

Abstract
Background aims
Mesenchymal stromal cells (MSC) are ideal candidates for regenerative and immunomodulatory therapies. The use of xenogeneic protein–free Good Manufacturing Practice–compliant growth media is a prerequisite for clinical MSC isolation and expansion. Human platelet lysate (HPL) has been efficiently implemented into MSC clinical manufacturing as a substitute for fetal bovine serum (FBS). Because the use of human-derived blood materials alleviates immunologic risks but not the transmission of blood-borne viruses, the aim of our study was to test an even safer alternative than HPL to FBS: HPL subjected to pathogen inactivation by psoralen (iHPL).
Methods
Bone marrow samples were plated and expanded in α-minimum essential medium with 10% of three culture supplements: HPL, iHPL and FBS, at the same time. MSC morphology, growth and immunophenotype were analyzed at each passage. Karyotype, tumorigenicity and sterility were analyzed at the third passage. Statistical analyses were performed.
Results
The MSCs cultivated in the three different culture conditions showed no significant differences in terms of fibroblast colony-forming unit number, immunophenotype or in their multipotent capacity. Conversely, the HPL/iHPL-MSCs were smaller, more numerous, had a higher proliferative potential and showed a higher Oct-3/4 and NANOG protein expression than did FBS-MSCs. Although HPL/iHPL-MSCs exhibit characteristics that may be attributable to a higher primitive stemness than FBS-MSCs, no tumorigenic mutations or karyotype modifications were observed.
Conclusions
We demonstrated that iHPL is safer than HPL and represents a good, Good Manufacturing Practice–compliant alternative to FBS for MSC clinical production that is even more advantageous in terms of cellular growth and stemness.
Key Words: Good Manufacturing Practice, human platelet lysate, inactivation, mesenchymal stromal cells, psoralen
Introduction
Rapid progress in the fields of biotechnology and medicine has led to the development of new treatments and innovative medicinal products. Among the latter, new cell-based medicinal products, containing viable human cells of autologous or allogeneic origin, have a high potential for cell-based therapies for various severe diseases. In particular, mesenchymal stromal cells (MSCs) can easily be isolated from bone marrow (BM), thanks to their capacity to adhere and proliferate and expand in culture 1, 2. MSCs are multipotent stem cells with high immunomodulant proprieties, and they produce multiple cytokines, growth factors and adhesion molecules, all important factors that influence the hematopoietic microenvironment 3, 4. These cells' particular characteristics and high plasticity make them relevant in the fields of cell therapy, tissue repair and in tissue engineering strategies as therapeutic products tailored to a number of clinical scenarios, from degenerative to post-traumatic diseases caused by damage or cell loss 5, 6. The increasing use of MSCs as advanced therapy medicinal products (ATMP) has led to production processes that need to meet Good Manufacturing Practice (GMP) 7, 8.
The regulatory context for advanced therapy medicinal products is set out in Regulation (EC) N. 1394/2007, which is designed to facilitate patient access to these products while guaranteeing the highest level of safety for patients (9).
To ensure product safety and efficacy, GMP guarantees that products are consistently produced and controlled at the quality standards required for their intended use, from collection to release, including cell harvesting, cell manipulation processes, the maximum number of cell passages, combination with other components of the product, filling, packaging and so forth. Although human MSCs are not highly immunogenic, when expanded in xenogeneic sera such as in fetal bovine serum (FBS), they are likely to generate immune responses in some patients after administration 10, 11. It has been shown that a single preparation of 108 human MSCs grown under standard conditions in FBS carries with it approximately 7–30 mg of FBS proteins (10). Thus, in view of clinical GMP production, the use of xenogenic serum is complicated because there is high lot-to-lot variability and it is associated with a risk of transmitting infectious agents and immunizing effects 12, 13.
Therefore, regulatory guidelines for GMP production, aimed at minimizing the use of FBS, used in most expansion protocols as a cell culturing medium supplement, have further reinforced intensive searches for safer media supplementation alternatives (14).
Interesting studies have evidenced the possibility of replacing FBS with autologous or allogeneic platelet lysate obtained from single-donor or pooled human serum or from platelets because they contain a plethora of growth-promoting factors. Moreover, human platelet lysate (HPL) is being established as a safe and efficient MSC culture supplement for robust MSC cultivation, thus offering certain advantages as a potential FBS substitute 15, 16, 17, 18. Although the use of human-derived blood materials alleviates the immunologic risks of FBS, the possibility of transmitting blood-borne viruses remains, especially when materials from multiple donors are pooled to provide a sufficient volume for therapeutic-scale MSC expansion and to limit individual donor variability. Pathogen inactivation (PI) technologies are aimed at enhancing blood safety through the inactivation of emerging pathogens, both known and as yet-unidentified ones that and not detected by current screening or testing protocols. Since 1990, significant progress has been made in pathogen inactivation technology, which at present is widely available in European blood services, with multiple CE (an abbreviation of Conformité Européenne, ie, “European Conformity”) marked products to treat both platelets and plasma for transfusion. The CE marking states that a product has been assessed before being placed on the market and meets EU safety, health and environmental protection requirements.
The European experience with pathogen-inactivated platelets and plasma now numbers millions of units, with a safety record that has been widely reported in the literature (19) and extensive validation studies, through the use of blood products with high titers of added bacteria, enveloped and non-enveloped viruses and protozoa, have been performed to prove their efficacy.
Therefore, the application of the PI technique on platelets to be used for the preparation of HPL to supplement culture medium for MSC expansion in a GMP setting appears to be highly desirable because it might obviate virus transmission problems.
In the present study, the PI process was performed with the use of Intercept Blood System technology (INTERCEPT; Cerus Europe BV, Amersfoort, the Netherlands), which uses a photoactive compound, a derivative of psoralen (Amotosalem) and long-wavelength ultraviolet (UVA) illumination. On exposure to UVA light, Amotosalem becomes reactive and forms a chemical cross-link that locksup the strands of RNA and DNA, blocking and inactivating the replication of viruses, bacteria and leukocytes in platelet concentrates 20, 21, 22.
In our study, to ensure that the inactivation process does not induce changes in the cells, we set up, in parallel, MSC cultures in FBS (the standard supplement), HPL (recently used in GMP conditions as a cell growth supplement) and iHPL (the same batch of HPL after inactivation). We then compared the effects of the three supplements on cell growth, immunophenotype, multipotent capacity, karyotype, tumorigenesis and stemness protein expression.
Methods
HPL preparation
Whole blood (450 ± 45 mL) collection from 60 healthy blood donors was performed in a triple-bag system (Fresenius Kabi, Bad Homburg, Germany) containing 63 mL of citrate-phosphate-dextrose as an anticoagulant. According to Italian laws and European guidelines (19), the routine testing of blood donors was performed for the following: ABO blood groups, irregular red blood cell antibodies and infectious markers (hepatitis B and C, human immunodeficiency virus 1–2 and Treponema pallidum). The blood units were centrifuged and separated into plasma, buffy coat and red blood cells by use of an automated blood component separator (Compomat G5, Fresenius Kabi).
For buffy coat–platelet concentrate (BC-PCs) preparation, four O-group buffy coats were pooled with AB-group plasma and were then centrifuged and automatically separated through a leukoreduction filter by use of the TACSI system (Terumo BCT Europe, N.V. Zaventem, Belgium).
Twelve BC-PCs were prepared and six experimental replicates were performed. Each replicate was prepared from two BC-PC units through the use of a pool and split process, resulting in six test units and six paired control units.
Samples were taken from each unit to evaluate that platelet concentration, platelet content and leucocyte contamination complied with specifications set for transfusional purposes and for use with the inactivation processing set.
Platelet concentration was measured with the use of a hematology analyzer (Sysmex XE-2100), and residual white blood cell contamination was measured by means of the flow cytometric method (BD Leukocount kit; BD Biosciences, San Jose, CA, USA).
