

Using phage-display to discover and isolate therapeutic antibodies
Keywords: phage display, therapeutic antibodies
Abstract
Phage display involves the expression of selected proteins on the surface of filamentous phage through fusion with phage coat protein, with the genetic sequence packaged within, linking phenotype to genotype selection. When combined with antibody libraries, phage display allows for rapid in vitro selection of antigen-specific antibodies and recovery of their corresponding coding sequence. Large non-immune and synthetic human libraries have been constructed as well as smaller immune libraries based on capturing a single individual’s immune repertoire. This completely in vitro process allows for isolation of antibodies against poorly immunogenic targets as well as those that cannot be obtained by animal immunization, thus further expanding the utility of the approach. Phage antibody display represents the first developed methodology for high throughput screening for human therapeutic antibody candidates. Recently, other methods have been developed for generation of fully human therapeutic antibodies, such as single B-cell screening, next-generation genome sequencing and transgenic mice with human germline B-cell genes. While each of these have their particular advantages, phage display has remained a key methodology for human antibody discovery due its in vitro process. Here, we review the continuing role of this technique alongside other developing technologies for therapeutic antibody discovery.
Introduction
Antibodies are one of primary effector molecules of the human immune system and bind their target antigen through their complementarity-determining regions (CDRs) displayed on the Fab (antigen-binding fragment) portion of the antibody (Fig. 1). The CDRs encode unique structural diversity and provide antibodies with the ability to recognize a wide variety of targets. The protective efficacy of an antibody can be achieved through binding alone if it results in neutralization of a function of the target or may require additional immune functions provided by the Fc (crystallizable fragment) portion of the antibody molecule with receptors found on immune cells or complement proteins in serum. Antibody Fc region functionality can trigger or augment a number of different host defence mechanisms including phagocytosis, release of cytotoxic or immunomodulatory biomolecules and the complement cascade and formation of the membrane attack complex. These properties potentially make antibodies extremely potent molecules for therapy and they have been developed into an important class of drugs for the treatment of numerous cancers, autoimmune and infectious diseases (1).
Fig. 1.
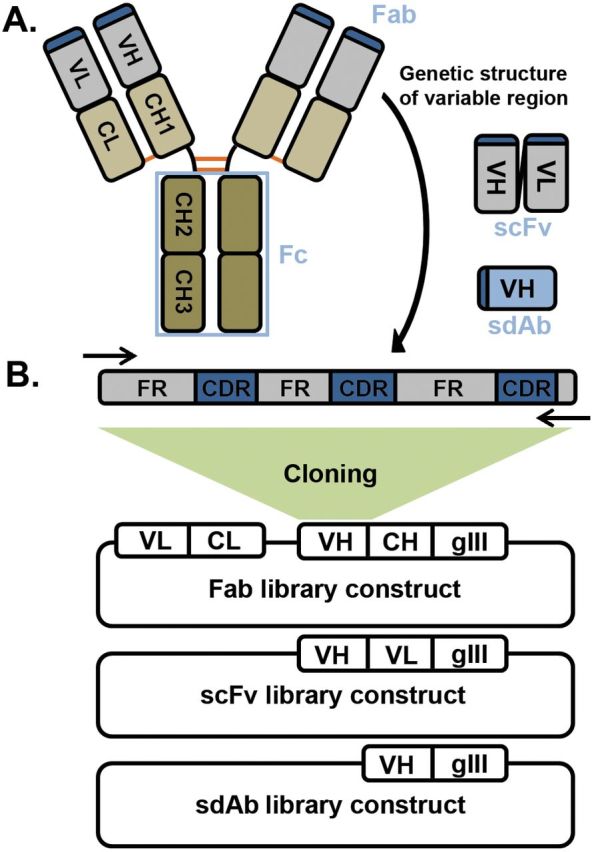
(A) Structure of a typical IgG molecule. Each antibody comprises two heavy and two light chains each of which have four and two immunoglobulin domains, respectively. The first domain is variable and determines specificity (VL and VH) while the second domain of the light chain (CL) and the second to fourth domains of the heavy (CH1-3) are constant across all antibodies of the same isotype. The light chain and first two domains of the heavy chain form the Fab, which is the portion expressed on the phage. The last two domains of the heavy chain form the Fc and are responsible for immune function through engagement of receptors on immune cells. Heavy and light chains are linked through a single disulfide bond (orange) between the CL and CH1 domains and the two heavy chains have multiple disulfide bonds at the hinge region between the CH1 and CH2. An scFv consists of just variable light and variable heavy domains joined by a flexible polypeptide linker while an sdAb as the name implies is only a single immunoglobulin (usually VH) domain which is sufficient for binding. (B) Variable domain genetic structure and construction of a natural phage display library. Each variable domain consists of three hypervariable CDRs interspersed between the more conserved FRs. The immunoglobulin domain folds such that the CDRs are brought together to form the antigen-binding surface at the tip of the Fab. Degenerate primers (arrows) are used to amplify the entire variable heavy and light chains (or alternatively variable and first constant domain) from a source of B cells and cloned in-frame with the phage coat protein (usually gene III) into E. coli to produce an Fab, scFv or sdAb library. The rest of the phage genome is supplied through replication defective helper phage to produce antibody-displaying phage.
Display of properly folded proteins on phage through fusion with phage coat proteins was initially conceived as a means to identify the sequence of the protein target which binds to an antibody (2). Ironically, one of the most common uses of phage display today, almost 30 years after its initially development, is to discover antibodies against selected protein targets. Originally developed by Greg Winter et al. at the MRC Laboratory of Molecular Biology and Richard Lerner, Carlos Barbas et al. at Scripps Research institute, the basic concept that a vast antibody repertoire could be displayed on phage with their genetic sequences packaged within has allowed for the robust screening of a variety of target antigens and rapid recovery of specific antibody sequences for recombinant expression (3–5).
Today, a wide variety of antibody phage display libraries exist, with the antibodies displayed either as Fabs, scFvs (single-chain variable fragments) or sdAbs (single-domain antibodies) in fusion with typically the gene III coat protein (6,7) (Fig. 1). The small size and solubility of the phage particle (up to 1013 particles/ml) has allowed repertoire sizes of up to 1011–12 to be efficiently displayed and manipulated, which improves the likelihood of finding suitable antibodies (8). These antibody sequences can be obtained from natural sources (animal or human) or synthetically constructed [usually a mix of pre-defined framework regions (FRs) with randomized sequences in the CDRs] and isolation of specific sequences is carried out by repeated enrichment against the target of interest.
