

Abstract
As a complementary approach to two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), multi-dimensional chromatography separation methods have been widely applied in all kinds of biological sample investigations. Multi-dimensional liquid chromatography (MDLC) coupled with bio-mass spectrometry (MS) is playing important roles in proteome research due to its high speed, high resolution and high sensitivity. Proteome analysis strategies mainly include bottom-up and top-down approaches which carry out biological sample separation based on peptide and protein levels, respectively. Electrophoretic methods combined with liquid chromatography like IEF-HPLC and HPLC-SDS-PAGE have been successful applied for protein separations. As for MDLC strategy, ion-exchange chromatography (IEX) together with reversed phase liquid chromatography (RPLC) is still a most widely used chromatography in proteome analysis, other chromatographic methods are also frequently used in protein pre-fractionations, while affinity chromatography is usually adopted for specific functional protein analysis. Recent MDLC technologies and applications to variety of proteome analysis have been achieved great development. A digest peptide-based approach as so-called “bottom-up” and intact protein-based approach “top-down” analysis of proteome samples were briefly reviewed in this paper. The diversity of combinations of different chromatography modes to set up MDLC systems was demonstrated and discussed. Novel developments of MDLC techniques such as high-abundance protein depletion and chromatography array were also included in this review.
Keywords: Multi-dimensional liquid chromatography, Proteomic analysis, Bottom-up and top-down approaches
1. Introduction
With the demands of proteome analysis, high-resolution and high-throughput peptide/protein-based separation technologies are undergoing great development. As an orthogonal highly resolving separation technique, two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), introduced more than a quarter century ago [1], [2] still plays a central role in many analysis of proteome samples such as cell lines, tissues or biological fluids. But the 2D-PAGE has some limitations in protein analysis for high- (>150 kDa) and low-molecular weight (<10 kDa) proteins as well as those of proteins with extreme pI values. Another limitation of 2D-PAGE is that it cannot be coupled on-line with MS to achieve a fast multi-dimensional protein separation and identification [3]. As a result, multi-dimensional liquid phase-based separation methods using different electrophoretic and chromatographic techniques have been greatly developed as complementary methodologies. The feasibility of multi-modular combinations of HPLC, isoelectric focusing (IEF), chromatofocusing (CF), capillary electrophoresis (CE) as well as combinations of different HPLC modes provides numerous options for the separation of protein complexes and peptides.
There are basically two strategies in multi-dimensional separation systems used in proteomic research, one is called “bottom-up” and the other is “top-down” approach. The “shotgun” approach is one of the typical “bottom-up” strategy which is based on reversed phased or multi-dimensional liquid chromatography separations of tryptic digests of a whole protein sample coupled on-line with electrospray mass spectrometry is excellent for identifying a large number of proteins [4], [5], [6], [7]. Such a strategy has made a great contribution in the research area of expression proteomics. While the “top-down” method is based on the separations of intact protein complexes, therefore it is able to get molecular information about the intact protein and may be advantageous for the detection of proteins’ post-translational modifications (PTMs) [8]. Multi-dimensional separation methods are carried out either on peptide or on protein level according to their prospective goals.
Many reviews on multi-dimensional separation strategies were published to provide a comprehensive view of the development and application of these technologies in proteome investigations [3], [9], [10], [11]. Here we have briefly reviewed recent development and application of multi-dimensional liquid phased chromatography in proteomic research. Chromatographic together with electrophoretic methods as well as MDLC systems are still the promising methodologies for proteome research. Improvement and innovation of multi-dimensional liquid-based separation methods have been made continuously for better settlement of various kinds of proteome investigations. MDLC strategies in either bottom-up or top-down approach for proteome researches will be mainly concerned. Novel usages of MDLC such as high-abundant protein depletion and chromatography array will also be discussed.
2. Bottom-up approach
The research of proteome is much more complicated than genome research mainly because of the complexity of proteome samples. As far as we know, no single chromatographic or electrophoretic procedure to date possesses the peak capacity required to resolve such a complex mixture into its individual components [10]. Nevertheless, multi-dimensional separation strategies still make it possible to resolve such problems. Among all these methods, peptide-based separation approach are of widely used since peptides possess greater solubility in a wider range of solvents and are hence easier to separate than proteins.
2.1. LC coupled with electrophoretic techniques
Electrophoretic methods are usually combined with chromatographic separation approaches because of the orthogonality, high resolution and compatibility with chromatography. Balgley et al. developed an on-line capillary isoelectric focusing combined with nano-reversed phase liquid chromatography in an automated and integrated platform for cell heterogeneity proteome research [12]. CIEF fractions were loading directly to trap columns packed with C18 RPLC by using two 6-port micro-valves. Ten micrograms of digested proteins extracted from glioblastoma multiforme tissue were loaded in CIEF–nano-RPLC-ESI-MS/MS run, and 6866 fully tryptic peptides corresponding to 1820 distinct proteins were identified. Compared with multi-dimensional HPLC runs like SCX–RPLC approach, CIEF–nano-RPLC method provides better resolution and requires less loading sample amount because of the ultrasensitive and excellent separation capability of capillary IEF. It is reported that the percentage of identified peptides present in more than one CIEF fraction is significantly less than that obtained from ion-exchange chromatography fractions [13]. Balgley et al. also utilize on-line CIEF–nano-RPLC-ESI-MS/MS for characterization of the human salivary proteome [14]. Micro-scale solution isoelectric focusing (μsol-IEF) coupled with analytical narrow-range 2D-PAGE has been successfully used to improve detection of low-abundance proteins [15]. Westman-Brinkmalm et al. investigated μsol-IEF as the pre-fractionation method prior to LC–MS/MS without cleaning up the buffer or detergent of the digested IEF fractions. This approach received better results than single LC–MS/MS separating approach and was also comparable to the results of μsol-IEF coupled with 2D-PAGE in analysis of proteins from a glioma cell line [16]. Aebersold's group reported the utilization of free-flow electrophoresis (FFE) as a pre-separation method before μRPLC-MS/MS instead of ion-exchange separation of peptides for protein analysis [17]. Some on-line LC-CE systems were also reviewed by Stroink et al. [18]. Further discussion will not be taken since this paperwork concerns mainly about MDLC systems.