Each control unit remained untreated. The test unit was sterile, docked to an INTERCEPT Large Volume processing set (CERUS Corporation, Concord, CA, USA) and processed according to the manufacturer's instructions. The component was treated with 3 J·cm−2 UVA light, in the presence of 150 μmol/L Amotosalen. After treatment, the BC-PC was transferred to the associated Compound Adsorption Device (CAD) bag and incubated for 16 h, with agitation at 22°C.
Samples were taken from each unit to evaluate platelet concentrations, recovery after inactivation treatment and sterility.
All BC-PCs were frozen at −35°C and thawed at 37°C three times to obtain platelet fragmentation and growth factor release. The BC-PCs were subsequently centrifuged at 5000g for 8 min to remove platelet bodies and collect the supernatant.
To improve standardization and reduce individual donor variations, all six untreated supernatant units (from a batch of 60 different donors) were pooled in a single HPL unit. The other six treated units were also pooled. Each batch of HPL was then divided into aliquots of 100–150 mL and frozen again at −35°C until use.
Each batch of HPL and iHPL was tested for the presence of endogenous and adventitious viruses. DNA extraction was performed starting with 1 mL of HPL and iHPL, followed by an extraction step with the use of the easy MAG automatic extractor (Biomérieux, Marcy l'Etoile, France) according to the manufacturer's instructions, and eluted in a final volume of 50 μL. For endogenous viruses, 5 μL of elute was added to 20 μL of an amplification mix to detect human cytomegalovirus and Epstein-Barr virus (EBV) (Q-CMV Real-Time Complete Kit, EBV Q-PCR Alert Kit, ELITech Group, Puteaux, France), following the manufacturer's instructions. Twenty microliters of elute was added to 20 μL of amplification mix to detect parvovirus B19 (Parvovirus B19 Elitè MGB Kit, ELITech Group), reconstituted following the manufacturer's instructions. RV15 OneStep ACE detection (Seegene, Seoul, Korea) was used according to the manufacturer's instructions to simultaneously detect 15 major respiratory viruses (respiratory syncytial virus-a; respiratory syncytial virus-b; influenza A; influenza B; adenovirus; coronavirus 229E/NL63; coronavirus OC43; parainfluenza 1, 2, 3, 4; rhinovirus A, B, C; bocavirus 1, 2, 3, 4; enterovirus and metapneumovirus).
Harvest and preparation of MSCs
BM cells were harvested from the iliac crest of adult or pediatric donors who underwent BM collection for a related patient after informed consent. We used a part of the BM initially dedicated to transplantation, or, when available, from an unfiltered BM collection bag (Baxter Healthcare Corporation, Deerfield, IL, USA) that was normally discarded before BM infusion. The bag was washed three times with phosphate-buffered saline ×1 and the cells were centrifuged, counted and plated directly in different culture media.
First, we tested HPL at a concentration of 10%, 7% or 5% with 20 IU/mL heparin (this avoids coagulation of the medium through clumping of the fibrinogen in the HPL) to find the best results in terms of cellular growth. To eliminate any traces of residual platelets, HPL was filtered by 0.2-μm filters before the use.
The whole BM sample was then equally split into three cellular culture conditions: α-minimum essential medium (α-MEM) (Sigma-Aldrich, LTO Irvine, Ayrshire, UK) containing 10% of (i) FBS, (ii) HPL and (iii) iHPL. The seeding density of the initial whole BM was at 10,000 cells/cm2, as previously reported (23).
After 7 days, the non-adherent cells were removed and discarded. The adherent cells were re-fed every 5–7 days and, when they reached confluence, were detached, counted by means of the Fast Read disposable chamber as previously described (24) and re-plated for a further 3–5 passages at 1000 cells/cm2. We considered MSCs at passage 1 (P1) the first time the cells were harvested and re-plated. The cellular condition was maintained from the cellular plating of whole BM and during the expansion process. We indicated the following passages with increasing numbers: P2, P3, and so on. On the basis of our experience and our previously reported data (23), we found that three passages suffice for clinical applications, and we analyzed the cells to P3.
Colony-forming unit fibroblasts
To quantify MSC precursors, we performed a colony-forming unit fibroblast (CFU-F) test. The BM cells were plated directly in α-MEM containing 10% FBS, HPL or iHPL at densities of 10,000 cells/cm2 or 100,000 cells/cm2 in a six-well plate. MSC clonogenic precursors were scored macroscopically after 7–10 days from seeding, and clusters of >50 cells were considered colonies. All experiments were performed in duplicate and by two different operators. The CFU-F means were expressed as fibroblastic clones obtained from 1 million BM cells (CFU-F/106 cells).
MSC cellular growth evaluation
To evaluate cellular growth, the cell growth rate was expressed in terms of population doubling (PD) by means of the formula (log N/log 2), where N is the cell number of the detached cells divided by the initial number of seeded cells and the expansion in terms of cumulative PD (cPD).
MSC cytofluorimetric analysis
To analyze the immunophenotype, flow cytometer analysis was performed on adherent cells at each passage. Briefly, 200,000 cells were incubated with the appropriate amount of antibody according the specific antibody titration as described in Rustichelli et al. (25) for 20 min with anti-CD90 fluorescein isothiocyanate (FITC), CD73 phycoerythrin, CD34 FITC, CD14 FITC, CD45 FITC (Becton Dickinson, San Jose, CA, USA), CD 105 antigen-presenting cells (APC) and CD146 APC (Miltenyi Biotec srl, Bologna, Italy). The labeled cells were acquired by means of FACScanto II (Becton Dickinson) with use of the DIVA software program. The percentage of positive cells was calculated through the use of cells stained with immunoglobulin FITC/phycoerythrin/APC as a negative control, and mean fluorescence intensity was analyzed on the positive cells.
MSC immunocytochemistry
To analyze the stemness protein expression, we performed immonocytochemical staining for Octamer-3/4, (Oct-3/4) and for Homeobox transcription factor Nanog (NANOG) according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). Briefly, the cells were fixed and permeabilized with methanol and acetone (1:1) at −20°C for 10 min. Non-specific binding was blocked with 5% normal horse serum in antibody diluent. The cells were incubated with goat anti-NANOG (1:50) and goat anti–Oct-3/4 antibodies (1:100) and then with the secondary antibody Alexa fluor 488–coupled anti-mouse (Molecular Probes, Eugene, OR, USA; 1:200). Incubation with the primary antibody was performed overnight at 4°C, whereas incubation with the secondary antibody was for 1 h at room temperature. The cells were examined under epifluorescence microscopy and analyzed with the use of AxioVision Rel 4.2 (Axiovert 200, Carl Zeiss, AG, Jena, Germany). Positive cells were counted and compared with the total cell counts labeled with 4′, 6-diamidino-2-phenylindole (DAPI, Molecular Probes).
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted with the use of RNeasy Plus Mini Kits (Qiagen, Austin, TX, USA). Reverse-transcription polymerase chain reaction (RT-PCR) was carried out with the use of the high-capacity complementary DNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. RNA and complementary DNA concentrations were measured with the use of a GeneQuant pro spectrometer (Amersham Biosciences, GE Healthcare, Pittsburgh, PA, USA). We performed RT-PCR to detect the transcripts for human Oct-4 and NANOG through the use of specific assays (assay ID: OCT-4 (POU2F2) Hs00231269_m1; NANOG: Hs04260366_g1; HPRT1:Hs02800695_m1) and TaqMan Universal PCR Master Mix (Applied Biosystems).
Comparative cycle threshold (ΔΔCT) experiments were performed according to the manufacturer's specifications in a total reaction volume of 25 μL. All experiments were performed in three replicates. To normalize the PCR results, we used hypoxanthine phosphoribosyltransferase 1 (HPRT1) as the housekeeping gene. We performed the Relative Quantification (RQ) method, which is based on the expression levels of a target gene versus a housekeeping gene to compare different RT-PCR experiments For each set of experiments, we analyzed Nanog and Oct-4 messenger RNA expression levels in MSCs obtained in three different culture conditions at P3, and the FBS-MSC condition was used as reference sample.
