

Abstract
A one-tube reverse transcription-polymerase chain reaction (RT-PCR) for absolute feline coronavirus (FCoV) quantitation was developed. The assay is based on the 5′ nuclease activity of the Thermus flavus (Tfl) polymerase and a fluorogenic probe which generates fluorescence when it is cleaved. The fluorogenic probe, also called TaqMan™ probe (Perkin Elmer, Foster City, USA), is an oligonucleotide designed to bind between the two PCR primers to the target cDNA and is labeled with a reporter and a quencher dye. In the intact probe, the quencher dye suppresses the fluorescence of the reporter dye by Förster-type energy transfer. During the polymerase extension steps the Tfl exonuclease activity cleaves the hybridised probe resulting in the generation of fluorescent emission of the reporter dye. The threshold cycle (CT value) indicates the increase of reporter fluorescence and is directly related to the initial amount of target cDNA or RNA, respectively. Fluorescence is monitored in real time after each cycle by a Perkin-Elmer ABI Prism® 7700 Sequence Detector. After completion of amplification, the CT values of the samples are calculated back to a standard curve, generated by amplification of diluted standard molecules. The one-tube RT-PCR described below allows precise quantitation, is highly sensitive, rapid (no separate reverse transcription step and no post-amplification steps), easy to handle, allows for a high sample throughput, shows a very good reproducibility, and can be executed with a low risk of contamination. The design of the primers–probe combination enables the detection of all known FCoV strains and is also useful for the detection of canine coronavirus, transmissible gastroenteritis virus and porcine respiratory coronavirus.
Keywords: FCoV, Viral RNA, Quantitation, One-tube RT-PCR, TaqMan™, Fluorogenic 5′ nuclease assay
1. Introduction
Feline coronavirus (FCoV) is known to be highly prevalent in the cat population, especially in catteries (Addie and Jarrett, 1992, Pedersen, 1995, Fehr et al., 1997). FCoV causes mild gastrointestinal and respiratory diseases mainly in kittens (Pedersen et al., 1981) and, it is hypothesised, due to mutations during intestinal replication (Pedersen, 1995, Vennema et al., 1995), feline infectious peritonitis (FIP). FIP is actually the most important fatal infectious disease in cats, ≈5–12% of seropositve cats develop lethal FIP (Addie and Jarrett, 1992, Fehr et al., 1997). The pathogenicity of FCoV leading to the FIP syndrome may be linked to increased replication due to a high FCoV load in a cat which can not be determined without a quantitative detection of the causative agent. The fact that FIP is still incurable emphasises that fighting FIP has to depend on preventive measures. Efficacious prevention can be accomplished by detection and separation of FCoV shedding from non shedding cats, resulting in the reduction of coronaviral load or even the elimination of FCoV from a cattery (Foley et al., 1997). Vaccination has been shown to be efficacious if the cats are FCoV naive at the time of vaccination (Fehr et al., 1997). Both strategies require a method to detect FCoV quantitatively, because only cats shedding FCoV at high level may transmit the virus to other cats (Foley et al., 1997).
It becomes evident that quantitation of coronaviral load in connection with the detection of FCoV shedders or the development of strategies for the prevention or elimination of FCoV in catteries will depend on PCR procedures that allow the reliable and fast analysis of large volumes of samples.
Several polymerase chain reaction (PCR) based methods have been suggested in detection of the positive-stranded FCoV RNA, but none of these were designed to be quantitative (Li and Scott, 1994, Herrewegh et al., 1995, Gamble et al., 1997). In addition, conventional PCR methods are time consuming due to several post-amplification steps, contain a certain risk of cross-contamination between the samples due to a separate labor-intensive reverse transcription (RT) step and a second PCR step in nested PCR systems, are limited in sensitivity and allow only relatively few samples to be processed at one time. The ideal method for FCoV detection should be quantitative, highly sensitive, fast, easy to handle, allow for a high sample throughput and contain a low risk of contamination.