2.2. Multi-dimensional LC separation methods
2.2.1. Hybrid-phase chromatography separations
Bi-phase column was first introduced by Yates et al. [19], [20], [21] as on-line multi-dimensional protein identification technology (MudPIT) where the SCX and RPLC materials are sequentially packed into a single micro-capillary column (Fig. 1 ). Similar procedures were reported for the separation of other biological fluids or cells [22], [23], [24], [25], [26]. Zeng et al. [27] reported an integrated column that consisted of one strong cation-exchange column and one reversed phase column. These two columns were connected directly to fulfill an on-line SCX–RPLC-MS/MS analysis of mouse liver proteins. Nine pH gradients generated from citric acid–trimethylamine buffer or 10 mM citric acid–ammonium hydroxide buffer were utilized instead of salt gradients which is commonly used in sample elution of SCX column so that SCX fractions can be loaded directly into RPLC column. The exclusion of salt removal steps in traditional SCX–RPLC on-line or off-line separation approach is the key point to achieve fast 2D-LC separation and MS/MS identification. On this basis Zeng's group developed a SCX and SAX (strong anion-exchange chromatography) combined pre-fractionation strategy called Yin-Yang multi-dimensional chromatography. It was used to provide an unbiased profiling of protein expression and phosphorylation of mouse liver tissue [28]. Peptides generated from SCX column with elution buffer at pH 2.5 were further separated by SAX column which also used a series of pH gradient buffer for gradient elution. Thereafter SAX fractions together with other SCX fractions were analyzed by nano-RPLC-MS/MS. This Yin-Yang MDLC was demonstrated to bare comprehensive separation ability towards peptide complexes with wide-spread pI values.
Fig. 1.
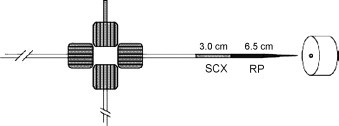
MudPIT electrospray interface including a biphasic micro-capillary column, packed with SCX and RP packing material connected to a micro-cross [19].
Jiang et al. used a SCX trap column instead of commonly used RP trap column for automated on-line nanoflow RPLC-MS/MS separations [29]. SCX trap column was employed to play the role of RP trap column to gain higher peak capacity. On the other hand, SCX trap column was coupled with nanoflow RPLC-MS/MS to fulfill multi-dimensional separations by introducing several volatile salt gradient steps to elute peptides bound to SCX trap column gradually. Another application of this SCX trap column system was investigated by Wang et al. in the same research group [30]. Since SCX trap column was used instead of RP trap column, good separation performance as well as good proteomic coverage was presented. Especially, basic and hydrophilic peptides with pI > 4.5 and GRAVY < −0.5 were found to be more efficiently analyzed.
Using tandem anion/cation-exchange columns can rapidly generate positively charged, negatively charged and neutral fractions [31], thus proteins or peptides with extensive pI values can be obtained and detected. To some extent, hybrid-phase columns like SCX–RPLC or RPLC with SCX trap column exhibit higher peak capacity and wider dynamic range than single-dimensional HPLC column. Nevertheless, the incompatibility of the elution conditions of different ion-exchange chromatography (IEX) as well as IEX/RPLC columns limits the use of these “hybrid” 2D-LC systems in comprehensive proteome separation. Only pH gradients or volatile salt gradients are allowed in these systems that will limit the separation ability of IEX columns.
2.2.2. Ion-exchange chromatography–RPLC
IEX together with RPLC has become an example of orthogonal 2D-LC analysis. The total peak capacity in this 2D separation can be greater than 5000 and high-sensitivity peptide identification may therefore be achieved because of increased resolution and the resultant decrease in peptide overlap [32]. Among various kinds of 2D-LC techniques, the combination of SCX mode separation in the first dimension and RP separation in the second dimension is the dominant separation technology of today. As a classical proteome research strategy, this SCX–RPLC approach provides thousands of biological information as a complement to traditional 2D-PAGE approach.
Fujjj et al. described a typical on-line SCX–RPLC workflow in their publication [33]. Stepwise salt elution using ammonium formate was employed for SCX separations and resulted fractions were subjected to RPLC-MS analysis after concentration and desalting. Vitali et al. [34] also used SCX–RPLC-MS/MS strategy to identify highly expressed proteins from Bifidobacterium infantis. A comprehensive view of the gene complement of B. infantis was obtained by this analysis. Digested proteins were first separated through a SCX column and the fractions were collected every minute. After drying and resuspending, fractions were further separated and identified through a home-made RPLC coupling with Q-TOF mass spectrometer. Ru et al. [35] run digested human urine sample 10 times using a MudPIT strategy for proteomic profiling. The relatively less amount of sample loading than 2D gel or gel-LC-based strategies was achieved. Nagalla et al. [36] investigated human cervical-vaginal fluid (CVF) proteome utilizing SCX–RPLC-MS/MS as well as one-dimensional gel electrophoresis analysis. A functional classification of human CVF proteome was made and compared with serum and amniotic fluid proteomes to provide a potential biomarker data foundation. Skipp et al. [37] did a research of Chlamydia trachomatis by a combination of 2-DE, MudPIT and SDS-PAGE-nano-RPLC-MS/MS strategies. Among these three approaches SDS-PAGE-nano-HPLC-MS/MS presented the best performance in detecting components of expressed proteome. Except for SCX–RPLC separation, McMullan et al. [38] used SAX column to separate soluble proteins of Geobacillus thermoleovorans T80 sub-proteome first, the fractions were collected, concentrated and digested into peptides to be further analyzed by RPLC-ESI-MS/MS. A total of 294 proteins were identified through this approach.
The technological developments of the coupling LC with MALDI make MALDI-MS/MS a powerful strategy in protein identification. Beeumen's group [39] adopted off-line 2D-LC coupled to MALDI-TOF-TOF mass spectrometry approach for myelin protein identification. Myelin protein extraction was digested and first separated through capillary SCX column and followed by nano-RPLC. RPLC fractions were spotted onto MALDI targets and analyzed by MALDI-TOF-TOF mass spectrometry. Less loading amount and better detection of proteins including membrane proteins, low-abundant proteins, and highly basic proteins were achieved and compared to 2D-PAGE approach. Hattan et al. [40] testified the feasibility of MALDI-MS/MS in coupling to HPLC for the analysis of whole cell lysates of wild-type yeast by three different LC workflows: SCX–RPLC, high-pH SAX–RPLC and RPLC (intact protein)-SCX–RPLC. The flexibility and reproducibility of the whole workflows were demonstrated through this work.
IEX–RPLC strategy plays a robust role in MDLC system. The combination of IEX–RPLC has the following characteristics: (1) IEX offers high sample capacity; (2) RPLC is excellently compatible with MS; (3) both modes provide high efficiency [41], [42]. The only limitation of this technique is the time required to achieve the separations. Since fractions of the first dimension must be separated further in the second dimensional RPLC gradient elution runs. The whole runs might require several days to complete [30].
