

Abstract
The interferon system provides a powerful and universal intracellular defense mechanism against viruses. Knockout mice defective in IFN signaling quickly succumb to all kinds of viral infections. Likewise, humans with genetic defects in interferon signaling die of viral disease at an early age. Among the known interferon-induced antiviral mechanisms, the Mx pathway is one of the most powerful. Mx proteins belong to the dynamin superfamily of large GTPases and have direct antiviral activity. They inhibit a wide range of viruses by blocking an early stage of the viral replication cycle. Likewise, the protein kinase R (PKR), and the 2–5 OAS/RNaseL system represent major antiviral pathways and have been extensively studied. Viruses, in turn, have evolved multiple strategies to escape the IFN system. They try to go undetected, suppress IFN synthesis, bind and neutralize secreted IFN molecules, block IFN signaling, or inhibit the action of IFN-induced antiviral proteins. Here, we summarize recent findings about the astonishing interplay of viruses with the IFN response pathway.
Keywords: Interferon, Mx, Interferon-stimulated gene products, Viral countermeasures, Interferon antagonists
1. Starting point: interferon-mediated inborn resistance to viruses in mice
Historically, mouse models of genetically determined resistance against viruses were useful to find antiviral factors involved in innate immunity. Some inbred mouse strains proved to be less susceptible to infection by specific viruses than others. In most cases, the degree of antiviral resistance was controlled by several genes, but occasionally a single gene was found to be responsible [1]. A good example is the inborn resistance against influenza and influenza-like viruses found in wild mice and some inbred mouse strains [2], [3], [4]. Forty-five years ago, Jean Lindenmann, co-discoverer of interferon with Alick Isaacs, described an inbred mouse strain which was unusually resistant when infected with doses of influenza A virus (FLUAV) that were lethal to ordinary laboratory mice [4]. Subsequent work revealed that this unusual resistance is brought about by a single gene, Mx1 (for orthomyxovirus resistance gene 1), localized on mouse chromosome 16 [5], and that the Mx1 protein has intrinsic antiviral activity [6], [7]. Unexpectedly, the Mx1 gene turned out to belong to the so-called interferon (IFN) responsive genes (ISGs) and is strictly regulated by type I (α and β) and type III (λ) IFNs [8], [9]. Gene expression is rapidly induced in viral infections through the action of virus-induced IFNs. In the absence of IFNs, the Mx gene is silent, making Mx transcripts or protein an excellent marker for type I IFN activity [10], [11].
In influenza virus-susceptible mice, the Mx1 gene is defective. Most inbred strains of mice carry nonfunctional Mx1 alleles [12]. Why intact Mx1 genes are absent in most inbred mouse strains remains unresolved. Most likely, the reason is a founder effect, suggesting that most laboratory mice share the distal part of chromosome 16 with a common ancestor mouse. Genetic defects present in laboratory mice but rarely in wild mice have been described for other gene loci. A single autosomal dominant gene locus, designated Flv/Wnv, is responsible for natural resistance of mice against infection with West Nile virus (WNV) and other flaviviruses. The gene was recently identified as Oas1b, a member of a large IFN-regulated gene family encoding 2′-5′-oligoadenylate synthetases (2–5 OAS) known to play an important role in antiviral defense [13], [14]. The intact Oas1b gene is again found in wild mice and some rare inbred strains but not in most laboratory strains which carry a nonsense mutation in the distal part of chromosome 5. In contrast to Mx1, comparisons of the mouse and human genomes did not reveal a direct equivalent of the mouse Oas1b gene in humans [15]. Additional examples of genetic resistance are known in mice in which single genes play a major role [16].
Here, we summarize recent advances in our understanding of some of these IFN-regulated defense mechanisms and discuss how viruses manage to counteract these restriction elements.
2. Transcriptional activation of IFN genes
Type I IFNs are produced by cells in direct response to virus infection and comprise a large number of IFN-α subspecies and a single IFN-β, as well as some additional family members [17], [18]. The recently discovered IFN-λ1, IFN-λ2, and IFN-λ3 (also termed IL-28A, IL-28B, and IL-29) are functionally similar to the type I IFNs but use distinct receptors to mediate their antiviral activity [19]. Conserved molecular signatures of viruses serve as “danger signals” which are recognized by specialized receptors of the host cell. These receptors are collectively called pattern recognition receptors (PRRs) because they recognize a diverse range of conserved pathogen-associated molecular patterns (PAMPs) found in infectious disease agents (Fig. 1 ). The main PAMP of viruses appear to be nucleic acids, such as double-stranded RNA (dsRNA) molecules [20] and specific structures at the 5′ end of certain viral ssRNA genomes which carry a 5′ triphosphate group [21], [22]. dsRNA or 5′ triphosphate moieties are usually not present on host RNA species and appear to provide an ideal recognition pattern for non-self [23].
Fig. 1.
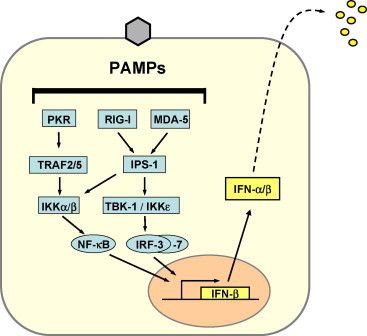
Type I IFN induction. PAMP recognition by intracellular PRRs leads to activation of the transcription factors NF-κB, IRF-3 and AP-1 (not shown). The cooperative action of these factors is required for full activation of the IFN-β promoter. IRF-3 is phosphorylated by the kinases TBK-1 or IKKɛ which in turn are activated by RIG-I or MDA5 via IPS-1. NF-κB is activated by the PKR pathway as well as by IPS-1. The IFN-induced IRF-7 later enhances IFN gene transcription, but is also involved in immediate early IFN-β transcription.
