

Highlights
-
•
Codon usage bias of porcine epidemic diarrhea virus is low.
-
•
Mutational bias and natural selection pressure influence codon usage bias of porcine epidemic diarrhea virus.
-
•
Natural selection plays an increasingly significant role during evolution of porcine epidemic diarrhea virus.
Keywords: Codon usage bias, Porcine epidemic diarrhea virus, Mutation bias, Natural selection
Abstract
Porcine epidemic diarrhea virus (PEDV) has been responsible for several recent outbreaks of porcine epidemic diarrhea (PED) and has caused great economic loss in the swine-raising industry. Considering the significance of PEDV, a systemic analysis was performed to study its codon usage patterns. The relative synonymous codon usage value of each codon revealed that codon usage bias exists and that PEDV tends to use codons that end in T. The mean ENC value of 47.91 indicates that the codon usage bias is low. However, we still wanted to identify the cause of this codon usage bias. A correlation analysis between the codon compositions (A3s, T3s, G3s, C3s, and GC3s), the ENC values, and the nucleotide contents (A%, T%, G%, C%, and GC%) indicated that mutational bias plays role in shaping the PEDV codon usage bias. This was further confirmed by a principal component analysis between the codon compositions and the axis values. Using the Gravy, Aroma, and CAI values, a role of natural selection in the PEDV codon usage pattern was also identified. Neutral analysis indicated that natural selection pressure plays a more important role than mutational bias in codon usage bias. Natural selection also plays an increasingly significant role during PEDV evolution. Additionally, gene function and geographic distribution also influence the codon usage bias to a degree.
1. Introduction
Porcine epidemic diarrhea (PED) was first identified in Belgium and the United Kingdom in 1971 (Rosenberg et al., 1977). PED is a great threat to swine health and production, and it has caused devastating diseases with a severe impact on the swine-raising industry (Huang et al., 2013, Lee and Lee, 2014, Puranaveja et al., 2009, Wang et al., 2014). PED is characterized by vomiting, watery diarrhea and dehydration and causes a high death rate among suckling piglets. PED has been reported in many countries since 1971, including China (Chen et al., 2008, Tian et al., 2013), Thailand (Puranaveja et al., 2009, Temeeyasen et al., 2014), South Korea (Lee and Lee, 2014, Park et al., 2013), and the United States (Huang et al., 2013, Wang et al., 2014). Outbreaks of PED were first identified in the United States between 2013 and 2014, and in less than one year, 2692 PED cases were found in 23 US states, which led to great economic losses (Huang et al., 2013, Wang et al., 2014). The genome of the causative agent was identified as being closely related to a strain from China (Wang et al., 2014). During 2013 and 2014, outbreaks of PED also occurred in South Korea, and genomic sequences of the South Korean isolated virus were shown to be similar to the US PED strains (Lee and Lee, 2014). The recent frequency in outbreaks and the great economic losses associated with PED indicate the importance of understanding this virus.
PED is caused by the porcine epidemic diarrhea virus (PEDV). The first PEDV isolate was named CV777 (Egberink et al., 1988, Huang et al., 2013, Rosenberg et al., 1977), and sequencing of its viral genome indicated that PEDV and the bat coronavirus have a common evolutionary precursor (Huang et al., 2013). The PEDV genome is composed of seven open reading frames (ORFs) (Egberink et al., 1988, Kocherhans et al., 2001). ORF1a and ORF1b encode the replicase proteins. The next five ORFs encode the viral proteins, which include the spike protein (S), the ORF3 protein (ORF3), the small membrane proteins (E), the membrane proteins (M), and the nucleocapsid protein (N).
Synonymous codons are not used randomly in the genomes of organisms. The same codon may be present in different genes in a single genome as well as in different parts of one specific gene (Fu, 2010, Li et al., 2012, Liu et al., 2010a, Zhou et al., 2010). This phenomenon is called codon usage bias. Codon usage bias has been confirmed in many viruses (Auewarakul et al., 2009, Fu, 2010, D’Andrea et al., 2011, Li et al., 2012, Liu et al., 2010a, Shi et al., 2013, Wang et al., 2011, Wong et al., 2010, Zhou et al., 2010). Synonymous codon usage bias is strong in some viruses, such as the hepatitis A virus (D’Andrea et al., 2011). Other viruses have weak codon usage bias, such as SARS (Zhao et al., 2008), classical swine fever virus (Tao et al., 2009), and H9N2 influenza A virus (Liu et al., 2010a). Codon usage bias is related to mutation bias, natural selection, gene function, gene length, tRNA abundance, and RNA structure. Codon usage bias is generally influenced by one of the two following biological pressures: (1) pressure from mutational bias and/or (2) pressure from natural selection (Fu, 2010). In some RNA and DNA viruses, mutational bias has been identified as playing a more important role, compared with natural selection, such as in foot-and-mouth virus (Zhou et al., 2010)and herpes virus (Fu, 2010). Natural selection has also been confirmed as being the dominant factor in some viruses such as Parvoviridae (Shi et al., 2013). The nucleotide content in the third codon position (A3s, T3s, G3s, C3s, GC3s, AT3s) is based on the codon usage bias (Wong et al., 2010). Many analytic methods for analyzing codon usage bias have been developed and are based on examining the nucleotide content of the third codon position. These methods have been shown to be effective in uncovering the codon usage bias of the genome and other significant information, such as the effective number of codons (Wright, 1990).