Here we report a fluorogenic probe-based one-tube FCoV RT-PCR assay based on the reverse transcription activity of avian myeloblastosis virus reverse transcriptase (AMV-RT) in combination with the polymerisation and 5′ nuclease activity of Thermus flavus (Tfl) polymerase which fulfills all these criteria. For absolute quantitation, a dilution series of FCoV RNA-standard is amplified with the samples. The assay is based on the reverse transcription and amplification of a portion of the FCoV 7b gene, which is known to be highly conserved among coronavirus isolates (Herrewegh et al., unpublished data).
A fluorogenic probe (a 25 base target cDNA specific oligonucleotide which is added to the RT-PCR mixture) is designed to hybridise to a sequence located between the PCR primers (Fig. 1 ). A fluorescent reporter dye FAM (6-carboxyfluorescein) is covalently attached to the 5′ end of the probe and a quencher dye TAMRA (6-carboxytetramethylrhodamine) is covalently attached to the 3′ end of the probe. The quencher dye absorbs the emission of the reporter dye by Förster-type energy transfer while the probe is intact (Förster, 1948). The 3′ end of the probe is phosphorylated to prevent extension by the Tfl during the polymerisation steps. While the reporter dye emission is quenched due to the physical proximity of the quencher dye in the intact probe, the 5′→3′ exonuclease activity of Tfl polymerase leads to cleavage of the probe in the extension phase of the PCR cycle. The effect is a dislocation of the reporter dye and, as a result of the decrease of the energy transfer, a rise of the reporter’s fluorescence. The resulting relative increase of the reporter dye’s fluorescencent emission is measured in the course of every PCR cycle using a Perkin Elmer ABI Prism 7700 Sequence Detector, which is a combination of thermal cycler and luminescence spectrometer. The data are transferred to a connected computer where calculations and graphics are demonstrated by a special software.
Fig. 1.
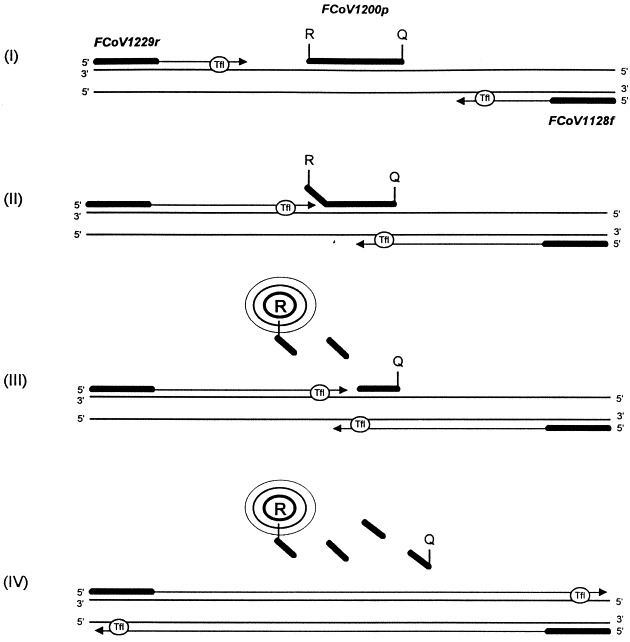
Principles of the 5′ nuclease fluorogenic assay for feline coronavirus.
2. Methods and materials
2.1. Primers and probe
Primers and probe sequences (Table 1 ) were designed with Perkin-Elmer Primer Express™ software. The probe sequence was selected in conformity to the manufacturers guidelines (Livak et al., 1995), which are briefly (a) a melting temperature of ≈5°C above the annealing temperature; (b) no complementarity to forward or reverse primer; (c) no self-complementarity; (d) not four or more identical nucleotides in a row, especially Gs; (e) no G at the 5′ end of the probe; and (f) a slightly increased melting temperature compared to that of the primers. The TaqMan™ fluorogenic probe was synthesised by Perkin-Elmer, Weiterstadt, Germany. The fluorescent reporter dye at the 5′ end of the FCoV probe was FAM (6-carboxyfluorescein) and the rhodamine quencher dye at the 3′ end was TAMRA (6-carboxytetramethylrhodamine). A phosphate group was added at the 3′ end of the probe to prevent extension during polymerisation.
