

Abstract
CIRE/mDC-SIGN is a C-type lectin we originally identified as a molecule differentially expressed by mouse dendritic cell (DC) populations. Immunostaining with a CIRE/mDC-SIGN-specific mAb revealed that CIRE/mDC-SIGN is indeed on the surface of some CD4+, CD4−8− DCs and plasmacytoid pre-DCs, but not on CD8+ DCs. It has been proposed that CIRE/mDC-SIGN is the functional orthologue of human DC-SIGN (hDC-SIGN), a molecule that both enhances T cell responses and facilitates antigen uptake. We assessed if CIRE/mDC-SIGN and hDC-SIGN exhibit functional similarities. CIRE/mDC-SIGN is down-regulated upon activation, but unlike hDC-SIGN, incubation with IL-4 and IL-13 did not enhance CIRE/mDC-SIGN expression, indicating differences in gene regulation. Like hDC-SIGN, CIRE/mDC-SIGN bound mannosylated residues. However, we could detect no role for CIRE/mDC-SIGN in T cell–DC interactions and the protein did not bind to pathogens known to interact with hDC-SIGN, including Leishmania mexicana, cytomegalovirus, HIV and lentiviral particles bearing the Ebolavirus glycoprotein. The binding of CIRE/mDC-SIGN to hDC-SIGN ligands was not rescued when CIRE/mDC-SIGN was engineered to express the stalk region of hDC-SIGN. We conclude that there are significant differences in the fine specificity of the C-type lectin domains of hDC-SIGN and CIRE/mDC-SIGN and that these two molecules may not be functional orthologues.
Keywords: cell-surface molecules, dendritic cells, rodents
Introduction
Antigen-presenting dendritic cells (DCs) are essential for initiating immune responses because of their unique ability to stimulate naive T lymphocytes (1). However, DCs are heterogeneous in both their ontogeny and function (2). In mouse spleen, there exist three major populations of conventional dendritic cells (cDCs): CD4+CD8− (CD4+ DC), CD4−CD8− (double negative [DN] DC) and CD4−CD8+ DC (CD8+ DC) (3). While there is debate over the haematological origins of these populations (4–6), they are functionally distinct. For example, CD8+ DCs are relatively poor stimulators of T cells (7, 8), and were originally proposed to have a regulatory function (9). However, if activated, CD8+ and CD8− DCs can both induce immune responses (10, 11), though they preferentially induce Th1 and Th2 type responses, respectively (12, 13). This is in part due to CD8+ DCs being more potent producers of IL-12 (14, 15). CD8+ DCs have a unique ability to take-up dying cells (16), and are especially adept at cross-presenting exogenous antigen on class I MHC (17, 18). Lymph node CD8+ DCs are the most efficient at presenting viral antigen and initiating T cell responses (19). Finally, DC populations occupy different micro-environments in lymphoid organs (15, 20), CD8+ DCs tend to concentrate in T cell areas, whereas CD8− DCs are found in marginal zones but migrate to T cell areas upon stimulation (21).
Molecules whose expression differs among these DC populations are of particular interest as they may underpin their functional differences. Using both genetic (22, 23) and immunophenotyping approaches (3, 24, 25), several such molecules have been identified and recently we cloned the cell-surface protein CIRE, which is expressed by DN DCs and CD4+ DCs, but not by CD8+ DCs (26).
CIRE is a type II membrane protein with a C-type lectin domain (CTLD) at the cell surface. The CIRE protein sequence shows a 57% identity with the human molecule DC-specific intercellular adhesion molecule (ICAM)-grabbing non-integrin (hDC-SIGN) (26, 27). Human DC-SIGN facilitates interactions with T cells by binding ICAM-3, and with endothelial cells by binding ICAM-2 (28, 29). It also plays a role in the capture, internalisation and presentation of foreign antigen (30). Notably, several pathogens interact with hDC-SIGN, including viruses such as HIV, human cytomegalovirus (HCMV) and Ebolavirus (EBOV), bacteria such as Mycobacterium and parasites such as Leishmania and the eggs of Schistosoma mansoni (31).
The sequence similarity, together with the cellular expression and genomic localisation data, suggest that CIRE is mouse DC-SIGN (26, 27) and therefore will be referred to as CIRE/mDC-SIGN. However, whether CIRE/mDC-SIGN and hDC-SIGN are indeed functional orthologues is open to question. The issue is clouded by gene duplication events in both the mouse and human genomes. In humans, there is a DC-SIGN homologue (DC-SIGNR or L-SIGN), which is just as similar to CIRE/mDC-SIGN as is hDC-SIGN (32). In mice, there are four CIRE/mDC-SIGN homologues whose CTLDs display a level of identity to hDC-SIGN as close as CIRE/mDC-SIGN itself (26, 27).
We have now generated a mAb to CIRE/mDC-SIGN, enabling us to characterise this molecule at the protein level. We confirm the differential expression of CIRE/mDC-SIGN in DC subsets and demonstrate that CIRE/mDC-SIGN is indeed a lectin able to bind mannosylated ligands in a calcium-dependent manner. However, CIRE/mDC-SIGN does not bind to any of the hDC-SIGN ligands tested, indicating that significant differences exist in the ligand-binding specificity of the two molecules. The function of CIRE/mDC-SIGN might therefore differ from that of hDC-SIGN.
Methods
Mice
C57BL/6J wehi, CBA/CaH mice and Wistar rats were bred under specific pathogen-free (SPF) conditions at the Walter and Eliza Hall Institute (WEHI). Germ-free C57BL/6J mice were bred at the WEHI facility and sacrificed within 12 h of arrival into our SPF holding facility.
Generating an anti-CIRE/mDC-SIGN mAb
A synthetic peptide was synthesised (H-MSKESTWYWVDGSPLTLSFMKYWSKC-NH2) and conjugated to keyhole limpet haemocyanin (KLH) (Mimotopes, Victoria, Australia). To generate mAbs, Wistar rats were immunised intra-peritoneally (i.p.) with 75 μg of KLH-conjugated peptide in CFA, boosted 5 weeks later with 50 μg of KLH-conjugated peptide in incomplete Freunds adjuvant and again 4 days prior to fusion intravenously and i.p. with 10 μg of KLH-conjugated peptide in aqueous solution. Hybridomas secreting specific mAbs were identified by flow cytometric analysis of supernatants using FLAG-CIRE/mDC-SIGN-CHO and Neo-CHO (26). Four clones, from several thousand screened, produced anti-CIRE/mDC-SIGN mAb, but only one clone, 5H10, remained stable in culture and continued to produce mAb.
Antibodies
The following fluorochrome-conjugated mAbs were used: anti-CD11c (N418)–allophycocyanin, –Cy5 or –FITC; anti-CIRE/mDC-SIGN (5H10)–biotin; anti-CD4 (GK1.5)–Alexa 594, anti-CD8 (53-6.7 or YTS 169.4)–FITC; anti-CD45RA (14.8)–FITC; isotype control IgG2a–biotin (PharMingen, San Diego, CA, USA); goat anti-rat–FITC antibody (Caltag, Burlingame, CA, USA); streptavidin–PE (PharMingen). To better visualise CIRE/mDC-SIGN on the DC surface, the amplification system Flow-Amp (Flow-Amp Systems, Cleveland, OH, USA) was used according to the manufacturer's guidelines. CIRE/mDC-SIGN staining was always after pre-incubation with rat Ig and 2.4G2 (10 min 4°C), and anti-CIRE/mDC-SIGN or isotype control mAb was then added into the pre-blocking mix. The anti-hDC-SIGN (AZN-1) supernatant was kindly donated by D. Hart (Mater Institute, Queensland, Australia).
Isolation of DCs, macrophages and peripheral blood monocytes
The isolation of DC sub-populations has been described (3, 33). Briefly, tissues were chopped, digested with collagenase and DNAase at room temperature and treated with EDTA. Low-density cells were enriched by density centrifugation. Non-DC-lineage cells were coated with mAb (KT3-1.1, T24/31.7, TER119, RB6-8C5, ID3) and then removed using immunomagnetic beads. Coating with RB6-8C5 mAb (anti-Gr-1) did not result in the depletion of plasmacytoid pre-dendritic cells (pDCs) (33). The remaining cells were stained with various combinations of fluorochrome-conjugated mAb and populations enriched for CD11c+CIRE/mDC-SIGN+ and CD11c+CIRE/mDC-SIGN− cells, or purified as CD11c+CD8+CD4−, CD11c+CD8−CD4+ and CD11c+CD8− CD4− or as CD11cint CD45RA+ and CD11chi and CD45RA−, all by sorting on a MoFlo Instrument (Cytomation Inc.). Due to the low level and frequency of CIRE/mDC-SIGN expression on DCs, the CD11c+CIRE/mDC-SIGN− DC purity was above 98%, but the CD11c+CIRE/mDC-SIGN+ DC purity was only 50–75%, the main contaminants being CD11c+CIRE/mDC-SIGN− DCs. To obtain blood mononuclear cells, mice were bled by cardiac puncture into tubes containing heparinized buffered saline solution. Mononuclear cells were isolated by density centrifugation using Histopaque 1.083 (Sigma, Castle Hill, Australia) and cells bearing CD3, Thy-1, Gr-1 and the erythrocyte marker TER119 removed by immunomagnetic bead depletion. Cells from the peritoneal cavity or the bone marrow were obtained by flushing with medium, and then removing erythrocytes with lysis buffer (0.099 mM EDTA disodium, 0.145 M NH4Cl, 0.012 M NaHCO3).