Induction of type I IFN gene expression is transcriptionally regulated and is best understood for IFN-β (Fig. 1). The IFN-β promoter has binding sites for several transcription factors which cooperate for maximal promoter activation. There is general agreement that interferon regulatory factor 3 (IRF-3) plays a central role [24]. IRF-3 needs to be phosphorylated to become active. The enzymes responsible for IRF-3 phosphoryation have recently been demonstrated to be the IKK-like kinases IKKɛ and TBK-1 [25], [26]. These kinases are activated by the RNA helicase RIG-1 and/or MDA5 [27], [28] and, presumably, some Toll-like receptors (TLR) [29], [30]. RIG-I and MDA5 bind dsRNA molecules and 5′ triphosphorylated ssRNAs in the cytoplasm of infected cells. Phosphorylated IRF-3 homo-dimerizes and moves into the nucleus where it recruits the transcriptional coactivator, CREB-binding protein (CBP), to initiate IFN-β mRNA synthesis [24]. In addition, NF-κB and ATF-2/cJUN (AP-1) are activated as a more general stress response. Together these transcription factors strongly upregulate IFN-β expression (Fig. 1).
A second IRF family member, IRF-7, is expressed in most cells at very low amounts. It needs to be induced by IFN to reach sufficient levels and is then activated by virus infection in much the same way as IRF-3 [31]. IRF-7 is part of a positive feedback loop leading to amplification of IFN gene expression. Activated IRF-7 cooperates with IRF-3 and stimulates expression of the numerous IFN-α genes leading to a broad IFN-α response [32]. In specialized IFN-α-producing cells, e.g. plasmacytoid dendritic cells, IRF-7 is constitutively present at high levels and is directly activated in response to signals from certain TLRs which stimulate immediate IFN-α synthesis [33], [34].
3. Interferon-induced antiviral pathways
The various IFN-α subspecies and the single IFN-β bind to and activate a common type I IFN receptor (IFNAR), whereas type III (λ) IFNs activate their cognate type III receptor. Both receptors signal to the nucleus through the so-called JAK-STAT pathway (Fig. 2 ). This pathway is well characterized [35] and will not be described here in detail.
Fig. 2.
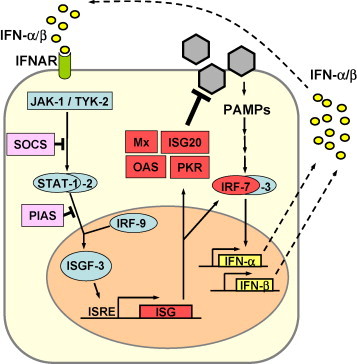
IFN signalling. IFN-α and IFN-β bind to the type I IFN receptor (IFNAR) and activate the expression of numerous ISGs via the JAK/STAT pathway. IRF-7 amplifies the IFN response by inducing the expression of several IFN-α subtypes. Mx, ISG20, OAS and PKR are examples of proteins with antiviral activity.
Type I and type III IFNs activate the expression of an overlapping set of more than 300 IFN-stimulated genes (ISGs) which have antiviral, antiproliferative, and immunomodulatory functions [36], [37]. Three IFN-induced enzyme systems represent major antiviral pathways and have been extensively studied. These include protein kinase R (PKR) [38], [39], the 2–5 OAS/RNaseL system [40] and the Mx GTPases [41]. Mice lacking one of these components show dramatically increased virus susceptibilities [42], [43]. Interestingly, cells from so-called triple knock-out mice lacking PKR, RNaseL and Mx1 are still capable of mounting a limited IFN-induced antiviral state, indicating that additional antiviral pathways exist [43].
Additional proteins with potentially important antiviral activities are ISG20 [44], P56 [45], [46], guanylate-binding protein-1 (GBP-1) [47] and promyelocytic leukemia protein (PML) [48]. ISG20 is an IFN-induced 3′-5′ exonuclease that specifically degrades ssRNA in vitro. Expression of ISG20 leads to a reduction in vesicular stomatitis virus (VSV) gene expression and blocks viral replication in cell culture [44]. P56 binds the eukaryotic initiation factor 3e (eIF3e) subunit of the eukaryotic translation initiation factor eIF3 and is likely to suppress viral as well as cellular RNA translation [46], [49]. GBP-1 belongs to the dynamin superfamily of large GTPases like Mx and has antiviral activity against VSV [47]. Finally, the PML protein is a TRIM family member (also called TRIM19) which shuttles between the cytoplasm and the nucleus where it forms a specialized subnuclear compartment known as nuclear domain-10 (ND10) or PML nuclear body. Overexpression of PML has been found to suppress replication of several viruses, including VSV, influenza A virus (FLUAV) [50], lymphocytic choriomeningitis virus [51] and human foamy retrovirus (HFV) [52]. Interestingly, cells from wild-type and PML knockout mice proved to be equally permissive for herpes simplex virus 1 (HSV-1) and FLUAV, suggesting that PML itself has no antiviral activity against these viruses [53], [54]. Rather, IFN responsiveness of cells may be influenced by PML and PML nuclear bodies, as suggested by recent findings [53].
4. Mx is a major player in IFN-induced host defense
All available evidence indicates that recovery from influenza virus infection in mice requires a functional Mx1 defense system [7]. Numerous experiments using Mx1-congenic or Mx1-transgenic mice demonstrated that the Mx1 system is indispensable for recovery from infection with otherwise deadly influenza viruses [55]. Moreover, it became clear that the course of disease observed in Mx1-positive mice reflects quite well the characteristics of an uncomplicated acute influenza virus infection in man, indicating that these animals better mimic the innate immune system of humans than standard laboratory mice. The human ortholog, called MxA, has a broad antiviral activity against a range of different viruses. MxA-sensitive viruses include members of the bunyaviruses, orthomyxoviruses, paramyxoviruses, rhabdoviruses, togaviruses, picornaviruses, reoviruses and Hepatitis B virus, a DNA virus with a genomic RNA intermediate [41]. The mechanism of action has so far been studied for a small number of viruses only and is still incompletely understood. In general, Mx proteins were found to bind to essential viral components and to block their functions. For example, the human MxA protein accumulates in the cytoplasm of IFN-treated cells and blocks replication of the infecting virus soon after cell entry. It has been shown to target the viral capsids by recognizing the major capsid component, the viral ribonucleoprotein of some orthomyxo- and bunyaviruses [56], [57], [58]. The protective power of the human MxA GTPase is best demonstrated in MxA-transgenic mice. Human MxA was sufficient to turn susceptible Mx1-negative mice into resistant animals [59]. Moreover, even the constitutive expression of MxA in otherwise IFN-nonresponsive IFNAR0/0 animals conferred full resistance against disease, thus highlighting the importance of the human Mx system for antiviral defense [59].