The defining attribute of all phage display libraries is the physical linking of antibody phenotype (specificity and affinity) with genotype (sequence) via the phage particle—this allows for in vitro selection on immobilized antigen or whole cells (Fig. 2). As this method by-passes the immune system, the usual factors that influence antibody development and limit diversity based on those that can be isolated in animals are not applicable. As such, antibodies can be isolated against targets that have low immunogenicity in animals (non-proteinaceous antigens), to regions of a protein not usually targeted due to the presence of immunodominant epitopes elsewhere, or to hydrophobic targets that are challenging for in vivo manipulation (9–11). As such, phage display offers high potential for discovery of novel therapeutic antibodies that cannot be generated naturally through in vivo immunization or infection. Furthermore, the ability to display synthetic repertoires devoid of any negative selection has made this technique a key method for the generation of potent fully human antibody therapeutics against self-antigens (12). Nixon et al. provide an in-depth review of the clinical development of such drugs (13).
Fig. 2.

General strategy for phage panning. Polyclonal phage expressing recombinant antibodies on their surface is applied to target antigen presented as either immobilized on a magnetic bead, polystyrene surface or on the surface of a whole cell. Phage carrying antigen-binding Fab bind and non-specific Fab are removed through stringent washing. Antigen-bound phage is eluted off either typically through pH change or protease digestion and re-infected into E. coli, from which a new library enriched for antigen-binding clones can be made. After several cycles, the library would be sufficiently enriched so that the individual clones can be isolated from E. coli stock, expressed as monoclonal phage, tested, sequenced and the specific antibodies expressed recombinantly.
Another key attribute of phage display is that selection can be far more rapid as compared to the natural immune response, and in our laboratory, we have been able to identify monoclonal antibodies (mAbs) in as short a period as 1 week (14). This is particularly useful for generation of antibodies against novel or genetically modified pathogens in an emergency outbreak scenario. In recent years, however, alternative antibody discovery systems have been developed, such as high-throughput testing of individual B cells and next-generation sequencing of B-cell repertoires. We review here the use of phage display against various types of targets, such as self-antigens, non-protein targets and infectious disease agents, in relation to on-going developments in both phage display and other antibody discovery technologies.
Targeting self-antigens: going beyond the boundaries of the immune system
In the human immune system, naive B cells targeting self-antigens are deleted by negative selection and immunization in a different host is therefore required to generate specific antibodies against human targets. However, the resultant antibodies, even after humanization by replacement of constant regions or of FRs with human equivalents, are usually more immunogenic than a human-derived antibody (15). While this can result in increased toxicity, the primary concern is the potential for reduced clinical efficacy of the therapy which can occur with development of a humoral immune response against the antibody.
For self-antigens, such as tumour cell markers, phage display is an ideal technique for the generation of fully human antibodies due to the absence of tolerance mechanisms. Semi-synthetic or synthetic libraries, whose sequences have not been negatively selected for any antigen, are usually the first choice. An additional advantage of these libraries is that design features can be incorporated to allow facile affinity maturation, such as CDRs flanked by restriction sites for easy replacement or appropriate consensus primers for targeting of specific CDRs for directed mutagenesis (16). Several fully human synthetic library-derived mAbs have been approved by the US Food and Drugs Administration or completed clinical trials. The first of these was adalimumab, an anti-TNF-α antibody developed by Cambridge Antibody Technologies for the treatment of rheumatic arthritis and Crohn’s disease (17,18). Subsequently, other antibodies were developed for the treatment of cancer and autoimmune disease such as necitumumab (non-small cell lung cancer), ramucirumab (gastrointestinal cancers) and belimumab (systemic lupus erythematosus) (12,19,20). Many others are currently at advanced stages of clinical development and may reach market approval within the next few years (21,22). As mAbs gain popularity as a form of alternative drug therapy, the use of phage displayed antibody libraries has also gained a reputation as the premier technology for antibody discovery.
Semi-synthetic libraries, which were the first to be made, typically consist of FRs sourced from naturally occurring antibodies and CDR regions synthesized from degenerate oligonucleotides (23,24). In many instances, to simplify construction, only the CDR3 region was fully synthetic. Subsequently, libraries with fully synthetic variable sequences were introduced, with the FR based on consensus sequences from natural antibodies but optimized for high expression on phage in Escherichia coli to reduce the effect of variable expression of the human framework sequences on selection. The first fully synthetic Human Combinatorial Antibody Libraries (HuCAL) generated to produce therapeutic antibodies was reported by Morphosys in 2000 with their first version of the HuCAL (16). Several versions of the HuCAL has since been constructed that incorporate additional features such as improved phage elution by disulphide bond cleavage, selection for in-frame clones, additional CDR3 loop length and diversity and optimization for expression in mammalian cell culture through removal of unwanted potential post-translational modification sites (25,26). These modifications have resulted in an increase in the number of antibodies isolated per target antigen. There are several antibodies generated from the Morphosys libraries currently in various stages of clinical development; a recent example being the potent anti-cancer activity of OMP-18R5, with activity in multiple preclinical human tumour models (27). This antibody targeting the Wnt pathway was isolated from the HuCAL GOLD library and is currently in Phase 1 clinical testing.
While synthetic libraries have allowed the development of human antibodies with reduced immunogenicity relative to humanized animal antibodies against self-antigens, the CDRs of these libraries can also be immunogenic as often the sequences are not naturally present in the human antibody repertoire (28). With this in mind, another approach to build synthetic libraries has been to generate novel variable region sequences by overlapping PCR of individual amplified germline CDRs with respective FR (29). While allowing generation of novel sequences that target self-antigens, it also prevents the generation of synthetic sequences that could contain high levels of T-cell epitopes and be strongly immunogenic. Indeed, antibodies obtained from this library had a proportion of T-cell epitopes no higher than that of naturally occurring antibodies. An antibody against ICAM-1 was isolated from this library and shown to have potent anti-myeloma activity in vivo (30).