Considering the recent increase in the prevalence of PEDV and its great threat to the swine industry, we chose to examine the codon usage bias of PEDV to better understand the codon usage pattern of the PEDV genome, the differences in codon usage between the PEDV and pig genomes, and the evolutionary pattern of PEDV codon usage.
2. Materials and methods
2.1. Sequence data
The complete genome sequences of the PEDV isolates were retrieved from the GenBank database (http://www.ncbi.nlm.nih.gov). To better understand PEDV codon usage bias, only the viruses with complete genomic information were included in our study. Detailed information about the 43 PEDV isolates, including their accession number, the time when they were isolated, and the country where they were isolated, is listed in the supplied materials (Table S1).To perform the phylogenetic analysis, the sequence data were compiled and edited using the DNASTAR software package (Madison, WI, USA). The edited data were then aligned using the BioEdit (version 7.0.9.0) sequence analysis program and the ClustalW method. The unrooted phylogenetic tree was constructed with the MEGA 4.0 software with the evolutionary pattern of the 43 PEDV isolates using the pairwise deletion model and calculated based on 1,000 replicates.
2.2. Nucleotide composition
The nucleotide content (A%, T%, G%, and C%) of each PEDV strain was analyzed using the MEGA 4.0 biosoftware for windows. The nucleotide composition of the third synonymous codon position of each codon (A3s, T3s, G3s, and C3s) was calculated using the CodonW program (version 1.4.2) (http://codonw.sourceforge.net//).
2.3. Codon usage indices
Relative synonymous codon usage (RSCU) values were first proposed in 1986 (Sharp and Li, 1986). The RSCU value is independent of the amino acid composition and has been used widely to estimate the codon usage bias between genes. A higher RSCU value means that the codon is used more frequently or has a higher codon usage bias. If the RSCU value of a specific codon is higher than 1.0, it is considered to have a positive codon usage bias. When the RSCU value is less than 1.0, it is considered to have a negative codon usage bias.
The effective number of codons (ENC) value is not influenced by the amino acid composition or the gene length. In the ENC analysis, an ENC value is given to each codon. The ENC value ranges from 20 to 61. In contrast to the RSCU value, a higher ENC value correlates to a weaker codon usage bias. If the codon of one gene is completely randomly and unbiased, then the expected ENC value (ENC∗) is calculated from GC3s (Sharp and Li, 1986):
The s value is the GC3s content of each codon. When the expected ENC value is plotted against the GC3s value, an expected curve is formed. A dot located on the curve is regarded as unbiased.
2.4. Principal component analysis
Principal component analysis (PCA) is a common statistical method used to explain the codon usage of a specific gene. In the analysis, the RSCU value of each codon is explained by a 59-dimension space and transformed into unrelated factors. In this model, PCA can determine the major variation from the RSCU value of each codon. Using both the PCA and correlation analysis, the factors influencing the codon usage bias can be effectively determined.
2.5. Codon adaptation index
Codon adaptation index (CAI) is one of the most widespread methods for analyzing codon usage bias due to the natural selection pressure. It represents the adaption of the virus to the host. The value of the CAI value is between 0 and 1. A higher CAI value indicates the stronger adaptation to the host. The codon usage pattern of the Sus scrofa is obtained from an online website (http://www.kazusa.or.jp/codon/) (Puigbo et al., 2008). To estimate the codon adaption of the PEDV to Sus scrofa, the CAI value is calculated using the CAIcal biosoftware (http://genomes.urv.es/CAIcal). In the analysis, the synonymous codon usage pattern of the viral host is deposited as reference and the CAI value of the PEDV is calculated after comparison with the reference from Sus scrofa. ORFs 1a/b is not included in the analysis because of the nucleotide length restrictions of the online tool.
2.6. Hydropathicity and aromaticity indices
The hydropathicity and aromaticity of a single gene product are thought to be the result of translation selection and according to the natural selection (Lobry and Gautier, 1994). In our study, the Gravy and Aroma score of each gene product was obtained using the CodonW program (version 1.4.2) to reflect the hydropathicity and aromaticity, respectively. A higher Gravy or Aroma score means a more hydrophobic or aromatic amino acid product, respectively.