5. Viral countermeasures
Viruses are known to block the IFN system at different levels, and different approaches are used by different viruses to accomplish this. An efficient strategy is used by vaccinia and other poxviruses which express soluble IFN-binding proteins to neutralize secreted IFN molecules [60], [61], [62], [63]. Most viruses have evolved multifunctional proteins which specifically target distinct components of the IFN signaling cascade. A large number of viral proteins with anti-IFN properties have been described in the past few years and have been extensively described in a number of recent reviews [64], [65], [66].
5.1. Viral subversion of IFN induction
To subvert innate immunity, many viruses interfere with one or several steps in the IFN induction pathway. The NS1 protein of influenza A virus binds to both dsRNA and ssRNA presumably by recognizing inter- or intramolecular dsRNA regions. Importantly, NS1 also associates with RIG-I in infected cells and seems to impair its signaling function [22], [67]. In contrast, the V protein of paramyxovirus SV5 has no apparent RNA-binding activity. It inhibits IFN induction by targeting the RIG-I-related RNA sensor MDA-5 [27], [68]. Next in line is the adaptor protein Cardif/IPS-1/MAVS/VISA which connects the RNA sensors RIG-I and MDA5 with the IRF-3 kinases TBK-1/IKK-ɛ [69], [70], [71], [72]. It is specifically cleaved by the NS3-4A protease of hepatitis C virus (HCV) and additional flaviviruses [70], [73], [74] (see also contribution of Michael Gale in this volume). Activation of IRF-3 by TBK-1 is prevented by the phosphoprotein P of Rabies virus [75] and the G1 glycoprotein of the hantavirus NY-1 [76]. IRF-3 itself is degraded by the NPro proteins of pestiviruses such as classical swine fever virus and of bovine viral diarrhea virus [77], [78], [79] via the proteasomal pathway [80], [81]. Also, the E6 protein of human papilloma virus 16 binds and inactivates IRF-3 [82], and the proteins ORF 3b, ORF 6 and N of SARS-coronavirus directly target IRF-3 [83] to inhibit IFN induction [84]. A sophisticated strategy to block IRF-3 is used by certain herpesviruses. Human herpes virus 8 (HHV-8), the causative agent of Kaposi sarcoma, expresses several IRF homologues, termed vIRFs, which exert a dominant-negative effect [85], [86], [87], [88], [89], [90], [91].
Many viruses which lytically infect the host cell simply prevent IFN synthesis by imposing a general block on host cell transcription. For example, the nonstructural NSs proteins of the Rift Valley Fever virus and Bunyamwera virus interfere with the basic transcription machinery [92], [93], [94], [95].
5.2. Targeting the effector proteins of the antiviral state
An efficient way to escape the IFN response is to directly inhibit the proteins that mediate the antiviral state. IFN-regulated PKR and 2–5 OAS are expressed in a latent, inactive form in uninfected cells. Both enzymes need to be activated by viral dsRNA. This requirement makes them vulnerable to IFN antagonists found in many viruses. Some viruses express RNA-binding proteins which are able to prevent the activation of PKR or the 2–5 OAS/RNaseL system by sequestering dsRNA molecules [96], [97], [98], [99], [100], [101], [102], [103]. An alternative strategy used by viruses is to encode small RNAs which compete with dsRNA for binding to PKR, thereby preventing activation. This is the case for adenoviruses [104], HCV [105], Epstein-Barr virus (EBV) [106], and HIV-1 [107]. Several viruses express proteins which either directly bind to or otherwise inactivate PKR. For example, the γ34.5 protein of HSV-1 triggers the dephosphorylation of eIF-2α, thus reverting the translational block established by PKR [108]. The E2 protein of HCV acts as pseudosubstrate for PKR [109], as does the Tat protein of HIV-1 [110] or the K3L protein of vaccinia virus [111]. Interestingly, FLUAV exploits a cellular pathway to block PKR in that it activates p58IPK, a cellular inhibitor of PKR [102] and NS1 to block PKR as well as the 2–5 OAS/RNaseL system [112], [113]. Poliovirus induces the degradation of PKR [114]. Many viruses also block the RNaseL pathway, either by expressing dsRNA-binding proteins (see above), or by other, more direct means. Encephalomyocarditis virus as well as HIV-1 induce the synthesis of RLI, a cellular RNaseL inhibitor [115], [116]. Infection with HSV-1 and HSV-2 activates the synthesis of 2′-5′-oligoadenylate derivatives which bind and prevent RNaseL activation [117]. The Poliovirus genome contains a conserved RNA structure which inhibits RNaseL [118]. The antiviral effect of IFN is inhibited in cells infected with RSV [119], [120], an effect most probably mediated by the viral NS1 and the NS2 proteins [121], [122], [123].
Certain viruses induce the disruption of PML nuclear bodies (also called ND10) by proteasome-dependent degradation of PML and Sp100 [124]. In HSV-1 infected cells, viral ICP0 accumulates in ND10 and induces the degradation of PML and Sp100, an activity which requires the E3 ligase activity of ICP0 [125], [126]. Similar disruptions of ND10 were observed in cells infected with CMV, EBV, HPV and adenoviruses [127]. It is conceivable that viruses disassemble these nuclear structures to get rid of antiviral components but sufficient data supporting this view are not yet available.
There is no evidence for a specific viral inhibitor of Mx proteins so far. Mx proteins are not posttranslationally modified and their activity is not modulated by dsRNA. Nevertheless, viruses have found means to subvert the Mx system.