Potential assay
To analyze the multipotent capacity, MSCs at P3 in each culture condition were cultured in osteogenic, adipogenic (STEMCELL Technologies, Vancouver, BC, Canada) and chondrogenic media (Lonza, Cologne, Germany) for 21 days, according to the manufacturer's instructions. Briefly, 5000 and 10,000 cells, for control samples and for differentiation experiments, were seeded in a six-well plate for osteogenesis and adipogenic culture conditions, respectively. Osteogenic differentiation was demonstrated by the accumulation of crystalline hydroxyapatite by Von Kossa staining and the adipogenic differentiation by the presence of intracellular lipid vesicles assessed by oil red O. MSC chondrogenic differentiation was obtained as previously described (2), and the differentiation was evaluated by alcian blue staining, which identifies the presence of hyaluronic acid and sialomucin.
MSC karyotype analysis
To exclude cytogenetic transformation during ex vivo expansion, MSCs at P3 were detached and then cultivated until approximately 80% of confluence was reached. Karyotype analysis was performed after the cells were arrested at the metaphase by incubation with Colcemid (Invitrogen Corporation, Grand Island, NY, USA). The cells were then maintained in a hypotonic solution (0.075 mol/L KCl), fixed with 3:1 methanol/acetic acid and stained with Giemsa, through the use of standard laboratory protocols for chromosome analysis. At least 50 cells at metaphase were analyzed with the use of MackType software (Nikon Corporation, Tokyo, Japan) according to the International System for Human Cytogenetic Nomenclature.
Tumorigenesis tests
To exclude potential tumorigenicity induced by the ex vivo expansion of MSCs, we performed soft agar tests.
The cells at P3 in each culture condition were harvested, washed and seeded at a density of 1000 cells/well in 24-well plates in duplicate. The test was performed with the use of 0.8% and 0.3% agar in α-MEM +10% HPL/iHPL/FBS (final volume/well = 1 mL), arranged, respectively, at the base and on the surface.
The cells were incubated for 21 days at 37°C in the presence of 5% CO2. After incubation, the colonies were counted with the use of the inverted microscope.
A primary line of osteosarcoma was used as the positive control, and a sample of whole BM from a healthy donor was used as the negative control.
Microbiological control and mycoplasma detection
For microbiological control, the samples (HPL, media and cellular supernatants) were tested through BacT/ALERT (bioMérieux, Durham, NC, USA) (26) by the Bacteriology Laboratory of the City of Science and Health of Turin, S. Anna Hospital.
We performed the mycoplasma detection test (Mycoplasma Detection Kit for conventional PCR, Minerva Biolabs, Berlin, Germany), with the use of an aliquot of supernatant at each passage during the expansion process and on an amount of each growth supplement (pure HPL, iHPL and FBS). Briefly, any mycoplasma contamination of the analyzed samples was detected through a semi-quantitative PCR reaction. As the positive control, we used a positive control DNA, provided by the kit. To monitor the success of the extraction procedure, we used the internal control DNA of Venor GeM. As a negative control, we used PCR-grade water.
Statistical analysis
Statistical analyses were performed with the use of SPSS 20 (IBM, Chicago, IL, USA).
First, we investigated whether the distributions of each group were normally distributed by use of the Shapiro-Wilk test. Because the distributions were not normal, we used the non-parametrical Friedman test to compare the different groups. We considered a significant difference if the P value was <0.05.
Results
HPL preparation and INTERCEPT treatment
Two batches of HPL, inactivated (i-HPL) and uninactivated (HPL), were prepared by pooling six paired leuko-reduced BC-PCs as described above. The average starting parameters of platelet content, volume and leukocyte content in the BC-PCs (Table I ) for PI treatment were in keeping with the recommended specifications for both the treatment of platelets with the INTERCEPT Large Volume processing system and for transfusion use.
Table I.
BC-PC characteristics before and after inactivation treatment.
| Large-volume set specifications (range) | Before inactivation (n = 6) | After inactivation (n = 6) | |
|---|---|---|---|
| Platelet content (×1011) | 2.5–7.0 | 2.5–3.3 | 2.1–2.8 |
| Volume (mL) | 255–390 | 307–321 | 286–309 |
| CAD agitation duration | 16–24 h | N/A | 16 |
| Leukocyte content (106/unit) | 1 | – | 0.03–0.84 |
Values are expressed as range of minimum and maximum values.
Compared with the paired untreated units, inactivated BC-PCs showed a lower platelet count as the result of the addition of amotosalen and the consequent dilution (Table II ). The multiple transfer steps, including the CAD incubation, reduced volume and platelet yield by up to 10%.
Table II.
Inactivated and uninactivated BC-PC characteristics.
| Inactivated BC-PCs | Uninactivated BC-PCs | |
|---|---|---|
| Volume (mL) | 299 ± 9.0 | 321 ± 8.9 |
| Platelet count (×103/μL) | 836 ± 85 | 895 ± 116 |
| Platelet content (×1011) | 2.5 ± 0.3 | 2.9 ± 0.3 |
| Leukocyte content (106/unit) | 0.18 ± 0.3 | 0.19 ± 0.3 |
Values are expressed as mean ± SD.
Sterility testing was negative in all BC-PC units.
In all HPL batches tested for endogenous and adventitious viruses, only one sample resulted as positive for parvovirus B19 DNA (HPL). All iHPLs were negative.
MSC harvest and preparation
Nineteen BM samples were collected from healthy donors: 10 were >18 years of age (age range: 39–50 years) and nine were <18 years (age range: 0.5–10 years). The study was carried out according to the Helsinki Declaration.
MSC isolation and expansion
Adherent cell clones were observed in all the samples after 7 days of culture, and an adherent monolayer was achieved over the following 10–12 days. HPL/iHPL-MSCs but not FBS-MSCs reached confluence 7 days after plating (P1). HPL-MSCs reached a mean ± standard deviation (SD) of cPDs after P3 of 6.88 ± 0.049, 8.27 ± 0.82 and 12.64 ± 1.11 when they were expanded in α-MEM + 5%, 7% and 10% HPL, respectively. At P3, the cPDs of 10% HPL-MSCs were significantly greater than those obtained by culturing the cells in α-MEM + 5%/7% HPL and α-MEM + 10% FBS (5.04 ± 0.19), used as the control (P = 0.029). On these bases, we chose a 10% HPL supplement for all the following experiments.
CFU-F potential and MSC morphology
After 7 days, the number of CFU-F/106 cells was significantly greater when the cells were plated at 10,000 cells/cm2 (low density) than when plated at 100,000 cells/cm2 (high density), independent of the cellular condition. The mean CFU-F number was 99.3 ± 11.07, 106.8 ± 12 and 99.5 ± 9.6 at a plating density of 10,000 cells/cm2 and 33.1 ± 4.75, 29.3 ± 14.7 and 30.9 ± 9.88 at a plating density of 100,000 cells/cm2, respectively, for HPL/iHPL/FBS MSCs (Figure 1 A). Although the absolute number of colonies counted in the three culture conditions was not significantly different, we observed that MSCs from CFU-Fs grown in HPL and iHPL were smaller than those from FBS CFU-Fs. Moreover, when CFU-Fs from HPL and iHPL-MSCs were detached and analyzed for cell numbers, they were more populated, smaller and denser (Figure 1B; a,b) and reached confluence faster (Figure 1B; d,e) than those cultured in FBS (Figure 1B; c,f).
Figure 1.