Table 1.
(a) Feline coronavirus (FCoV) primers and probe for the FCoV fluorogenic assay; (b) position of primers and probe in the sequence of the FCoV 7b gene
| Primer or probe |
Sequence (5′→3′) |
Length of fragment (bp) |
|||
| (a) | |||||
| Forward primer FcoV1128f | GAT TTG ATT TGG CAA TGC TAG ATT T | ||||
| Reverse primer FcoV1229r | AAC AAT CAC TAG ATC CAG ACG TTA GCT | 102 | |||
| Probe FCoV1200p | aTCC ATT GTT GGC TCG TCA TAG CGG Ab | ||||
| (b) | |||||
| 1001 | GGCAACCCGA | TGTCTAAAAC | TGGTCTTTCC | GAGGAATTAC | GGGTCATCGC |
| 1051 | GCTGCCTACT | CTTGTACAGA | ATGGTAAGCA | CGTGTAATAG | GAGGTACAAG |
| FCoV1128→ | |||||
| 1101 | CAACCCTATT | GCATATTAGG | AAGTTTAGAT | TTGATTTGGC | AATGCTAGAT |
| ←FCoV1200p | |||||
| 1151 | TTAGTAATTT | AGAGAAGTTT | AAAGATCCGC | TATGACGAGC | CAACAATGGA |
| ←FCoV1229r | |||||
| 1201 | AGAGCTAACG | TCTGGATCTA | GTGATTGTTT | AAAATGTAAA ATTGTTTGAA | |
| 1251 | AATTTTCCTT | TTGATAGTGA | TG | ||
Reporter dye (FAM) labeled nucleotide.
Quencher dye (TAMRA) labeled nucleotide.
The primer pair for the FCoV fluorogenic probe was selected by paying attention that no loop or dimer formation with the other primers and a melting temperature of ≈59°C. Because all FCoV strains should be detectable, probe and primers were designed to have a high cross-reactivity with all known FCoV strains. Additionally, this approach should also be efficient for the detection of canine coronavirus (CCV), transmissible gastroenteritis virus (TGE) and porcine respiratory coronavirus (PRCV). A compilation of sequence comparisons and the cross-reactivity of our FCoV fluorogenic approach with different coronaviruses is shown in Table 2. The Genetics Computer Group (GCG, University of Wisconsin) sequence analysis package was used for sequence analysis.
Table 2.
Conformity of the feline coronavirus (FCoV) fluorogenic assay sequence with the sequence of canine coronavirus (CCV), transmissible gastroenteritis virus (TGEV), porcine respiratory coronavirus (PRCV), and human coronavirus 229E (HCV); the nonconformity between TaqMan™ and HCV sequence decreases the risk of contamination due to shedded human coronaviruses
| FCoV | GATTTGATTT | GGCAATGCTA | GATTTAGTAA | TTTAGAGAAG | T..TTAAAGATC | |
| CCV | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – .. – – – – – – – – – | |
| TGEV | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – .. – – – – – – – – – | |
| PRCV | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – .. – – – – – – – – – | |
| HCV | T–GG– – –ACG | CTAGTATAAC | TCA– – –CA– – | –G–GCT–G– – | –AA–C– – – – – –. | |
| FCoV | CGCTATGACG | AGCCAACAAT | GGAAGAGCTA | ACGTCTGGAT | CTAGTGATTG | TT |
| CCV | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – |
| TGEV | – – – – –C– – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – |
| PRCV | – – – – –C– – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – – – – – – – – – | – – |
| HCV | – – –AT– – – – – | – – – – – – – – – – | – – – – – – – –C– | GTCAT–T–TC | T–GAGACC–A | –C |
2.2. Feline coronavirus standard for absolute quantitation
Standard RNA templates were created as follows: complementary DNA (cDNA) from an American FCoV strain was amplified by PCR with primers FCoV1128f and FCoV1229r. The fragment was separated on a 2% agarose gel, excised, isolated from the gel and ligated into the polylinker of a pT7-blue vector (Novagen, Madison, WI). The recombinant plasmid (named pT7-StFCoV) was transformed into E. coli (NovaBlue, Novagen) and a positive colony was amplified in LB-medium containing ampicillin. The plasmids were purified (Birnboim and Doly, 1979), linearised, purified by gel electrophoresis and extracted from the gel using a DEAE membrane (Dretzer et al., 1981). The standard plasmids were quantified by spectrophotometrical analysis. Verification and orientation of the sequence was verified by sequencing. The amount of 1.5 μg of the linearised plasmid was used to perform an in vitro transcription with T7 polymerase (Boehringer Mannheim, Mannheim, Germany) at 