6. Viral escape from the Mx response
The mouse Mx1 protein inhibits a very early step of the influenza virus multiplication cycle. It blocks primary transcription of the incoming viral genome, a process which is performed by the associated viral polymerase. Since the virus can not transcribe and replicate its genome in the presence of Mx, generation of Mx escape mutants is virtually impossible. Therefore, a prime strategy of Mx-sensitive orthomyxoviruses is to suppress IFN production in the vertebrate host, thereby avoiding Mx expression in potential target cells. The influenza-like Thogoto virus (THOV) was recently shown to have an accessory protein with IFN-antagonistic activity. The sixth genomic segment of THOV encodes two transcripts: a spliced mRNA that codes for the matrix (M) protein and an unspliced mRNA that encodes a C-terminally extended M protein, named ML [128]. Recombinant mutant viruses were generated that lacked ML. These ML-deficient viruses were strong IFN inducers but showed otherwise no obvious growth deficits in IFN-defective cells or animals [129]. In IFN-competent Mx1-positive mice, however, the mutant virus devoid of ML was highly attenuated. In contrast, wild-type virus expressing ML were able to grow in such animals because ML was blocking IFN production [130]. Interestingly, ML inhibits the transcriptional activity of IRF-3 which is required for IFN gene expression. Recent data show that the ML protein of THOV interferes with IRF-3 dimerization and recruitment of the transcriptional coactivator CBP by activated IRF-3 [132]. The IFN-specific transcription factor IRF-3 is known to be a central player in IFN gene expression and, not surprisingly, is affected by viral proteins from many unrelated viruses (see above).
Another strategy of orthomyxoviruses is to escape from Mx by fast growth. In collaboration with Adolfo Garcia-Sastre at the Mount Sinai School of Medicine in New York, our group recently studied an exceptional influenza A virus strain that is highly pathogenic for Mx1-positive mice [131]. This virus acquired a number of virulence-enhancing mutations during passage in Mx1 mice. Interestingly, the highly virulent virus was still susceptible to the antiviral action of mouse Mx1 protein. Mx1-positive mice, but not Mx1-deficient mice were highly resistant to the virus if pretreated with IFN shortly before infection (Fig. 3 ). Also, the highly virulent virus was not a better inhibitor of IFN production than an ordinary influenza virus, indicating that the IFN antagonistic function of its NS1 protein did not make the difference. A series of experiments demonstrated that the highly virulent virus was able to replicate much faster in mouse lung than a normal mouse-adapted virus. It grew to about 1000 fold higher titers within the first 24 h post infection, as compared to standard virus. We concluded that the virus had a head start due to an unusually high virus multiplication speed and could outrun the establishment of the IFN-mediated antiviral Mx response. In other words, inducible host defense mechanisms can be overcome if the pathogen multiplies extremely fast. It is conceivable that highly pathogenic viruses may often profit from the fact that the IFN system requires time to become active. It remains to be seen whether a similar evasion strategy is used by other successful influenza A viruses such as the Asian H5N1 viruses or the pandemic strain of 1918.
Fig. 3.
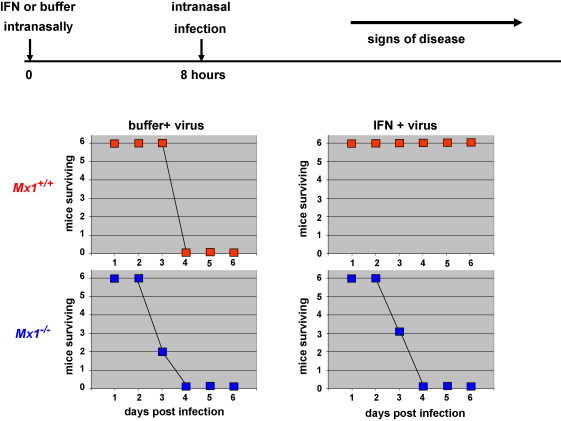
IFN protects Mx1-positive but not Mx1-negative mice from lethal influenza. Mx1-positive (Mx1+/+) and Mx1-negative mice (Mx1−/−) on a congenic C57BL/6 genetic background were treated with either buffer or 5 × 105 units of IFN. Ten hours later the mice were infected with 1000 pfu (equivalent to 100 LD50) of highly virulent influenza A virus and their health status was recorded for up to 14 days (for details see Ref. [131])
7. Concluding remarks
The interplay between viruses and the IFN system is an interesting facet of the virus–host relationship and reflects an ongoing evolutionary race between the two genetic systems. Emerging viruses have to constantly adapt to guarantee successful trans-species transmission in new hosts. Pandemic influenza A viruses presumably display a number of evasion mechanisms, including the surprisingly simple measure of fast growth. Our present knowledge of the IFN system and viral escape strategies is still limited. Future research should provide a better insight into the intricate interplay between viruses and the innate immune defenses of the host.
Conflict of interest statement
None.
Acknowledgements
We thank Peter Staeheli for critically reading the manuscript. Our own work described in the text was supported be grants from the Deutsche Forschungsgemeinschaft.
Biographies

Otto Haller received his M.D. in 1972 and worked until 1975 as a Postdoctoral Fellow in the group of Prof. Jean Lindenmann (the co-discoverer of interferon together with Alick Isaacs) at the Institute of Medical Microbiology, University of Zürich, Switzerland. He was a Research Associate at the University of Uppsala, Sweden, and an Assistant Professor at the University of Zürich and the Rockefeller University, New York. In 1989, he was appointed Full Professor in the Faculty of Medicine and Director of the Department of Virology, University of Freiburg, Germany. He is a member of the German Academy of Natural Scientists Leopoldina, past President of the German/Swiss/Austrian Society for Virology (GfV) and acting President of the International Society of Interferon and Cytokine Research (ISICR). He was awarded the Swiss Cancer Prize for early work on natural killer (NK) cells, the Latsis Prize of the Swiss National Science Foundation for his work on genetic control of host resistance against influenza viruses, and the 1998 Milstein Award of ISICR for his work on Mx. The focus of his research interest is virus-host cell interactions, viral pathogenesis, and innate immunity.