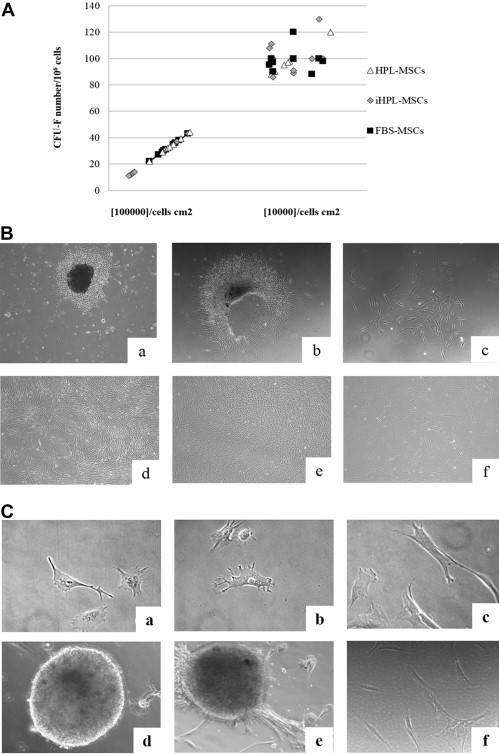
HPL and iHPL-MSCs showed differences in CFU-F and cellular morphology compared with FBS-MSCs. (A) CFU-F numbers of HPL (white triangle), iHPL (gray diamond) and FBS (black square)-MSCs plated at seeding densities of 10,000 cells/cm2 and 100,000 cells/cm2. Each symbol represents an experiment (n = 9), and no significant differences were observed. (B) Representative phase images at ×5 magnification of CFU-Fs in the three different conditions: HPL (a) and iHPL (b)-MSC CFU-Fs were denser, more homogeneous and populated with cells than were FBS-CFU-Fs (c). Moreover, HPL and iHPL-MSCs (d, e) reached confluence faster than did FBS (f)-MSCs. (C) Representative phase images at ×40 magnification show the MSCs in the three different culture conditions after 3 and 12 h from plating: HPL and iHPL-MSCs emitted prominent extensions (a, b) and formed spherical structures similar to embryoid bodies (d, e). FBS-MSCs show a polygonal morphology with large, jagged cytoplasm (c) and did not form three-dimensional structures (f).
We also observed that HPL/iHPL-MSCs appeared smaller, had easily visible nucleoli and were more homogeneous in their morphological characteristics (Figure 1C; a,b) than were FBS-MSCs. FBS-MSCs but were larger and showed a polygonal morphology with a large, jagged cytoplasm (Figure 1C; c). Furthermore, when HPL/iHPL-MSCs were detached and re-plated, after 12 h of cell culture, they tended to reform colonies, emit prominent extensions morphologically similar to pseudopodia and, interestingly, create tri-dimensional structures similar to embryoid bodies (Figure 1C; d,e), which we had not observed in FBS-MSCs (Figure 1C; f).
MSC expansion and immunophenotype
Cellular growth analysis showed that cPDs at the third passage were 11.16 ± 1.01, 11.00 ± 0.98 and 7.16 ± 1.10 (mean ± SD), respectively, in HPL, iHPL and FBS-MSCs. HPL and iHPL-MSCs values showed a strong correlation (0.099 with P < 0.001), and significant statistical differences were observed on the cellular growth in terms of cPDs of HPL/iHPL-MSCs compared with FBS-MSCs at P2 and P3 (P = 0.00041 and P = 0.00032, respectively), as shown in Figure 2 , which shows the cPDs of the three passages of all BM-MSCs cultured in each condition with the relative means and standard deviations.
Figure 2.
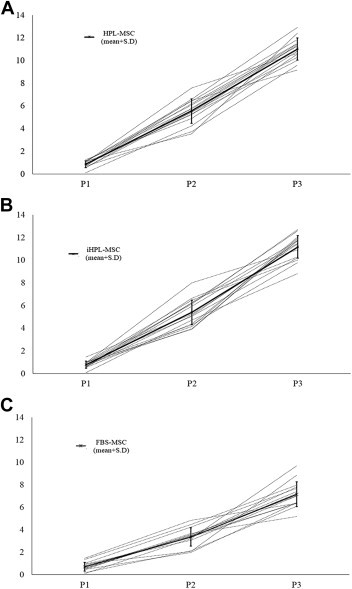
HPL and iHPL-MSCs show higher proliferative potential than do FBS-MSCs. (A–C) Cumulative PD of HPL, iHPL and FBS-MSCs (dashes). Results are shown as the cPD value at the first three passages of each 16 independent experiments in the three culture conditions. Mean ± SD values are represented in each graph as a thick black line. HPL and iHPL-MSCs show a higher proliferative potential than do FBS-MSCs from the second passage of culture.
During the first three passages, MSCs were analyzed at each passage for the expression of CD45, CD34 and CD14, hematopoietic surface antigens; CD90 (a membrane glycoprotein, also called Thy-1), used as a stem cell marker; CD105 (endoglin); CD73 (Ecto-5-prime-nucleotidase) and CD146 (cell surface glycoprotein MUC18). At the first passage, MSCs isolated from whole BM and cultured in α-MEM + 10% HPL, iHPL and FBS were CD45-, CD34- and CD14-negative, with an antigen expression of <5%, whereas they showed a high expression of CD90, CD73, CD105 and CD146. In all culture conditions, during the expansion time, the MSCs were negative for hematopoietic antigens, whereas at each passage, they expressed high percentages of CD90, CD73, CD105 and CD146. No statistical differences were observed among the three different media. Table III shows the mean percentage of antigen expression, which was analyzed at the 1st, 2nd and 3rd passages on the cells cultured with three different supplemented media. The fluorescence means of the positive markers at the third passage showed a slight increase for CD90 marker and a very slight increase for CD73 in HPL and iHPL-MSC compared with FBS-MSCs. In particular, HPL, iHPL and FBS-MSCs showed, respectively, a fluorescent mean intensity for CD90 of 37,301.14 ± 7569.091; 31,360.14 ± 5628.379 and 25,385.86 ± 4410.403. For CD73, the mean intensities were 16,834.86 ± 10,988.55, 17,940.29 ± 11,108.43, 16,054.43 ± 10,132.85 for CD105, 3143.71 ± 2110.57, 3962.57 ± 2782, 3455.14 ± 1659.26, respectively. However, these differences were not statistically significant. In all the markers, we concluded that there was a strong correlation between the values (<0.098 with P < 0.001) obtained in HPL and iHPL, thus demonstrating that PI does not interfere with the MSC immunophenotype.
Table III.
Immunophenotypic analysis of HPL-MSCs versus iHPL-MSCs versus FBS-MSCs.
| HPL-MSCs |
iHPL-MSCs |
FBS-MSCs |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Passage | P1 | P2 | P3 | P1 | P2 | P3 | P1 | P2 | P3 |
| CD90 | 99.1 ± 1.4 | 100 ± 0.1 | 99.6 ± 0.1 | 98.5 ± 2.5 | 97.8 ± 3.8 | 99 ± 0.9 | 98.8 ± 0.6 | 99 ± 0.4 | 98.9 ± 0.5 |
| CD73 | 99.1 ± 1.0 | 98.5 ± 1.4 | 97.9 ± 0.1 | 98.8 ± 1.0 | 99.1 ± 1.0 | 99.1 ± 1.0 | 97.4 ± 1.6 | 95.5 ± 3.1 | 98.7 ± 0.7 |
| CD105 | 93.9 ± 2.1 | 95.7 ± 2.7 | 92.7 ± 6.4 | 92 ± 6.6 | 88 ± 5.2 | 95.9 ± 4.6 | 95 ± 4.3 | 91.6 ± 5.3 | 98.1 ± 1.6 |
| CD34/45/14 | 1.4 ± 1.0 | 0.44 ± 0.1 | 0.4 ± 0.3 | 1.8 ± 0.6 | 0.6 ± 0.6 | 0.3 ± 0.8 | 1.8 ± 1.7 | 0.5 ± 0.2 | 0.4 ± 0.4 |
| CD146 | 92.5 ± 1.9 | 93.8 ± 2.3 | 92.2 ± 1.9 | 85.2 ± 6.0 | 91.7 ± 6.2 | 93 ± 1.1 | 94.2 ± 5.0 | 94.7 ± 2.7 | 90.8 ± 4.0 |
Values are expressed as mean percentage ± SD of positive cells for MSC antigen expression. Cells were analyzed at the 1st, 2nd and 3rd passages for each experimental condition (n = 16). No significant differences were observed.