2.7. Neutral evolution analysis
The neutral analysis is used to estimate the varying role of mutational pressure and natural selection in the PEDV. In the analysis, the P12 (GC12s) value of the synonymous codon is plotted against its P3 (GC3s) value (Sueoka, 1988). To study the evolutional characteristics of mutational pressure and natural selection in the PEDV, the P12 or the P3 value is plotted against the evolutionary time. The evolutionary speed of the mutational pressure and natural selection pressure is expressed as the slope of a simple regression line.
2.8. Statistical analysis
The correlation analysis was carried out using the statistical software SPSS (version 19.0) with the Spearman’s rank method.
3. Results
3.1. Compositional properties of PEDV
The composition properties of the 43 PEDV strains were analyzed and listed in Table 1 . The results indicate that all of the PEDV strains were A/T rich and C/G poor. The most abundant base is T, and then subsequently A, G, and C. The SD value was very small for all of the four nucleotides with A having the lowest SD value. This indicates that the base content varied little between the PEDV strains, especially with respect to the A nucleotide.
Table 1.
The nucleotide content of the 43 PEDV strains.
| Nucleotide content |
||||
|---|---|---|---|---|
| T% | C% | A% | G% | |
| KF840537 | 33.4 | 19.2 | 24.8 | 22.7 |
| KJ158152 | 33.4 | 19.0 | 24.8 | 22.8 |
| KJ662670 | 33.5 | 19.1 | 24.8 | 22.6 |
| KJ584361 | 33.4 | 19.1 | 24.8 | 22.6 |
| KJ778615 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF761675 | 33.5 | 19.1 | 24.8 | 22.7 |
| KJ623926 | 33.4 | 19.1 | 24.8 | 22.7 |
| KJ196348 | 33.4 | 19.1 | 24.8 | 22.7 |
| KJ408801 | 33.5 | 19.1 | 24.8 | 22.6 |
| KJ020932 | 33.4 | 19.1 | 24.8 | 22.7 |
| KJ399978 | 33.4 | 19.1 | 24.8 | 22.6 |
| KF650370 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF650373 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF468752 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF468753 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF468754 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF384500 | 33.4 | 19.1 | 24.8 | 22.7 |
| KF272920 | 33.5 | 19.1 | 24.8 | 22.6 |
| KC189944 | 33.3 | 19.1 | 24.7 | 22.9 |
| KC140102 | 33.5 | 19.0 | 24.8 | 22.6 |
| KC210147 | 33.4 | 19.1 | 24.8 | 22.7 |
| KC196276 | 33.4 | 19.2 | 24.8 | 22.7 |
| JX560761 | 33.4 | 19.0 | 24.8 | 22.8 |
| JX647847 | 33.4 | 19.1 | 24.8 | 22.7 |
| JX524137 | 33.5 | 19.1 | 24.8 | 22.6 |
| JX489155 | 33.4 | 19.1 | 24.8 | 22.7 |
| JX261936 | 33.5 | 19.0 | 24.8 | 22.7 |
| JX188454 | 33.4 | 19.1 | 24.8 | 22.7 |
| JX112709 | 33.5 | 19.0 | 24.8 | 22.7 |
| JX088695 | 33.4 | 19.1 | 24.8 | 22.7 |
| JQ023161 | 33.4 | 19.1 | 24.7 | 22.8 |
| JN825712 | 33.4 | 19.1 | 24.8 | 22.7 |
| JQ282909 | 33.4 | 19.2 | 24.8 | 22.7 |
| JN547228 | 33.4 | 19.1 | 24.8 | 22.7 |
| GU937797 | 33.2 | 19.2 | 24.8 | 22.8 |
| EF185992 | 33.2 | 19.2 | 24.8 | 22.8 |
| AF353511 | 33.2 | 19.2 | 24.7 | 22.8 |
| KF452322 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF452323 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF804028 | 33.5 | 19.1 | 24.8 | 22.6 |
| KF267450 | 33.5 | 19.1 | 24.8 | 22.6 |
| KC210145 | 33.4 | 19.2 | 24.8 | 22.6 |
| KC210146 | 33.4 | 19.1 | 24.8 | 22.8 |
| Average ± SD value | 33.43 ± 0.08 | 19.10 ± 0.05 | 24.79 ± 0.03 | 22.68 ± 0.08 |
3.2. Synonymous codon usage in PEDV
The RSCU value of each synonymous codon in the PEDV genome was calculated (Table 2 ). The results indicate that codon usage bias exists in the coding sequences of the PEDV genome. The most frequently used codon is GUU (valine, 18822 times). The least frequently used codon is CGG (arginine, 534 times). Among the hydrophobic amino acids, the most frequently used codon is GUU (valine) and the least common is CCG (proline, 728 times). Among the hydrophilic amino acids, the most and least frequently used codons are GGU (glycine, 16151 times) and CGG (arginine). It was also observed that some of the synonymous codons that code for a single amino acid had similar RSCU values. For example, the CUC and CUG codons that encode leucine and the GUC and GUG that encode valine had similar RCSU values indicating they were used with similar frequency. It was also observed that the 18 most frequently used codons for each amino acid ended in either a U (T) or a G. Among the 18 codons, 15 codons ended with a U (T)-ended and 3 codons ended with a G, which further confirmed the codon usage bias in the synonymous codon usage pattern in the PEDV.