Georg Kochs received his Ph.D. in 1988 from the Faculty of Biology at the University of Freiburg, Germany. As a postdoctoral fellow he worked in basic research at the Institute of Biochemistry and the Goedecke AG, Freiburg and joined the group of Otto Haller in 1993. Since 1998, he is an independent research group leader in the Department of Virology, Freiburg, Germany. His work is focused on the biochemistry and antiviral function of the Mx proteins and the molecular biology as well as virulence of orthomyxoviruses. He has published over 50 research papers and review articles.

Friedemann Weber received his M.Sc. in 1993 from the Department of Microbiology and his Ph.D. in 1997 from the Department of Virology at the University of Freiburg, Germany. He was an EMBO Long Term Postdoctoral Fellow at the Institute of Virology in Glasgow, UK, and is currently a research group leader in the Department of Virology, Freiburg, Germany. In 2003, he has received the Milstein Young Investigator Award of ISICR and in 2007 he will be awarded the “Löffler-Frosch-Preis” of the Gesellschaft für Virologie (GfV). He is interested in the interaction of highly pathogenic RNA viruses with the interferon system, with particular emphasis on interferon-inducing viral structures, their intracellular receptors, and viral escape strategies. He has published over 50 research papers, review articles and book chapters.
References
- 1.Guenet J.L., Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 2003;19:24–31. doi: 10.1016/s0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 2.Haller O. Inborn resistance of mice to orthomyxoviruses. Curr Top Microbiol Immunol. 1981;92:25–52. doi: 10.1007/978-3-642-68069-4_3. [DOI] [PubMed] [Google Scholar]
- 3.Haller O., Acklin M., Staeheli P. Influenza virus resistance of wild mice: wild-type and mutant Mx alleles occur at comparable frequencies. J Interferon Res. 1987;7:647–656. doi: 10.1089/jir.1987.7.647. [DOI] [PubMed] [Google Scholar]
- 4.Lindenmann J. Resistance of mice to mouse-adapted influenza A virus. Virology. 1962;16:203–204. doi: 10.1016/0042-6822(62)90297-0. [DOI] [PubMed] [Google Scholar]
- 5.Reeves R.H., O’Hara B.F., Pavan W.J., Gearhart J.D., Haller O. Genetic mapping of the Mx influenza virus resistance gene within the region of mouse chromosome 16 that is homologous to human chromosome 21. J Virol. 1988;62:4372–4375. doi: 10.1128/jvi.62.11.4372-4375.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnheiter H., Skuntz S., Noteborn M., Chang S., Meier E. Transgenic mice with intracellular immunity to influenza virus. Cell. 1990;62:51–61. doi: 10.1016/0092-8674(90)90239-b. [DOI] [PubMed] [Google Scholar]
- 7.Staeheli P., Haller O., Boll W., Lindenmann J., Weissmann C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell. 1986;44:147–158. doi: 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 8.Haller O., Arnheiter H., Lindenmann J., Gresser I. Host gene influences sensitivity to interferon action selectively for influenza virus. Nature. 1980;283:660–662. doi: 10.1038/283660a0. [DOI] [PubMed] [Google Scholar]
- 9.Holzinger D., Jorns C., Stertz S. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol. 2007;81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonelli G., Simeoni E., Turriziani O. Correlation of interferon-induced expression of MxA mRNA in peripheral blood mononuclear cells with the response of patients with chronic active hepatitis C to IFN-alpha therapy. J Interferon Cytokine Res. 1999;19:243–251. doi: 10.1089/107999099314171. [DOI] [PubMed] [Google Scholar]
- 11.Roers A., Hochkeppel H.K., Horisberger M.A., Hovanessian A., Haller O. MxA gene expression after live virus vaccination: a sensitive marker for endogenous type I interferon. J Infect Dis. 1994;169:807–813. doi: 10.1093/infdis/169.4.807. [DOI] [PubMed] [Google Scholar]
- 12.Staeheli P., Grob R., Meier E., Sutcliffe J.G., Haller O. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol. 1988;8:4518–4523. doi: 10.1128/mcb.8.10.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perelygin A.A., Scherbik S.V., Zhulin I.B., Stockman B.M., Li Y., Brinton M.A. Positional cloning of the murine flavivirus resistance gene. Proc Natl Acad Sci USA. 2002;99:9322–9327. doi: 10.1073/pnas.142287799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mashimo T., Lucas M., Simon-Chazottes D. A nonsense mutation in the gene encoding 2′-5′-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc Natl Acad Sci USA. 2002;99:11311–11316. doi: 10.1073/pnas.172195399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brinton M.A., Perelygin A.A. Genetic resistance to flaviviruses. Adv Virus Res. 2003;60:43–85. doi: 10.1016/s0065-3527(03)60002-3. [DOI] [PubMed] [Google Scholar]
- 16.Casanova J.L., Schurr E., Abel L., Skamene E. Forward genetics of infectious diseases: immunological impact. Trends Immunol. 2002;23:469–472. doi: 10.1016/s1471-4906(02)02289-5. [DOI] [PubMed] [Google Scholar]
- 17.van Pesch V., Lanaya H., Renauld J.C., Michiels T. Characterization of the murine alpha interferon gene family. J Virol. 2004;78:8219–8228. doi: 10.1128/JVI.78.15.8219-8228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts R.M., Ezashi T., Rosenfeld C.S., Ealy A.D., Kubisch H.M. Evolution of the interferon tau genes and their promoters, and maternal-trophoblast interactions in control of their expression. Reprod Suppl. 2003;61:239–251. [PubMed] [Google Scholar]
- 19.Ank N., West H., Paludan S.R. IFN-lambda: novel antiviral cytokines. J Interferon Cytokine Res. 2006;26:373–379. doi: 10.1089/jir.2006.26.373. [DOI] [PubMed] [Google Scholar]
- 20.Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornung V., Ellegast J., Kim S. 5′-Triphosphate RNA Is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 22.Pichlmair A., Schulz O., Tan C.P. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′ phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 23.Bowie A.G., Fitzgerald K.A. RIG-I: tri-ing to discriminate between self and non-self RNA. Trends Immunol. 2007;28:147–150. doi: 10.1016/j.it.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 25.Fitzgerald K.A., McWhirter S.M., Faia K.L. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 26.Sharma S., TenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 27.Andrejeva J., Childs K.S., Young D.F. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoneyama M., Kikuchi M., Natsukawa T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 29.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 30.Uematsu S., Akira S. Toll-like receptors and type I interferons. J Biol Chem. 2007;282:15319–15323. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- 31.tenOever B.R., Sharma S., Zou W. Activation of TBK1 and IKK epsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol. 2004;78:10636–10649. doi: 10.1128/JVI.78.19.10636-10649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy D.E., Marie I., Prakash A. Ringing the interferon alarm: differential regulation of gene expression at the interface between innate and adaptive immunity. Curr Opin Immunol. 2003;15:52–58. doi: 10.1016/s0952-7915(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 33.Honda K., Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T., Sato S., Ishii K.J. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004 doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 35.Levy D.E., Darnell J.E., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 36.de Veer M.J., Holko M., Frevel M. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 37.Der S.D., Zhou A., Williams B.R., Silverman R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia M.A., Gil J., Ventoso I. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams B.R. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 40.Silverman R.H. Fascination with 2-5A-dependent RNase: a unique enzyme that functions in interferon action. J Interferon Res. 1994;14:101–104. doi: 10.1089/jir.1994.14.101. [DOI] [PubMed] [Google Scholar]
- 41.Haller O., Kochs G. Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic. 2002;3:710–717. doi: 10.1034/j.1600-0854.2002.31003.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhou A., Paranjape J., Brown T.L. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou A., Paranjape J.M., Der S.D., Williams B.R., Silverman R.H. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology. 1999;258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- 44.Espert L., Degols G., Gongora C. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J Biol Chem. 2003;278:16151–16158. doi: 10.1074/jbc.M209628200. [DOI] [PubMed] [Google Scholar]
- 45.Guo J., Hui D.J., Merrick W.C., Sen G.C. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J. 2000;19:6891–6899. doi: 10.1093/emboj/19.24.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hui D.J., Bhasker C.R., Merrick W.C., Sen G.C. Viral stress-inducible protein p56 inhibits translation by blocking the interaction of eIF3 with the ternary complex eIF2.GTP.Met-tRNAi. J Biol Chem. 2003;278:39477–39482. doi: 10.1074/jbc.M305038200. [DOI] [PubMed] [Google Scholar]
- 47.Anderson S.L., Carton J.M., Lou J., Xing L., Rubin B.Y. Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology. 1999;256:8–14. doi: 10.1006/viro.1999.9614. [DOI] [PubMed] [Google Scholar]
- 48.Regad T., Chelbi-Alix M.K. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene. 2001;20:7274–7286. doi: 10.1038/sj.onc.1204854. [DOI] [PubMed] [Google Scholar]
- 49.Wang C., Pflugheber J., Sumpter R., Jr. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chelbi-Alix M.K., Quignon F., Pelicano L., Koken M.H., de The H. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J Virol. 1998;72:1043–1051. doi: 10.1128/jvi.72.2.1043-1051.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Djavani M., Rodas J., Lukashevich I.S. Role of the promyelocytic leukemia protein PML in the interferon sensitivity of lymphocytic choriomeningitis virus. J Virol. 2001;75:6204–6208. doi: 10.1128/JVI.75.13.6204-6208.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Regad T., Saib A., Lallemand-Breitenbach V., Pandolfi P.P., de The H., Chelbi-Alix M.K. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 2001;20:3495–3505. doi: 10.1093/emboj/20.13.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chee A.V., Lopez P., Pandolfi P.P., Roizman B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol. 2003;77:7101–7105. doi: 10.1128/JVI.77.12.7101-7105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Engelhardt O.G., Sirma H., Pandolfi P.P., Haller O. Mx1 GTPase accumulates in distinct nuclear domains and inhibits influenza A virus in cells that lack promyelocytic leukaemia protein nuclear bodies. J Gen Virol. 2004;85:2315–2326. doi: 10.1099/vir.0.79795-0. [DOI] [PubMed] [Google Scholar]
- 55.Arnheiter H., Frese M., Kambadur R., Meier E., Haller O. Mx transgenic mice–animal models of health. Curr Top Microbiol Immunol. 1996;206:119–147. doi: 10.1007/978-3-642-85208-4_8. [DOI] [PubMed] [Google Scholar]
- 56.Kochs G., Haller O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc Natl Acad Sci USA. 1999;96:2082–2086. doi: 10.1073/pnas.96.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kochs G., Janzen C.H., Hohenberg H., Haller O. Antivirally active MxA protein sequesters LaCrosse virus nucleocapsid protein into perinuclear complexes. Proc Natl Acad Sci USA. 2002;99:3153–3158. doi: 10.1073/pnas.052430399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reichelt M., Stertz S., Krijnse-Locker J., Haller O., Kochs G. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic. 2004;5:772–784. doi: 10.1111/j.1600-0854.2004.00219.x. [DOI] [PubMed] [Google Scholar]
- 59.Hefti H.P., Frese M., Landis H. Human MxA protein protects mice lacking a functional alpha/beta interferon system against LaCrosse virus and other lethal viral infections. J Virol. 1999;73:6984–6991. doi: 10.1128/jvi.73.8.6984-6991.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alcami A., Smith G.L. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J Virol. 1995;69:4633–4639. doi: 10.1128/jvi.69.8.4633-4639.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alcami A., Symons J.A., Smith G.L. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol. 2000;74:11230–11239. doi: 10.1128/jvi.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puehler F., Weining K.C., Symons J.A., Smith G.L., Staeheli P. Vaccinia virus-encoded cytokine receptor binds and neutralizes chicken interferon-gamma. Virology. 1998;248:231–240. doi: 10.1006/viro.1998.9278. [DOI] [PubMed] [Google Scholar]