37°C for 2 h. After digestion with RNase free DNase, the resulting RNA transcripts were purified with phenol-chloroform and precipitated with isopropanol. The RNA transcripts were disolved in diethylpyrocarbonate (DEPC) treated water and quantified by spectrophotometrical analysis. The dilutions of the standard plasmid were carried out in TE buffer (pH 7.6) containing 30 μg carrier RNA (transfer RNA from E. coli, Sigma, Buchs, Switzerland) /ml. Endpoint dilution amplification was used to confirm the calculated copy number. The diluted RNA stock was aliquoted and frozen immediately at −70°C. Each aliquot was used only once for a fluorogenic RT-PCR.
2.3. Fluorogenic reverse transcription-polymerase chain reaction
The 50 μl PCR mixture for one reaction contained 7 μl optimised single-buffer 5×(Access RT-PCR system, Promega, Madison, WI), 1.5 μl PCR buffer 10×(500 mM KCl, 100 mM Tris–HCl (pH 8.3), passive reference dye ROX, which is part of the Perkin-Elmer TaqMan® buffer A), 2 mM MgSO4, 500 μM deoxynucleotide triphosphates, 0.1% Triton X-100 (t-octylphenoxypolyethoxyethanol, Sigma, Buchs, Switzerland), 1 μM of each primer, 200 nM of fluorogenic probe, 5 U of AMV reverse transcriptase/reaction, 5 U of Tfl polymerase/reaction and 10 μl of diluted standard or template RNA. After a reverse transcription step of 45 min at 48°C and a denaturation step of 2 min at 94°C, 45 cycles, each 1 min at 64°C, 2 min at 68°C, and 30 s at 94°C, followed. Reverse transcription and amplification were carried out in a single tube in an ABI Prism® 7700 Sequence Detector without modifying or moving the samples between RT and PCR.
2.4. Feline coronavirus strains and other coronaviruses
To evaluate the cross-reactivity of this FCoV RT-PCR assay, several FCoV strains and isolates from coronaviruses of other species than the cat were tested (Table 5). Cell culture natants were used without being modified whereas faeces samples and ascites fluid were diluted 1:10 and treated at 95°C for 10 min to eliminate RT-PCR impeding factors.
Table 5.
Cross-reactivity of the feline coronavirus (FCoV) fluorogenic assay with several FCoV strains and coronaviruses from other species
| Isolate |
Kind of sample |
Result |
| Feline coronavirus strains (FCoV) | ||
| FIPV 204859 | Cell culture supernatant | Detectable |
| FIPV UCD 1 VP II | Cell culture supernatant | Detectable |
| UCD 5 | Cell culture supernatant | Detectable |
| FIPV UCD 8 | Ascites fluid | Detectable |
| FeCV UCD 1 | Faeces suspension | Detectable |
| FeCV RM | Faeces suspension | Detectable |
| Canine coronavirus (CCV) | Cell culture supernatant | Detectable |
| Transmissible gastroenteritis virus (TGEV) | Cell culture supernatant | Detectable |
| Porcine respiratory coronavirus (PRCV) | Cell culture supernatant | Detectable |
| Porcine epidemic diarrhoea virus (PEDV) | ||
| Isolate 1 | Cell culture supernatant | Not detectable |
| Isolate 2 | Cell culture supernatant | Not detectable |
| Isolate 3 | Cell culture supernatant | Not detectable |
| Human coronavirus (HCV) strain 229E | ||
| Isolate 1 | Cell culture supernatant | Not detectable |
| Isolate 2 | Cell culture supernatant | Not detectable |
2.5. Reverse transcription-polymerase chain reaction data analysis
During amplification, fluorescence intensity of FAM, TAMRA and ROX in each tube of a 96-well plate is measured by the ABI Prism 7700 Sequence Detector. Fluorescence is monitored after each cycle when running the ‘real time’ mode or only at the end point of PCR running the ‘plate read only’ mode. The ‘real time’ mode is accordingly used for quantitative PCR, the ‘plate read only’ mode for qualitative results. Data are transferred to a connected computer and saved. Calculations and graphic presentation of the generation of fluorescence are carried out by the Sequence Detector Software (SDS), which is part of the ABI Prism 7700 Sequence Detection system. For statistical analysis data were transferred into a Microsoft Excel worksheet.