Over the years, semi-synthetic and fully synthetic libraries have shown utility beyond targeting of self-antigens, as these libraries are also capable of generating antibodies of high affinity against a wide range of target antigens (16,24,25). One such high-affinity antibody which was recently approved for clinical use is raxibacumab for the treatment of inhalational anthrax (12). Extremely high diversity synthetic libraries have also been generated with the goal of providing higher affinity antibodies than currently approved therapeutic antibodies, thus circumventing the need for further improvement by affinity maturation. Hoet et al. at Dyax Corp reported the isolation of very high-affinity antibodies through synthetic diversity introduced into the key antigen-contact sites of CDR1 and CDR2 within the heavy chain in combination with naturally occurring diversity in the heavy chain CDR3 and light chains from donors with autoimmune diseases (31). Their human antibody phage display libraries have produced multiple drug candidates currently in different phases of clinical development.
Non-protein targets: carbohydrates, lipids and post-translationally modified proteins
While antibodies are typically raised against proteins, the first therapeutic antibodies in clinical use in the early 20th century, prior to the discovery of antibiotics, were actually polyclonal preparations that recognized the carbohydrate coat of pathogenic bacteria such as Neisseria meningitidis and Streptococcus pneumoniae. These were raised in animals against inactivated whole bacteria for treatment of severe bacterial infections and each preparation was effective only on bacterial strains with homologous carbohydrate structures (32,33). This lead to a realization that antibody recognition of the coat structure was an important correlate for efficacy, and the functional classification of bacteria into serotypes for proper matching of antibody and bacterial strain. The requirement for multiple polyclonal antibodies for treatment of even one bacterial species due to large numbers of serotypes was one of the reasons for the switch to more broadly acting antibiotics once they became available (33). Nonetheless, their effective use is an indication of the therapeutic potential of non-protein targets.
To generate high-affinity antibodies against such targets, a strong immune response is required which is usually stimulated through conjugation with a protein carrier, delivery with an adjuvant such as lipid A or as an antigen complex with proteins such as whole inactivated bacteria (34–37). Even so, the antibodies obtained can often be low affinity IgM due to the inability to engage the mechanisms required for affinity maturation and class switching, hence limiting the utility of animal immunization for antibody generation (36,38). On the other hand, phage display has been used to produce anti-carbohydrate antibodies of reasonable affinity in the low nanomolar range (39). In particular, a phage library constructed with the heavy chain CDR3 enriched for basic residues to improve binding to negatively charged carbohydrates produced anti-carbohydrate antibodies that had relatively high affinity (KD ≈ 50nM) and excellent specificity (40). Although no anti-carbohydrate antibodies have been approved for routine clinical use, such antibodies have been shown to be able to target both carbohydrate-rich tumour antigen and bacterial lipopolysaccharides, the latter having been used on occasion to treat severe cases of sepsis (41,42).
Anti-lipid/lipoprotein antibodies are fairly uncommon in nature and in humans, they are usually associated with metabolic, autoimmune and neurodegenerative diseases; perhaps unsurprisingly given the key role lipids play in these processes. Cerebrospinal fluid and serum antibodies against myelin basic protein, sulfatide and oxidized lipids have been found in patients with multiple sclerosis, and serum antibodies against neuronal gangliosides are directly implicated in the pathology of the peripheral neuropathy Guillain–Barré syndrome (43–45). Anti-oxidized low-density lipoprotein antibodies on the other hand may play a role in the development of atherosclerosis (46). Isolation of such antibodies via phage display and the use of recombinant versions in animal models of disease may help determine their pathological role and potentially reverse its effects (47).
For example, in anti-phospholipid syndrome, antibodies against phospholipid-binding proteins such as β2-glycoprotein I are considered to be a major driver of pathology through inappropriate complement fixation. The use of phage display to obtain human antibodies of the same specificity has enabled the development of blocking antibodies that are defective in fixing complement which were shown to inhibit the effect of endogenous antibodies in animal models (48). In another example, with certain animal models of atherosclerosis, a high-titre antibody response to the malondialdehyde low-density lipoprotein immunogen also appears to correlate with reduced disease severity, suggesting that such antibodies could be used therapeutically (49). Phage display can also be used to develop anti-idiotypic antibodies; these have been shown to have therapeutic potential by binding and blocking the effects of naturally occurring autoantibodies (50).
Using phage display, we and others have also been able to generate anti-lipid antibodies even against pure lipids completely insoluble in aqueous solution, through enrichment against crystalline microdroplets formed following evaporation of organic solvents (9,10,51). While having potential for use as diagnostic reagents to identify targeted lipids in crude mixtures, it remains unclear whether such antibodies can recognize lipids in their natural biological environment and the therapeutic potential of anti-lipid antibodies generated through such modalities remains an open question.
Another type of target uniquely suited to phage display is proteins with post-translational modifications such as glycosylation or phosphorylation. In such cases, the approach is to engineer a specific recognition domain into a particular CDR region which significantly increases the likelihood that the recovered antibody will recognize the modification in addition to the protein epitope (52). For recognition of phosphorylated peptides, a phosphate-binding motif from a naturally occurring antibody was used to generate a mutant antibody library from which antibodies binding specifically to either phosphoserine, phosphotyrosine or phosphothreonine were identified. The CDR amino acids responsible for phosphorylated amino acid recognition were identified and then used to generate a second set of libraries which preserved these structures but had the rest of the CDR randomized; these libraries were then used to screen phosphorylated peptide targets. A similar strategy was used to identify glycan-binding antibodies based on a motif isolated from the anti-HIV antibody 2G12, which binds the HIV-1 glycoprotein gp120 primarily through its glycosylation (53).
Advantages of antibody selection in vitro
A key advantage of phage display is the in vitro nature of the antibody identification process, as this allows for selection methodologies not possible with in vivo antibody generation. By carefully controlling the selection and screening conditions on immobilized antigen, for example, by the presentation of specific antigen conformations or the inclusion of competitors for direct selection towards specific targets or epitopes, the generation of antibodies can be biased towards desired regions of the target (54–56). Moreover, binding affinity and on and off rates of recovered antibodies can be optimized independently according to the selection strategy employed, enabling tailoring to the desired biomedical application (57,58). This can be further enhanced during recombinant expression as full antibodies where various immunoglobulin isotypes and constructs with alternative functionality can be easily generated.