- 63.Symons J.A., Alcami A., Smith G.L. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81:551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Sastre A., Biron C.A. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 65.Haller O., Kochs G., Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology. 2006;344:119–130. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hengel H., Koszinowski U.H., Conzelmann K.K. Viruses know it all: new insights into IFN networks. Trends Immunol. 2005;26:396–401. doi: 10.1016/j.it.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 67.Mibayashi M., Martinez-Sobrido L., Loo Y.M., Cardenas W.B., Gale M., Jr., Garcia-Sastre A. Inhibition of retinoic acid-inducible gene-I-mediated induction of interferon-{beta} by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Childs K., Stock N., Ross C. Mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 69.Kawai T., Takahashi K., Sato S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 70.Meylan E., Curran J., Hofmann K. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 71.Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 72.Xu L.G., Wang Y.Y., Han K.J., Li L.Y., Zhai Z., Shu H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:1–14. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 73.Chen Z., Benureau Y., Rijnbrand R. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J Virol. 2007;81:964–976. doi: 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin R., Lacoste J., Nakhaei P. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brzozka K., Finke S., Conzelmann K.K. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol. 2005;79:7673–7681. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alff P.J., Gavrilovskaya I.N., Gorbunova E. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J Virol. 2006;80:9676–9686. doi: 10.1128/JVI.00508-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bauhofer O., Summerfield A., McCullough K.C., Ruggli N. Role of double-stranded RNA and Npro of classical swine fever virus in the activation of monocyte-derived dendritic cells. Virology. 2005;343:93–105. doi: 10.1016/j.virol.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 78.La Rocca S.A., Herbert R.J., Crooke H., Drew T.W., Wileman T.E., Powell P.P. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, Npro. J Virol. 2005;79:7239–7247. doi: 10.1128/JVI.79.11.7239-7247.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ruggli N., Bird B.H., Liu L., Bauhofer O., Tratschin J.D., Hofmann M.A. N(pro) of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-alpha/beta induction. Virology. 2005;340:265–276. doi: 10.1016/j.virol.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 80.Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N. Npro of classical swine fever virus interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol 2007; in press. [DOI] [PMC free article] [PubMed]
- 81.Hilton L., Moganeradj K., Zhang G. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol. 2006;80:11723–11732. doi: 10.1128/JVI.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ronco L.V., Karpova A.Y., Vidal M., Howley P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Sars coronavirus proteins Orf 3b, Orf 6, and nucleocapsid function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spiegel M., Pichlmair A., Martinez-Sobrido L. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol. 2005;79:2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burysek L., Yeow W.S., Lubyova B. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J Virol. 1999;73:7334–7342. doi: 10.1128/jvi.73.9.7334-7342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Burysek L., Yeow W.S., Pitha P.M. Unique properties of a second human herpesvirus 8-encoded interferon regulatory factor (vIRF-2) J Hum Virol. 1999;2:19–32. [PubMed] [Google Scholar]
- 87.Fuld S., Cunningham C., Klucher K., Davison A.J., Blackbourn D.J. Inhibition of interferon signaling by the Kaposi's sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J Virol. 2006;80:3092–3097. doi: 10.1128/JVI.80.6.3092-3097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li M., Lee H., Guo J. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor. J Virol. 1998;72:5433–5440. doi: 10.1128/jvi.72.7.5433-5440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lubyova B., Pitha P.M. Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors. J Virol. 2000;74:8194–8201. doi: 10.1128/jvi.74.17.8194-8201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lubyova B., Kellum M.J., Frisancho A.J., Pitha P.M. Kaposi's sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J Biol Chem. 2004;279:7643–7654. doi: 10.1074/jbc.M309485200. [DOI] [PubMed] [Google Scholar]
- 91.Zimring J.C., Goodbourn S., Offermann M.K. Human herpesvirus 8 encodes an interferon regulatory factor (IRF) homolog that represses IRF-1-mediated transcription. J Virol. 1998;72:701–707. doi: 10.1128/jvi.72.1.701-707.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bouloy M., Janzen C., Vialat P. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J Virol. 2001;75:1371–1377. doi: 10.1128/JVI.75.3.1371-1377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Billecocq A., Spiegel M., Vialat P. NSs protein of Rift Valley Fever Virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004;78:9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Le May N., Dubaele S., De Santis L.P., Billecocq A., Bouloy M., Egly J.M. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell. 2004;116:541–550. doi: 10.1016/s0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- 95.Thomas D., Blakqori G., Wagner V. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J Biol Chem. 2004;279:31471–31477. doi: 10.1074/jbc.M400938200. [DOI] [PubMed] [Google Scholar]
- 96.Cardenas W.B., Loo Y.M., Gale M., Jr. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Child S.J., Hanson L.K., Brown C.E., Janzen D.M., Geballe A.P. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J Virol. 2006;80:10173–10180. doi: 10.1128/JVI.00905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Garcia-Sastre A., Egorov A., Matassov D. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 99.Garcia-Sastre A. Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology. 2001;279:375–384. doi: 10.1006/viro.2000.0756. [DOI] [PubMed] [Google Scholar]