Running the ‘real time’ mode, the SDS software offers two possibilities for data analysis, ΔR n and C T (threshold cycle value) calculation. The ΔR n calculation mode is based on the R n value, which is the ratio of the fluorescence signal between the reporter dye and the passive reference ROX. During PCR, R n increases due to the cleavage of a probe with every copy of target that is produced, until the reaction reaches its plateau. ΔR n represents the normalised reporter signal minus the base line signal established in the first few cycles of PCR. Like R n, ΔR n increases during PCR as amplicon copy number increases until the reaction approaches a plateau.
The threshold cycle (C T) represents the PCR cycle at which an increase in reporter fluorescence above a base line signal can first be detected. A threshold of 10 S.D. above the base line is used to determine the C T values. Because of a correlation between C T value and starting copy number of the template, the C T value is used for quantitation of the initial template number in the samples. The Sequence Detection software generates a standard curve of C T versus starting copy number for all standards and then determines the starting copy number of unknowns by interpolation.
2.6. Standard sequencing
The specificity of the fluorogenic RT-PCR was confirmed by sequencing the plasmid pT7-StFCoV by the chain termination method (Microsynth, Balgach, Switzerland). Two sequencing attempts showed right insert and right orientation of the insert in the plasmid.
3. Results
The analytical sensitivity of the fluorogenic RT-PCR assay was 10–100 times greater than the conventional nested RT-PCR (Herrewegh et al., 1995). A dilution series of a modified life FCoV vaccine (Primucell-FIP™) was prepared and tested in triplicate by fluorogenic RT-PCR and by nested RT-PCR. Nested RT-PCR products were evaluated by agarose gel electrophoresis. Whereas the conventional nested RT-PCR detected the FCoV down to a 1:107 dilution (2 of 3 positive), the fluorogenic RT-PCR detected the whole virus even to a 1:109 dilution (1 of 3 positive) (Table 3 ).
Table 3.
Sensitivity of feline coronavirus (FCoV) fluorogenic assay and nested FCoV reaction transcription-polymerase chain reaction (RT-PCR)
| Dilution |
Fluorogenic assay |
Nested RT-PCR |
||||
| 1/104 | + | + | + | + | + | + |
| 1/105 | + | + | + | + | + | + |
| 1/106 | + | + | + | + | + | + |
| 1/107 | + | + | + | + | + | − |
| 1/108 | − | + | + | − | − | − |
| 1/109 | + | − | − | − | − | − |
| 1/1010 | − | − | − | − | − | − |
| 1/1011 | − | − | − | − | − | − |
To determine the reproducibility of the fluorogenic RT-PCR dilutions of the standard (50, 500, and 5000 molecules of standard RNA) and viral RNA (1:106, 1:105, and 1:104 dilutions of a modified life FCoV vaccine) were prepared. The precision within-run was tested by pipetting the same dilutions ten times on the same 96-well reaction plate, the precision from run-to-run by running the same dilutions in ten different RT-PCRs. In all RT-PCRs all standard samples and samples with viral RNA were detected. The coefficient of variation (in %) of the precision within-run and from run-to-run was in the range of 0.57–1.50 and 1.16–3.42, respectively (Table 4 ).