DC activation
Isolated DCs (2 × 106 cells ml−1) were cultured in 24-well plates for 18–20 h in modified RPMI-1640 medium containing 10% FCS, antibiotics, 10−4 M 2-mercaptoethanol, granulocyte macrophage colony-stimulating factor (GM-CSF) (50 U ml−1) and CpG-1668 (0.5 μM) (GeneWorks, Adelaide, Australia) or in 96-well flat-bottom plates with murine recombinant IL-4 (Immunex) and IL-13 (R&D Systems, Minneapolis, MN, USA).
Mixed leucocyte reaction
CD4 or CD8 T cells were isolated from lymph node cell suspensions by coating irrelevant cells with mAb (anti-erythrocytes, TER119; anti-B220, RA3-6B2; anti-Gr-1, RB6-6CS; Mac-1, M1/70 and anti-CD8, 53.6-7 or anti-CD4, GK1.5), and removing coated cells using IgG-coupled magnetic beads (Dynabeads, Dynal) at a 1:10 cell-to-bead ratio; purity was >95%. DCs were isolated and sorted as CD11c+CIRE/mDC-SIGN+ and CD11c+CIRE/mDC-SIGN− cells. Varying numbers of DCs were incubated for 3–4 days with 20 000 CD4 or CD8 T cells in V-bottom 96-well plates in modified RPMI-1640 medium containing 10% FCS, antibiotics and 10−4 M 2-mercaptoethanol. Cultures were pulsed at the end of the incubation period with 1 μCi per well of [3H]thymidine for 6 h, harvested onto glass-fibre filters and thymidine incorporation was counted by liquid scintillation. Five replicates of all cultures were done.
Generating constructs
CIRE/mDC-SIGN and hDC-SIGN cDNAs were cloned into the EcoRV restriction site of the pIRES-Neo plasmid (Clonetech, Heidelberg, Germany). Briefly, CIRE/mDC-SIGN cDNA was amplified from previously isolated clones (26) using the PCR and Pwo polymerase (Roche, Mannheim, Germany) under standard amplification conditions: 25 cycles: 94°, 30 s; 55°, 30 s; 72°, 1 min. The primers used in this reaction were: CIRE/mDC-SIGN-For primer 5′-TAG TAG ATA TCG GCG CGC CTC ACT TGC TAG GGC AGG A and -Rev primer 5′-TAG TAG ATA TCG GCG CGC CTG AAA CAT GAG TGA TTC TAA G. Similarly, hDC-SIGN was amplified from cDNA derived from human DCs, using primers designed from published sequences (34): hDC-SIGN-For primer 5′-TAG TAG ATA TCT GGG GTG ACA TGA GTG AC and DC-SIGN-Rev primer 5′-TAG TAG ATA TCT ACG CAG GAG GGG GGT TT. The CIRE/mDC-SIGN/hDC-SIGN hybrid molecules were generated using an overlapping PCR strategy. To generate CIRE/mDC-SIGN/h-CTLD, cDNA encoding the cytoplasmic domains, transmembrane domain and hinge region of CIRE/mDC-SIGN was amplified by PCR using the CIRE/mDC-SIGN-For primer, plus the CIRE/mDC-SIGN/hCTLD-Rev primer 5′-TGT CCA TTC CCA GGG ACA GGA GCG GCA CAG TCG AT. The first 17 nt of this primer correspond to sequence encoding the C-terminus of the CIRE/mDC-SIGN hinge region, whereas the last 17 nt encode the N-terminus of the hDC-SIGN CTLD. Similarly, cDNA corresponding to the hDC-SIGN CTLD was amplified with the hDC-SIGN-Rev primer and the CIRE/mDC-SIGN/hCTLD-For primer ATC GAC TGT GCC GCT CCT GTC CCT GGG AAT GGA CA, whose sequence is the reverse and complement of the CIRE/mDC-SIGN/hCTLD-Rev primer. The cDNAs encoding the CIRE/mDC-SIGN cytoplasmic, transmembrane and hinge regions and the DC-SIGN CTLD were purified by conventional methods, and joined by PCR using the CIRE/mDC-SIGN-For and hDC-SIGN-Rev primers. The resulting cDNA was then cloned into the EcoRV site of pIRES-Neo. A parallel strategy was used to generate the hDC-SIGN/mCTLD hybrid. Here cDNA encoding the hDC-SIGN cytoplasmic, transmembrane and hinge regions was amplified using the hDC-SIGN-For primer and the DC-SIGN/mCTLD-Rev primer 5′-CGT CCA GTC CCA GGG GCA GGG GTG GCA CAG GCG TT. Similarly, cDNA encoding the mouse CIRE/mDC-SIGN CTLD was amplified using the CIRE/mDC-SIGN-Rev primer and primer whose sequence was the reverse and complement of DC-SIGN/mCTLD-Rev, namely DC-SIGN/mCTLD-For primer 5′-AAC GCC TGT GCC ACC CCT GCC CCT GGG ACT GGA CG. Again, the two resulting cDNA fragments were joined in an overlapping PCR using the hDC-SIGN-For and CIRE/mDC-SIGN-Rev primers. Mouse SIGNR1 was sub-cloned into the expression plasmid pcDNA3.1Zeo (Invitrogen, CA, USA) and has the AU1 tag fused to the carboxy terminal (35).
Transfection
CHO-KI (CHO) cells were transfected with the pIRES-Neo plasmid containing the neomycin phosphotransferase gene plus inserted gene or with the control pCI-neo plasmid (Promega, Annandale, NSW, Australia) only, using the FuGENE 6 Transfection Reagent (Roche, Indianapolis, IN, USA) according to the manufacturer's guidelines. Transfectants were allowed to recover for 24 h before selection with 750 μg ml−1 G418 (Geneticin, GIBCO). Mouse SIGNR1 transfectants were selected with Zeocin (250 μg ml−1) (Invitrogen). All tranfectants were stained with specific antibodies [anti-human DC-SIGN (120507; R&D Systems); anti-CIRE (5H10) and biotinylated AU1 mAb (AU1; Covance, CA, USA], visualised with the appropriate secondary reagent and sorted on a MoFlo Instrument (Cytomation Inc.)
Binding affinity of CIRE/mDC-SIGN
Adherent CHO-K1 cells expressing FLAG-CIRE/mDC-SIGN, CIRE/mDC-SIGN, mouse SIGNR1 or the neomycin resistance gene only were made into single-cell suspension by a brief incubation with 0.01 M EDTA/PBS and then washed twice with 5% FCS–RPMI-1640. Cells (105) were re-suspended in various dilutions of mannan (Sigma, Castle Hill, NSW, Australia) in an ice-cold buffered balanced salt solution containing 2% FCS, Ca2+ and Mg2+, or alternatively, in a similar solution where 5 mM EDTA substituted for Ca2+ and Mg2+, then incubated with mannosylated FITC-conjugated BSA (10 μg ml−1) (Sigma) for 20 min at 37°C. Binding was visualised by flow cytometry.
Parasites and parasite-binding assays
Leishmania promastigotes (World Health Organization's Reference Centre for Leishmaniases, Jerusalem, Israel) were maintained by passaging in athymic nude mice. Promastigotes were grown in M199 medium (Invitrogen) containing 10% FCS and 2 mM glutamine. Parasites were cultured for no longer than 6 weeks to assure virulence. The Leishmania parasites used were a cloned line of Leishmania major V121 (MHOM/IL/67/JERICHO II) and Leishmania mexicana (MNYC/BZ/62/M379). Transfected cells were plated out on glass cover slips in 24-well trays at a density of 5 × 104 cells per well and incubated at 37°C for 24 h before infection. Cells in duplicate wells were infected with promastigotes at a multiplicity of infection of 5:1. Infection was allowed to proceed for 24 h at 33°C for L. mexicana and 37°C for L. major, before free parasites were removed by washing, and cells fixed in methanol and stained with Giemsa. The percent infected cells or cells with attached parasites and the number of parasites attached or internalised in each cell were determined after counting at least 400 cells in each of duplicate samples. In some experiments, the cells were infected with L. major amastigotes obtained from infected CBA/N athymic nude mice as described (36).
HCMV-binding assay
HCMV strain AD169 and MCMV strain K181-Perth were incubated, at multiplicities of infection ranging from 50 to 1, with 5 × 104 CHO transfectants for 1 h at 4°C in PBS/0.1% BSA/1 mM CaCl2/2 mM MgCl2, as described previously (37). The percentage of cells to which AD169 and K181 attached was determined by flow cytometric analysis using an anti-HCMV gB mAb (clone 1-M-12, Biodesign) and an anti-MCMV gH mAb (clone 8D1.22A, kindly provided by L. Loh, University of Saskatchewan, Saskatchewan, Canada), respectively. A FITC-conjugated anti-mouse antibody was used to detect binding. CHO cells transfected with the neomycin resistance gene alone were included in all the assays as a negative control.
