

Abstract
Mammalian ortheoreoviruses are currently being investigated as novel cancer therapeutics, but the cellular mechanisms that regulate susceptibility to reovirus oncolysis remain poorly understood. In this study, we present evidence that virion disassembly is a key determinant of reovirus oncolysis. To penetrate cell membranes and initiate infection, the outermost capsid proteins of reovirus must be proteolyzed to generate a disassembled particle called an infectious subviral particle (ISVP). In fibroblasts, this process is mediated by the endo/lysosomal proteases cathepsins B and L. We have analyzed the early events of infection in reovirus-susceptible and -resistant cells. We find that, in contrast to susceptible glioma cells and Ras-transformed NIH3T3 cells, reovirus-resistant cancer cells and untransformed NIH3T3 cells restrict virion uncoating and subsequent gene expression. Disassembly-restrictive cells support reovirus infection, as in vitro-generated ISVPs establish productive infection, and pretreatment with poly(I:C) does not prevent infection in cancer cells. We find that the level of active cathepsin B and L is increased in tumors and that disassembly-restrictive glioma cells support reovirus oncolysis when grown as a tumor in vivo. Together, these results provide a model in which proteolytic disassembly of reovirus is a critical determinant of susceptibility to reovirus oncolysis.
Introduction
Reoviruses are members of the Reoviridae family of non-enveloped, double-stranded RNA (dsRNA) viruses. The mammalian reovirus, commonly found in stagnant water and sewage, infects a wide range of species including chimpanzees, monkeys, pigs, cattle, cats, sheep, mice, and humans.1 , 2 , 3 Because of its ubiquity in the natural environment, up to 50% of adults have had a previous exposure to reovirus as evidenced by anti-reovirus antibodies.3 In immunocompetent animals, reovirus infection produces few clinical symptoms and the virus is not linked to any known human disease (hence the designation “orphan”).4 , 5
Reovirus is an enteric virus that is transmitted mainly through the fecal-oral route and principally infects the gastrointestinal and respiratory tracts of mammals.6 To gain entry into the target cells of its host organism, reovirus virions must undergo proteolytic disassembly (uncoating). Uncoating results in the degradation of the outermost capsid protein σ3 and the subsequent cleavage of the underlying viral protein μ1/μ1C to generate an infectious subviral particle (ISVP).7 , 8 This processing gives the subvirion particle the capacity to penetrate the endosome/lysosome or the plasma membrane and thereby gain entry to the cytoplasm where reovirus replication can occur.9 , 10 , 11 Work in animal models suggests that, during natural enteric infections, σ3 is proteolytically degraded extracellularly within the intestinal lumen.12 , 13 In cell culture, however, reovirus takes advantage of endosomal and lysosomal proteases for its proteolytic conversion into ISVP.11 , 14 The necessity of reovirus uncoating for infectivity in vivo and in vitro is further demonstrated by the capacity of protease inhibitors to block enteric infection in mice and restrict reovirus uncoating and infection in cell culture.13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21
Recently, reovirus type-3 Dearing has been investigated as an oncolytic agent against a variety of human cancers (reviewed in ref. 22). The oncolytic properties of reovirus are also being investigated in clinical trials23 , 24 (http://www.oncolyticsbiotech.com). These studies revealed that reovirus infects all but a few cancer cells while leaving normal cells relatively unaffected. Atlhough it has been proposed that reovirus requires an activated Ras signaling pathway in order to mediate oncolysis,25 , 26 , 27 other studies have demonstrated that active Ras alone does not control susceptibility of cells to reovirus infection.28 , 29 , 30 Several cell lines, including the highly susceptible L929 mouse fibroblasts, are very susceptible to reovirus despite low levels of Ras activity (T.A. and P.A.F., unpublished results). Furthermore, we have observed that the addition of proteases into the culture medium or direct infection with ISVPs of restrictive cells can render them permissive for reovirus infection.31 , 32 These observations suggest that the protease-mediated disassembly of reovirus' capsid may determine whether or not a cell or tumor is permissive for reovirus infection and that this may be independent of elevated Ras activity.