Table 2.
The synonymous codon usage pattern in the PEDV. The bolded and italic are the preferentially used codons and RSCU values for the PEDV.
| AA | Codon | RSCU/number | Codon | RSCU/number | |
|---|---|---|---|---|---|
| Phe | UUU | 1.39/15220 | Tyr | UAU | 1.33/11489 |
| UUC | 0.61/6708 | UAC | 0.67/5828 | ||
| Asn | AAU | 1.28/13756 | Cys | UGU | 1.38/8730 |
| AAC | 0.727754 | UGC | 0.62/3882 | ||
| Lys | AAA | 0.72/7319 | Arg | CGU | 2.20/4973 |
| AAG | 1.28/13029 | CGC | 1.16/2619 | ||
| Leu | UUA | 0.75/4267 | CGA | 0.35/783 | |
| UUG | 1.61/9167 | CGG | 0.24/534 | ||
| CUU | 1.93/10979 | AGA | 1.12/2537 | ||
| CUC | 0.61/3451 | AGG | 0.92/2086 | ||
| CUA | 0.49/2804 | Ser | UCU | 1.93/9243 | |
| CUG | 0.61/3464 | UCC | 0.68/3264 | ||
| Pro | CCU | 1.93/6630 | UCA | 1.03/4925 | |
| CCC | 0.54/1859 | UCG | 0.27/1290 | ||
| CCA | 1.32/4532 | AGU | 1.42/6792 | ||
| CCG | 0.21/728 | AGC | 0.67/3220 | ||
| Thr | ACU | 1.87/11090 | Asp | GAU | 1.26/13288 |
| ACC | 0.68/4022 | GAC | 0.74/7789 | ||
| ACA | 1.14/6773 | Glu | GAA | 0.92/6829 | |
| ACG | 0.32/1895 | GAG | 1.08/8092 | ||
| Val | GUU | 2.02/18822 | Gly | GGU | 2.47/16151 |
| GUC | 0.74/6924 | GGC | 0.94/6152 | ||
| GUA | 0.50/4698 | GGA | 0.43/2818 | ||
| GUG | 0.74/6875 | GGG | 0.16/1073 | ||
| Ala | GCU | 1.93/9243 | Ile | AUU | 1.78/11072 |
| GCC | 0.70/4448 | AUC | 0.64/4010 | ||
| GCA | 1.11/7047 | AUA | 0.58/3595 | ||
| GCG | 0.27/1732 | His | CAU | 1.40/5194 | |
| Gln | CAA | 0.90/5143 | CAC | 0.60/2228 | |
| CAG | 1.10/6253 | Met | AUG | 1.00/6766 | |
To estimate the degree of codon usage bias in the PEDV genome, the ENC value of the 43 strains was calculated. The average ENC value was 47.91 ± 0.13. This high ENC value (>45) indicated that the PEDV had a low codon usage bias.
3.3. The influence of mutational bias on PEDV codon usage bias
The ENC value of the coding region of the PEDV was plotted against the GC3s value (Fig. 1 ). The results show that the ENC values of 43 PEDV strains were clustered together with few changes between them. This indicates that the ENC value changed little between different strains, which was in accordance with the small ENC SD value. The ENC value was not located on the expected curve, revealing that the PEDV genome has the characteristics of codon usage bias. Some other factors, such as mutational bias and natural selection pressure, might contribute to the codon usage bias observed in the PEDV.
Fig. 1.
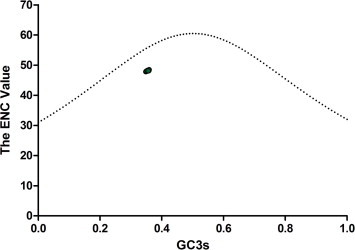
Plot of the ENC value versus the GC3s for the PEDV. The dotted line is composed of the expected ENC values. The ENC values of the PEDV strains are marked with green fixed circles.
To investigate the possible influence of mutational pressure on the codon usage bias in the PEDV, the correlation analysis was performed between the codon compositions (A3s, T3s, G3s, C3s, and GC3s), the ENC values and nucleotide compositions (A%, T%, G%, C%, and GC%) (Table 3 ). The results indicate that most of the codon compositions correlated with the nucleotide compositions. Additionally, the ENC value always correlated with the nucleotide compositions and the P values were all less than 0.01. These results confirmed the codon usage bias of the PEDV was influenced by the nucleotide compositions, and hence by mutational bias.
Table 3.