Another screening modality available to phage display is the targeting of antigen presented on the surface of whole cells or tissue. This is especially useful for membrane-bound antigen such as cell surface receptors and transmembrane proteins whose structure is not easily recapitulated by free antigen and is particularly useful for tumour-specific surface antigens. However, this approach is complicated by the diversity of antigens on the cell surface which may result in selection of phage against common surface epitopes (59). One means to overcome this is to leverage on unique biological properties of the target antigen. For example, signalling receptors such as HER2, epidermal growth factor receptor and p75 neurotrophin receptor, cycle from the cell surface to the interior and recovery of phage from the cell interior has allowed specific phage enrichment (60–62). However, most cell types will carry multiple membrane proteins capable of internalizing bound phage. An alternative and complementary means for selection is to pan against alternating cell types expressing the same target antigen to prevent selection of common surface antigens; this can be achieved through the use of mammalian or yeast expression vectors for expression of recombinant antigen on a suitable host cell (62,63).
In addition to these in vitro techniques, phage library approaches with in vivo binding modalities have also been explored. First described for organ targeting using peptide phage libraries where the library was injected intravenously into mice and the phage were subsequently rescued from the brain and kidneys (64), the technique has also been successfully carried out in tumour-bearing mice (65). In recent years, similar methodologies have also been employed with antibody phage libraries not only to identify potentially therapeutic antibodies but also for determining the accessibility of a given target (66), further expanding the boundaries of phage display technology.
Today, almost all widely accessible commercial libraries are based on non-immune repertoires of high complexity to enable use in the selection of antibodies against a virtually unlimited number of target antigens. The latest state-of-art libraries reach complexities of up to 1011–1012 clones and thus may even exceed the size of individual human cell repertoires (67,68). While high-affinity antibodies can be isolated from these libraries, occasionally the affinity of the obtained antibodies is low as the genes are obtained from naive B cells that have not yet undergone affinity maturation. In such cases, where the affinity required for therapeutic efficacy is higher than that of the antibody obtained, in vitro affinity maturation may be performed. This is often accomplished by introducing random or specific mutations in the CDRs or the exchange of whole heavy or light chain domains by chain shuffling (69–73). Recently, Li et al. described an alternative phage display approach for affinity maturation of a natural human antibody MSL-109 targeting human cytomegalovirus (CMV), where each of the CDRs was allowed to vary in only one amino acid residue at a time (69). Higher affinity antibodies were isolated which had improved CMV neutralization potency when expressed as Fab fragments in bacterial cells.
In another novel application of in vitro phage library screening, single antibodies capable of dual epitope recognition have also been developed. Carried out by Bostrom et al., a subset of solvent exposed residues of the light chain CDRs of the anti-HER2 antibody Herceptin were randomized to generate a highly diverse combinatorial phage library (1010 particles) (74). By subjecting the library to selection for VEGF binding while maintaining HER2 binding, dual antigen-binding clones were identified which inhibited both VEGF and Erb-B2-mediated cell proliferation in vitro and tumour progression in mouse models (74). Such ‘two-in-one’ antibodies may represent a paradigm shift and provide exciting new opportunities for therapeutics.
Generating fully human antibodies against pathogens with phage display
In contrast to lipids, carbohydrates and self-antigens, high-affinity antibodies against pathogen-derived protein antigens are easily generated through immunization or during the course of a natural infection (75,76). Present at high levels during the course of the immune response due to secretion from short-lived plasma cells and plasma blasts, lower levels are maintained in the serum by secretion from long-lived plasma cells and usually have therapeutic or prophylactic efficacy. This was put to good use in the pre-antibiotic era through serum therapy, where patients were given pathogen-specific polyclonal serum raised in animals (33). More recently, human-derived antibodies have been shown to be efficacious against a number of highly pathogenic diseases such as severe acute respiratory syndrome (SARS) coronavirus, Ebola and H5N1 avian influenza (77–80). Recovery of such naturally occurring antibodies from convalescent patients is clearly of prime importance for developing drugs against pathogens to which there are no other effective treatments. In addition, they provide a wealth of information on the humoral immune response, its duration and maintenance over time, and how it can be effective, or in some cases, ineffective, against infection (81,82).
To capture these antibodies, immune libraries can be constructed relatively quickly, allowing identification of the high-affinity antibodies that can be used to generate diagnostic reagents and also tested for their therapeutic potential. In addition, novel in vitro screening strategies can be employed to isolate rare broadly neutralizing antibodies, as has been highlighted in the hunt for antibodies against pandemic influenza. Kashyap et al. reported the generation of combinatorial antibody libraries from the bone marrow of five survivors of a H5N1 avian influenza outbreak in Turkey yielding >300 unique antibodies against H5N1 viral antigens, several of which were shown to have broadly neutralizing properties across various H5 subtypes (83). Similarly, Throsby et al., using combinatorial display libraries constructed from human memory B cells elicited by seasonal influenza vaccination, isolated 13 antibodies exhibiting broad heterosubtypic neutralizing activity against antigenically diverse H1, H2, H5, H6, H8 and H9 influenza subtypes (84). The most potent antibody was also protective in mice when given before and after lethal H5N1 or H1N1 challenge and the epitope was localized to the highly conserved membrane-proximal stem of the HA1 and HA2 subunits of hemagglutinin (84,85).
Using a combination of panning strategies against the hemagglutinin from several antigenically distinct H5N1 isolates to bias-selection of antibodies towards more conserved domains of the protein, we were able to isolate similar antibodies to that described by Throsby et al. from a non-immune Fab phage display library (11). The selection of such antibodies from even non-immune phage displayed libraries highlights the potential of these readily available libraries for the development of therapeutically relevant antibodies against emerging pathogens of high concern and may enable more rapid development of therapies against novel emergent pathogens in the future.
Phage display in comparison with other methodologies for human antibody discovery
The main competition for phage libraries in the area of cancer and autoimmune disease therapeutics where self-antigens are the primary target are transgenic mice whose immunoglobulin germline genes have been replaced with human equivalents. This technology, developed in the 1990s, has been driven mainly by two biotech companies—Mederax and Abgenix—which licence the HuMab and XenoMouse strains, respectively (86–88). During the course of V(D)J recombination, these reassemble into fully human antibodies although they are carried on murine B cells and the mice can generate functional immune responses (including somatic hypermutation) during immunization, from which antibody-secreting hybridomas can be made. Some of these antibodies are already approved for clinical use and more are in the clinical pipeline (12,89).