Enhancement of HIV-1 transmission and Ebolavirus glycoprotein-mediated infection
Ebolavirus glycoprotein (EBOV-GP)-bearing lentiviral pseudotypes and replication competent HIV-1 NL4-3 harbouring the luciferase gene in place of nef (NL4-3luc) were generated as described (38, 39). In brief, 293T cells were either transiently co-transfected with pNL4-3 E−R− Luc (40) and an expression plasmid for EBOV-GP of the Zaire subspecies (for generation of EBOV-GP-bearing pseudotypes), or transfected with NL4-3luc (for generation of replication competent HIV-1 NL4-3 reporter virus), using the calcium phosphate method. The culture medium was replenished after 16 h and then harvested 48 h post-transfection. The supernatants were passed through 0.2-μm filters and stored at −80°C. Lectin-mediated enhancement of viral infection was assessed employing lectin expressing CHO transfectants. To analyse the impact of lectin expression on EBOV-GP-driven infection, the CHO cell lines [CHO cells are permissive to EBOV-GP-dependent entry (41)] were seeded in 96-well plates and spin infected with EBOV-GP-bearing pseudotypes at 2000 r.p.m. for 2.5 h as described (42). After overnight incubation, the infection medium was replaced by fresh culture medium and cells cultivated for 3 days before cells were lysed and luciferase activities in cell lysate determined using a commercially available kit (Promega, Madison, WI, USA). To assess lectin-mediated enhancement of HIV-1 transmission, the CHO cell lines (which are not permissive to HIV-1 infection) were seeded in 96-well plates, incubated with HIV-1 NL4-3luc for 2 h at 37°C, washed with fresh culture medium and co-cultivated with receptor-positive CEMx174 5.25 M7 cells (43). Luciferase activities in cell lysates were determined 3 days after the start of the co-culture.
Western blot analysis of chimeric molecules expressed in CHO cells
Cell lysates from parental CHO cells or CHO cells expressing hDC-SIGN, or the chimeric molecules (CIRE/mDC-SIGN lectin fused to hDC-SIGN stalk and hDC-SIGN lectin fused to CIRE/mDC-SIGN stalk) and CIRE/mDC-SIGN, were separated on SDS-PAGE under reducing and non-reducing conditions. For reducing conditions, samples were re-suspended in buffer (62.5 mM Tris–HCl pH 6.8, 10% glycerol, 4% SDS and bromophenol blue) containing 5% β-mercaptoethanol and boiled (5 min) and vortexed several times, whereas the non-reduced samples were lysed in buffer without β-mercaptoethanol. Samples were transferred onto mobilon-P polyvinylidene difluoride membrane (Millipore) according to the manufacturer's instruction. Membranes were blocked with 2% skim milk, then probed with mouse anti-human DC-SIGN mAb (120507; R&D Systems) and anti-CIRE/mDC-SIGN (LWC06; eBiosciences, San Diego, CA, USA) and revealed using donkey anti-mouse–HRP antibody (Chemicon International, Boronia, Australia) and anti-rat–HRP (Amersham Life Science), respectively. The membranes were developed with Super Signal West Pico Chemiluminescent Substrate (Pierce, Rockford), according to manufacturer's guidelines.
Results
Surface expression of CIRE/mDC-SIGN on different cell types
We previously expressed FLAG-tagged CIRE/mDC-SIGN on the surface of CHO cells (26), but since the FLAG peptide could theoretically alter the conformation of the lectin, we generated a new CIRE/mDC-SIGN construct that coded only for the native protein. A new mAb (5H10) we raised against CIRE/mDC-SIGN recognised both CIRE/mDC-SIGN and FLAG-CIRE/mDC-SIGN, but not the neo-control transfectant (Fig. 1A). It is theoretically possible that this anti-CIRE/mDC-SIGN mAb cross-reacted with the mouse DC-SIGN siblings (SIGNR1-4), despite the fact that the immunogenic peptide selected was chosen from a region of genetic variability. Since mAb 5H10 reacted with splenocytes (see below) and only mouse SIGNR1 is expressed in the spleen and on macrophages (27, 44–47), we determined whether this mAb cross-reacted with mouse SIGNR1. SIGNR1 expression was detected with an antibody against the antigenic AU1 tag, but not by mAb 5H10, indicating that 5H10 does not recognise mouse SIGNR1 (Fig. 1B).
Fig. 1.
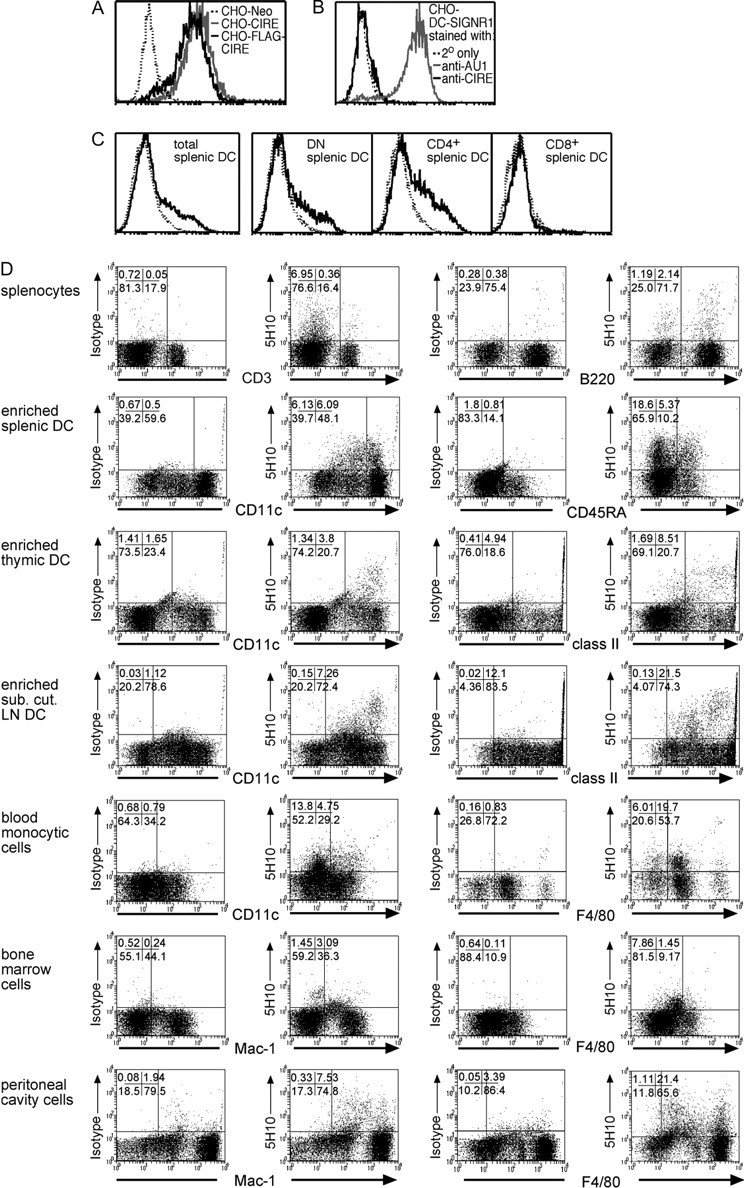
Cytofluorometric analysis of CIRE/mDC-SIGN expression. (A) CHO cells expressing FLAG-CIRE/mDC-SIGN (CHO-FLAG-CIRE), CIRE/mDC-SIGN (CHO-CIRE) or the neomycin resistance gene alone (Neo-CHO) were stained with anti-CIRE/mDC-SIGN mAb and counterstained with goat anti-rat FITC antibody. (B) CHO expressing mouse SIGNR1 (where mouse SIGNR1 was genetically fused to express the AU1 sequence) was stained with 5H10. (C) Splenic DCs from SPF mice were isolated and identified by high forward light scatter and staining with anti-CD11c-Cy5. DCs were further stained with anti-CD4–Alexa and anti-CD8–FITC. Only in the samples depicted in panel C was the staining intensity for CIRE/mDC-SIGN amplified using Flow-Amp. The continuous line represents the anti-CIRE/mDC-SIGN staining on gated cells and the dotted line represents staining of isotype control antibody. Histographs are on a four-decade logarithmic scale. (D) Bone marrow cells, blood monocytes, peritoneal cavity cells, splenocytes, splenic cDCs, splenic pDCs, LNs–DCs and thymic DCs were isolated from germ-free mice. DCs were stained using anti-CD11c–FITC, anti-CD45RA–FITC or anti-class II (M5/114-FITC). Other cell types were stained with anti-F4/80–FITC, anti-CD3 (KT3-1.1-FITC), anti-B220 (RA3-6B2-FITC) and anti-Mac-1 (M1/70-FITC). Cells were stained using anti-CIRE/mDC-SIGN–biotin mAb, then detected in flow cytometry using streptavidin–PE. The CIRE/mDC-SIGN staining seen on transfectants (panel A) and primary cells (panel D) was not amplified using Flow-Amp. Percentages of cells in respective quadrants are displayed in the upper left corner.