Pluripotency marker expression analysis
When HPL/iHPL-MSCs were detached and re-plated, they tended to form aggregates of cells similar in structure to embryoid bodies, which were not seen in FBS-MSCs; we therefore analyzed embryonic stem cell markers such as Oct-3/4 and NANOG by immune-cytochemical technique and RT-PCR. We observed positivity of these markers at both protein and molecular expression levels. In particular, we observed that the intensity of Oct-3/4 and Nanog nuclear protein expression was higher in HPL/iHPL-MSCs compared with FBS-MSCs, in terms of number and expression intensity. As shown in Figure 3 , in the cellular structures formed by HPL/iHPL-MSCs, the fluorescence is more concentrated in the nucleus (Figure 3A; a,b, and Figure 3B; g,h) than that observed in isolated cells, in which both nuclear and perinuclear distribution appeared (Figure 3A; c, and Figure 3B; i). By counting the positive cells, we found that HPL-MSCs were 100% ± 0% and 100% ± 0%, iHPL-MSCs were 100% ± 0% and 97% ± 3.1% and FBS-MSCs were 89% ± 6.6% and 82% ± 13%, respectively, for Oct-3/4 and NANOG protein expression. These data were confirmed by RT-PCR in which in all analyzed samples, we observed that HPL and iHPL induced a higher Oct-3/4 and Nanog gene expression compared with FBS (Oct-3/4 mean RQ values: 4.2 ± 2.7, 4.6 ± 2.2 and 1 ± 0; NANOG mean RQ values: 1.8 ± 0.6, 1.9 ± 1.6 and 1 ± 0, respectively, for HPL, iHPL and FBS-MSCs). The right panel in Figure 3A, B shows a representative experiment of MSCs isolated and expanded from the same BM sample in three different cellular conditions. The Friedman test, for both protein and messenger RNA expression, showed a significant statistical difference for Oct-3/4 (P = 0.050 and P = 0.022, respectively).
Figure 3.
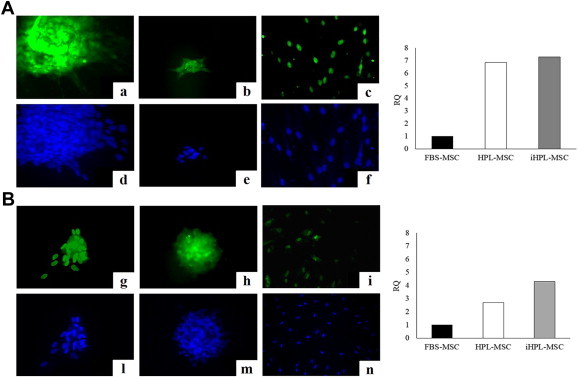
HPL and iHPL-MSCs show more embryonic stem cell marker proteins and messenger RNA expression than do FBS-MSCs. (Panel A for Oct-3/4 and Panel B for NANOG) On left side of the panel, immunocytochemistry analysis of embryonic markers in HPL (left column), iHPL (middle column) and FBS (right column)-MSCs (×40 magnification); on the right side of the panel, a representative experiment of RT-PCR: Oct-3/4 (a–c) and NANOG (g–i) were more expressed in HPL and iHPL-MSCs compared with FBS-MSCs in terms of both protein and messenger RNA. For immunocytochemistry, figures are representative of one of three experiments, and DAPI staining was used to evidence the nucleus of each analyzed cell (d–f, l–n). For RT-PCR, five experiments were conducted, but both panels show the most representative experiment.
MSC differentiation potential assay
The MSCs obtained in the three different conditions showed multipotent capacity as all samples at P3 differentiated into osteoblasts, adipocytes and chondrocytes, as shown in Figure 4 .
Figure 4.
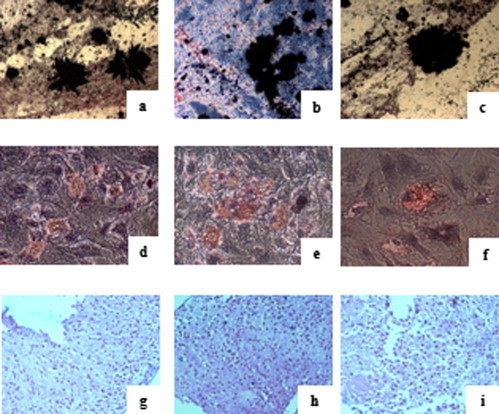
MSC differentiation potential assay after 3 weeks of specific induction in MSCs in the three different conditions. Von Kossa staining (a–c) shows the presence of calcium oxalates in osteoblasts, oil red O shows intracytoplasmatic vacuoles in adipocytes (d–f) and alcian blue (g–i) shows the hyaluronic acid for chondrocytes, respectively, in HPL (left column), iHPL (middle column) and FBS (right column)-MSCs (n = 19).
MSC karyotype analysis and tumorigenesis tests
At P3, the cells expanded in α-MEM + 10% HPL, iHPL and FBS showed no chromosome modifications or tumorigenic transformations (Figure 5 ).
Figure 5.
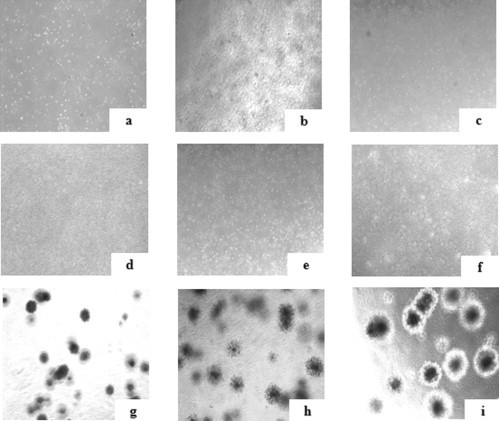
Soft agar assay to exclude tumorigenesis potential in MSCs in the three different conditions (n = 3). HPL, iHPL and FBS-MSCs (a–c) did not form colonies, nor did whole BM used as a negative control (d–f) compared with primary osteosarcoma tumor cells used as a positive control (g–i).
MSC viability and microbiological control
At each passage, viability was >95% in all cell culture conditions, and all microbiological analyses, including the mycoplasma test, were negative.
Discussion
This study shows that HPL is a safe and efficient MSC culture supplement for robust MSC culture, thus offering certain advantages compared with FBS, especially in terms of cell growth and stemness maintenance. Moreover, it represents a good GMP-compliant alternative to animal serum for MSC clinical production, confirming recent data reported in the literature 15, 27, 28.
HPL preparation was performed according to blood bank procedures, in which a high standardization is strongly recommended. The development of HPL batches from multiple sources (ie, from 60 different donors), makes HPL a virtually standardized medium supplement in both a growth factor concentration and in inter-donor variability.
Moreover, because the risk of transmission of infective agents not routinely tested, or for which no tests are available remains, HPL quality and safety had to be greatly improved. This was done with photochemical treatment by amotosalem and UVA, a technology that is efficient against the vast majority of known pathogens and that might also prevent the transmission of unknown pathogens 20, 29, 30, 31, 32
INTERCEPT technology is routinely used for PI of platelets and plasma for clinical use because it inactivates a wide spectrum of bacteria, viruses, and parasites as well as contaminating leukocytes 33, 34, 35, 36, 37. Because the INTERCEPT process and nucleic acids targeting PI technology is not, however, effective against prion diseases, the risk of prion transmission by treated lysates remains, even though precautionary measures such as donor selection and leucocyte depletion may be taken to obviate the problem.