One disadvantage of these first-generation strains appears to be the use of human constant regions, which has been suggested to affect downstream signalling in B cells and result in defective immune responses against particular antigens; newer strains of transgenic mice have been developed that have only the variable regions replaced and utilize murine constant regions (90). Currently, more antibodies in clinical use or trials are derived from transgenic mice as compared to phage display, but this may not be an accurate comparison of their utility as the technology for transgenic mice was developed several years ahead of the first truly diverse synthetic antibody libraries (12). Additionally, as antibody generation is governed by the rules of the immune response, their utility for generating high-affinity antibodies against epitopes common to humans and mice, non-protein targets, or antibodies with novel binding characteristics such as those achieved by in vitro selection under non-physiological conditions, is limited.
For isolation of antibodies from human subjects, while phage display was the first technique suitable for the screening of large numbers of B cells, in recent years, additional techniques of similar robustness and efficacy have been developed. These fall into two main categories: analysis of single B cells through in vitro immortalization, activation into antibody-secreting cells, or recovery of their individual antibody transcripts; or alternatively, high-throughput analysis of polyclonal antibodies. The latter approach can be carried out at either the proteome or transcriptome level through mass spectrometry of digested peptides or Next Generation Sequencing of harvested B-cell mRNA, respectively. (80,91–94). Both these techniques have one main advantage over phage display; they allow elucidation of the heavy and light chain pairings and frequencies of the B cells of interest. As such, they may be more useful for characterizing the dynamics of humoral immune responses (95). On the other hand, they require significant investment in specialist equipment, such as a high-speed fluorescence-activated cell sorter (FACS) or Next Generation Sequencer, as well as robotic handling equipment if large numbers of cells are to be screened (77).
Phage display does not require any such equipment and can be performed with basic microbiological and molecular biology apparatus with the exception of an electroporator if high efficiency cloning is required (67). Another advantage is that a phage library once created is usually sufficient for numerous screenings and can be stored almost indefinitely, while the various omics procedures involve destruction of the sample and single B-cell cultures, unlike murine hybridomas, do not allow for indefinite antibody production (96). Even antibodies produced by B cells immortalized with Epstein–Barr virus can gradually lose affinity due to re-activation of somatic hypermutation (P. A. MacAry, unpublished data). Hence, while these techniques require a finely honed screening strategy only available with well-characterized pathogens, phage display may be more suited towards investigation of poorly understood or novel pathogens where multiple screening targets and conditions need to be investigated. In addition, side-by-side comparison of phage display and Next Generation sequencing produced completely different sets of antibodies that were attributed to the poor expression of particular antibodies on phage. Nevertheless, the antibodies produced by either technique had comparable specificity and affinity (97).
The future for phage display
While for some applications, such as isolation of antibodies from patients, phage display may be complemented or substituted with other techniques, an area where phage display remains critical is for directed evolution of antibodies for increased stability under non-physiological conditions of pH and temperature—such selection is simply not possible in vivo. Neither are other in vitro alternatives such as cell surface display (mammalian, yeast or bacterial) or ribosome display feasible due to their lower resistance to high temperatures or extreme pH. Phage can tolerate temperatures of up to 60°C and a pH range of 2–11 (98). Table 1 provides a summary comparison of the advantages and disadvantages of these various in vitro techniques.
Table 1.
Comparison of phage display with other in vitro antibody screening techniques
| Phage display | Ribosome display (99) | Yeast display (100) | Single B cell screening (101) | |
|---|---|---|---|---|
| Source | Immune or non-immune donor/synthetic | Immune donor | ||
| Screening size | Up to 1011 | Up to 1014 | Up to 109 | Up to 106 |
| Antibody format | ScFv/Fab/sdAb | ScFv | Fab or IgG | IgG |
| Pros | Robust. Requires minimal instrumentations. Non-physiological and stringent selection conditions possible due to phage stability at pH/temperature extremes. | Cell-free expression and high concentration of ribosome particles permits extremely large practical library sizes. Compulsory PCR step convenient for mutagenesis. | Eukaryotic expression permits proper disulphide bridge formation and N-linked glycosylation. Direct quantitative monoclonal analysis by flow cytometry. | Isolation of naturally paired VH-VL immunoglobulin genes that have gone through affinity maturation. |
| Cons | Lacks post-translational modification of antibody fragments. | Requires optimization due to relative instability of RNA– ribosomal complex. | Smaller library size compared to phage/ribosome display. | Labour intensive and technically challenging. Smallest library size. |
| Affinity maturation | Can be performed in vitro. | Can be used to select for high- affinity binders. | Can be used to select for high-affinity binders during FACS sorting. | Not required. |
Phage display is also likely to remain useful for discovery of antibodies against non-protein targets, evolution of dual-binding antibodies and for affinity maturation, due to the limitations of the natural immune system. However, antibodies against non-protein targets such as lipids and carbohydrates remain a fairly unexplored area, partially due to the lack of validated therapeutic targets aside from bacterial membrane surface carbohydrates. One area that could be of particular interest is lipids involved in autoimmune and neurodegenerative disorders, for which there is some evidence that antibodies could have both protective and pathological roles and which a therapeutic antibody designed to suppress immune function, e.g. of the IgG4 isotype, could have benefits.
For regular protein antigens, there may still be one particular niche for phage display based on its strengths: the rapid development of therapeutic antibodies against emerging diseases or a genetically modified pathogen in an emergency situation. Antibodies are a natural and well-studied component of the immune system and use of a naturally derived antibody should have minimal side effects and thus be ideal for emergency use. In such a situation, rapid antibody isolation is critical. Phage display allows for the screening of an extremely large collection of antibodies of up to 1011 sequences, far more than that possible by Next Generation sequencing or single B-cell analysis, thus improving the chance of isolating rare antibodies. Even if an immune library is required, rapid construction and screening is possible using cloning protocols such as MEGAWHOP which do not require prior digestion of PCR immunoglobulin sequences and new panning strategies that enable effective enrichment over only one cycle of selection (102,103). In addition, vectors have been developed that can express antibodies both on phage as well as in mammalian cell culture as full length IgG through the use of intron-mediated differential splicing without any need for sub-cloning, which enables rapid high-throughput expression analysis of the full length antibody (104). As such, phage display is likely to remain a key technique for antibody discovery for the foreseeable future.
Funding
Future System and Technology Directorate (FSTD), Ministry of Defence, Singapore to B.J.H and the Singapore National Research Foundation (NRF370062-HUJ-NUS) to P.A.M.
Conflict of interest statement: The authors declared no conflicts of interest.