In this study we investigate the requirement for reovirus uncoating in the glioma cell lines U87 and U118 as well as in the NIH3T3 cell line and its Ras-transformed derivatives. We demonstrate a lack of effective reovirus disassembly and penetration in the reovirus-resistant glioma U118 and the non-transformed NIH3T3 cell lines. Protease-generated ISVP particles efficiently infect these disassembly-restrictive cells and other reovirus-resistant cancer cells. We also find that synthetic dsRNA does not protect transformed cells from infection. These results support a model in which the requirement for proteolytic uncoating determines the susceptibility of cancer cells to reovirus oncolysis. Our finding that reovirus can productively infect and mediate in vivo oncolysis of tumors that restrict disassembly in vitro suggests that the microenvironment of tumors and the cellular environment of cancer cells, perhaps through elevated cathepsin B and L activity, facilitate the extracellular conversion of reovirus virions into particles that can penetrate cell membranes and mediate oncolysis.
Results
Reovirus disassembly is ineffective in resistant U118 glioma and NIH3T3 cell lines
Reovirus infects the majority of glioma cell lines, and only a few lines are resistant to reovirus infection.33 , 34 Until now, reovirus disassembly has not been addressed as a factor in the resistance of glioma cells to reovirus oncolysis. To examine reovirus capsid disassembly in susceptible and resistant cells, [35S]-labeled reovirus particles were exposed to the glioma cell lines U87 and U118, the Ras-transformed NIH3T3 (NIH3T3-Ras), and the empty vector pBabe-transfected NIH3T3 (NIH3T3). We found that [35S]-labeled reovirus is proteolytically processed in the susceptible cell lines but not in resistant cell lines. Removal of σ3 and cleavage of μ1C to δ, indicative of ISVP formation, can be readily observed 3 hours post infection (h.p.i.) in the U87 cells but not in the reovirus-resistant U118 cells even up to 24 h.p.i. (Figure 1a ). Processing, although minimal when compared to the susceptible glioma U87 and the L929 cells, was also observed in the Ras-transformed NIH3T3 cells but not in the NIH3T3 controls (Figure 1b). Reovirus disassembly has previously been reported to be inhibited when cells are exposed to the cysteine protease inhibitor E64.8 , 16 , 17 , 18 , 19 To verify that this compound could also block reovirus uncoating in our susceptible cells, permissive cells (U87, NIH3T3-Ras, and L929) were exposed to E64 (100 μmol/l) 1 hour before [35S]-labeled reovirus exposure as just described. We found that the degradation of σ3 protein and cleavage of μ1C into δ was completely inhibited in the presence of E64 up to 24 hours after exposure to the virus (Figure 1c). Similar results were obtained in a subsequent experiment by Western blotting with a specific anti-μ1 reovirus antibody that recognizes both μ1C and δ (Figure 1d). These results demonstrate that the proteolytic conversion of reovirus virions into ISVPs is deficient in cells resistant to reovirus infection (U118 glioma and the non-transformed NIH3T3 cell line) and that such ineffective reovirus uncoating could explain, in part, the resistance of these cells to infection and oncolysis.
Figure 1.
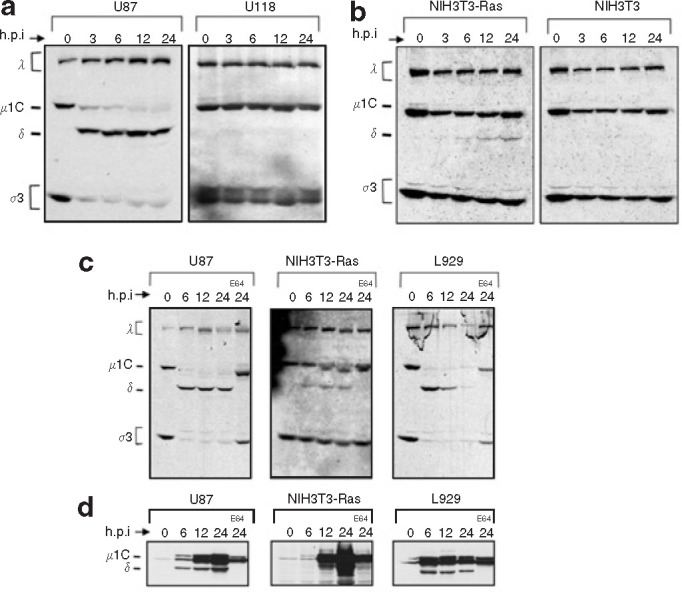
Evaluation of the proteolytic processing of reovirus in susceptible and resistant cell lines. (a) [35S]-labeled reovirus particles at 25,000 counts per minute were exposed to the glioma cell lines U87 and U118 as well as (b) Ras-transformed NIH3T3 (NIH3T3-Ras), and empty vector pBabe NIH3T3 (NIH3T3). Radiolabeled virus was allowed to bind at 4°C for 1 hour and then cells were returned to 37°C; cells were subsequently lysed at the defined time point after infection. Lysates were cleared of debris by centrifugation, and supernatants were submitted to sodium dodecyl sulfate polyacrylamide gel electrophoresis SDS-PAGE followed by autoradiography. (c) Reovirus proteolysis in susceptible cells treated with the E64 protease inhibitor (Sigma) in vitro and processed as described. Cells were exposed to 100 μmol/l of E64 for 1 hour before reovirus infection. After binding, E64 was again added to the cells to a final concentration of 100 μmol/l. (d) Western blot using a specific anti-μ1 reovirus antibody that detects both μ1C and δ was performed on a replicate of the experiment performed in c but using non-radioactive reovirions. h.p.i., hours post infection.