Splenic DC subsets were then investigated for surface expression of CIRE/mDC-SIGN. Only a small proportion of total cDCs expressed CIRE/mDC-SIGN. This protein was only found on a fraction of CD4+ and DN DCs, but not on CD8+ DCs (Fig. 1C). Thus, the protein expression of CIRE/mDC-SIGN agreed with the expression profile predicted by the reverse transcription (RT)–PCR analysis (26). Interestingly, CIRE/mDC-SIGN was also expressed on a large proportion of pDCs (33; Fig. 2A).
Fig. 2.

CIRE/mDC-SIGN is down-regulated upon activation and is not up-regulated by IL-4 and IL-13. (A) Isolated splenic cDCs (98% < CD11hiCD45RA−) and pDCs (98% < CD11cint CD45RA+) from SPF mice were purified by cell sorting and cultured in the presence of GM-CSF or GM-CSF and CpG-1668. The continuous line represents the anti-CIRE/mDC-SIGN staining and the dotted line is the background staining of an isotype control antibody. Histographs are on a logarithmic scale. The data are representative of minimum five independent experiments. (B) Freshly isolated DCs (>90% CD11c+) from SPF mice were cultured in the presence of GM-CSF plus various concentrations of IL-4 or IL-13 for 18 h. The level of CIRE expression was determined by flow cytometry. Open bars represent the control staining with an isotype-matched mAb. Data presented are representative of a minimum of four independent experiments.
Next we determined whether CIRE/mDC-SIGN was expressed on other cell types. Bone marrow cells, peritoneal cells, splenocytes, blood monocytes, T and B cells and various organ-derived DCs were assessed for their expression of CIRE/mDC-SIGN. Splenic DCs consistently contained a CIRE/mDC-SIGN+ sub-population. However, unseparated cells derived from bone marrow, blood or lymphoid organs of our standard laboratory mice produced conflicting results, with staining for CIRE/mDC-SIGN varying from undetectable levels up to 25% of the cells analysed (data not shown).
Since we had already shown by RT–PCR that CIRE/mDC-SIGN is down-regulated upon activation (26), we reasoned that the mice might be exposed to undefined pathogens in our holding rooms, and this may have activated their immune system. Thus, germ-free mice were obtained and sacrificed within 12 h of entering the animal holding facilities. Indeed, germ-free mice consistently displayed a significant proportion of CIRE/mDC-SIGN+ cells in the bone marrow, peritoneal cavity, spleen and blood (Fig. 1D). The majority of positive cells were myeloid (monocytes, macrophages) since lymphocytes were almost all negative. While T cells did not express CIRE/mDC-SIGN, a minority of splenocytes (2%), which were CD19+ and presumed to be B cells, expressed this protein (data not shown). Interestingly, the proportion of cDCs and pDCs expressing CIRE/mDC-SIGN differed between lymphoid compartments (Fig. 1D); 10–20% splenic DCs were positive for CIRE/mDC-SIGN, while only 7–9% of lymph node DCs and 2% of thymic DCs were positive for this marker. The reason for this variation remains to be elucidated.
To determine whether CIRE/mDC-SIGN protein was down-regulated upon activation, as predicted by RT–PCR, cDCs and pDCs were isolated and stained before and after overnight incubation with GM-CSF or GM-CSF and CpG. While only a small fraction of freshly isolated cDCs (5–10%) from normal B6 mice were CIRE/mDC-SIGN+, up to 50% of pDCs expressed this marker. Upon activation, the expression of CIRE/mDC-SIGN on both cDCs and pDCs was completely lost (Fig. 2A). Since purified CIRE/mDC-SIGN+ and CIRE/mDC-SIGN− cells had comparable survival rates in cell culture (data not shown), the disappearance of CIRE/mDC-SIGN+ cells reflected down-regulation rather than preferential cell death. It has been reported that incubation with IL-4 (48) and IL-13 (49) increases the expression levels of hDC-SIGN. However, when freshly isolated DCs from SPF mice were incubated with various concentrations of IL-4 or IL-13, there was no up-regulation of CIRE/mDC-SIGN (Fig. 2B). This discrepancy could reflect the different cell types used; we used freshly isolated splenic DCs not monocyte-derived DCs or macrophages or could be due to genuine differences in regulation of gene expression.
Binding of mannose by CIRE/mDC-SIGN
Sequence analysis of CIRE/mDC-SIGN predicts a binding of mannosylated residues similar to hDC-SIGN. To establish whether CIRE/mDC-SIGN had a mannose-binding specificity, CHO cells expressing FLAG-CIRE/mDC-SIGN, CIRE/mDC-SIGN, mouse SIGNR1 or the neomycin resistance gene alone were incubated with mannosylated FITC-conjugated BSA. FLAG-CIRE/mDC-SIGN and CIRE/mDC-SIGN CHO cells clearly bound mannosylated FITC-conjugated BSA (Fig. 3A). Furthermore, this binding was inhibited by mannan (Fig. 3A) in a dose-dependent manner (Fig. 3B), and by EDTA (Fig. 3A and B), showing that CIRE/mDC-SIGN, like hDC-SIGN, bound ligands in a mannose-specific and Ca2+-dependent fashion. However, binding of the mannosylated FITC-conjugated BSA was not inhibited by the mAb 5H10, indicating this mAb does not neutralise the CIRE/mDC-SIGN lectin-binding site (data not shown). Interestingly, while CIRE/mDC-SIGN bounds α-D-mannosylated FITC-conjugated BSA, FITC–ovalbumin peptide containing mainly (Man)nGlcNAcGlcNAc-Asn failed to bind CIRE/mDC-SIGN (50). The reason for this discrepancy is not clear but may reflect difference in the substrate (type or level of mannosylation). Importantly, both human DC-SIGN and mouse SIGNR1 also bind mannosylated FITC-conjugated BSA in a Ca2+-dependent manner (Fig. 3C), indicating that other lectins are capable of binding this substrate.
Fig. 3.
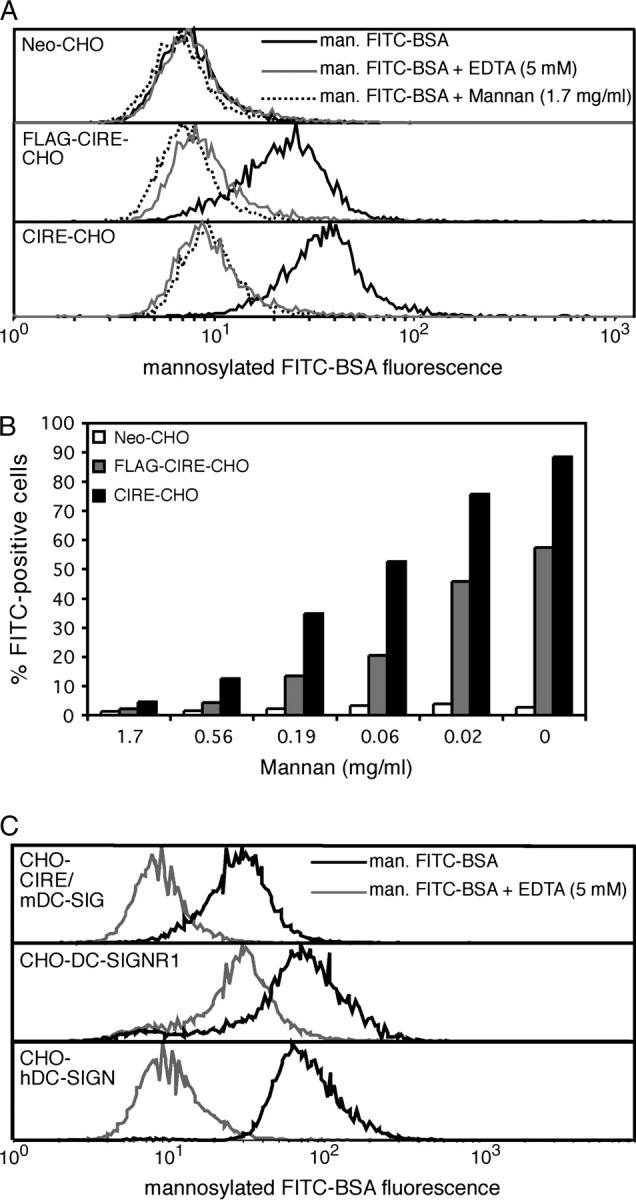
CHO cells expressing CIRE/mDC-SIGN bind mannosylated protein in a calcium-dependent, mannan-inhibitable manner. CHO cells (105) expressing FLAG-CIRE/mDC-SIGN (FLAG-CIRE-CHO), CIRE/mDC-SIGN (CIRE-CHO) or neomycin resistance gene only (Neo-CHO) were re-suspended in various dilutions of mannan in ice-cold medium containing 2% FCS and Ca2+ and Mg2+ or in medium where the Ca2+ and Mg2+ were replaced by 5 mM EDTA. (A) Mannan and EDTA inhibit the binding of mannosylated FITC-conjugated BSA to CIRE/mDC-SIGN. (B) Mannan inhibits the binding of mannosylated FITC-conjugated BSA by CIRE/mDC-SIGN in a dose-dependent manner. Data are representative of a minimum of five independent experiments. (C) Human DC-SIGN and mouse SIGNR1 also bind mannosylated FITC-conjugated BSA in a Ca2+-dependent manner. Cells were then incubated with mannosylated FITC-conjugated BSA and fluorescence levels quantitated by flow cytometry. Data are representative of two independent experiments.