2.2.3. Miscellaneous two-dimensional chromatography techniques
Some other 2D-LC modes, size exclusion chromatography (SEC) coupled with RPLC as MDLC separation systems, were also reported [43], [44], [45] and used successfully in proteomic analysis of yeast [46], immunoglobulin fusion proteins [47], cytochrome b 6 f complex [48] and complex protein or peptide mixtures. Rieux et al. [49] developed a multi-dimensional separation set-up with a restricted access material (RAM), a reversed phase trap column, to remove albumin for analysis of peptide neurotransmitter. RAM is used in chromatography for the separation of low-molecular-weight analytes from matrix components like albumin based on the combination of size exclusion and adsorption. High efficiency removal of albumin (99.7–99.8%) was realized through this separation set-up.
Qian et al. [50] developed an automatic immobilized metal affinity chromatography (IMAC) capillary RPLC-ESI-MS/MS system, by which all procedures needed in phosphopeptide analysis including IMAC enrichment, RPLC separation, and nanospray MS/MS, can be done automatically under the control of the MassLynx program. The platform was applied to the identification of phosphorylation sites of recombinant human telomeric repeat binding factor 1 treated with kinase in vitro, and two phosphorylation sites are defined.
Affinity column for high-abundance proteins depletion in human plasma are widely investigated. Novotny's group [51] made a lectin microcolumn to investigate glycoproteins in human serum. Serum sample after six highest abundant proteins depletion was further preconcentrated through a lectin column for glycoproteins. The trapped glycoproteins were eluted and analyzed utilizing LC–MS. About 271 glycoproteins were identified.
2.2.4. Three-dimensional chromatography techniques
Two-dimensional chromatography is not enough to separate all peptides in complicated proteome samples. Ultra high-resolution/peak capacity is always important to identify more proteins, especially those low-abundance proteins [12]. Although organelle proteome analysis is an efficient way to identify more proteins as did by Zhang's group for nuclear proteome in C57 mouse liver tissue [52], a pre-fractionation are usually demanded for low-abundance protein identifications. A three-dimensional (3D) chromatography in coupling to MS/MS system was reported using SEC as pre-fractionation prior to on-line SCX–RPLC-ESI-MS/MS [53]. Normal human liver protein extracts was tryptic digested before and after SEC pre-separation (Fig. 2 ). This SEC–SCX–RPLC method has been demonstrated to have great improvements in both proteome coverage and protein identification confidence in comparing to 2D-LC–MS. A lot of mid- and low-abundance proteins as well as proteins with extreme MW or pI were found in this work. Both pre- and post-proteolytic separations were studied to reveal that 3D chromatography was more effective for complicated proteome analysis. Both strategies were recommended to use for complicated samples, and post-proteolytic procedure was more efficient in protein identifications. Conrads et al. [54] used WAX or WCX columns as pre-fractionation step prior to SCX–RPLC-MS/MS analysis for mouse serum proteome research. Additional pre-fractionation step enhanced the throughput and resolution of MDLC system, therefore made it possible to detect more proteins especially mid- and low-abundance proteins in biological samples.
Fig. 2.
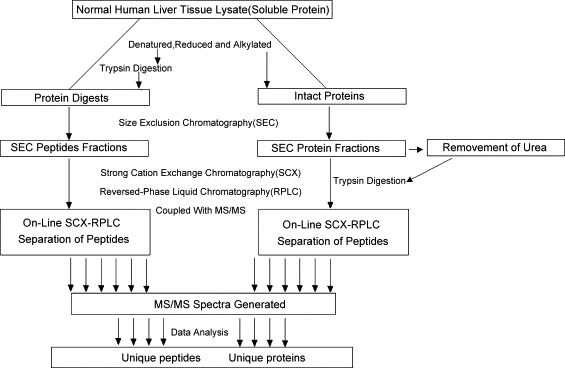
Diagram of SEC–SCX–RPLC approach. The flow of experimental information starting from cell lysate through data analysis is shown. Two NHL global protein samples were prepared, either digested or non-digested, and subjected to different SEC, removing urea, tryptic digestion of each protein fraction, and strong cation-exchange chromatography. The subsequent 240 peptide fractions were then analyzed via RPLC-MS/MS, resulting in spectra that were searched against IPI database using MASCOT [53].
Three-dimensional separation techniques are used in cases where 2D approaches do not give satisfactory results. However, multi-dimensional chromatography as used in proteomics fractionation generally does not exceed three dimensions due to the dramatic increases of fractions to be managed and analyzed [55]. Also dilution effect of fractionations makes it harsh to carry out all dimensional separations.
Actually as a bottom-up approach, shotgun strategy has been well demonstrated to process moderate/high peak capacity and resolution as well as the compatibility with MS. MDLC techniques have been used widely due to their complete automation, low cost, high sensitivity and so on. These are the main reasons for multi-dimensional proteins identification technology widely applied to current analysis of mouse liver plasma membrane proteins [56], mitochondria proteome of human fetal liver [57], subcellular fractions from rat liver [58], SARS associated corona virus proteins [59], etc. Moreover, bottom-up approach can also be used for biomarker discovery or functional proteome research and proteome expression profiling. In conclusion, multi-dimensional separation of peptides combined with mass spectrometry is an important and prospective area of proteomic research.
3. Top-down approach
One of the disadvantages in separating thousands and even millions of digest peptides is the increased complexity of samples. Although peptide level proteome technologies offer better resolution and less sample discrimination than 2D-PAGE, bottom-up approaches provide very limited “true” molecular information of intact proteins, particularly for those proteins with post-translational modifications [60]. While utilizing top-down approach, the overall sample complexity can be dramatically reduced, and quantitative results and the PTMs can be potentially obtained. A large portion of disease biomarkers believes to be low-abundance proteins. Peptides of the low-abundant proteins are always covered by plenty of other peptides in shotgun approach. Therefore, top-down approach has the great potential to enhance the proteomic research in biomarker discovery.
3.1. LC coupled with electrophoretic techniques
As a basic separation mode in capillary electrophoresis, CIEF not only offers a rapid and high-resolution protein separation, but also affords a concentration factor of hundreds of times during focusing. Furthermore, CIEF provides a direct capillary interface for on-line combination with nano-RPLC in an integrated and multi-dimensional separation platform for resolving complex protein mixtures [61]. An on-line combination of CIEF to nano-RPLC-ESI-MS/MS was developed by Lee's group for intact yeast protein characterization [62]. Soluble fractions of intact protein from S. cereviseae were separated to nine fractions which were subsequently parked to C18 trap columns to fulfill on-line connection and separation with nano-RPLC-ESI-MS/MS. A total of 534 proteins with molecular weight between 5 and 70 kDa were identified from this intact protein-based approach. Liquid IEF combined with capillary LC separations were also reported [70]. This will be further discussed in Section 3.3.