The correlation between the codon compositions (A3s, T3s, G3s, C3s, and GC3s), the ENC values, nucleotide compositions (A%, T%, G%, C%, and GC%), the first axis values, the second axis values, the Gravy values, and the Aroma values of the PEDV.
| A% | T% | C% | G% | GC% | 1st axis | 2nd axis | Gravy | Aroma | |
|---|---|---|---|---|---|---|---|---|---|
| A3s | 0.472⁎⁎ | 0.501⁎⁎ | 0.058 | −0.604⁎⁎ | −0.587⁎⁎ | −0.566⁎⁎ | −0.686⁎⁎ | −0.482⁎⁎ | 0.105 |
| T3s | 0.351⁎ | 0.834⁎⁎ | −0.346⁎ | −0.751⁎⁎ | −0.796⁎⁎ | −0.805⁎⁎ | −0.718⁎⁎ | −0.333⁎ | 0.510⁎⁎ |
| C3s | −0.331⁎ | −0.755⁎⁎ | 0.691⁎⁎ | 0.468⁎⁎ | 0.805⁎⁎ | 0.797⁎⁎ | 0.408⁎⁎ | 0.054 | −0.362⁎ |
| G3s | −0.407⁎⁎ | −0.569⁎⁎ | −0.091 | 0.734⁎⁎ | 0.565⁎⁎ | 0.539⁎⁎ | 0.835⁎⁎ | 0.625⁎⁎ | −0.252 |
| GC3s | −0.403⁎⁎ | −0.810⁎⁎ | 0.293 | 0.708⁎⁎ | 0.816⁎⁎ | 0.780⁎⁎ | 0.739⁎⁎ | 0.415⁎⁎ | −0.337⁎ |
| ENC | −0.309⁎ | −0.756⁎⁎ | 0.603⁎⁎ | 0.484⁎⁎ | 0.829⁎⁎ | 0.810⁎⁎ | 0.393⁎ | 0.162 | −0.310⁎ |
0.01 < P < 0.05.
P < 0.01.
The PCA analysis revealed the first principal axis covered 39.90% of the total variation and that the second principal axis covered 22.66% of the total variation. The third and fourth axes accounted for 9.13% and 7.54% of the total variation, respectively. This indicates that the first and second axes were responsible for the main change in the variation of the RSCU value, namely the trend of codon usage bias. The first and second axes were then plotted against each other (Fig. 2 ). Once again, all the data points were clustered around the origin and did not diverge too much from one other. This reflects the relatively little change in the codon usage bias between the different strains. The correlation analysis between the codon compositions and the first axis value and the second axis value revealed these compositions were correlated or significantly correlated (Table 3). This also proved that the mutational bias contributed to the PEDV codon usage bias.
Fig. 2.
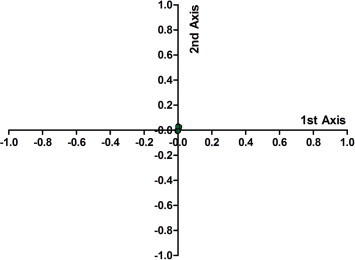
The first two axes in the correspondence analysis based on the RSCU values of PEDV.
3.4. The influence of natural selection on PEDV codon usage bias
To investigate the influence of natural selection pressure on the PEDV codon usage bias, correlation analysis was completed using the Gravy and Aroma values and the codon compositions (Table 3).The results indicate that the Gravy value is correlated with the A3s, T3s, G3s, and GC3s and that the Aroma value is correlated with the T3s, C3s, GC3s, and the ENC value. This confirmed that natural selection influenced the PEDV codon usage bias.
The codon usage pattern of Sus scrofa is available online at http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=9823. It was found that most of the preferred codons used to encode for each amino acid in the host and viral genomes was different, except for the codons for glutamic acid, glutamine, and lysine. However, the mean CAI value of the PEDV adaption to pig was 0.612 ± 0.055 (P < 0.05), which indicates that natural selection contributed to the PEDV codon usage bias.
3.5. Natural selection dominates over mutational bias in codon usage pattern
To distinguish the roles of natural selection and mutational bias in shaping the PEDV codon usage pattern, the P12 value (the mean value of GC1s and GC2s) was plotted against the P3 value (GC3s) (Fig. 3 A). In the analysis, the P12 value was correlated with the P3 value (r = 0.3917, P = 0.0113). The correlation coefficient was 0.2580 ± 0.097. The correlation coefficient shows that the relative neutrality is 25.80%, or that the relative constraint of P3 (0% constraint) is 74.20%. This result confirmed that natural selection pressure dominated over the mutational bias in the PEDV coding sequences.
Fig. 3.
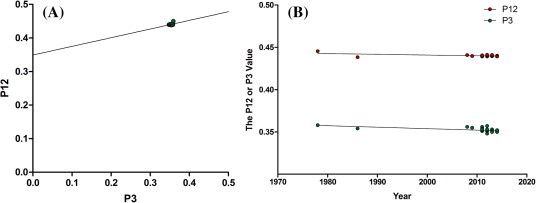
(A) The neutral analysis plot of P12 against P3. (B) The evolutional analysis of the P12 and P3 values. The solid line represents the regression line.