In resistant cell lines, reovirus binds, accumulates within lysosomes, but is not efficiently transcribed
To investigate whether the underlying cause of the defective disassembly in resistant cells reflects differences in the binding and entry of the virus, we used radiolabeled reovirus to compare the binding efficiency of the permissive and resistant cells for the virus. Cells were incubated with [35S]-labeled reovirus for 1 hour at 4°C before collecting the cell lysate for quantitative measurements by scintillation counting. We found that different amount of reovirus could bind to resistant and susceptible cells (Figure 2a ). L929 cells were the most efficient at binding reovirus, followed by the highly susceptible glioma cell line U87 and NIH3T3 cells. The reovirus resistant U118 glioma cells had the lowest binding for reovirus. Although inferior binding with the glioma U118 and the NIH3T3 cells may reduce the permissiveness of these cells to reovirus infection, virions do bind onto these cells. Moreover, the binding measured for the resistant NIH3T3 cells was the same as for the susceptible Ras-transformed NIH3T3, further suggesting that the level of binding on these cells does not mediate differential susceptibility. E64 treatment did not affect the level of reovirus binding in any cell line tested, which excludes the possibility of reduced susceptibility via interference with binding.
Figure 2.

Assessment of reovirus binding, internalization, and viral transcription in susceptible and resistant cell lines. (a) [35S]-labeled reovirus was allowed to bind on cells in minimal medium for 1 hour at 4°C. Cells were washed twice in phosphate-buffered saline and lysed in a sonification buffer. Samples were subjected to scintillation counting, which was performed in triplicate. (b) Cy3-labeled reovirus at a multiplicity (MOI) of infection of 5,000 was added for 24 hours to cells grown on glass slides, after which cells were fixed in 4% formaldehyde. Immunofluorescence with the lysosomal marker lysosomal associated membrane protein 2 (LAMP2) antibody followed by secondary fluorescein isothiocyanate (FITC) was performed; slides were then mounted with a 4′,6-diamidino-2-phenylindole (DAPI) mounting medium (VECTOR) and photographed by multiple acquisition with a fluorescent Zeiss microscope (magnification ×400). (c) Total RNA from cells were purified using the RNeasy RNA extraction kit; equal RNA amounts were subjected to Northern blotting using a digoxygenin-labeled probe against the positive strand of reovirus S1 transcripts. ISVP, infectious subviral particle.
Since reovirions bind to both resistant and susceptible cells, we next evaluated whether reovirus accumulated in lysosomes within our disassembly-restrictive cells. Previous studies have described how reovirus capsid disassembly is necessary to perforate either the endosomal/lysosomal or the plasma membrane and thus to penetrate the cytoplasm.9 , 10 , 11 In order to do this, we labeled reovirus with Cy3-maleimide monofunctional dye and exposed resistant and susceptible cells grown on glass slides to this labeled reovirus. Immunofluorescence was then performed to detect lysosomal associated membrane protein 2, frequently used as a functional marker of lysosomes.35 Our results show that, in the susceptible U87 glioma cells and in the Ras-transformed NIH3T3 cells, the Cy3-labeled reovirus is present not only within areas where the lysosomal associated membrane protein 2 staining is found but also appears diffusely throughout the cytoplasm, suggesting that perforation of the lysosome and penetration of the virus occurred in these cells. On the other hand, in resistant cells and E64 disassembly inhibited susceptible cells, a punctuate Cy3 fluorescence pattern was observed that was restricted to the same compartment as the lysosomal associated membrane protein 2, indicating lysosomal localization (Figure 2b).