3.2. Multi-dimensional LC separation methods
Xia et al. [63] developed a strategy for human plasma proteome research. Pre-fractionation of human plasma proteins using on-line strong cation-exchange chromatography and reversed phase liquid chromatography (SCX–RPLC) was performed and followed by trypsin digestion and LC–MS/MS identification. A total of 1292 distinct proteins were successfully identified, among which some proteins known to be present in serum at <10 ng/mL were detected. Hood et al. [54] developed a MDLC strategy that did not involve depletion of the high-abundance serum proteins. This involved the use of tandem anion- and cation-exchange chromatography followed by tryptic digestion and μRPLC-MS/MS, resulting in the identification of 4567 unique proteins. These proteins represent approximately 16% of all mouse proteins; and 16 of the 34 proteins involved in the β-catenim/Wnt signaling pathway could be identified. Zabrouskov et al. [64] utilized SEC and SAX to fractionate chloroplast proteins prior to Fourier transform mass spectrometry analysis of the intact proteins. Sharma et al. [65] investigated lysates from S. oneidensis using weak anion-exchange chromatography combined with on-line RPLC-FT-ICR mass spectrometer. A set of 715 intact proteins was detected using this protocol.
3.3. Novel and comprehensive usages of MDLC techniques
Depletion of high-abundant proteins is always the bottleneck in the area of biomarker discovery. Some new strategies of high-abundant protein depletion were proposed in addition to widely used immuno-affinity depletions. Gradiflow [66] is capable of separating proteins of different molecular weight and pI values to achieve the depletion of albumin in human plasma. Wasinger et al. [67] adopted Gradiflow strategy as the pre-fractionation step prior to 2D-LC–MS/MS analysis of human plasma proteome. Zhang's group [68] developed a novel strategy of multi-dimensional chromatography separation in intact protein level for the depletion of high-abundant proteins existed in rat liver tissue extraction, as shown in Fig. 3 . Protein samples were first separated with a SCX column and fractions were collected every 2 min. Then SCX fractions were loaded onto RPLC column, respectively. Peak intensity of RPLC chromatogram detected by UV absorption detection was used to distinguish high-abundant proteins from mid- and low-abundant proteins. RPLC fractions with peak intensity above 0.1AU were taken as high-abundant proteins and depleted. After separations, fractions of high-abundance proteins were tryptically digested and MS/MS analyzed, whereas other fractions defined as mid- and low-abundant proteins were pooled together and submitted for further RPLC-MS/MS analysis (Fig. 4 ). The number of identified proteins was increased about three times in comparing to a 2D-LC due to the depletion of high-abundant proteins. This strategy was demonstrated to be more universal and low cost for proteome analysis.
Fig. 3.
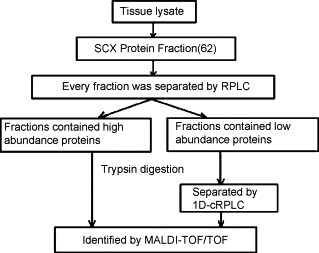
Schematic outline of the separation of the tissue lysate proteins [68].
Fig. 4.
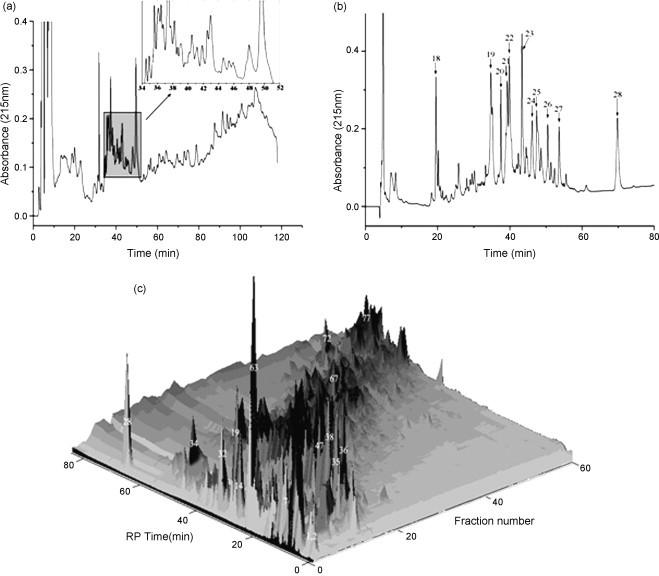
(a) Separation chromatogram of rat liver tissue lysate proteins by 1D SCX. Fractions were collected every 2 min automatically from 3 to 127 min, and collection time of fraction was demonstrated in the magnified illustration. (b) The RPLC elution profile of SCX fraction 3. (c) 3D display of SCX–RPLC separation of rat liver proteins sample. In total, 62 fractions were obtained from the first dimensional separation of SCX, which were separated by RPLC further. Positions of part high-abundance proteins were as labeled [68].
Variety of combination of pre-fractionations for intact proteins are capable to gain better resolutions and result in better analysis for samples of complexity and wide-span of dynamic range. Qian et al. compared five approaches for the characterization of human serum proteome [69]. Six proteins with highest abundance were depleted and the serum was further analyzed using different combination of separation methods such as chromatography/2D-PAGE, on-line or off-line LC–LC–MS spectrum. Different approaches bring up different information of sample's proteome. Meanwhile, selection of separation strategies can improve system resolution effectively. Barnea et al. [70] evaluated three pre-fractionation methods including SCX chromatography, SDS-PAGE and liquid phase IEF prior to 1D or 2D capillary LC separation. Hoffman et al. [11] evaluated comprehensive multi-dimensional separation strategies. Utilizing more than two dimensions of fractionation can effectively profile the low-abundance proteins in plasma proteome research. When the goal of proteome analysis is to find as many proteins as possible, the use of different pre-fractionation methods are quite necessary. Boyes et al. [71] integrated LC and PAGE methods for human serum proteome analysis. High protein recovery was maintained in pre-fractionation step so as to realize unbiased and reproducible analysis results. LC coupled with PAGE was also evaluated by Marcus et al. in analysis of membrane protein from mouse brain [72]. One-dimensional RPLC separation, 2D SCX/RPLC separation, PAGE/SCX/RPLC separation and 2D-PAGE strategies were assessed in this work. MDLC strategy seems to be less discriminating for hydrophobic membrane proteins and it is a complementary to 2D-PAGE for profiling of many proteome samples.