3.6. The evolutional analysis of codon usage pattern
To understand the evolutional pattern of the PEDV codon usage, the P12 value and the P3 value were plotted against the evolutional time from 1978 to 2014, respectively (Fig. 3B). Both the P12 value and the P3 value were found to be negatively correlated with the time (for P12, r = −0.5152 and P = 0.0008; for P3, r = −0.5597 and P = 0.0002). The changing rates of the P12 value and P3 value were −1.763 ± 0.429 × 10−4 and −0.816 ± 0.223 × 10−4 bases per year, respectively. The results suggest that the GC content at all codon three positions decreased as the PEDV evolved. Additionally, the rate of change of the GC3s was about twice of that of the GC12s, indicating that compared with the mutational bias, that the natural selection pressure played an increasingly role in shaping the PEDV codon usage pattern.
3.7. The codon usage pattern is also influenced by other factors
The gene function, the genomic group, and the geographic distribution were included in our study to investigate their possible roles in the PEDV codon usage pattern (Fig. 4 ). The average ENC value of the coding sequences from ORF1 a/b, S, ORF3, E, M, and N is shown in Fig. 4A. The differences between the ENC values between the six genes were analyzed by one-way ANOVA. It was observed that the differences between the average ENC values of the different genes were significant (P < 0.01). The most biased gene was the ORF1 a/b gene and the least biased gene was the M gene. This revealed that gene function contributed the codon usage bias to a degree.
Fig. 4.
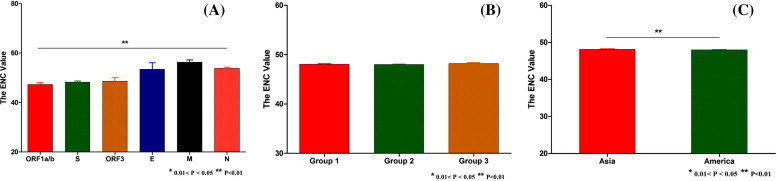
The influence of gene function, the genogroup, and the geographic distribution on the codon usage bias of the PEDV.
The phylogenetic analysis of the 43 PEDV strains showed that they were clustered into three main genogroups (Fig. S1). Most of the strains were clustered in group1 and the least were found in group2. Fig. 4B demonstrates that the average ENC value is equal between the three genomic groups. However, using the statistics method, the P value was shown to be beyond the confidence interval. Accordingly, this indicates that the ENC value was not influenced by the genogroups.
Finally, we determined the possible role of geographic distribution on the PEDV codon usage (Fig. 4C). Most of the studied strains were isolated from either Asia or America. Only one strain (CV777) was isolated from the UK (Rosenberg et al., 1977); this strain was not included our analysis. Fig. 4C shows that even though the average ENC value of the strains from Asia and America are similar, that they could still be distinguished from one other using the student’s t-test (P = 0.0062). This result suggests that the geographic distribution also influenced the PEDV codon usage.
4. Discussion
Epidemiological surveillance of the PEDV indicates that the virus has been circulating through the swine population since 1971 (Puranaveja et al., 2009, Rosenberg et al., 1977, Wang et al., 2014). Since 2013, outbreaks caused by the PEDV have been reported in several countries, including China, South Korea, and America (Huang et al., 2013, Wang et al., 2014). PEDV had not been isolated from the United States until May 2013 (Huang et al., 2013, Wang et al., 2014). Sequence comparison and phylogenetic analyses indicate that the recent prevalence of the PEDV may have been caused by the international transport of pigs (Huang et al., 2013). To further understand the genomics of the PEDV, codon usage analysis of the PEDV was performed in our study. Codon usage analysis is an effective and well-established method to investigate the codon usage patterns of different organisms. Understanding the extent and causes of codon usage bias is essential to research focused on viral evolution and transmission. To date, only a limited number of studies have focused on synonymous codon usage in animal viruses. Among the swine viral diseases, codon usage bias has been analyzed in porcine reproductive and respiratory virus (Liu et al., 2010b), foot-and-mouth disease virus (Zhou et al., 2010), classical swine fever virus (Tao et al., 2009), and porcine circovirus (Chen et al., 2014).
