kayet A., Datta D., Das A., Dasgupta S., Pathak T. 1,5-Disubstituted 1,2,3-triazole linked disaccharides: metal-free syntheses and screening of a new class of ribonuclease A inhibitors. Bioorg Med Chem. 2018;26:455–462. doi: 10.1016/j.bmc.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 156.Kozarski M., Kubacka D., Wojtczak B.A., Kasprzyk R., Baranowski M.R., Kowalska J. 7-Methylguanosine monophosphate analogues with 5′-(1,2,3-triazoyl) moiety: synthesis and evaluation as the inhibitors of cNIIIB nucleotidase. Bioorg Med Chem. 2018;26:191–199. doi: 10.1016/j.bmc.2017.11.032. [DOI] [PubMed] [Google Scholar]
- 157.Peterson K., Kumar R., Stenström O., et al. Systematic tuning of fluoro-galectin-3 interactions provides thiodigalactoside derivatives with single-digit nM affinity and high selectivity. J Med Chem. 2018;61:1164–1175. doi: 10.1021/acs.jmedchem.7b01626. [DOI] [PubMed] [Google Scholar]
- 158.Lu K., Cai L., Zhang X., et al. Design, synthesis, and biological evaluation of novel substituted benzamide derivatives bearing a 1,2,3-triazole moiety as potent human dihydroorotate dehydrogenase inhibitors. Bioorg Chem. 2018;76:528–537. doi: 10.1016/j.bioorg.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 159.Syeda S.S., Sánchez G., Hong K.H., Hawkinson J.E., Georg G.I., Blanco G. Design, synthesis, and in vitro and in vivo evaluation of ouabain analogues as potent and selective Na, K-ATPase α4 isoform inhibitors for male contraception. J Med Chem. 2018;61:1800–1820. doi: 10.1021/acs.jmedchem.7b00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Thomson C.G., Le Grand D., Dowling M., et al. Development of autotaxin inhibitors: a series of zinc binding triazoles. Bioorg Med Chem Lett. 2018;28:2279–2284. doi: 10.1016/j.bmcl.2018.05.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.