Since it is proposed that reovirus transcription is not initiated until the virus has been disassembled and its core has entered the cytoplasm of cells,36 we next addressed whether reovirus transcription occurs in our susceptible and resistant cells, using Northern blotting as a quantitative approach. S1 reovirus transcripts 24 h.p.i. were detected only in the susceptible U87 and Ras-transformed NIH3T3 cells and were blocked when these cells were treated with E64. However, when cells were exposed to ISVPs, the presence of S1 transcripts were detected in both resistant and susceptible cells even in the presence of E64 (Figure 2c). These results are consistent with a lack of penetration of reovirus particles in the restrictive U118 glioma cell line and in the non-transformed NIH3T3 cells.
ISVPs infect reovirus–resistant cancer and non-transformed cell lines
We next examined whether infection with in vitro-generated ISVPs could bypass this disassembly block and result in a productive infection in the resistant cells. According to the model of the molecular basis of reovirus oncolysis,25 Ras signaling in susceptible cancer cells overcomes a translational block of reovirus transcripts found in resistant, non-transformed cells. Therefore, this model would predict that infection by ISVP particles should also be restrained in the resistant NIH3T3 and U118 cell lines through a translation block. In contrast, we found that ISVPs efficiently infected both the resistant glioma cell line U118 and the non-transformed NIH3T3 cells (Figure 3a ). Whereas reovirus protein synthesis from reovirion infection was apparent in the susceptible cell lines U87 and Ras-transformed NIH3T3—though not when these cells were treated with the disassembly inhibitor E64 (or the weak base ammonium chloride (data not shown))—viral protein synthesis following ISVP infection occurred in the resistant U118 and NIH3T3 cell lines as well as the susceptible cell lines even when treated with E64. Similar results were obtained by immunofluorescence using a rabbit anti-reovirus type-3 antibody followed by FITC–conjugated goat anti-rabbit IgG to detect positive reovirus infection (green fluorescent cells) (Figure 3b). Increased viral progeny was also measured in conditions where productive reovirus infection was observed (Figure 3c). We next examined the viability of the resistant and susceptible cells following reovirus or ISVP infection with or without treatment with E64 as described previously (Figure 3d). We found that the protease inhibitor E64 efficiently prevented oncolysis 48 h.p.i. of our susceptible cancer cells infected with reovirus but not with ISVPs. ISVP infection also resulted in a significant decrease in viability of the reovirus-resistant glioma U118 cells. The non-transformed NIH3T3 cells generally remained viable at 48 h.p.i.
Figure 3.

Infection of susceptible and resistant cell lines by proteolytically processed reovirus particles (infectious subviral particle or ISVPs). (a) Cells with or without treatment with the protease inhibitor E64 (100 μmol/l) 1 hour before reovirus exposure were pulse-labeled with [35S]methionine for 6 hours at 18 hours post infection (h.p.i.) with an MOI of 20 of reovirus or ISVPs (reovirions processed in 200 μg/ml of chymotrypsin for 30 minutes at 37°C). Cells were lysed; then reovirus proteins were immunoprecipitated from part of the lysate using rabbit polyclonal anti-reovirus antibodies and analyzed by SDS-PAGE and autoradiography. (b) Evaluation of reovirus infection using the same infection procedure as described previously but measured at 24 h.p.i. by immunofluorescence using a rabbit anti-reovirus type-3 antibody followed by FITC–conjugated goat anti-rabbit IgG to detect positive reovirus infection (green fluorescing cells). (c) Viral progeny production 48 h.p.i. with the foregoing treatments. Cells grown in 6-well plates were infected with reovirus at an MOI of 20; they were then assayed for progeny virus production by plaque titration on L929 cells following three rounds of freeze-thaw and serial dilution of the supernatants. All titration experiments were repeated in triplicate. (d) Cells grown to 50% confluence were infected with reovirus or ISVP at an MOI of 20. Cell viability was measured at 48 h.p.i. by MTT assay, and metabolically active cells were quantified by scanning the plates at the 595-nm reference wavelength in a microtiter plate reader. A MTT reagent without cells was used as background control. The experiments were performed in triplicate. (e) Evaluation of ISVP infection of other reovirus resistant cells—the glioma U343, the Burkitt lymphoma cell line Daudi, and the normal human foreskin fibroblast HS68—as measured either by immunofluorescence (left panel), metabolic SDS-PAGE as in a (middle panel), or MTT/WST-1 for viability (bottom panel) and proteolytic reovirus processing (right panel).