DCs expressing CIRE/mDC-SIGN are not endowed with enhanced stimulatory capacity
Human DC-SIGN has been proposed to facilitate the interaction between T cells and DCs thereby supporting primary immune responses (28). To test whether mouse DCs expressing CIRE/mDC-SIGN could induce a more potent proliferative response than their CIRE/mDC-SIGN− counterparts, DC subsets were purified based on their expression of CIRE/mDC-SIGN and incubated with allogeneic T cells. Since the anti-CIRE/mDC-SIGN mAb does not block binding of mannosylated BSA (data not shown), we assumed that the CIRE/mDC-SIGN molecule is still free to interact with its natural ligand partner. However, the expression of CIRE/mDC-SIGN did not correlate with any enhanced CD8 T cell or CD4 T cell proliferative response (Fig. 4).
Fig. 4.
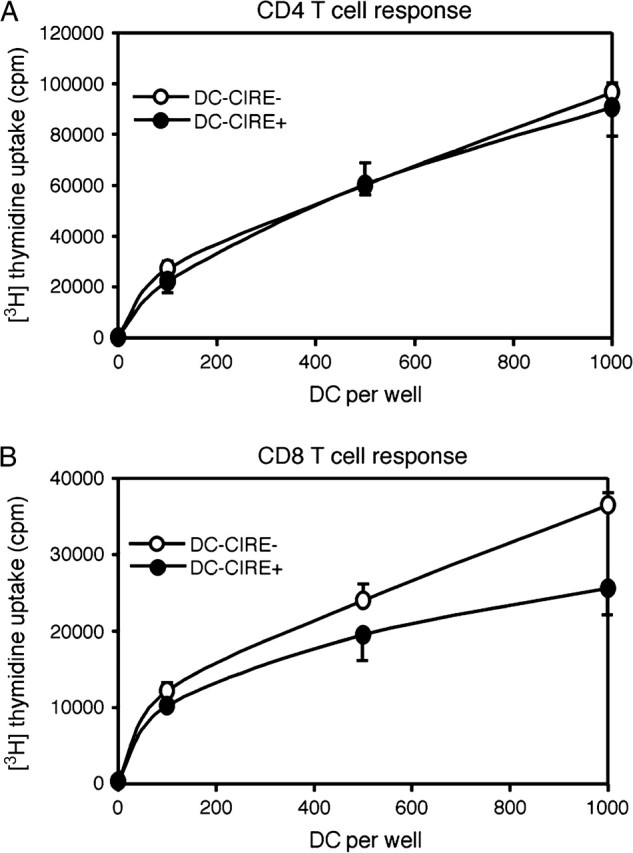
CIRE/mDC-SIGN+ DC does not preferentially activate T cells into proliferation. DCs from SPF mice were isolated and then sorted based on their expression of CD11c+CIRE/mDC-SIGN+ (purity: 75%) and CD11c+CIRE/mDC-SIGN− (purity: 98%). DCs were incubated with 20 000 CD4 or CD8 T cells for 4 and 3 days, respectively, then pulsed with 1 μCi per well of [3H]thymidine for 6 h, harvested onto glass-fibre filters and thymidine incorporation was counted by liquid scintillation. All cultures were performed in quintets and are representative of two independent experiments.
Lack of pathogen binding by CIRE/mDC-SIGN
Human DC-SIGN has been shown to bind many pathogens (31). To determine whether the same pathogens can bind CIRE/mDC-SIGN, CHO cells were transfected with vectors that expressed CIRE/mDC-SIGN, hDC-SIGN or the neomycin resistance cassette. The levels of CIRE/mDC-SIGN and hDC-SIGN on the surface of CHO transfectants were measured by flow cytometry (Fig. 5). While it appears that CHO cells express significantly higher levels of hDC-SIGN than CIRE/mDC-SIGN, it is important to note that a direct comparison cannot be drawn: expression of both proteins was detected upon staining with different primary mAbs (mouse-derived AZN-1 for hDC-SIGN and rat-derived 5H10 for mDC-SIGN/CIRE) and corresponding secondary antibodies. However, it is obvious from the use of identical reagents that the levels of CIRE/mDC-SIGN on the cell surface of the transfected CHO cells were significantly higher than ever seen on the surface of native DC (data not shown and Fig. 1A and C). Surprisingly, CHO cells expressing CIRE/mDC-SIGN did not bind HIV-1, EBOV-GP-bearing pseudotypes, HCMV, MCMV or Leishmania (Fig. 5), indicating that DCs expressing CIRE/mDC-SIGN should also lack this capacity. By contrast, CHO cells expressing hDC-SIGN did bind these pathogens (Fig. 5) in agreement with previous studies (34, 37, 39, 51–53).
Fig. 5.

CIRE/mDC-SIGN does not bind pathogens known to interact with hDC-SIGN. CHO cells were co-transfected with the neomycin resistance gene plus the cDNA coding for CIRE/mDC-SIGN, hDC-SIGN or the chimeric molecules (CIRE/mDC-SIGN lectin fused to hDC-SIGN stalk and hDC-SIGN lectin fused to CIRE/mDC-SIGN stalk). Transfectants were stained with 5H10 (anti-CIRE) or AZN-1 (anti-DC-SIGN) and counterstained with anti-rat–PE and anti-mouse–FITC, respectively. The level of fluorescence was determined by flow cytometry. Filled histograph denotes the background staining of control transfectants, lacking the binding site of the primary mAb. Continuous line represents the levels of CIRE/mDC-SIGN (A) and hDC-SIGN (B). Dotted line represents the levels of CIRE/mDC-SIGN-lectin fused to hDC-SIGN stalk (A) and hDC-SIGN lectin fused to CIRE/mDC-SIGN stalk (B). The above expression profiles of the CHO transfectants are representative of the expression pattern seen in the independently executed experiments seen in Fig. 5(C–F). CHO cells expressing CIRE/mDC-SIGN, hDC-SIGN or the chimeric molecules (CIRE/mDC-SIGN lectin fused to hDC-SIGN stalk and hDC-SIGN lectin fused to CIRE/mDC-SIGN stalk) or the neomycin resistance gene were tested for interaction with various pathogens. (C) Leishmania mexicana promastigotes were added at a multiplicity of infection of 5:1. Infection proceeded for 24 h at 33°C, before free parasites were removed, cells fixed in methanol and stained with Giemsa. The percent infected cells or cells with attached parasites were counted. The mean ± SEM of two independent experiments is shown. (D) Lentiviral pseudotypes bearing the EBOV-GP. The indicated CHO cell lines were inoculated with a luciferase reporter virus bearing EBOV-GP and luciferase activities in cell lysates were assessed 3 days after infection. The average of two independent experiments performed in quadruplicates is shown. Error bars indicate SEM. (E) HIV-1. Lectin-mediated HIV transmission was assessed by incubating transfectants with a HIV-1 NL4-3 variant harbouring the luciferase gene in place of nef, washing with culture medium and co-cultivation with receptor-positive target cells. Luciferase activities in cell lysates were determined 3 days after the start of the co-cultivation. The average ± SEM from two independent experiments carried out in quadruplicates is presented. (F) HCMV. Strain AD169 was added and incubated (1 h at 4°C in PBS/0.1% BSA/1 mM CaCl2/2 mM MgCl2) and percentage of transfectants to which AD169 attached was determined by FACS analysis using an anti-HCMV gB mAb. The data are presented as the mean ± SEM pooled from two independent experiments.
The influence of the stalk on the pathogen-binding properties of CIRE/mDC-SIGN and hDC-SIGN
To further investigate the pathogen-binding differences between CIRE/mDC-SIGN and hDC-SIGN, we focused on a fundamental difference between these two molecules. hDC-SIGN has a long stalk with seven tandem repeats, proposed to form an alpha helix and allow tetramerisation (54), while CIRE/mDC-SIGN has a short stalk with no tandem repeats. To determine the relative importance of the stalk and the lectin domain in facilitating binding, two chimeric molecules were constructed. The first contained the hDC-SIGN lectin domain fused to the stalk and cytosolic section of CIRE/mDC-SIGN, the second contained the CIRE/mDC-SIGN lectin domain fused to the stalk and cytosolic section of hDC-SIGN. hDC-SIGN, CIRE/mDC-SIGN and the chimeras were efficiently expressed at the cell surface (Fig. 5A and B). Again, while direct comparison between the levels of CIRE/mDC-SIGN, hDC-SIGN and the chimeras cannot be drawn, it is clear that the cell-surface levels of CIRE/mDC-SIGN and of the chimera containing the CIRE/mDC-SIGN lectin are significantly higher that seen under physiological conditions on DCs (Fig. 1A versus 1B).