Table 4.
Coefficient of variation (in %) of precision within-run and precision from run-to-run
| Standard RNA (copies) |
Complete virus (dilution) |
|||||
| 50 |
500 |
5000 |
1:106 |
1:105 |
1:104 |
|
| CV within-run | 1.50 | 0.88 | 0.69 | 0.57 | 0.91 | 0.59 |
| CV from run-to-run | 3.42 | 2.10 | 1.70 | 2.82 | 2.09 | 1.16 |
To assess the risk of cross-contamination, either by PCR products of previous reactions or during pipetting on the 96-well plate, 120 no template controls were included between the samples for reproducibility testing. To decrease the risk of cross-contamination, pipetting was performed in a laminar flow hood. The 96-well plates used in the 7700 ABI Prism system might generate a problem of cross-contamination because pipetting has to be done with the PCR tubes open. All the wells used are only closed with rows of linked caps after pipetting. However, none of the 120 no template controls gave a positive signal indicative for cross-contamination on the 96-well plate. The risk of cross-contamination can therefore be considered as very low, when pipetting is done in a laminar flow hood.
To verify that the amplification efficiency of the FCoV standard RNA transcripts is equal to that of the same sequence within the viral genome, the efficiencies of amplification of both templates has to be shown to be approximately equal. A very sensitive method to compare the efficiencies of amplification is to compare the slope of the standard curve of a standard RNA dilution series to the slope of the standard curve of a dilution series of complete virus particles. The efficiencies can be considered as equal if the difference of the slopes (Δs) is smaller than 0.1. In this approach, the Δs value was 0.06 (Fig. 2 ), which proves that these standard RNA transcripts are reliable for FCoV quantification.
Fig. 2.
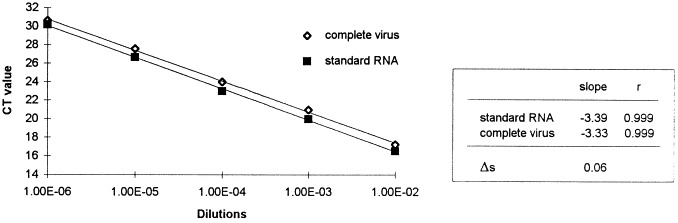
Amplification efficiencies of standard RNA and complete virus.
Several isolates were used to determine the cross-reactivity of this RT-PCR assay. The expected results, based on the sequence comparisons, were confirmed: all tested FCoV isolates as well as CCV, TGEV and PRCV were detectable, HCV 229E and PEDV were not detectable (Table 5 ).
4. Discussion
PCR is an extremely sensitive method for the detection of specific nucleic acid sequences. A disadvantage of conventional PCR is that it is a very time-consuming process, especially if the template is RNA and a reverse transcription step is needed or if a second round of amplification is necessary to achieve the required sensitivity. Furthermore, such steps increase the risk of inaccuracy and contamination. Several RT-PCR assays have been developed to detect FCoV (Li and Scott, 1994, Herrewegh et al., 1995, Gamble et al., 1997). Two of those assays are nested PCRs and in all of those ethidium bromide staining and UV light transillumination of electrophoretically separated PCR products is necessary to detect PCR products. A further big disadvantage of all those RT-PCRs is that none of them is able to quantitate FCoV.
The one-tube fluorogenic RT-PCR assay for FCoV quantitation described here is fast, simple and reliable. The basis of this assay is a fluorogenic oligonucleotide, called TaqMan™ probe, which is added to the RT-PCR mix. This fluorogenic probe is cleaved during the PCR polymerisation steps due to a 5′ nuclease activity of the Tfl polymerase. While the unhybridised probe does not contribute significantly to the signal, the cleavage of the probe leads to the generation of fluorescence. This fluorescence is measured during each cycle by a luminescence spectrometer (ABI Prism 7700) and transferred to a connected computer, where graphics and calculations, for example standard curves or initial amounts of template in the samples, are automatically performed. The use of such a fluorogenic probe increases the level of specificity of the assay. This assay is a one-tube RT-PCR, where all ingredients for RT and PCR are mixed and RT-PCR is performed without additional handling or reopening of the tubes. No post amplification steps are required and calculation of the initial amount of viral particles is done by the software, resulting in a very short processing and assay time. RT-PCR testing of ≈90 samples, including pipetting, can be performed on a 96-well plate including some non template controls and standard dilutions in <6 h. The standard described, a cloned part of the highly conserved FCoV 7b gene, allows absolute quantitation of FCoV in clinical samples.