References
- 1. Chan C. E., Chan A. H., Hanson B. J., Ooi E. E. 2009. The use of antibodies in the treatment of infectious diseases. Singapore Med. J. 50:663. [PubMed] [Google Scholar]
- 2. Smith G. P. 1985. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228:1315. [DOI] [PubMed] [Google Scholar]
- 3. McCafferty J., Griffiths A. D., Winter G., Chiswell D. J. 1990. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348:552. [DOI] [PubMed] [Google Scholar]
- 4. Huse W. D., Sastry L., Iverson S. A., et al. 1989. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 246:1275. [DOI] [PubMed] [Google Scholar]
- 5. Barbas C. F., 3rd, Kang A. S., Lerner R. A., Benkovic S. J. 1991. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc. Natl Acad. Sci. USA 88:7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoogenboom H. R. 2005. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 23:1105. [DOI] [PubMed] [Google Scholar]
- 7. Arbabi-Ghahroudi M., MacKenzie R., Tanha J. 2009. Selection of non-aggregating VH binders from synthetic VH phage-display libraries. Methods Mol. Biol. 525:187. [DOI] [PubMed] [Google Scholar]
- 8. Ponsel D., Neugebauer J., Ladetzki-Baehs K., Tissot K. 2011. High affinity, developability and functional size: the holy grail of combinatorial antibody library generation. Molecules 16:3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan C. E., Zhao B. Z., Cazenave-Gassiot A., et al. 2013. Novel phage display-derived mycolic acid-specific antibodies with potential for tuberculosis diagnosis. J. Lipid Res. 54:2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Islam M. O., Lim Y. T., Chan C. E., et al. 2012. Generation and characterization of a novel recombinant antibody against 15-ketocholestane isolated by phage-display. Int. J. Mol. Sci. 13:4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lim A. P., Chan C. E., Wong S. K., Chan A. H., Ooi E. E., Hanson B. J. 2008. Neutralizing human monoclonal antibody against H5N1 influenza HA selected from a Fab-phage display library. Virol. J. 5:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nelson A. L., Dhimolea E., Reichert J. M. 2010. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9:767. [DOI] [PubMed] [Google Scholar]
- 13. Nixon A. E., Sexton D. J., Ladner R. C. 2014. Drugs derived from phage display: From candidate identification to clinical practice. MAbs 6:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan C. E., Chan A. H., Lim A. P., Hanson B. J. 2011. Comparison of the efficiency of antibody selection from semi-synthetic scFv and non-immune Fab phage display libraries against protein targets for rapid development of diagnostic immunoassays. J. Immunol. Methods 373:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hwang W. Y., Foote J. 2005. Immunogenicity of engineered antibodies. Methods 36:3. [DOI] [PubMed] [Google Scholar]
- 16. Knappik A., Ge L., Honegger A., et al. 2000. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J. Mol. Biol. 296:57. [DOI] [PubMed] [Google Scholar]
- 17. Jespers L. S., Roberts A., Mahler S. M., Winter G., Hoogenboom H. R. 1994. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology (N. Y.) 12:899. [DOI] [PubMed] [Google Scholar]
- 18. Lorenz H. M. 2002. Technology evaluation: adalimumab, Abbott laboratories. Curr. Opin. Mol. Ther. 4:185. [PubMed] [Google Scholar]
- 19. Reichert J. M. 2011. Antibody-based therapeutics to watch in 2011. MAbs 3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dantas-Barbosa C., de Macedo Brigido M., Maranhao A. Q. 2012. Antibody phage display libraries: contributions to oncology. Int. J. Mol. Sci. 13:5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bradbury A. R., Sidhu S., Dübel S., McCafferty J. 2011. Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotechnol. 29:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rothe A., Hosse R. J., Power B. E. 2006. In vitro display technologies reveal novel biopharmaceutics. FASEB J. 20:1599. [DOI] [PubMed] [Google Scholar]
- 23. de Kruif J., Boel E., Logtenberg T. 1995. Selection and application of human single chain Fv antibody fragments from a semi-synthetic phage antibody display library with designed CDR3 regions. J. Mol. Biol. 248:97. [DOI] [PubMed] [Google Scholar]
- 24. Griffiths A. D., Williams S. C., Hartley O., et al. 1994. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 13:3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prassler J., Thiel S., Pracht C., et al. 2011. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 413:261. [DOI] [PubMed] [Google Scholar]
- 26. Rothe C., Urlinger S., Löhning C., et al. 2008. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J. Mol. Biol. 376:1182. [DOI] [PubMed] [Google Scholar]
- 27. Gurney A., Axelrod F., Bond C. J., et al. 2012. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl Acad. Sci. USA 109:11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harding F. A., Stickler M. M., Razo J., DuBridge R. B. 2010. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs 2:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Söderlind E., Strandberg L., Jirholt P., et al. 2000. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat. Biotechnol. 18:852. [DOI] [PubMed] [Google Scholar]
- 30. Veitonmäki N., Hansson M., Zhan F., et al. 2013. A human ICAM-1 antibody isolated by a function-first approach has potent macrophage-dependent antimyeloma activity in vivo. Cancer Cell 23:502. [DOI] [PubMed] [Google Scholar]
- 31. Hoet R. M., Cohen E. H., Kent R. B., et al. 2005. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 23:344. [DOI] [PubMed] [Google Scholar]
- 32. Pier G. B. 2007. Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. 297:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Casadevall A., Scharff M. D. 1994. Serum therapy revisited: animal models of infection and development of passive antibody therapy. Antimicrob. Agents Chemother. 38:1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Singh K. V., Kaur J., Varshney G. C., Raje M., Suri C. R. 2004. Synthesis and characterization of hapten-protein conjugates for antibody production against small molecules. Bioconjug. Chem. 15:168. [DOI] [PubMed] [Google Scholar]
- 35. Mäkelä P. H. 2003. Conjugate vaccines–a breakthrough in vaccine development. Southeast Asian J. Trop. Med. Public Health 34:249. [PubMed] [Google Scholar]
- 36. Harrison B. A., Fernández H., Chandan V., et al. 2011. Characterization and functional activity of murine monoclonal antibodies specific for α1,6-glucan chain of Helicobacter pylori lipopolysaccharide. Helicobacter 16:459. [DOI] [PubMed] [Google Scholar]