The mannose-capped lipophosphoglycan of L. mexicana has previously been shown to bind hDC-SIGN (53). We therefore tested the various CHO transfectants for ability to bind L. mexicana promastigotes on incubation at a multiplicity of infection of 5:1 for 24 h. This assay only gave a maximum of 15% of cells showing parasite attachment. However, it was clear that the only cells binding the organisms were those expressing the hDC-SIGN lectin domain (i.e. CHO cells expressing hDC-SIGN and those expressing the chimeric molecule containing the lectin domain of hDC-SIGN and the stalk of CIRE/mDC-SIGN) (Fig. 5C). These results indicated that the hDC-SIGN lectin was the only essential domain for this interaction. An additional point of interest was that neither promastigotes nor amastigotes of L. major were able to bind to hDC-SIGN, indicating a potentially important host and parasite specificity for this host–pathogen interaction (data not shown).
Human DC-SIGN has been shown to bind many viruses, including EBOV (39, 51), HIV (52) and HCMV (37). In accordance with this, EBOV-GP-driven infection of CHO cells expressing hDC-SIGN was at least two logs above control cells (Fig. 5D). Pre-incubation with mannan strongly reduced EBOV-GP-driven infection of DC-SIGN-expressing cells but not of control cells, indicating that the enhancement of infection was mannose specific and, thus, indicative of hDC-SIGN engagement (data not shown). In contrast, stable expression of CIRE/mDC-SIGN by CHO cells did not enhance infection and this defect could not be rescued by the introduction of the hDC-SIGN stalk (Fig. 5F). Moreover, the hDC-SIGN chimera containing the CIRE/mDC-SIGN stalk also failed to enhance infection. The results with HIV-1 and HCMV were comparable to those obtained with EBOV-GP-bearing pseudotypes. Thus, HCMV bound to CHO cells expressing hDC-SIGN but no specific binding was observed with cells expressing CIRE/mDC-SIGN or chimeric lectins (Fig. 5F). Similarly, only hDC-SIGN-expressing cells transmitted a replication competent HIV-1 reporter virus to permissive cells with appreciable efficiency and in a mannan-sensitive manner, while transmission by cells positive for CIRE/mDC-SIGN or chimeric lectins was inefficient (Fig. 5E and data not shown). Binding of MCMV to CIRE/mDC-SIGN was not observed after introduction of the hDC-SIGN stalk (data not shown).
To determine whether the stalk of hDC-SIGN did indeed facilitate multimerisation when fused to CIRE/mouse DC-SIGN, or conversely whether the stalk of CIRE/mouse DC-SIGN abolished tetramerisation, we conducted western blot analysis to estimate the sizes of the chimeric proteins expressed by the various transfectants. Proteins in the cell lysate of parental CHO-KI cells that do not express mouse or human DC-SIGN are not detected with the anti-CIRE/mDC-SIGN and anti-human DC-SIGN mAb (Fig. 6; lane 1). DC-SIGN is a 46-kDa protein containing one N-linked glycosylation site, which is utilised (38). In accordance with this, under reducing conditions, the predominant product of DC-SIGN (lane 2) resolved at 46 kDa with a minor product detected at ∼44 kDa (presumed to be non-glycosylated protein). In contrast, under non-reducing conditions, an additional DC-SIGN (lane 2) product was detected at ∼180 kDa, corresponding to the expected tetramer (184 kDa). The chimeric protein of CIRE/mDC-SIGN lectin fused to hDC-SIGN stalk is expected to be 44 kDa (lane 3) and, accordingly, under reducing conditions a band of 44 kDa was detected. Interestingly, under non-reducing conditions, the predominant product runs at ∼180 kDa, the expected size (176 kDa) of the tertramerised protein, and only a minor monomeric product is detected at 44 kDa. The second chimera (hDC-SIGN lectin fused to CIRE/mDC-SIGN stalk) was expected to be 29 kDa and under reducing conditions a band is indeed detected at 29 kDa (lane 4). However, regardless of whether the cell lysates were run under reducing or non-reducing conditions, no multimer was detected, consistent with the notion that the CIRE/mDC-SIGN stalk fails to allow multimerisation. In accordance with this, CIRE/mDC-SIGN (27 kDa) failed to multimerise, since an oligomerisation product of CIRE/mDC-SIGN was not detected under non-reducing or reducing conditions.
Fig. 6.

The chimeric molecule containing the CIRE/mDC-SIGN lectin domain fused to human DC-SIGN stalk appears to form multimers similarly to hDC-SIGN. Cell lysates from parental CHO cells (lane 1) or CHO cells expressing hDC-SIGN (lane 2), or the chimeric molecules [CIRE/mDC-SIGN lectin fused to hDC-SIGN stalk (lane 3) and hDC-SIGN lectin fused to CIRE/mDC-SIGN stalk (lane 4)] and CIRE/mDC-SIGN (lane 5) were separated on SDS-PAGE (10%) under reducing and non-reducing conditions, probed with mouse anti-human DC-SIGN mAb and anti-CIRE/mDC-SIGN and then revealed using anti-rat–HRP and anti-mouse–HRP antibody.
Thus, the stalk of hDC-SIGN does not endow CIRE/mDC-SIGN with the capacity to bind any of the tested viruses, arguing that the mouse lectin has a different binding specificity compared with hDC-SIGN. Furthermore, replacing the stalk of hDC-SIGN with that of CIRE/mDC-SIGN abolished binding to three of the four pathogens analysed, suggesting that hDC-SIGN does indeed require the oligomerisation and spatial orientation facilitated by its long stalk in order to functionally interact with many ligands.
Discussion
The development of a mAb against CIRE/mDC-SIGN has enabled us to study CIRE/mDC-SIGN expression at the protein level. Our initial attempt to determine CIRE/mDC-SIGN expression on various leukocytes harvested from mice produced variable results. However, cells from germ-free mice with restricted exposure to conventional mouse holding room conditions showed higher and more reproducible CIRE/mDC-SIGN expression. This suggests that CIRE/mDC-SIGN expression is exquisitely sensitive to activation or mild inflammation induced by microbial exposure. These results are in keeping with our experimental data showing that activation of cDCs or pDCs results in complete down-regulation of CIRE/mDC-SIGN expression (26).
Using the CIRE/mDC-SIGN mAb, we confirmed at the protein level that among cDCs, CIRE/mDC-SIGN is expressed by CD4+, DN DCs but not CD8+ DCs (26). CIRE/mDC-SIGN is also strongly expressed by a proportion of splenic pDCs (33), macrophages and monocytes. There is little expression of CIRE/mDC-SIGN on lymphocytes, except perhaps on a very minor subset of B cells.
It has been proposed that mouse CIRE/mDC-SIGN is the orthologue of hDC-SIGN. Complicating this proposition is the fact that in the mouse there are at least five closely related C-type lectins (CIRE/mDC-SIGN, mSIGNR1—4), all sharing comparable amino acid identity with the two human C-type lectins, hDC-SIGN and hDC-SIGNR (26, 27). However, since like human DC-SIGN, CIRE/mDC-SIGN is the only mouse DC-SIGN gene that is closely juxtaposed to the CD23 gene, and is the only mouse lectin expressed in DCs, it was concluded to be the hDC-SIGN orthologue. In this report, using a mAb against mouse CIRE/mDC-SIGN, we show that its expression pattern has some similarity to that of hDC-SIGN. Mouse CIRE/mDC-SIGN is expressed on freshly isolated splenic, thymic and lymph node DCs; unfortunately, these tissue sources have not been used for isolation of human DCs. However, CIRE/mDC-SIGN can be detected on monocyte-derived DCs (data not shown) and macrophages and hDC-SIGN is expressed on monocyte-derived DCs, monocyte-derived Langerhans cells, CD1alo dermal DCs and macrophage subsets (28, 49, 55–57). In the mouse, CIRE/mDC-SIGN is also on the cell surface of spleen pDCs; notably, there are reports of its expression on human pDCs, but the data are conflicting (49, 55). Interestingly, mouse blood monocytes express CIRE/mDC-SIGN, whereas human monocytes do not express DC-SIGN (28, 55, 56). There maybe some parallels between the down-regulation of CIRE/mDC-SIGN on freshly isolated mouse DCs when activated with CpG (Fig. 2) LPS, IFN-γ and poly-IC (data not shown) and the reduction observed when human monocyte-derived DCs are ‘matured’ with factors such as LPS, prostaglandin E2 and tumour necrosis factor-α (28, 55). However, there are clearly differences in gene regulation in response to IL-4 and IL-13. Freshly isolated mouse DCs do not up-regulate CIRE/mDC-SIGN in the presence of IL-4 and IL-13, whereas hDC-SIGN, on monocyte-derived DCs and macrophages, is up-regulated in the presence of IL-4 and IL-13 (48, 49). It remains to be seen whether this is a difference between the different types of DCs studied, or whether it is a species difference between mouse and man.