The INTERCEPT Blood System is a Class III medical device with a registration that required regulatory review of preclinical and clinical data. This system is routinely used in many blood centers in Europe, the Middle East and many other countries, with at least 700,000 INTERCEPT-treated units transfused world-wide 29, 31. The manufacturer has supported an extensive program of post-market surveillance and hemovigilance studies to monitor the introduction of INTERCEPT products in Europe. In studies of >30,000 treated platelet units and >30,000 treated plasma units to date in a broad patient population, INTERCEPT platelets and plasma have a safety profile comparable with conventional components. The efficacy and safety of INTERCEPT platelets have been established in 11 trials and >1000 patients (38).
Moreover, the recent Swiss hemovigilance data, reporting 2 years of routine transfusion of 62,500 inactivated platelet concentrates, support the improvement safety profile and the successful prevention of septic transfusion reactions after the introduction of the pathogen inactivation technique on platelet components (39).
INTERCEPT-treated platelets have received additional country-specific regulatory approvals in France, Germany and Switzerland (19). A new application of this widely used technique might be a new niche for blood banks. The latter, in fact, could use this method to provide safer products for clinical cell culture and transplantation and, by doing so, increase safety for patients.
To our knowledge, no data have been reported about PI on platelet lysate for MSC expansion. Although a recent study on the use of PI-HS compared with uninactivated pathogen–PI-HS (40) showed that PI does not exert a negative impact on human islets of Langerhans, MSCs, T cells or cell lines and may even have a positive effect by the downregulation of inflammatory mediators induced by DNA or RNA strands released from damaged cells. We sought to compare simultaneously HPL and iHPL with FBS to exclude abnormalities in MSC isolation and expansion caused by the inactivation itself.
The literature has widely reported the efficacy of psoralen-UVA inactivation through the use of blood products with high titers of added bacteria, enveloped and non-enveloped viruses and protozoa. Although demonstration of the inactivation of contaminating pathogens was beyond the scope of our study, we did however a number of viruses, that are not required by the Regulatory Authority and not routinely tested by the Transfusional Center, batches of HPL and iHPL to show that PI can inactivate viruses. The negativity of almost all of the viruses tested showed that a good selection of donors by the Transfusion Center is a fundamental requirement to have safe initial blood components. We only had positivity for Parvovirus B19, which is a virus that can be transmitted from asymptomatic blood donors to the recipients of their blood components (41). High rates of seroconversion, as well as a few cases of symptomatic illnesses and hypoplastic anemia, have been described in patients receiving clotting factor concentrates that were derived from large plasma pools. The virus is relatively heat-stable, and it resists treatment with solvents and detergents. B19 DNA was found by PCR in plasma-derived clotting factor concentrates from various manufacturers and treated with the use of different virus-inactivation methods by various investigators (42). Clinically evident transfusion-transmitted B19 infection is, however, infrequent, even in susceptible hosts such as human immunodeficiency virus–infected hemophilia patients receiving clotting factor concentrates. The fact that the only positive HPL batch was negative after inactivation shows that we also found the PI process to be efficient and safer.
In our hands, INTERCEPT treatment on HPL resulted in a 10% loss of platelets, but these data confirmed those reported by Wagner et al. (43) and did not affect cell culture behavior.
PI treatment may have traces of psoralens, which may contaminate iHPL. With the use of the CAD, the levels of residual amotosalem are greatly reduced and the psoralen concentration in platelets is approximately 0.5 μmol/L, which is the approximate concentration in the iHP. We reduced this concentration 10-fold through the use of HPL at a concentration of 10% in the cell culture. Although we did not test the presence of psoralen in MSCs expanded in HPL, we consider the psoralen traces in our MSC clinical product to be absolutely negligible and safe. This conclusion is in light of the fact that that during extracorporeal photochemotherapy, which is used in variants of cutaneous T-cell lymphomas, graft-versus-host disease, systemic sclerosis, in solid organ transplant rejection and Crohn disease (44), a leukocyte fraction from the peripheral blood is exposed to approximately 40–50 mmol/L of photo-activated 8-methoxypsoralen and reinfused into the patient without collateral effects. However, regarding transfusional purposes, INTERCEPT products are contraindicated in patients with a history of allergic response to psoralens; the certificate of analysis for MSCs isolated and expanded in GMP should state that traces of psoralens might be present so that they are not used in psoralen-allergic patients.
We found that HPL and iHPL had similar characteristics. They were more advantageous, in terms of cellular growth, than was FBS and did not interfere with mesenchymal phenotypes because they did not modify the mesenchymal marker molecule expression, Furthermore, HPL and iHPL caused no chromosomal alterations or karyotype modifications in cells expanded up to the third passage. Interestingly, when whole BM was seeded in HPL and iHPL, no significant differences in the CFU-F number were observed compared with FBS-MSC, although they did appear to be denser and richer in terms of cells/colony. This observation was confirmed by the number of detached cells at each passage because HPL and iHPL-MSCs showed higher proliferative potential. Moreover, they were smaller and more morphologically homogeneous than were FBS-MSC. Differences in their CFU-F potential and colony morphology may be representative and possibly predictive of cell fate and function. A recent report (52) showed that CFU-Fs can be analyzed by combining two different pieces of information: diameter and density, which are properties related to and affected by cell proliferation, and mobility and differentiation potential. On these bases, small and dense colonies, which we observed in HPL and iHPL-MSCs, represent the BM-MSC fraction with more immature stem cells. These data suggest a higher clonal expansion of HPL/iHPL-MSCs that might be predictive of greater stemness than that of FBS-MSCs.
In one batch of HPL and iHPL, we tested the presence of growth factors such as platelet-derived growth factor and fibroblast growth factor. We did not observe any differences in terms of quantity (data not shown), but we maintain that the growth-promoting effect of HPL and iHPL is probably due to growth factors or specific compounds that are released from the platelet fraction.
Another important aspect observed in both HPL or iHPL was the formation of spherical structures similar to embryoid bodies, which, together with the pseudopodia emission, suggested that HPL/iHPL-MSCs had a more primitive stemness than did FBS-MSC, as shown in MSCs isolated from neonatal tissues compared with adult tissue (45). As reported in the literature, the pseudopodia emission may suggest a trend of the cells to migrate and consequently to repopulate damaged tissues and organs with potential implications for regenerative medicine (46). The entity of this occurrence is higher in HPL/iHPL-MSCs compared with that in FBS-MSCs.
To verify whether these morphological and proliferative characteristics of the HPL/iHPL-MSCs might be linked to more stemness than FBS-MSC, we analyzed the presence of pluripotentency markers such as Oct-3/4 and NANOG, which we had already observed in MSCs isolated from neonatal tissue such as the amniotic fluid (47). Oct-3/4 is a key transcription factor for the pluripotent and self-renewing phenotypes of undifferentiated embryonic stem cells (48). NANOG, a homeodomain protein present in pluripotent human cells, plays a critical role in the regulation of the cell fate of the pluripotent inner cell mass during embryonic development, maintaining the pluripotent epiblast and preventing differentiation to the primitive endoderm (49). NANOG is thought to function in concert with other factors such as Oct-3/4. Interestingly, the spheroid structures that were observed in HPL and iHPL, not only during the seeding but also when the MSCs were detached and replaced in the passages after the 1st, were highly positive for Oct-3/4 and NANOG. Moreover, during expansion, HPL and iHPL induce a selection of more primitive MSCs compared with that in FBS.