The precision within-run and from run-to-run was evaluated with dilutions of the standard RNA transcripts and dilutions of a modified life vaccine (Primucell-FIP™). The coefficient of variation of the precision within-run and from run-to-run was in the range of 0.57–1.50% and 1.16–3.42%, respectively. Thus, the reproducibility in this assay is better than by conventional RT-PCR, where, for example, the precision within-run is in the range of 2–7% (Wang et al., 1989, Kinoshita et al., 1992, Nagano and Kelly, 1994).
The analytical sensitivity of the assay described above was 10–100 times greater than that of the conventional nested PCR. This sensitivity combined with the capability of quantitation makes this assay a valuable tool for FCoV research and diagnosis of FCoV infection.
The risk of contamination has been shown to be very low when pipetting is done in a laminar flow hood and when the different working steps were strictly separated. The fact that the RT-PCR tubes are never opened during the PCR process is an advantage of the fluorogenic RT-PCR, and reduces the risk of contamination.
The standard template was shown to be useful for quantitation of the complete viral RNA, based on the amplification efficiency which was shown to be approximately equal for both amplicons.
This FCoV assay was designed to detect all strains of FCoV. It should also be useful for the detection of other coronaviruses, such as canine coronavirus (CCV), transmissible gastroenteritis virus (TGEV) and porcine respiratory coronavirus (PRCV), but not human coronavirus (HCV 229E), because a considerable part of infectious respiratory diseases in humans is caused by HCV 229E and a detection of this strain might lead to a risk of contamination due to HCV 229E possibly shed by the people handling the samples and reagents. The FCoV sequence selected in the present study has, as desired, a high conformity to CCV, TGEV and PRCV but only a low conformity to HCV 229E. PEDV might not be detectable, because it is reported that no 7b gene is existing (Bridgen et al., 1993, Singh et al., submitted for publication). All FCoV strains tested and several coronaviruses of other species than the cat were detected by the assay revealing its wide cross-reactivity. HCV 229E and PEDV were in fact not detectable.
These results show that our one-tube fluorogenic RT-PCR for FCoV quantitation allows absolute quantitation, is highly sensitive, reliable, rapid, easy to handle, enables a high sample throughput, and contains a low risk of contamination. These characteristics make this assay an excellent tool for the detection of FCoV.
Acknowledgements
Support for this project was provided by the Union Bank of Switzerland on behalf of a customer and a grant from Pfizer Animal Health. HCV 229E, PEDV, and PRCV isolates were kindly provided by Professor M. Ackermann, Institute of Virology, School of Vetinary Medicine, University of Zurich. We thank Dr R. Wicki, Ph.D., Perkin Elmer, for technical support with the ABI Prism 7700 and Dr. M. Koller, Catalys, Switzerland for assistance concerning the Promega Access RT-PCR system.