Chromatography used as pre-gel electrophoresis is quite effective to reduce sample complexity. Ottens et al. [73] presented a dual-phase ion-exchange chromatography as a pre-fractionation method prior to polyacrylamide gel electrophoresis/reversed phase liquid chromatography tandem MS spectrometry. The dual-phase ion-exchange chromatography column named CAX contained both cation- and anion-exchange media together to achieve a better recovery of proteins than single-phase IEX columns. Extract proteins from 1 mg cerebellum and cortex tissues were loaded to the column, respectively and the fractions were collected every minute and submitted to further analysis by SDS-PAGE for comparison of cerebellum and cortex proteomics. More than twice of the differential protein spots were observed through CAX-PAGE-RPLC-MS/MS approach. Hanash's group [74] have implemented an orthogonal three-dimensional intact protein analysis system coupled with protein tagging and immuno-depletion of abundant proteins to quantitatively profile the human plasma proteome. After immuno-depletion, dyed and mixed proteins were subsequently separated by IEF, RPLC and SDS-PAGE. Differences in the abundance of resolved proteins were determined based on dye ratios. This strategy was applied to profile the plasma proteome for changes with acute graft versus host disease (GVHD), and the allogeneic bone marrow transplantation. Using capillary HPLC ESI Q-TOF MS, 75 proteins were identified in the micro- to femtomolar ranges that exhibited quantitative differences between the pre- and post-GVHD samples. Kakisaka et al. [75] investigated the aberrant expression of plasma proteins in patients with pancreatic cancer by multi-dimensional liquid chromatography and 2D fluorescence two-dimensional difference gel electrophoresis (DIGE). Six high-abundance plasma proteins were depleted by use of an immuno-affinity column, and low-abundance ones were separated into five fractions by anion-exchange chromatography. The fractionated plasma proteins were subjected to 2D-DIGE with highly sensitive fluorescent dyes. Finally 33 different proteins between non-cancer bearing healthy donors and patients with pancreatic cancer were found including 27 up-regulated and 6 down-regulated in cancer. The up-regulation of leucine-rich alpha-2-glycoprotein (LRG) was not previously identified in pancreatic cancer. It has a clinical significance as a cancer biomarker.
4. Chromatography array technology
Array-based electrophoretic strategies such as protein chips and capillary electrophoresis array have been successfully applied in genome and proteome research with their high throughput and fast analysis time. Chromatography array technology has some challenges because of the complexity and high requirements for HPLC systems. However, many progresses have been made toward this area. Horn et al. [76] presented a high-throughput procedure for proteome research of native constituents. Human serum sample was first separated with SEC column in a mild condition and collected in 96-well microplate. Ninety-six anion-exchange columns (AEC) were arranged just as 96-well microplate to separate SEC fractions in a parallel way. All fractions were submitted for quantitative assay and MS identification. The use of microplate as a medium made it possible to fulfill a high-throughput array separation. This can even increase the system resolution by the adoption of a third separation dimension. Our group developed a capillary array RPLC-based multi-dimensional separation systems coupled with MALDI-TOF-MS/MS for high-throughput proteome research [77], [78]. It includes a SCX column as the first dimension and 10 parallel capillary RPLC columns as the second dimension [77]. The schematic diagram was shown in Fig. 5 . Multiple channel interface was fabricated. Digested proteins extracted from liver cancer tissue were analyzed by the system. Over 1202 proteins were found with only one-tenth time needed in common 2D-LC system. An automated and optimized system was further developed [78]. Capillary SCX chromatographic column was used as the first dimension and 18 parallel capillary RPLC columns were integrated as the second separation dimension (Fig. 6 ). Peptides bound to the SCX phase were eluted using gradient elution. Effluents were sequentially transferred onto each subset of precolumns. After salt fractionation peptide fractions were concurrently back-flushed from the precolumns and separated simultaneously with 18 capillary RP columns. LC effluents were directly deposited onto the MALDI target plates for subsequent MALDI experiments. An 18-fold increase in throughput compared with serial-based 2D-LC system was realized by this new system. The effectiveness of array-based MDLC/MS platform for separation and identification of a complex proteome sample demonstrated that it would be a powerful high-throughput way for large-scale proteome research.
Fig. 5.
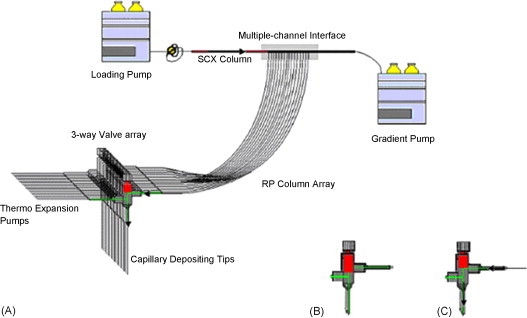
(A) Schematic diagram of column array-based 2D-LC system, (B) post-column micro-valve was open, and (C) post-column micro-valve were closed [77].
Fig. 6.
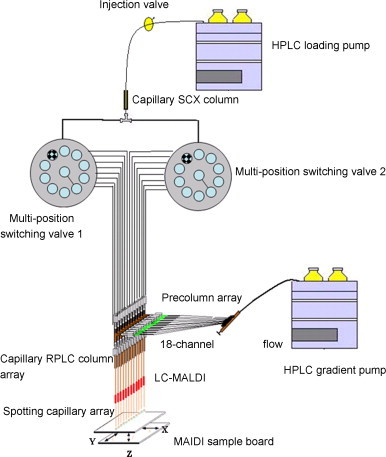
Schematic diagram of the array-based 2D-LC–MS/MS system [78].
Except for all the discussion and review above, MDLC chromatography separation science has been also used for protein–protein interaction [79], traditional Chinese medicine research [80], [81], [82], [83], [84], [85], etc. The effects for chromatography separation conditions are also studied. García studied the effect of mobile phase additives on sensitivity in the analysis of peptides and proteins by HPLC-ESI-MS [86]. Wang et al. evaluated various chromatography conditions including alkyl chain length in the stationary phase, capillary column temperature, and ion-pairing agent, on intact protein separations for top-down proteomics [87]. While Millea et al. investigated the effect of protein mixture separation using linear and step gradients in the first dimension of IEC-RPLC strategy [88]. Comprehensive multi-dimensional liquid chromatography techniques were summarized in a review published lately [89].