- 37. Lu Z., Rynkiewicz M. J., Yang C. Y., et al. 2013. The binding sites of monoclonal antibodies to the non-reducing end of Francisella tularensis O-antigen accommodate mainly the terminal saccharide. Immunology 140:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swartz G. M., Jr, Gentry M. K., Amende L. M., Blanchette-Mackie E. J., Alving C. R. 1988. Antibodies to cholesterol. Proc. Natl Acad. Sci. USA 85:1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang W., Matsumoto-Takasaki A., Kusada Y., et al. 2007. Isolation and characterization of phage-displayed single chain antibodies recognizing nonreducing terminal mannose residues. 2. Expression, purification, and characterization of recombinant single chain antibodies. Biochemistry 46:263. [DOI] [PubMed] [Google Scholar]
- 40. Schoonbroodt S., Steukers M., Viswanathan M., et al. 2008. Engineering antibody heavy chain CDR3 to create a phage display Fab library rich in antibodies that bind charged carbohydrates. J. Immunol. 181:6213. [DOI] [PubMed] [Google Scholar]
- 41. Sakai K., Yuasa N., Tsukamoto K., et al. 2010. Isolation and characterization of antibodies against three consecutive Tn-antigen clusters from a phage library displaying human single-chain variable fragments. J. Biochem. 147:809. [DOI] [PubMed] [Google Scholar]
- 42. Ziegler E. J., Fisher C. J., Jr, Sprung C. L., et al. 1991. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N. Engl. J. Med. 324:429. [DOI] [PubMed] [Google Scholar]
- 43. Kanter J. L., Narayana S., Ho P. P., et al. 2006. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat. Med. 12:138. [DOI] [PubMed] [Google Scholar]
- 44. Willison H. J., Yuki N. 2002. Peripheral neuropathies and anti-glycolipid antibodies. Brain 125(Pt 12):2591. [DOI] [PubMed] [Google Scholar]
- 45. Gabibov A. G., Belogurov A. A., Jr, Lomakin Y. A., et al. 2011. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J. 25:4211. [DOI] [PubMed] [Google Scholar]
- 46. Shaw P. X., Hörkkö S., Tsimikas S., et al. 2001. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler. Thromb. Vasc. Biol. 21:1333. [DOI] [PubMed] [Google Scholar]
- 47. Cabiedes J., Cabral A. R. 2009. Anti-beta(2)GP-I and anti-prothrombin antibodies generated by phage display. Methods Mol. Biol. 562:61. [DOI] [PubMed] [Google Scholar]
- 48. Agostinis C., Durigutto P., Sblattero D., et al. 2014. A non-complement-fixing antibody to β2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood 123:3478. [DOI] [PubMed] [Google Scholar]
- 49. George J., Afek A., Gilburd B., et al. 1998. Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis 138:147. [DOI] [PubMed] [Google Scholar]
- 50. Mimouni D., Blank M., Payne A. S., et al. 2010. Efficacy of intravenous immunoglobulin (IVIG) affinity-purified anti-desmoglein anti-idiotypic antibodies in the treatment of an experimental model of pemphigus vulgaris. Clin. Exp. Immunol. 162:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsuruta L. R., Tomioka Y., Hishinuma T., et al. 2003. Characterization of 11-dehydro-thromboxane B2 recombinant antibody obtained by phage display technology. Prostaglandins Leukot. Essent. Fatty Acids 68:273. [DOI] [PubMed] [Google Scholar]
- 52. Koerber J. T., Thomsen N. D., Hannigan B. T., Degrado W. F., Wells J. A. 2013. Nature-inspired design of motif-specific antibody scaffolds. Nat. Biotechnol. 31:916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stewart A., Liu Y., Lai J. R. 2012. A strategy for phage display selection of functional domain-exchanged immunoglobulin scaffolds with high affinity for glycan targets. J. Immunol. Methods 376:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanna P. P., Williamson R. A., De Logu A., Bloom F. E., Burton D. R. 1995. Directed selection of recombinant human monoclonal antibodies to herpes simplex virus glycoproteins from phage display libraries. Proc. Natl Acad. Sci. USA 92:6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Osbourn J. K., Earnshaw J. C., Johnson K. S., Parmentier M., Timmermans V., McCafferty J. 1998. Directed selection of MIP-1 alpha neutralizing CCR5 antibodies from a phage display human antibody library. Nat. Biotechnol. 16:778. [DOI] [PubMed] [Google Scholar]
- 56. Parsons H. L., Earnshaw J. C., Wilton J., et al. 1996. Directing phage selections towards specific epitopes. Protein Eng. 9:1043. [DOI] [PubMed] [Google Scholar]
- 57. Lu D., Shen J., Vil M. D., et al. 2003. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem. 278:43496. [DOI] [PubMed] [Google Scholar]
- 58. Yuan B., Schulz P., Liu R., Sierks M. R. 2006. Improved affinity selection using phage display technology and off-rate based selection. Electron. J. Biotechnol. 9. [Google Scholar]
- 59. Hoogenboom H. R., Lutgerink J. T., Pelsers M. M., et al. 1999. Selection-dominant and nonaccessible epitopes on cell-surface receptors revealed by cell-panning with a large phage antibody library. Eur. J. Biochem. 260:774. [DOI] [PubMed] [Google Scholar]
- 60. Tani H., Osbourn J. K., Walker E. H., Rush R. A., Ferguson I. A. 2013. A novel in vivo method for isolating antibodies from a phage display library by neuronal retrograde transport selectively yields antibodies against p75(NTR.). MAbs 5:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Becerril B., Poul M. A., Marks J. D. 1999. Toward selection of internalizing antibodies from phage libraries. Biochem. Biophys. Res. Commun. 255:386. [DOI] [PubMed] [Google Scholar]
- 62. Heitner T., Moor A., Garrison J. L., Marks C., Hasan T., Marks J. D. 2001. Selection of cell binding and internalizing epidermal growth factor receptor antibodies from a phage display library. J. Immunol. Methods 248:17. [DOI] [PubMed] [Google Scholar]
- 63. Zhou Y., Zou H., Zhang S., Marks J. D. 2010. Internalizing cancer antibodies from phage libraries selected on tumor cells and yeast-displayed tumor antigens. J. Mol. Biol. 404:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pasqualini R., Ruoslahti E. 1996. Organ targeting in vivo using phage display peptide libraries. Nature 380:364. [DOI] [PubMed] [Google Scholar]
- 65. Staquicini F. I., Cardó-Vila M., Kolonin M. G., et al. 2011. Vascular ligand-receptor mapping by direct combinatorial selection in cancer patients. Proc. Natl Acad. Sci. USA 108:18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sánchez-Martín D., Martínez-Torrecuadrada J., Teesalu T., et al. 2013. Proteasome activator complex PA28 identified as an accessible target in prostate cancer by in vivo selection of human antibodies. Proc. Natl Acad. Sci. USA 110:13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Haard H. J. 2002. Construction of large naïve Fab libraries. Methods Mol. Biol. 178:87. [DOI] [PubMed] [Google Scholar]