- 100.Hartman A.L., Dover J.E., Towner J.S., Nichol S.T. Reverse genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J Virol. 2006;80:6430–6440. doi: 10.1128/JVI.00044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 102.Lee T.G., Tomita J., Hovanessian A.G., Katze M.G. Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of Mr 68,000 from influenza virus-infected cells. Proc Natl Acad Sci USA. 1990;87:6208–6212. doi: 10.1073/pnas.87.16.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Valchanova R.S., Picard-Maureau M., Budt M., Brune W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J Virol. 2006;80:10181–10190. doi: 10.1128/JVI.00908-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mathews M.B., Shenk T. Adenovirus virus-associated RNA and translation control. J Virol. 1991;65:5657–5662. doi: 10.1128/jvi.65.11.5657-5662.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vyas J., Elia A., Clemens M.J. Inhibition of the protein kinase PKR by the internal ribosome entry site of hepatitis C virus genomic RNA. RNA. 2003;9:858–870. doi: 10.1261/rna.5330503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elia A., Laing K.G., Schofield A., Tilleray V.J., Clemens M.J. Regulation of the double-stranded RNA-dependent protein kinase PKR by RNAs encoded by a repeated sequence in the Epstein-Barr virus genome. Nucleic Acids Res. 1996;24:4471–4478. doi: 10.1093/nar/24.22.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gunnery S., Rice A.P., Robertson H.D., Mathews M.B. Tat-responsive region RNA of human immunodeficiency virus 1 can prevent activation of the double-stranded-RNA-activated protein kinase. Proc Natl Acad Sci USA. 1990;87:8687–8691. doi: 10.1073/pnas.87.22.8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.He B., Gross M., Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Taylor D.R., Shi S.T., Romano P.R., Barber G.N., Lai M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 110.Roy S., Katze M.G., Parkin N.T., Edery I., Hovanessian A.G., Sonenberg N. Control of the interferon-induced 68-kilodalton protein kinase by the HIV-1 tat gene product. Science. 1990;247:1216–1219. doi: 10.1126/science.2180064. [DOI] [PubMed] [Google Scholar]
- 111.Davies M.V., Elroy-Stein O., Jagus R., Moss B., Kaufman R.J. The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J Virol. 1992;66:1943–1950. doi: 10.1128/jvi.66.4.1943-1950.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li S., Min J.Y., Krug R.M., Sen G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology. 2006;349:13–21. doi: 10.1016/j.virol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 113.Min J.Y., Krug R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci USA. 2006;103:7100–7105. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Black T.L., Barber G.N., Katze M.G. Degradation of the interferon-induced 68,000-M(r) protein kinase by poliovirus requires RNA. J Virol. 1993;67:791–800. doi: 10.1128/jvi.67.2.791-800.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Martinand C., Montavon C., Salehzada T., Silhol M., Lebleu B., Bisbal C. RNase L inhibitor is induced during human immunodeficiency virus type 1 infection and down regulates the 2-5A/RNase L pathway in human T cells. J Virol. 1999;73:290–296. doi: 10.1128/jvi.73.1.290-296.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martinand C., Salehzada T., Silhol M., Lebleu B., Bisbal C. RNase L inhibitor (RLI) antisense constructions block partially the down regulation of the 2-5A/RNase L pathway in encephalomyocarditis-virus-(EMCV)-infected cells. Eur J Biochem. 1998;254:248–255. doi: 10.1046/j.1432-1327.1998.2540248.x. [DOI] [PubMed] [Google Scholar]
- 117.Cayley P.J., Davies J.A., McCullagh K.G., Kerr I.M. Activation of the ppp(A2′p)nA system in interferon-treated, herpes simplex virus-infected cells and evidence for novel inhibitors of the ppp(A2′p)nA-dependent RNase. Eur J Biochem. 1984;143:165–174. doi: 10.1111/j.1432-1033.1984.tb08355.x. [DOI] [PubMed] [Google Scholar]
- 118.Han J.Q., Townsend H.L., Jha B.K., Paranjape J.M., Silverman R.H., Barton D.J. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J Virol. 2007;81:5561–5572. doi: 10.1128/JVI.01857-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Atreya P.L., Kulkarni S. Respiratory syncytial virus strain A2 is resistant to the antiviral effects of type I interferons and human MxA. Virology. 1999;261:227–241. doi: 10.1006/viro.1999.9835. [DOI] [PubMed] [Google Scholar]
- 120.Young D.F., Didcock L., Goodbourn S., Randall R.E. Paramyxoviridae use distinct virus-specific mechanisms to circumvent the interferon response. Virology. 2000;269:383–390. doi: 10.1006/viro.2000.0240. [DOI] [PubMed] [Google Scholar]
- 121.Schlender J., Bossert B., Buchholz U., Conzelmann K.K. Bovine respiratory syncytial virus nonstructural proteins NS1 and NS2 cooperatively antagonize alpha/beta interferon-induced antiviral response. J Virol. 2000;74:8234–8242. doi: 10.1128/jvi.74.18.8234-8242.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Spann K.M., Tran K.C., Chi B., Rabin R.L., Collins P.L. Suppression of the induction of alpha, beta, and gamma interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J Virol. 2004;78:4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wright P.F., Karron R.A., Madhi S.A. The interferon antagonist NS2 protein of respiratory syncytial virus is an important virulence determinant for humans. J Infect Dis. 2006;193:573–581. doi: 10.1086/499600. [DOI] [PubMed] [Google Scholar]
- 124.Moller A., Schmitz M.L. Viruses as hijackers of PML nuclear bodies. Arch Immunol Ther Exp (Warsz) 2003;51:295–300. [PubMed] [Google Scholar]
- 125.Boutell C., Sadis S., Everett R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol. 2002;76:841–850. doi: 10.1128/JVI.76.2.841-850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Van Sant C., Hagglund R., Lopez P., Roizman B. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc Natl Acad Sci USA. 2001;98:8815–8820. doi: 10.1073/pnas.161283098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Muller S., Dejean A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J Virol. 1999;73:5137–5143. doi: 10.1128/jvi.73.6.5137-5143.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kochs G., Weber F., Gruber S., Delvendahl A., Leitz C., Haller O. Thogoto virus matrix protein is encoded by a spliced mRNA. J Virol. 2000;74:10785–10789. doi: 10.1128/jvi.74.22.10785-10789.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hagmaier K., Jennings S., Buse J., Weber F., Kochs G. Novel gene product of Thogoto virus segment 6 codes for an interferon antagonist. J Virol. 2003;77:2747–2752. doi: 10.1128/JVI.77.4.2747-2752.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pichlmair A., Buse J., Jennings S., Haller O., Kochs G., Staeheli P. Thogoto virus lacking interferon-antagonistic protein ML is strongly attenuated in newborn Mx1-positive but not Mx1-negative mice. J Virol. 2004;78:11422–11424. doi: 10.1128/JVI.78.20.11422-11424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grimm D., Staeheli P., Hufbauer M. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc Natl Acad Sci USA. 2007;104:6806–6811. doi: 10.1073/pnas.0701849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jennings S., Martinez-Sobrido L., Garcia-Sastre A., Weber F., Kochs G. Thogoto virus ML protein suppresses IRF3 function. Virology. 2005;331:63–72. doi: 10.1016/j.virol.2004.10.015. [DOI] [PubMed] [Google Scholar]