5. Concluding remarks
Multi-dimensional liquid chromatography has been widely used in all kinds of biological samples. MDLC coupled with MS is playing important roles in proteome analysis. The MDLC analytical strategies mainly include bottom-up and top-down approaches. Electrophoretic methods combined with variety of liquid chromatography and MDLC also have been successful applied for protein analysis. New progressive strategies using MDLC techniques in high-abundance protein depletions and chromatography array were also perspective in the future proteome analysis. Some prospective applications using MDLC strategies for protein–protein interaction studies [79], biological sample analysis and traditional Chinese medicine analysis [80], [81], [82], [83], [84], [85] also have great significance in analytical science.
Acknowledgements
This paper was supported by the 973-Project: 2007CB914100/3, 863-Project: 2006AA02A308, Project GZ364, and by Shanghai Leading Academic Discipline Project, B109.
Footnotes
This paper is part of a Special Issue dedicated to the 50th anniversary of Journal of Chromatography.
References
- 1.Klose J. Hum. Genet. 1975;26:231. [Google Scholar]
- 2.O’Farrell P.H. J. Biol. Chem. 1975;250:4007. [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H., Hanash S. J. Chromatogr. B. 2003;787:11. doi: 10.1016/s1570-0232(02)00335-5. [DOI] [PubMed] [Google Scholar]
- 4.McCormack A.L., Schieltz D.M., Goode B., Yang S., Barnes G., Drubin D., Yates J.R., III Anal. Chem. 1997;69:767. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 5.Wolters D.A., Washburn M.P., Yates J.R., III Anal. Chem. 2001;73:5683. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 6.Washburn M.P., Ulaszek R., Deciu C., Schieltz D.M., Yates J.R., III Anal. Chem. 2002;74:1650. doi: 10.1021/ac015704l. [DOI] [PubMed] [Google Scholar]
- 7.Gygi S.P., Rist B., Griffin T.J., Eng J., Aebersold R. J. Protein Res. 2002;1:47. doi: 10.1021/pr015509n. [DOI] [PubMed] [Google Scholar]
- 8.VerBerkmoes N.C., Bundy J.L., Hauser L., Asano K.G., Razumovskaya J., Larimer F., Hettich R.L., Stephenson J.L., Jr. J. Proteome Res. 2002;1:239. doi: 10.1021/pr025508a. [DOI] [PubMed] [Google Scholar]
- 9.Lescuyer P., Hochstrasser D.F., Sanchez J.C. Electrophoresis. 2004;25:1125. doi: 10.1002/elps.200305792. [DOI] [PubMed] [Google Scholar]
- 10.Issaq H.J., Chan K.C., Janini G.M., Conrads T.P., Veenstra T.D. J. Chromatogr. B. 2005;817:35. doi: 10.1016/j.jchromb.2004.07.042. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman S.A., Joo Won-A., Echan L.A., Speicher D.W. J. Chromatogr. B. 2007;849:43. doi: 10.1016/j.jchromb.2006.10.069. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y., Rudnick P.A., Evans E.L., Li J., Zhuang Z., DeVoe D.L., Lee C.S., Balgley B.M. Anal. Chem. 2005;77:6549. doi: 10.1021/ac050491b. [DOI] [PubMed] [Google Scholar]
- 13.Guo T., Lee C.S., Wang W., DeVoe D.L., Balgley B.M. Electrophoresis. 2006;27:3523. doi: 10.1002/elps.200600094. [DOI] [PubMed] [Google Scholar]
- 14.Guo T., Rudnick P.A., Wang W., Lee C.S., DeVoe D.L., Balgley B.M. J. Proteome Res. 2006;5:1469. doi: 10.1021/pr060065m. [DOI] [PubMed] [Google Scholar]
- 15.Zhu Y., Lubman D.M. Electrophoresis. 2004;25:949. doi: 10.1002/elps.200305779. [DOI] [PubMed] [Google Scholar]
- 16.Westman-Brinkmalm A., Karisson G., Brive L.M., Hedberg-Fogel K., Persson R., Karlsson H., Ekman R., Blennow K. Rapid Commun. Mass. Spectrom. 2005;19:3651. doi: 10.1002/rcm.2237. [DOI] [PubMed] [Google Scholar]
- 17.Brunner E., Ye M., Weber G., Eckerskorn C., Aebersold R. J. Proteome Res. 2006;5:2241. doi: 10.1021/pr0600632. [DOI] [PubMed] [Google Scholar]
- 18.Stroink T., Ortiz M.C., Bult A., Lingeman H., de Jong G.J., Underberg W.J.M. J. Chromatogr. B. 2005;817:49. doi: 10.1016/j.jchromb.2004.11.057. [DOI] [PubMed] [Google Scholar]
- 19.Link A.J., Eng J., Schieltz D.M., Carmack E., Mize G., Morris D.R., Garvik B.M., Yates J.R., III Nat. Biotechnol. 1999;17:676. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 20.Harry J.L., Wilkins M.R., Herbert B.R., Packer N.H., Gooley A.A., Williams K.L. Electrophoresis. 2000;21:1071. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1071::AID-ELPS1071>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 21.Washburn M.P., Walters D., Yates J.R., III Nat. Biotechnol. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 22.Peng J., Schartz D., Elias J.E., Thoreen C.C., Cheng D., Marsischky G., Roelfs J., Finley D., Gygi S.P. Nat. Biotechnol. 2003;21:921. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 23.Tirumalai R.S., Chan K.C., Prieto D.A., Issaq H.J., Conrads T.P., Veenstra T.D. Mol. Cell. Proteomics. 2003;2:1096. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Blonder J., Conrads T.P., Terunuma A., Chan K.C., Yee C., Lucas D.A., Yu L.-R., Issaq H.J., Schaefer C.F., Buetow K.H., Vogel J.C., Veenstra T.D. Proteomics. 2004;4:31. [Google Scholar]