The availability of a CIRE/mDC-SIGN mAb enabled us to sort both transfected and ex vivo-purified CIRE/mDC-SIGN-expressing cells, and to begin addressing the question of CIRE/mDC-SIGN function. The key issue to be addressed was whether CIRE/mDC-SIGN is indeed a functional orthologue of hDC-SIGN. Human DC-SIGN binds ICAM-3 and has been proposed to help initiate interactions between DCs and T cells (28), though this observation has been challenged recently (57). In an allogeneic mixed leukocyte reaction, both CIRE/mDC-SIGN+ DCs and CIRE/mDC-SIGN− DCs induced comparable levels of T cell proliferation, arguing that this lectin in the mouse plays, at best, only a minor role in T cell activation and aligns our finding with that of Granelli-Piperno et al. (57). Another role proposed for hDC-SIGN is antigen capture and presentation (30, 31). A similar function has been proposed for CIRE/mDC-SIGN (50) and more directly shown for the homologue mSIGNR1, a C-type lectin expressed in marginal zone macrophages, which takes up blood-borne polysaccharide antigens (44–47). Our data showing that CIRE/mDC-SIGN binds mannosylated FITC–BSA in a calcium-dependent manner (Fig. 3) demonstrates that this molecule is also a C-type lectin with mannose-binding specificity. However, despite the common mannose-binding function, and despite the fact that CIRE/mDC-SIGN transfectants expressed significantly higher levels of CIRE/mDC-SIGN than physiologically found on mouse DCs, they did not interact appreciably with HIV-1, virion-associated EBOV-GP, HCMV or L. mexicana, pathogens that have been documented to interact with hDC-SIGN (34, 37, 39, 51–53).
In order to reconcile this discrepancy, we focused on the region that differs most significantly between the human and mouse molecule, namely the stalk region that supports the carbohydrate recognition domain (CRD). In hDC-SIGN, the stalk has seven and a half tandem repeats that give rise to an alpha helix, which is involved in the tetramerisation of the CRD (54). The oligomerisation caused by the stalk has two functions. The first function is to create a unique spatial arrangement between the individual CRDs and thus contribute to their ligand specificity. In the case of hDC-SIGN, it has been shown that this lectin specifically interacts with the trimannose motif in high mannose oligosaccharides and that these carbohydrates need to be spaced some distance apart to interact with multiple CRDs (53, 54, 58–60). This differs from the mannose-binding receptor, which binds terminal mannose residues (54). The second function of oligomerisation is to facilitate multivalent interaction and thus enhance avidity (54, 61). In the mouse, CIRE/mDC-SIGN does not have the tandem repeats or motifs that allow tetramer formation and we reasoned that this deficiency could contribute to its inability to bind the pathogens that hDC-SIGN bound. However, when the N-terminus of hDC-SIGN was fused to the mouse CIRE/mDC-SIGN CRD, the chimeric molecule failed to bind the pathogens tested despite having the capacity to multimerise (Fig. 6), arguing that the mouse CRD, even when endowed with the oligomerisation domain, is unable to recognise the same ligands as hDC-SIGN. When the hDC-SIGN CRD was fused to the CIRE/mDC-SIGN stalk, and thus lost its capacity to oligomerise (Fig. 6), it could no longer bind HIV-1, HCMV or EBOV-GP-bearing pseudotypes, suggesting that multivalent engagement is indeed required for binding. Indeed, the requirement for multimerisation of hDC-SIGN for the binding of HIV-1 envelope protein has been previously reported (62). Thus, for the binding of viruses, CIRE/mDC-SIGN differs from its human orthologue with respect to the function of the both CRD and the stalk region. Interestingly, the chimeric molecule containing the human DC-SIGN lectin fused to the CIRE/mDC-SIGN stalk, could still bind L. mexicana promastigotes, indicating that this interaction is independent of the tetramerisation of hDC-SIGN.
To conclude, it is clear that CIRE/mDC-SIGN and hDC-SIGN are highly related molecules. They share highly significant identity at the amino acid level, and both CTLDs do have the capacity to bind mannosylated molecules. Their pattern of expression is similar with both molecules expressed predominantly on DCs and other myeloid cells. Moreover, their evolutionary relatedness is evidenced by their chromosomal location, with both genes being tightly linked to CD23, in the mouse and human genomes. However, we consistently failed to demonstrate binding of mouse CIRE/mDC-SIGN to any of the hDC-SIGN ligands tested. Supporting our observation, CIRE/mDC-SIGN failed to bind Candida albicans and Escherichia coli (50), whereas hDC-SIGN has been shown to bind these pathogens (63, 64). Furthermore, even among the closely related CIRE/mDC-SIGN and mouse SIGNR1-4 lectins, there are distinct recognition patterns for microbes despite the conserved EPN (mannose recognition) motif (50). Thus, the CTLDs of hDC-SIGN and CIRE/mDC-SIGN, although endowed with similar carbohydrate specificities, differ in their abilities to interact with pathogens, suggesting that the fine specificity of the CTLDs of hDC-SIGN and CIRE/mDC-SIGN are significantly different and that they may not be simple functional orthologues. The question whether there are other yet to be defined, functional similarities between these two molecules will only be resolved by the identification of CIRE/mDC-SIGN-specific ligands.
Acknowledgments
We thank N. Landau for CEMx174 5.25 M7 cells and S. Becker for the EBOV-GP expression plasmid. This work was supported by the Australian National Health and Medical Research Council and the Australia Government's Cooperative Research Centres Program. T.G. and S.P. were supported by the Deutsche Forschungsgemeinschaft, SFB466.
Abbreviations
- cDCs
conventional dendritic cell
- CRD
carbohydrate recognition domain
- CTLD
C-type lectin domain
- DC
dendritic cell
- DN
double negative
- EBOV-GP
Ebolavirus glycoprotein
- GM-CSF
granulocyte macrophage colony-stimulating factor
- HCMV
human cytomegalovirus
- ICAM
intercellular adhesion molecule
- i.p.
intra-peritoneally
- KLH
keyhole limpet haemocyanin
- MCMV
mouse cytomegalovirus
- pDC
plasmacytoid pre-dendritic cell
- RT
reverse transcription
- SPF
specific pathogen free
- WEHI
Walter and Eliza Hall Institute
References
- 1. Steinman R. M. 1991. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9:271. [DOI] [PubMed] [Google Scholar]
- 2. Shortman K. and Liu Y. J. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151. [DOI] [PubMed] [Google Scholar]
- 3. Vremec D., Pooley J., Hochrein H., Wu L., and Shortman K. 2000. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164:2978. [DOI] [PubMed] [Google Scholar]
- 4. Shortman K. and Wu L. 2001. Parentage and heritage of dendritic cells. Blood 97:3325. [DOI] [PubMed] [Google Scholar]
- 5. Traver D., Akashi K., Manz M. et al.2000. Development of CD8alpha-positive dendritic cells from a common myeloid progenitor. Science 290:2152. [DOI] [PubMed] [Google Scholar]
- 6. Martin P., del Hoyo G. M., Anjuere F. et al.2000. Concept of lymphoid versus myeloid dendritic cell lineages revisited: both CD8alpha(−) and CD8alpha(+) dendritic cells are generated from CD4(low) lymphoid-committed precursors. Blood 96:2511. [PubMed] [Google Scholar]
- 7. Kronin V., Winkel K., Suss G. et al.1996. A subclass of dendritic cells regulates the response of naive CD8 T cells by limiting their IL-2 production. J. Immunol. 157:3819. [PubMed] [Google Scholar]
- 8. Suss G. and Shortman K. 1996. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 183:1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fazekas de St Groth B. 1998. The evolution of self-tolerance: a new cell arises to meet the challenge of self-reactivity. Immunol. Today 19:448. [DOI] [PubMed] [Google Scholar]
- 10. Ruedl C. and Bachmann M. F. 1999. CTL priming by CD8(+) and CD8(−) dendritic cells in vivo. Eur. J. Immunol. 29:3762. [DOI] [PubMed] [Google Scholar]
- 11. Schlecht G., Leclerc C. and Dadaglio G. 2001. Induction of CTL and nonpolarized Th cell responses by CD8alpha(+) and CD8alpha(−) dendritic cells. J. Immunol. 167:4215. [DOI] [PubMed] [Google Scholar]
- 12. Maldonada-Lopez R., De Smedt T., Pajak B. et al.1999. Role of CD8alpha+ and CD8alpha− dendritic cells in the induction of primary immune responses in vivo. J. Leukoc. Biol. 66:242. [DOI] [PubMed] [Google Scholar]
- 13. Pulendran B., Smith J. L., Caspary G. et al.1999. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl Acad. Sci. 96:1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hochrein H., Shortman K., Vremec D., Scott B., Hertzog P. and O'Keeffe M. 2001. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J. Immunol. 166:5448. [DOI] [PubMed] [Google Scholar]
- 15. Pulendran B., Lingappa J., Kennedy M. K. et al.1997. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J. Immunol. 159:2222. [PubMed] [Google Scholar]