To investigate the factors leading to the PEDV codon usage pattern, several systemic analytical methods were performed in our study. First, the RSCU value of the PEDV strains was calculated. The results indicate that codon usage bias exists and that the PEDV preferred codons ending in (U)T. The codon usage bias was further confirmed by the mean ENC value of 47.91. For comparison, the mean ENC value for the following other studied viruses is: (1) foot and mouth disease virus (mean ENC = 51.42) (Zhou et al., 2010), (2) H5N1 influenza A virus (mean ENC = 50.91) (Auewarakul et al., 2009), (3) duck enteritis virus (mean ENC = 52.17) (Jia et al., 2009), (4) SARS (mean ENC = 48.99) (Zhao et al., 2008), (5) classical swine fever virus (mean ENC = 51.7) (Tao et al., 2009), and (6) Hepatitis A virus (mean ENC = 39.78) (D’Andrea et al., 2011). An ENC value greater than 45 is considered to represent a low codon usage bias. The mean ENC value for PEDV was a little higher than most viruses, and the value is also higher than 45. Accordingly, the codon usage bias of PEDV is comparatively low. A low biased codon usage pattern may allow the virus exploit several codons for each amino acid, and accordingly is beneficial for viral replication in the host cells. Additionally, the existence of the codon usage bias in PEDV was identified in the ENC plotted-GC3s analysis. As shown in Fig. 1, the data points representing the ENC value for each PEDV strain were lower than the expected curve. When the PEDV codons are completely random with bias present, all of the data points lie upon the expected curve. This reveals that other factors also influence the PEDV codon usage pattern. Mutational bias and natural selection pressures have been shown to be the two main forces involved in shaping the synonymous codon usage pattern of RNA viruses. To examine the possible role of mutational bias in the codon usage pattern, we performed a correlation analysis between the nucleotide content and the codon composition. The strong correlation between these two variables (except in the C% and A3s, G3s, and GC3s) showed that mutational bias contributed to the codon usage pattern. A significant correlation was shown between the ENC values and the nucleotide content (Table 3), which also reveals the importance of the mutational bias. The role of mutation bias was further demonstrated by the PCA analysis. Using the PCA analysis, we found that the first and second axes values were significantly correlated with the nucleotide content. A weak codon usage bias may be caused by natural selection when the viruses try to adapt to the host cell. Thus, we then investigated the role of natural selection in shaping the PEDV codon usage pattern by the investigating the relationships between the Gravy value, Aroma value and nucleotide content, and the high CAI value compared with the pig genome codon usage pattern. Next, the natural selection pressure was proven as being more important than mutation bias in neutral analysis. In addition, gene function and geographic distribution were also identified as influencing factors in shaping the PEDV codon usage pattern, while the genomic group had no affect on the PEDV codon usage bias. Finally, we identified several factors that contribute to the PEDV codon usage bias.
In summary, our study identified that the PEDV codon usage pattern is low. Two main factors, mutational bias and natural selection pressure, contribute to the codon usage pattern with the latter playing a more critical role. In the evolutionary process, natural selection pressure plays an increasingly role. In addition, other factors, such as gene function and geographic distribution, also influence the codon usage bias to some degree. These systemic analyses on the PEDV codon usage may be beneficial to further studies examining this important pig pathogen.
Acknowledgments
This work was supported by the Science and Technology Planning Project of Guangdong Province (2010A020102003), the Key Program for Scientific and Technological Innovations of Higher Education Institutes in Guangdong Province (cxzd117).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.meegid.2014.09.004.
Appendix A. Supplementary data
References
- Auewarakul P., Chatsurachai S., Kongchanagul A., Kanrai P., Upala S., Suriyaphol P., Puthavathana P. Codon volatility of hemagglutinin genes of H5N1 avian influenza viruses from different clades. Virus Genes. 2009;38:404–407. doi: 10.1007/s11262-009-0349-y. [DOI] [PubMed] [Google Scholar]
- Chen J.F., Sun D.B., Wang C.B., Shi H.Y., Cui X.C., Liu S.W., Qiu H.J., Feng L. Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes. 2008;36:355–364. doi: 10.1007/s11262-007-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Sun J., Tong X., Xu J., Deng H., Jiang Z., Jiang C., Duan J., Li J., Zhou P., Wang C. First analysis of synonymous codon usage in porcine circovirus. Arch. Virol. 2014;159:2145–2151. doi: 10.1007/s00705-014-2015-5. [DOI] [PubMed] [Google Scholar]
- D’Andrea L., Pinto R.M., Bosch A., Musto H., Cristina J. A detailed comparative analysis on the overall codon usage patterns in hepatitis A virus. Virus Res. 2011;157:19–24. doi: 10.1016/j.virusres.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egberink H.F., Ederveen J., Callebaut P., Horzinek M.C. Characterization of the structural proteins of porcine epizootic diarrhea virus, strain CV777. Am. J. Vet. Res. 1988;49:1320–1324. [PubMed] [Google Scholar]