- 25.Blonder J., Stiles B.G., Hale M.L., Conrads T.P., Lucas D.A., Issaq H.J., Veenstra T.D. Electrophoresis. 2004;25:1307. doi: 10.1002/elps.200405891. [DOI] [PubMed] [Google Scholar]
- 26.Yu L.-R., Conrads T.P., Uo T., Issaq H.J., Morrison R.S., Veenstra T.D. J. Proteome Res. 2004;3:469. doi: 10.1021/pr034090t. [DOI] [PubMed] [Google Scholar]
- 27.Dai J., Shieh C.H., Sheng Q., Zhou H., Zeng R. Anal. Chem. 2005;77:5793. doi: 10.1021/ac050251w. [DOI] [PubMed] [Google Scholar]
- 28.Dai J., Jin W., Sheng Q., Shieh C., Wu J., Zeng R. J. Proteome Res. 2007;6:250. doi: 10.1021/pr0604155. [DOI] [PubMed] [Google Scholar]
- 29.Jiang X., Feng S., Tian R., Han G., Jiang X., Ye M., Zou H. Proteomics. 2007;7:528. doi: 10.1002/pmic.200600661. [DOI] [PubMed] [Google Scholar]
- 30.Wang F., Jiang X., Feng S., Tian R., Jiang X., Han G., Liu H., Ye M., Zou H. J. Chromatogr. A. 2007;1171:56. doi: 10.1016/j.chroma.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 31.Nice E.C., Rothacker J., Weinstock J., Lim L., Catimel B. J. Chromatogr. A. 2007;1168:190. doi: 10.1016/j.chroma.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y., Xiang R., Horváth C., Wilkins J.A. J. Chromatogr. A. 2004;1053:27. [PubMed] [Google Scholar]
- 33.Fujii K., Nakano T., Hike H., Usui F., Banbo Y., Tojo H., Nishimura T. J. Chromatogr. A. 2004;1057:107. doi: 10.1016/j.chroma.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 34.Vitali B., Wasinger V., Brigidi P., Guilhaus M. Proteomics. 2005;5:1859. doi: 10.1002/pmic.200401080. [DOI] [PubMed] [Google Scholar]
- 35.Ru Q.C., Katenhusen R.A., Zhu L.A., Silberman J., Yang S., Orchard T.J., Brzeski H., Liebman M., Ellsworth D.L. J. Chromatogr. A. 2006;1111:166. doi: 10.1016/j.chroma.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 36.Dasari S., Pereira L., Reddy A.P., Michaels J.A., Lu X., Jacob T., Thomas A., Rodland M., Roberts C.T., Gravett M.G., Nagalla S.R. J. Proteome Res. 2007;6:1258. doi: 10.1021/pr0605419. [DOI] [PubMed] [Google Scholar]
- 37.Skipp P., Robinson J., O’Connor C.D., Clarke I.N. Proteomics. 2005;5:1558. doi: 10.1002/pmic.200401044. [DOI] [PubMed] [Google Scholar]
- 38.Graham R.L.J., Pollock C.E., Ternan N.G., McMullan G. J. Proteome Res. 2006;5:822. doi: 10.1021/pr0504642. [DOI] [PubMed] [Google Scholar]
- 39.Vanrobaeys F., Coster R.V., Dhondt G., Devreese B., Beeumen J.V. J. Proteome Res. 2005;4:2283. doi: 10.1021/pr050205c. [DOI] [PubMed] [Google Scholar]
- 40.Hattan S.J., Marchese J., Khainovski N., Martin S., Juhasz P. J. Proteome Res. 2005;4:1931. doi: 10.1021/pr050099e. [DOI] [PubMed] [Google Scholar]
- 41.Shen Y.F., Smith R.D. Electrophoresis. 2002;23:3106. doi: 10.1002/1522-2683(200209)23:18<3106::AID-ELPS3106>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 42.Opiteck G.J., Lewis K.C., Jorgenson J.W., Anderegg R.J. Anal. Chem. 1997;69:1518. doi: 10.1021/ac961155l. [DOI] [PubMed] [Google Scholar]
- 43.Opiteck G., Jorgenson J., Anderegg R. Anal. Chem. 1997;69:2283. doi: 10.1021/ac961156d. [DOI] [PubMed] [Google Scholar]
- 44.Opiteck G., Ramirez S., Jorgenson J., Moseley M., III Anal. Biochem. 1998;258:349. doi: 10.1006/abio.1998.2588. [DOI] [PubMed] [Google Scholar]
- 45.Bedani F., Kok W., Janssen H. J. Chromatogr. A. 2006;1133:126. doi: 10.1016/j.chroma.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 46.Gao J., Opiteck G.J., Friedrichs M.S., Dongre A.R., Hefta S.R. J. Proteome Res. 2003;2:643. doi: 10.1021/pr034038x. [DOI] [PubMed] [Google Scholar]
- 47.Nemeth-Cawley J.F., Tangarone B.S., Rouse J.C. J. Proteome Res. 2003;2:495. doi: 10.1021/pr034008u. [DOI] [PubMed] [Google Scholar]
- 48.Whitelegge J.P., Zhang H.M., Aguilera R., Taylor R.M., Cramer W.A. Mol. Cell. Proteomics. 2002;1:816. doi: 10.1074/mcp.m200045-mcp200. [DOI] [PubMed] [Google Scholar]
- 49.Rieux L., Bischoff R., Verpoorte E., Niederländer H.A.G. J. Chromatogr. A. 2007;1149:169. doi: 10.1016/j.chroma.2007.02.118. [DOI] [PubMed] [Google Scholar]
- 50.Wang J., Jiang Y., Jiang H., Cai Y., Qian X. Proteomics. 2006;6:404. doi: 10.1002/pmic.200500223. [DOI] [PubMed] [Google Scholar]
- 51.Madera M., Mechref Y., Klouckova I., Novotny M.V. J. Proteome Res. 2006;5:2348. doi: 10.1021/pr060169x. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J., Xu X., Shen H., Zhang X. J. Sep. Sci. 2006;29:2635. doi: 10.1002/jssc.200600065. [DOI] [PubMed] [Google Scholar]
- 53.Zhang J., Xu X., Gao M., Yang P., Zhang X. Proteomics. 2007;7:500. doi: 10.1002/pmic.200500880. [DOI] [PubMed] [Google Scholar]
- 54.Hood B.L., Zhou M., Chan K.C., Lucas D.A., Kim G.J., Issaq H.J., Veenstra T.D., Conrads T.P. J. Proteome Res. 2005;4:1561. doi: 10.1021/pr050107r. [DOI] [PubMed] [Google Scholar]
- 55.Guerrier L., Lomas L., Boschetti E. J. Chromatogr. A. 2005;1073:25. doi: 10.1016/j.chroma.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Cao R., Zhang L., Nie S., Wang X., Liang S. Chin. J. Biochem. Mol. Biol. 2005;21:134. [Google Scholar]
- 57.Zhang Y., Shi R., Meng Q., Wang J. Chin. J. Anal. Chem. 2005;33:1371. [Google Scholar]