References
- Addie D.D., Jarrett O. A study of naturally occurring feline coronavirus infections in kittens. Vet. Rec. 1992;130(7):133–137. doi: 10.1136/vr.130.7.133. [DOI] [PubMed] [Google Scholar]
- Birnboim H.C., Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7(6):1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgen A., Duarte M., Tobler K., Laude H., Ackermann M. Sequence determination of the nucleocapsid protein gene of the porcine epidemic diarrhoea virus confirms that this virus is a coronavirus related to human coronavirus 229E and porcine transmissible gastroenteritis virus. J. Gen. Virol. 1993;74(Pt 9):1795–1804. doi: 10.1099/0022-1317-74-9-1795. [DOI] [PubMed] [Google Scholar]
- Dretzer G., Bellard M., Sassone-Corsi P., Chambon P. A reliable method for the recovery of DNA fragments from agarose and acrylamide gels. Anal. Biochem. 1981;112(2):295–298. doi: 10.1016/0003-2697(81)90296-7. [DOI] [PubMed] [Google Scholar]
- Fehr D., Holznagel E., Bolla S., Hauser B., Herrewegh A.A.P.M., Horziner M.C., Lutz H. Placebo-controlled evaluation of a modified life virus vaccine against feline infectious peritonitis: safety and efficacy under field conditions. Vaccine. 1997;15(10):1101–1109. doi: 10.1016/S0264-410X(97)00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley J.E., Poland A., Carlson J., Pedersen N.C. Patterns of feline coronavirus infection and faecal shedding from cats in multiple-cat environments. J. Am. Vet. Med. Assoc. 1997;210(9):1307–1312. [PubMed] [Google Scholar]
- Förster V.T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phy. 1948;2:55–57. [Google Scholar]
- Gamble D.A., Lobbiani A., Gramegna M., Moore L.E., Colucci G. Development of a nested PCR assay for detection of feline infectious peritonitis virus in clinical specimens. J. Clin. Microbiol. 1997;35(3):673–675. doi: 10.1128/jcm.35.3.673-675.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A.P.M., de Groot R.J., Cepica A., Egberink H.F., Horzinek M.C., Rottier P.J. Detection of feline coronavirus RNA in faeces, tissues, and body fluids of naturally infected cats by reverse transcriptase PCR. J. Clin. Microbiol. 1995;33(3):684–689. doi: 10.1128/jcm.33.3.684-689.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh, A.A.P.M., Vennema, H., Horzinek, M.C., Rottier, P.J.M., de Groot, R.J. (unpublished data).
- Kinoshita T., Imamura J., Nagai H., Shimotohno K. Quantification of gene expression over a wide range by the polymerase chain reaction. Anal. Biochem. 1992;206(2):231–235. doi: 10.1016/0003-2697(92)90358-e. [DOI] [PubMed] [Google Scholar]
- Li X., Scott F.W. Detection of feline coronaviruses in cell cultures and in fresh and fixed feline tissues using polymerase chain reaction. Vet. Microbiol. 1994;42(1):65–77. doi: 10.1016/0378-1135(94)90078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K., Marmaro, J., Flood, S., Elmer, P., December 1995, Guidelines for designing TaqMan™ fluorogenic probes for 5′ nuclease assays, Perkin Elmer applied biosystems research news.
- Nagano M., Kelly P.A. Tissue distribution and regulation of rat prolactin receptor gene expression; quantitative analysis by polymerase chain reaction. J. Biol. Chem. 1994;269(18):13337–13345. [PubMed] [Google Scholar]
- Pedersen N.C. An overview of feline enteric coronavirus and infectious peritonitis virus infections. Feline Pract. 1995;23:7–22. [Google Scholar]
- Pedersen N.C., Boyle J.F., Floyd K. Infection studies in kittens, using feline infectious peritonitis virus propagated in cell culture. Am. J. Vet. Res. 1981;42(3):363–367. [PubMed] [Google Scholar]
- Singh, M., Tobler, K., Ackermann, M. A novel internal ORF product expressed from a polycistronic mRNA of porcine epidemic diarrhoea virus does not contribute to viral virulence (submitted for publication).
- Vennema H., Poland A., Hawkins K.F., Pedersen N.C. A comparison of the genomes of FECVs and FIPVs and what they tell us about the relationships between feline coronaviruses and their evolution. Feline Prac. 1995;23(3):40–44. [Google Scholar]
- Wang A.M., Doyle M.V., Mark D.F. Quantitation of mRNA by the polymerase chain reaction (published erratum appears in Proc. Natl. Acad. Sci. USA, 1990 April 87 (7), 2865) Proc. Natl. Acad. Sci. USA. 1989;86(24):9717–9721. doi: 10.1073/pnas.86.24.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]