- Fu M. Codon usage bias in herpesvirus. Arch. Virol. 2010;155:391–396. doi: 10.1007/s00705-010-0597-0. [DOI] [PubMed] [Google Scholar]
- Huang Y.W., Dickerman A.W., Pineyro P., Li L., Fang L., Kiehne R., Opriessnig T., Meng X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio. 2013;4:e00737–00713. doi: 10.1128/mBio.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R., Cheng A., Wang M., Xin H., Guo Y., Zhu D., Qi X., Zhao L., Ge H., Chen X. Analysis of synonymous codon usage in the UL24 gene of duck enteritis virus. Virus Genes. 2009;38:96–103. doi: 10.1007/s11262-008-0295-0. [DOI] [PubMed] [Google Scholar]
- Kocherhans R., Bridgen A., Ackermann M., Tobler K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23:137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Lee C. Outbreak-related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerg. Infect. Dis. 2014;20:1223–1226. doi: 10.3201/eid2007.140294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Zhao Z., Chen J., Wang B., Li Z., Li J., Cai M. Characterization of synonymous codon usage bias in the pseudorabies virus US1 gene. Virol. Sin. 2012;27:303–315. doi: 10.1007/s12250-012-3270-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wu C., Chen A.Y. Codon usage bias and recombination events for neuraminidase and hemagglutinin genes in Chinese isolates of influenza A virus subtype H9N2. Arch. Virol. 2010;155:685–693. doi: 10.1007/s00705-010-0631-2. [DOI] [PubMed] [Google Scholar]
- Liu Y.S., Zhou J.H., Chen H.T., Ma L.N., Ding Y.Z., Wang M., Zhang J. Analysis of synonymous codon usage in porcine reproductive and respiratory syndrome virus. Infect. Genet. Evol. 2010;10:797–803. doi: 10.1016/j.meegid.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry J.R., Gautier C. Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res. 1994;22:3174–3180. doi: 10.1093/nar/22.15.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Song D.S., Park B.K. Molecular epidemiology and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field isolates in Korea. Arch. Virol. 2013;158:1533–1541. doi: 10.1007/s00705-013-1651-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbo P., Bravo I.G., Garcia-Vallve S. CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct. 2008;3:38. doi: 10.1186/1745-6150-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puranaveja S., Poolperm P., Lertwatcharasarakul P., Kesdaengsakonwut S., Boonsoongnern A., Urairong K., Kitikoon P., Choojai P., Kedkovid R., Teankum K., Thanawongnuwech R. Chinese-like strain of porcine epidemic diarrhea virus, Thailand. Emerg. Infect. Dis. 2009;15:1112–1115. doi: 10.3201/eid1507.081256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M.L., Koplan J.P., Wachsmuth I.K., Wells J.G., Gangarosa E.J., Guerrant R.L., Sack D.A. Epidemic diarrhea at Crater Lake from enterotoxigenic Escherichia coli. A large waterborne outbreak. Ann. Intern. Med. 1977;86:714–718. doi: 10.7326/0003-4819-86-6-714. [DOI] [PubMed] [Google Scholar]
- Sharp P.M., Li W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- Shi S.L., Jiang Y.R., Liu Y.Q., Xia R.X., Qin L. Selective pressure dominates the synonymous codon usage in parvoviridae. Virus Genes. 2013;46:10–19. doi: 10.1007/s11262-012-0818-6. [DOI] [PubMed] [Google Scholar]
- Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA. 1988;85:2653–2657. doi: 10.1073/pnas.85.8.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P., Dai L., Luo M., Tang F., Tien P., Pan Z. Analysis of synonymous codon usage in classical swine fever virus. Virus Genes. 2009;38:104–112. doi: 10.1007/s11262-008-0296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temeeyasen G., Srijangwad A., Tripipat T., Tipsombatboon P., Piriyapongsa J., Phoolcharoen W., Chuanasa T., Tantituvanont A., Nilubol D. Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infect. Genet. Evol. 2014;21:205–213. doi: 10.1016/j.meegid.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Tian Y., Yu Z., Cheng K., Liu Y., Huang J., Xin Y., Li Y., Fan S., Wang T., Huang G., Feng N., Yang Z., Yang S., Gao Y., Xia X. Molecular characterization and phylogenetic analysis of new variants of the porcine epidemic diarrhea virus in Gansu, China in 2012. Viruses. 2013;5:1991–2004. doi: 10.3390/v5081991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Byrum B., Zhang Y. New variant of porcine epidemic diarrhea virus, United States, 2014. Emerg. Infect. Dis. 2014;20:917–919. doi: 10.3201/eid2005.140195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Liu Y.S., Zhou J.H., Chen H.T., Ma L.N., Ding Y.Z., Liu W.Q., Gu Y.X., Zhang J. Analysis of codon usage in Newcastle disease virus. Virus Genes. 2011;42:245–253. doi: 10.1007/s11262-011-0574-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E.H., Smith D.K., Rabadan R., Peiris M., Poon L.L. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 2010;10:253. doi: 10.1186/1471-2148-10-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Zhao S., Zhang Q., Liu X., Wang X., Zhang H., Wu Y., Jiang F. Analysis of synonymous codon usage in 11 human bocavirus isolates. Biosystems. 2008;92:207–214. doi: 10.1016/j.biosystems.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.H., Zhang J., Chen H.T., Ma L.N., Liu Y.S. Analysis of synonymous codon usage in foot-and-mouth disease virus. Vet. Res. Commun. 2010;34:393–404. doi: 10.1007/s11259-010-9359-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.