- 16. Iyoda T., Shimoyama S., Liu K. et al.2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pooley J., Heath W. R. and Shortman K. 2001. Intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells but cross-presented to CD8+ T cells by CD8+ dendritic cells. J. Immunol. 166:5327. [DOI] [PubMed] [Google Scholar]
- 18. den Haan J. M., Lehar S. M. and Bevan M. J. 2000. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allan R. S., Smith C. M., Belz G. T. et al.2003. Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science 301:1925. [DOI] [PubMed] [Google Scholar]
- 20. Steinman R. M., Pack M. and Inaba K. 1997. Dendritic cells in the T-cell areas of lymphoid organs. Immunol. Rev. 156:25. [DOI] [PubMed] [Google Scholar]
- 21. Reis e Sousa C., Hieny S., Scharton-Kersten T. et al.1997. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 186:1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caminschi I., Lucas K. M., O'Keeffe M. A. et al.2001. Molecular cloning of F4/80-like-receptor, a seven-span membrane protein expressed differentially by dendritic cell and monocyte-macrophage subpopulations. J. Immunol. 167:3570. [DOI] [PubMed] [Google Scholar]
- 23. Edwards A. D., Chaussabel D., Tomlinson S., Schulz O., Sher A. and Reis E. S. C. 2003. Relationships among murine CD11c(high) dendritic cell subsets as revealed by baseline gene expression patterns. J. Immunol. 171:47. [DOI] [PubMed] [Google Scholar]
- 24. Vremec D. and Shortman K. 1997. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159:565. [PubMed] [Google Scholar]
- 25. McLellan A. D., Kapp M., Eggert A. et al.2002. Anatomic location and T-cell stimulatory functions of mouse dendritic cell subsets defined by CD4 and CD8 expression. Blood 99:2084. [DOI] [PubMed] [Google Scholar]
- 26. Caminschi I., Lucas K. M., O'Keeffe M. A. et al.2001. Molecular cloning of a C-type lectin superfamily protein differentially expressed by CD8alpha(−) splenic dendritic cells. Mol. Immunol. 38:365. [DOI] [PubMed] [Google Scholar]
- 27. Park C. G., Takahara K., Umemoto E. et al.2001. Five mouse homologues of the human dendritic cell C-type lectin, DC-SIGN. Int. Immunol. 13:1283. [DOI] [PubMed] [Google Scholar]
- 28. Geijtenbeek T. B., Torensma R., van Vliet S. J. et al.2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 100:575. [DOI] [PubMed] [Google Scholar]
- 29. Geijtenbeek T. B., Krooshoop D. J., Bleijs D. A. et al.2000. DC-SIGN-ICAM-2 interaction mediates dendritic cell trafficking. Nat. Immunol. 1:353. [DOI] [PubMed] [Google Scholar]
- 30. Engering A., Geijtenbeek T. B., van Vliet S. J. et al.2002. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J. Immunol. 168:2118. [DOI] [PubMed] [Google Scholar]
- 31. van Kooyk Y. and Geijtenbeek T. B. 2003. DC-SIGN: escape mechanism for pathogens. Nat. Rev. Immunol. 3:697. [DOI] [PubMed] [Google Scholar]
- 32. Soilleux E. J., Barten R. and Trowsdale J. 2000. DC-SIGN; a related gene, DC-SIGNR; and CD23 form a cluster on 19p13. J. Immunol. 165:2937. [DOI] [PubMed] [Google Scholar]
- 33. O'Keeffe M., Hochrein H., Vremec D. et al.2002. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J. Exp. Med. 196:1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Curtis B. M., Scharnowske S. and Watson A. J. 1992. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl Acad. Sci. USA 89:8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marzi A., Gramberg T., Simmons G. et al.2004. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 78:12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glaser T. A., Wells S. J., Spithill T. W., Pettit J. M., Humphris D. C. and Mukada A. J. 1990. Leishmania major and L. donovani: a method for rapid purification of amastigotes. Exp. Parasitol. 71:343. [DOI] [PubMed] [Google Scholar]
- 37. Halary F., Amara A., Lortat-Jacob H. et al.2002. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity 17:653. [DOI] [PubMed] [Google Scholar]
- 38. Pohlmann S., Baribaud F., Lee B. et al.2001. DC-SIGN interactions with human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus. J. Virol. 75:4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simmons G., Reeves J. D., Grogan C. C. et al.2003. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305:115. [DOI] [PubMed] [Google Scholar]
- 40. Connor R. I., Chen B. K., Choe S. and Landau N. R. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935. [DOI] [PubMed] [Google Scholar]
- 41. Wool-Lewis R. J. and Bates P. 1998. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J. Virol. 72:3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Doherty U., Swiggard W. J. and Malim M. H. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74:10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hsu M., Harouse J. M., Gettie A., Buckner C., Blanchard J. and Cheng-Mayer C. 2003. Increased mucosal transmission but not enhanced pathogenicity of the CCR5-tropic, simian AIDS-inducing simian/human immunodeficiency virus SHIV(SF162P3) maps to envelope gp120. J. Virol. 77:989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baribaud F., Pohlmann S., Sparwasser T. et al.2001. Functional and antigenic characterization of human, rhesus macaque, pigtailed macaque, and murine DC-SIGN. J. Virol. 75:10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Geijtenbeek T. B., Groot P. C., Nolte M. A. et al.2002. Marginal zone macrophages express a murine homologue of DC-SIGN that captures blood-borne antigens in vivo. Blood 100:2908. [DOI] [PubMed] [Google Scholar]
- 46. Kang Y. S., Kim J. Y., Bruening S. A. et al.2004. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc. Natl Acad. Sci. USA 101:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang Y. S., Yamazaki S., Iyoda T. et al.2003. SIGN-R1, a novel C-type lectin expressed by marginal zone macrophages in spleen, mediates uptake of the polysaccharide dextran. Int. Immunol. 15:177. [DOI] [PubMed] [Google Scholar]
- 48. Relloso M., Puig-Kroger A., Pello O. M. et al.2002. DC-SIGN (CD209) expression is IL-4 dependent and is negatively regulated by IFN, TGF-beta, and anti-inflammatory agents. J. Immunol. 168:2634. [DOI] [PubMed] [Google Scholar]
- 49. Soilleux E. J., Morris L. S., Leslie G. et al.2002. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J. Leukoc. Biol. 71:445. [PubMed] [Google Scholar]
- 50. Takahara K., Yashima Y., Omatsu Y. et al.2004. Functional comparison of the mouse DC-SIGN, SIGNR1, SIGNR3 and Langerin, C-type lectins. Int. Immunol. 16:819. [DOI] [PubMed] [Google Scholar]
- 51. Alvarez C. P., Lasala F., Carrillo J., Muniz O., Corbi A. L. and Delgado R. 2002. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76:6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Geijtenbeek T. B., Kwon D. S., Torensma R. et al.2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587. [DOI] [PubMed] [Google Scholar]
- 53. Appelmelk B. J., van Die I., van Vliet S. J. et al.2003. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J. Immunol. 170:1635. [DOI] [PubMed] [Google Scholar]
- 54. Mitchell D. A., Fadden A. J. and Drickamer K. 2001. A novel mechanism of carbohydrate recognition by the C-type lectins DC-SIGN and DC-SIGNR. Subunit organization and binding to multivalent ligands. J. Biol. Chem. 276:28939. [DOI] [PubMed] [Google Scholar]
- 55. Turville S. G., Cameron P. U., Handley A. et al.2002. Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 3:975. [DOI] [PubMed] [Google Scholar]
- 56. Turville S. G., Arthos J., Donald K. M. et al.2001. HIV gp120 receptors on human dendritic cells. Blood 98:2482. [DOI] [PubMed] [Google Scholar]
- 57. Granelli-Piperno A., Pritsker A., Pack M. et al.2005. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin/CD209 is abundant on macrophages in the normal human lymph node and is not required for dendritic cell stimulation of the mixed leukocyte reaction. J. Immunol. 175:4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin G., Simmons G., Pohlmann S. et al.2003. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J. Virol. 77:1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feinberg H., Mitchell D. A., Drickamer K. and Weis W. I. 2001. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 294:2163. [DOI] [PubMed] [Google Scholar]
- 60. Guo Y., Feinberg H., Conroy E. et al.2004. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 11:591. [DOI] [PubMed] [Google Scholar]
- 61. Weis W. I., Taylor M. E. and Drickamer K. 1998. The C-type lectin superfamily in the immune system. Immunol. Rev. 163:19. [DOI] [PubMed] [Google Scholar]
- 62. Bernhard O. K., Lai J., Wilkinson J., Sheil M. M. and Cunningham A. L. 2004. Proteomic analysis of DC-SIGN on dendritic cells detects tetramers required for ligand binding but no association with CD4. J. Biol. Chem. 279:51828. [DOI] [PubMed] [Google Scholar]
- 63. Cambi A., Gijzen K., de Vries J. M. et al.2003. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 33:532. [DOI] [PubMed] [Google Scholar]
- 64. Klena J., Zhang P., Schwartz O., Hull S. and Chen T. 2005. The core lipopolysaccharide of Escherichia coli is a ligand for the dendritic-cell-specific intercellular adhesion molecule nonintegrin CD209 receptor. J. Bacteriol. 187:1710. [DOI] [PMC free article] [PubMed] [Google Scholar]