- 68. Lloyd C., Lowe D., Edwards B., et al. 2009. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 22:159. [DOI] [PubMed] [Google Scholar]
- 69. Li B., Fouts A. E., Stengel K., et al. 2014. In vitro affinity maturation of a natural human antibody overcomes a barrier to in vivo affinity maturation. MAbs 6:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thompson J., Pope T., Tung J. S., et al. 1996. Affinity maturation of a high-affinity human monoclonal antibody against the third hypervariable loop of human immunodeficiency virus: use of phage display to improve affinity and broaden strain reactivity. J. Mol. Biol. 256:77. [DOI] [PubMed] [Google Scholar]
- 71. Valjakka J., Hemminki A., Niemi S., Söderlund H., Takkinen K., Rouvinen J. 2002. Crystal structure of an in vitro affinity- and specificity-matured anti-testosterone Fab in complex with testosterone. Improved affinity results from small structural changes within the variable domains. J. Biol. Chem. 277:44021. [DOI] [PubMed] [Google Scholar]
- 72. Ho M., Pastan I. 2009. In vitro antibody affinity maturation targeting germline hotspots. Methods Mol. Biol. 525:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Marks J. D. 2004. Antibody affinity maturation by chain shuffling. Methods Mol. Biol. 248:327. [DOI] [PubMed] [Google Scholar]
- 74. Bostrom J., Yu S. F., Kan D., et al. 2009. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 323:1610. [DOI] [PubMed] [Google Scholar]
- 75. Blanchard-Rohner G., Pulickal A. S., Jol-van der Zijde C. M., Snape M. D., Pollard A. J. 2009. Appearance of peripheral blood plasma cells and memory B cells in a primary and secondary immune response in humans. Blood 114:4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Amanna I. J., Carlson N. E., Slifka M. K. 2007. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 357:1903. [DOI] [PubMed] [Google Scholar]
- 77. Corti D., Voss J., Gamblin S. J., et al. 2011. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333:850. [DOI] [PubMed] [Google Scholar]
- 78. ter Meulen J., van den Brink E. N., Poon L. L., et al. 2006. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 3:e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maruyama T., Rodriguez L. L., Jahrling P. B., et al. 1999. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J. Virol. 73:6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Traggiai E., Becker S., Subbarao K., et al. 2004. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat. Med. 10:871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Beltramello M., Williams K. L., Simmons C. P., et al. 2010. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yu X., Tsibane T., McGraw P. A., et al. 2008. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 455:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kashyap A. K., Steel J., Oner A. F., et al. 2008. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc. Natl Acad. Sci. USA 105:5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Throsby M., van den Brink E., Jongeneelen M., et al. 2008. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 3:e3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ekiert D. C., Bhabha G., Elsliger M. A., et al. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mendez M. J., Green L. L., Corvalan J. R., et al. 1997. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat. Genet. 15:146. [DOI] [PubMed] [Google Scholar]
- 87. Lonberg N., Taylor L. D., Harding F. A., et al. 1994. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 368:856. [DOI] [PubMed] [Google Scholar]
- 88. Tomizuka K., Yoshida H., Uejima H., et al. 1997. Functional expression and germline transmission of a human chromosome fragment in chimaeric mice. Nat. Genet. 16:133. [DOI] [PubMed] [Google Scholar]
- 89. Lonberg N. 2005. Human antibodies from transgenic animals. Nat. Biotechnol. 23:1117. [DOI] [PubMed] [Google Scholar]
- 90. Murphy A. J., Macdonald L. E., Stevens S., et al. 2014. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc. Natl Acad. Sci. USA 111:5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Reddy S. T., Ge X., Miklos A. E., et al. 2010. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat. Biotechnol. 28:965. [DOI] [PubMed] [Google Scholar]
- 92. Wrammert J., Smith K., Miller J., et al. 2008. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Franz B., May K. F., Jr, Dranoff G., Wucherpfennig K. 2011. Ex vivo characterization and isolation of rare memory B cells with antigen tetramers. Blood 118:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jin A., Ozawa T., Tajiri K., Obata T., Kishi H., Muraguchi A. 2011. Rapid isolation of antigen-specific antibody-secreting cells using a chip-based immunospot array. Nat. Protoc. 6:668. [DOI] [PubMed] [Google Scholar]
- 95. Ogunniyi A. O., Thomas B. A., Politano T. J., et al. 2014. Profiling human antibody responses by integrated single-cell analysis. Vaccine 32:2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jourdan M., Caraux A., De Vos J., et al. 2009. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 114:5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saggy I., Wine Y., Shefet-Carasso L., Nahary L., Georgiou G., Benhar I. 2012. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein Eng. Des. Sel. 25:539. [DOI] [PubMed] [Google Scholar]
- 98. Wörn A., Plückthun A. 2001. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 305:989. [DOI] [PubMed] [Google Scholar]
- 99. Dufner P., Jermutus L., Minter R. R. 2006. Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 24:523. [DOI] [PubMed] [Google Scholar]
- 100. Boder E. T., Raeeszadeh-Sarmazdeh M., Price J. V. 2012. Engineering antibodies by yeast display. Arch. Biochem. Biophys. 526:99. [DOI] [PubMed] [Google Scholar]
- 101. Tiller T. 2011. Single B cell antibody technologies. N. Biotechnol. 28:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Maaß A., Heiseler T., Maaß F., et al. 2014. A general strategy for antibody library screening via conversion of transient target binding into permanent reporter deposition. Protein Eng. Des. Sel. 27:41. [DOI] [PubMed] [Google Scholar]
- 103. Miyazaki K. 2011. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol. 498:399. [DOI] [PubMed] [Google Scholar]
- 104. Tesar D., Hötzel I. 2013. A dual host vector for Fab phage display and expression of native IgG in mammalian cells. Protein Eng. Des. Sel. 26:655. [DOI] [PubMed] [Google Scholar]