- 58.Jiang X., Zhou H., Zhang L., Sheng Q. Mol. Cell. Proteomics. 2004;3(5):441. doi: 10.1074/mcp.M300117-MCP200. [DOI] [PubMed] [Google Scholar]
- 59.Zeng R., Ruan H., Jiang X., Zhou H. J. Proteome Res. 2004;3:549. doi: 10.1021/pr034111j. [DOI] [PubMed] [Google Scholar]
- 60.Mann M., Jensen O.N. Nat. Biotechnol. 2003;2:255. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 61.Chen J., Balgley B.M., DeVoe D.L., Lee C.S. Anal. Chem. 2003;75:3145l. doi: 10.1021/ac034014+. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y., Balgley B.M., Rudnick P.A., Evans E.L., DeVoe D.L., Lee C.S. J. Proteome Res. 2005;4:36. doi: 10.1021/pr049876l. [DOI] [PubMed] [Google Scholar]
- 63.Jin W., Dai J., Li S., Xia Q. J. Proteome Res. 2005;4:613. doi: 10.1021/pr049761h. [DOI] [PubMed] [Google Scholar]
- 64.Zabrouskov V., Giacomelli L., van Wijk K.J., McLafferty F.W. Mol. Cell. Proteomics. 2003;2:1253. doi: 10.1074/mcp.M300069-MCP200. [DOI] [PubMed] [Google Scholar]
- 65.Sharma S., Simpson D.C., Tolic N., Jaitly N., Mayampurath A.M., Smith R.D., Pasa-Tolic L. J. Proteome Res. 2007;6:602. doi: 10.1021/pr060354a. [DOI] [PubMed] [Google Scholar]
- 66.Rothemund D.L., Locke V.L., Liew A., Thomas T.M., Wasinger V., Rylatt D.B. Proteomics. 2003;3:279. doi: 10.1002/pmic.200390041. [DOI] [PubMed] [Google Scholar]
- 67.Wasinger V.C., Locke V.L., Raftery M.J., Larance M., Rothemund D., Liew A., Bate I., Guilhaus M. Proteomics. 2005;5:3397. doi: 10.1002/pmic.200401160. [DOI] [PubMed] [Google Scholar]
- 68.Gao M., Zhang J., Deng C., Yang P., Zhang X. J. Proteome Res. 2006;5:2853. doi: 10.1021/pr0602186. [DOI] [PubMed] [Google Scholar]
- 69.Li X., Gong Y., Wang Y., Wu S., Cai Y., He P., Lu Z., Ying W., Zhang Y., Jiao L., He H., Zhang Z., He F., Zhao X., Qian X. Proteomics. 2005;5:3423. doi: 10.1002/pmic.200401226. [DOI] [PubMed] [Google Scholar]
- 70.Barnea E., Sorkin R., Ziv T., Beer I., Admon A. Proteomics. 2005;5:3367. doi: 10.1002/pmic.200401221. [DOI] [PubMed] [Google Scholar]
- 71.Martosella J., Zolotarjova N., Liu H., Nicol G., Boyes B.E. J. Proteome Res. 2005;4:1522. doi: 10.1021/pr050088l. [DOI] [PubMed] [Google Scholar]
- 72.Lohaus C., Nolte A., Blüggel M., Scheer C., Klose J., Gobom J., Schü A., Wiebringhaus T., Meyer H.E., Marcus K. J. Proteome Res. 2007;6:105. doi: 10.1021/pr060247g. [DOI] [PubMed] [Google Scholar]
- 73.Ottens A.K., Kobeissy F.H., Wolper R.A., Haskins W.E., Hayes R.L., Denslow N.D., Wang K.K.W. Anal. Chem. 2005;77:4836. doi: 10.1021/ac050478r. [DOI] [PubMed] [Google Scholar]
- 74.Wang H., Clouthier S.G., Galchev V., Misek D.E., Duffner U., Min C., Zhao R., Tra J., Omenn G.S., Ferrara J.L.M., Hanash S.M. Mol. Cell. Proteomics. 2005;4:618. doi: 10.1074/mcp.M400126-MCP200. [DOI] [PubMed] [Google Scholar]
- 75.Kakisaka T., Kondo T., Okano T., Fujii K., Honda K., Endo M., Tsuchida A., Aoki T., Itoi T., Moriyasu F., Yamada T., Kato H., Nishimura T., Todo S., Hirohashi S. J. Chromatogr. B. 2007;852:257. doi: 10.1016/j.jchromb.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horn A., Kreusch S., Bublitz R., Hoppe H., Cumme G.A., Schulze M., Moore T., Ditze G., Rhode H. Proteomics. 2006;6:559. doi: 10.1002/pmic.200500142. [DOI] [PubMed] [Google Scholar]
- 77.Liu C., Zhang X. J. Chromatogr. A. 2007;1139:191. doi: 10.1016/j.chroma.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 78.Gu X., Deng C., Yan G., Zhang X. J. Proteome Res. 2006;5:3186. doi: 10.1021/pr0602592. [DOI] [PubMed] [Google Scholar]
- 79.Babusiak M., Man P., Sutak R., Petrak J., Vyoral D. Proteomics. 2005;5:340. doi: 10.1002/pmic.200400935. [DOI] [PubMed] [Google Scholar]
- 80.Yang F., Quan J., Zhang T., Ito Y. J. Chromatogr. A. 1998;803:298. doi: 10.1016/s0021-9673(97)01273-9. [DOI] [PubMed] [Google Scholar]
- 81.Sheng L., Wang Y., Ma R., Ding G., Zhou H. Chin. J. Nat. Med. 2003;1:61. [Google Scholar]
- 82.Chen X., Kong L., Su X., Fu H. J. Chromatogr. A. 2004;1040:169. doi: 10.1016/j.chroma.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 83.Chen X., Hu L., Su X., Kong L. J. Sep. Sci. 2006;29:881. doi: 10.1002/jssc.200500442. [DOI] [PubMed] [Google Scholar]
- 84.Chen X., Kong L., Su X., Pan C. J. Chromatogr. A. 2005;1089:87. doi: 10.1016/j.chroma.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 85.Hu L., Chen X., Kong L., Su X. J. Chromatogr. A. 2005;1092:191. doi: 10.1016/j.chroma.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 86.García M.C. J. Chromatogr. B. 2005;825:111. doi: 10.1016/j.jchromb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y., Balgley B.M., Rudnick P.A., Lee C.S. J. Chromatogr. A. 2005;1073:35. doi: 10.1016/j.chroma.2004.08.140. [DOI] [PubMed] [Google Scholar]
- 88.Millea K.M., Kass I.J., Cohen S.A., Krull I.S., Gebler J.C., Berger S.J. J. Chromatogr. A. 2005;1079:287. doi: 10.1016/j.chroma.2005.04.048. [DOI] [PubMed] [Google Scholar]
- 89.Dugo P., Cacciola F., Kumm T., Dugo G., Mondello L. J. Chromatogr. A. 2008;1184:353. doi: 10.1016/j.chroma.2007.06.074. [DOI] [PubMed] [Google Scholar]