Although HPL/iHPL-MSCs exhibit increased cell growth, in terms of cPD, and characteristics attributable to higher primitive stemness than FBS-MSCs, we demonstrated that neither the HPL nor iHPL caused in vitro tumorigenic mutations or karyotype modifications in cells expanded up to the 3rd step. These data are in accordance (only for HPL) with that reported in the literature about GMP-compliant isolation and expansion of BM-MSC 50, 51, 52. Moreover, we previously reported that by plating whole BM at a low cellular density, it was possible to expand high numbers of MSCs for clinical use (23). In the present study, we further showed that, independent of the culture condition, the number of CFU-F/106 cells was significantly greater when the whole BM cells were plated at a low density, compared with cells plated at a high density. These results emphasized the advantage of the low seeding density of whole BM to isolate MSCs for large-scale use. The use of HPL as an alternative to FBS to isolate and expand MSC confirmed that it is possible to obtain a number of MSCs for clinical doses and further reduce manufacturing time, limiting the passage or reducing the starting volume of BM. Finally, we demonstrated that PI treatment did not modify the characteristics of HPL, made it safer and more suitable for MSC isolation and expansion for clinical use and might be a requirement for GMP MSC expansion.
Acknowledgments
We are grateful to Andrew Martin Garvey for editorial assistance and to Alessandra Mandese for data management.
Disclosure of interests: The authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
References
- 1.Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Mareschi K., Biasin E., Piacibello W., Aglietta M., Madon E., Fagioli F. Isolation of human mesenchymal stem cells: bone marrow versus umbilical cord blood. Haematologica. 2001;86:1099–1100. [PubMed] [Google Scholar]
- 3.Aggarwal S., Pittenger M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 4.Prockop D.J., Oh J.Y. Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther. 2012;20:14–20. doi: 10.1038/mt.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazzini L., Mareschi K., Ferrero I., Vassallo E., Oliveri G., Boccaletti R. Autologous mesenchymal stem cells: clinical applications in amyotrophic lateral sclerosis. Neurol Res. 2006;28:523–526. doi: 10.1179/016164106X116791. [DOI] [PubMed] [Google Scholar]
- 6.Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10:709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 7.Sensebé L., Bourin P. Producing MSC according GMP: process and controls. Biomed Mater Eng. 2008;18:173–177. [PubMed] [Google Scholar]
- 8.Fekete N., Rojewski M.T., Fürst D., Kreja L., Ignatius A., Dausend J. GMP-compliant isolation and large-scale expansion of bone marrow-derived MSC. PLoS One. 2012;7:e43255. doi: 10.1371/journal.pone.0043255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Regulation (EC) No. 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products.
- 10.Spees J.L., Gregory C.A., Singh H., Tucker H.A., Peister A., Lynch P.J. Internalized antigens must be removed to prepare hypoimmunogenic mesenchymal stem cells for cell and gene therapy. Mol Ther. 2004;9:747–756. doi: 10.1016/j.ymthe.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 11.Horwitz E.M., Gordon P.L., Koo W.K., Marx J.C., Neel M.D., McNall R.Y. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932–8937. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundin M., Ringdén O., Sundberg B., Nava S., Götherström C., Le Blanc K. No alloantibodies against mesenchymal stromal cells, but presence of anti-fetal calf serum antibodies, after transplantation in allogeneic hematopoietic stem cell recipients. Haematologica. 2007;92:1208–1215. doi: 10.3324/haematol.11446. [DOI] [PubMed] [Google Scholar]
- 13.Muller I., Kordowich S., Holzwarth C., Spano C., Isensee G., Staiber A. Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy. 2006;8:437–444. doi: 10.1080/14653240600920782. [DOI] [PubMed] [Google Scholar]
- 14.Halme D.G., Kessler D.A. FDA regulation of stem-cell-based therapies. N Engl J Med. 2006;355:1730–1735. doi: 10.1056/NEJMhpr063086. [DOI] [PubMed] [Google Scholar]
- 15.Fekete N., Gadelorge M., Fürst D., Maurer C., Dausend J., Fleury-Cappellesso S. Platelet lysate from whole blood-derived pooled platelet concentrates and apheresis-derived platelet concentrates for the isolation and expansion of human bone marrow mesenchymal stromal cells: production process, content and identification of active components. Cytotherapy. 2012;14:540–554. doi: 10.3109/14653249.2012.655420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernardi M., Albiero E., Alghisi A., Chieregato K., Lievore C., Madeo D. Production of human platelet lysate by use of ultrasound for ex vivo expansion of human bone marrow-derived mesenchymal stromal cells. Cytotherapy. 2013;15:920–929. doi: 10.1016/j.jcyt.2013.01.219. [DOI] [PubMed] [Google Scholar]
- 17.Schallmoser K., Bartmann C., Rohde E., Reinisch A., Kashofer K., Stadelmeyer E. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion. 2007;47:1436–1446. doi: 10.1111/j.1537-2995.2007.01220.x. [DOI] [PubMed] [Google Scholar]
- 18.Trojahn Kølle S.F., Oliveri R.S., Glovinski P.V., Kirchhoff M., Mathiasen A.B., Elberg J.J. Pooled human platelet lysate versus fetal bovine serum—investigating the proliferation rate, chromosome stability and angiogenic potential of human adipose tissue-derived stem cells intended for clinical use. Cytotherapy. 2013;15:1086–1097. doi: 10.1016/j.jcyt.2013.01.217. [DOI] [PubMed] [Google Scholar]
- 19.Council of Europe, European Committee (Partial Agreement) on Blood Transfusion (CD-P-TS), European Directorate for the Quality of Medicines & HealthCare of the Council of Europe (EDQM), Symposium on Implementation of Pathogen Reduction Technologies for Blood Components. September 2010.
- 20.Tice R.R., Gatehouse D., Kirkland D., Speit G. The pathogen reduction treatment of platelets with S-59 HCl (Amotosalen) plus ultraviolet A light: genotoxicity profile and hazard assessment. Mutat Res. 2007;630:50–68. doi: 10.1016/j.mrgentox.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Lin L., Cook D.N., Wiesehahn G.P., Alfonso R., Behrman B., Cimino G.D. Photochemical inactivation of viruses and bacteria in platelet concentrates by use of a novel psoralen and long-wavelength ultraviolet light. Transfusion. 1997;37:423–435. doi: 10.1046/j.1537-2995.1997.37497265344.x. [DOI] [PubMed] [Google Scholar]
- 22.van Rhenen D., Gulliksson H., Cazenave J.P., Pamphilon D., Ljungman P., Klüter H. Transfusion of pooled buffy coat platelet components prepared with photochemical pathogen inactivation treatment: the euroSPRITE trial. Blood. 2003;101:2426–2433. doi: 10.1182/blood-2002-03-0932. [DOI] [PubMed] [Google Scholar]
- 23.Mareschi K., Rustichelli D., Calabrese R., Gunetti M., Sanavio F., Castiglia S. Multipotent mesenchymal stromal stem cell expansion by plating whole bone marrow at a low cellular density: a more advantageous method for clinical use. Stem Cells Int. 2012;2012:920581. doi: 10.1155/2012/920581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gunetti M., Castiglia S., Rustichelli D., Mareschi K., Sanavio F., Muraro M. Validation of analytical methods in GMP: the disposable Fast Read 102(R) device, an alternative practical approach for cell counting. J Transl Med. 2012;10:112. doi: 10.1186/1479-5876-10-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rustichelli D., Castiglia S., Gunetti M., Mareschi K., Signorino E., Muraro M. Validation of analytical methods in compliance with good manufacturing practice: a practical approach. J Transl Med. 2013;11:197. doi: 10.1186/1479-5876-11-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorpe T.C., Wilson M.L., Turner J.E., DiGuiseppi J.L., Willert M., Mirrett S. BacT/Alert: an automated colorimetric microbial detection system. J Clin Microbiol. 1990;28:1608–1612. doi: 10.1128/jcm.28.7.1608-1612.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capelli C., Domenghini M., Borleri G., Bellavita P., Poma R., Carobbio A. Human platelet lysate allows expansion and clinical grade production of mesenchymal stromal cells from small samples of bone marrow aspirates or marrow filter washouts. Bone Marrow Transplant. 2007;40:785–791. doi: 10.1038/sj.bmt.1705798. [DOI] [PubMed] [Google Scholar]
- 28.Schallmoser K., Rohde E., Bartmann C., Obenauf A.C., Reinisch A., Strunk D. Platelet-derived growth factors for GMP-compliant propagation of mesenchymal stromal cells. Biomed Mater Eng. 2009;19:271–276. doi: 10.3233/BME-2009-0591. [DOI] [PubMed] [Google Scholar]
- 29.Irsch J., Lin L. Pathogen Inactivation of Platelet and Plasma Blood Components for Transfusion Using the INTERCEPT Blood System™. Transfus Med Hemother. 2011;38:19–31. doi: 10.1159/000323937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allain J.P., Bianco C., Blajchman M.A., Brecher M.E., Busch M., Leiby D. Protecting the blood supply from emerging pathogens: the role of pathogen inactivation. Transfus Med Rev. 2005;19:110–126. doi: 10.1016/j.tmrv.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prowse C.V. Component pathogen inactivation: a critical review. Vox Sang. 2013;104:183–199. doi: 10.1111/j.1423-0410.2012.01662.x. [DOI] [PubMed] [Google Scholar]
- 32.Tsetsarkin K.A., Sampson-Johannes A., Sawyer L., Kinsey J., Higgs S., Vanlandingham D.L. Photochemical inactivation of chikungunya virus in human apheresis platelet components by amotosalen and UVA light. Am J Trop Med Hyg. 2013;88:1163–1169. doi: 10.4269/ajtmh.12-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grellier P., Benach J., Labaied M., Charneau S., Gil H., Monsalve G. Photochemical inactivation with amotosalen and long-wavelength ultraviolet light of Plasmodium and Babesia in platelet and plasma components. Transfusion. 2008;48:1676–1684. doi: 10.1111/j.1537-2995.2007.01762.x. [DOI] [PubMed] [Google Scholar]
- 34.Lin L., Hanson C.V., Alter H.J., Jauvin V., Bernard K.A., Murthy K.K. Inactivation of viruses in platelet concentrates by photochemical treatment with amotosalen and long-wavelength ultraviolet light. Transfusion. 2005;45:580–590. doi: 10.1111/j.0041-1132.2005.04316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nussbaumer W., Allerstorfer D., Allersdorfer D., Grabmer C., Rheinschmidt M., Lin L. Prevention of transfusion of platelet components contaminated with low levels of bacteria: a comparison of bacteria culture and pathogen inactivation methods. Transfusion. 2007;47:1125–1133. doi: 10.1111/j.1537-2995.2007.01247.x. [DOI] [PubMed] [Google Scholar]
- 36.Rasonglès P., Angelini-Tibert M.F., Simon P., Currie C., Isola H., Kientz D. Transfusion of platelet components prepared with photochemical pathogen inactivation treatment during a Chikungunya virus epidemic in Ile de La Réunion. Transfusion. 2009;49:1083–1091. doi: 10.1111/j.1537-2995.2009.02111.x. [DOI] [PubMed] [Google Scholar]
- 37.Grass J.A., Hei D.J., Metchette K., Cimino G.D., Wiesehahn G.P., Corash L. Inactivation of leukocytes in platelet concentrates by photochemical treatment with psoralen plus UVA. Blood. 1998;91:2180–2188. [PubMed] [Google Scholar]
- 38.Infanti L., Stebler C., Job S., Ruesch M., Gratwohl A., Irsch J. Pathogen-inactivation of platelet components with the INTERCEPT Blood System™: a cohort study. Transfus Apher Sci. 2011;45:175–181. doi: 10.1016/j.transci.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Rueesch M. JM, Stoller R, Mansouri Taleghani M. Two years experience with pathogen inactivation for all platelet concentrates in Switzerland. 2013. [DOI] [PMC free article] [PubMed]
- 40.Ståhle M., Carlsson B., Le Blanc K., Korsgren O., Knutson F. Photochemical pathogen inactivation of human serum enables its large-scale application in clinical cell transplantation. Vox Sang. 2010;98(3 Pt 1):e364–e365. doi: 10.1111/j.1423-0410.2009.01257.x. [DOI] [PubMed] [Google Scholar]
- 41.Azzi A., Morfini M., Mannucci P.M. The transfusion-associated transmission of parvovirus B19. Transfus Med Rev. 1999;13:194–204. doi: 10.1016/s0887-7963(99)80033-9. [DOI] [PubMed] [Google Scholar]
- 42.Saldanha J., Minor P. Detection of human parvovirus B19 DNA in plasma pools and blood products derived from these pools: implications for efficiency and consistency of removal of B19 DNA during manufacture. Br J Haematol. 1996;93:714–719. doi: 10.1046/j.1365-2141.1996.d01-1679.x. [DOI] [PubMed] [Google Scholar]
- 43.Wagner S.J., Skripchenko A., Myrup A., Awatefe H., Thompson-Montgomery D., Moroff G. Evaluation of in vitro storage properties of prestorage pooled whole blood-derived platelets suspended in 100 percent plasma and treated with amotosalen and long-wavelength ultraviolet light. Transfusion. 2009;49:704–710. doi: 10.1111/j.1537-2995.2008.02040.x. [DOI] [PubMed] [Google Scholar]
- 44.Trautinger F., Just U., Knobler R. Photopheresis (extracorporeal photochemotherapy) Photochem Photobiol Sci. 2013;12:22–28. doi: 10.1039/c2pp25144b. [DOI] [PubMed] [Google Scholar]
- 45.Campagnoli C., Roberts I.A., Kumar S., Bennett P.R., Bellantuono I., Fisk N.M. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98:2396–2402. doi: 10.1182/blood.v98.8.2396. [DOI] [PubMed] [Google Scholar]
- 46.Langer H.F., Stellos K., Steingen C., Froihofer A., Schönberger T., Krämer B. Platelet derived bFGF mediates vascular integrative mechanisms of mesenchymal stem cells in vitro. J Mol Cell Cardiol. 2009;47:315–325. doi: 10.1016/j.yjmcc.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 47.Mareschi K., Rustichelli D., Comunanza V., De Fazio R., Cravero C., Morterra G. Multipotent mesenchymal stem cells from amniotic fluid originate neural precursors with functional voltage-gated sodium channels. Cytotherapy. 2009;11:534–547. doi: 10.1080/14653240902974024. [DOI] [PubMed] [Google Scholar]
- 48.Kehler J., Tolkunova E., Koschorz B., Pesce M., Gentile L., Boiani M. Oct4 is required for primordial germ cell survival. EMBO Rep. 2004;5:1078–1083. doi: 10.1038/sj.embor.7400279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chambers I., Colby D., Robertson M., Nichols J., Lee S., Tweedie S. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 50.Lucchini G., Introna M., Dander E., Rovelli A., Balduzzi A., Bonanomi S. Platelet-lysate-expanded mesenchymal stromal cells as a salvage therapy for severe resistant graft-versus-host disease in a pediatric population. Biol Blood Marrow Transplant. 2010;16:1293–1301. doi: 10.1016/j.bbmt.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 51.Sensebé L., Bourin P., Tarte K. Good manufacturing practices production of mesenchymal stem/stromal cells. Hum Gene Ther. 2011;22:19–26. doi: 10.1089/hum.2010.197. [DOI] [PubMed] [Google Scholar]
- 52.Hanley P.J., Mei Z., da Graca Cabreira-Hansen M., Klis M., Li W., Zhao Y. Manufacturing mesenchymal stromal cells for phase I clinical trials. Cytotherapy. 2013;15:416–422. doi: 10.1016/j.jcyt.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]