Finally, to determine whether the ability of a cell to process reovirus is a key determinant of infectivity in a broader range of cells, we tested other reovirus-resistant cells: the glioma U343, the Burkitt lymphoma cell line Daudi, and the normal human foreskin fibroblast HS68. In all cases we found that infection with ISVPs—as measured by immunofluorescence or metabolic SDS-PAGE to detect viral protein synthesis and by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazoium bromide (MTT) or 4-(3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio)-1,3-benzene disulfonate (WST-1) assay to measure viability—also resulted in positive infection and cell lysis (Figure 3e). Restriction in reovirus disassembly was also observed in these cells (Figure 3e, right panel). Given that ISVPs readily infect both susceptible and disassembly-restrictive cancer and normal cells, these results further suggest that the critical factor of resistance is at the level of reovirus penetration into the cytoplasm.
Reovirus protein synthesis is not inhibited by synthetic dsRNA treatment in transformed cell line
It has been proposed25 that resistance to reovirus infection is the direct result of inhibition of viral translation. In contrast, our data using ISVPs show that “resistant” cells (i.e., resistant to infection with virions) support a productive infection; ISVPs bypass the limitations of viral entry, uncoating, and penetration into the cytoplasm. To further examine the proposed viral translation block in resistant cells, we assessed whether pretreatment of cells before reovirus infection with synthetic dsRNA (poly(I:C) (20 μg/ml)), a potent activator of the dsRNA-dependent pathways of interferon and other anti-viral mechanisms, could inhibit viral protein synthesis. We found that viral protein synthesis was reduced in the non-transformed cells (NIH3T3 and HS68 cells) that were pretreated with poly(I:C) and infected with ISVPs. However, exposure of dsRNA did not prevent reovirus/ISVP infection of the susceptible transformed cells (U87 glioma, Ras-transformed NIH3T3, and the Burkitt lymphoma Raji cell lines). In addition, ISVP infection of the reovirus resistant glioma U118 and Burkitt lymphoma Daudi was not prevented by poly(I:C) treatment (Figure 4a ). Moreover, reovirus exposure to NIH3T3 cells 6 hours before ISVP infection did not result in the inhibition of viral protein synthesis that synthetic dsRNA provided (Figure 4b). Reovirus may be insufficient to stimulate an anti-viral response against ISVP infection in NIH3T3 cells owing to a lack of penetration by the virus. These data support a general model whereby cancer cells—but not normal cells—have defects in their anti-viral mechanism.37 They also suggest that, in reovirus-resistant cancer cells, the foremost factor in the resistance of these transformed cells is at the level of reovirus disassembly and not at the level of a viral messenger RNA translation block. Normal cells may benefit from the additional level of protection provided by an activated anti-viral pathway, but it appears that their primary resistance to reovirus resides in the inability to efficiently uncoat reovirus.
Figure 4.

Susceptible and resistant cell lines exposed to synthetic dsRNA before reovirus ISVP infection. (a) Cells with or without 20 μg/ml of synthetic dsRNA poly(I:C) treatment 6 hours before reovirus exposure were pulse-labeled with [35S]methionine for 6 hours at 18 hours after infection with a multiplicity of infection (MOI) of 20 of reovirus or ISVPs (reovirions processed in 200 μg/ml of chymotrypsin for 30 minutes at 37°C). Cells were lysed, and reovirus proteins were immunoprecipitated from the lysate using rabbit polyclonal anti-reovirus antibodies and then analyzed by SDS-PAGE and autoradiography. (b) NIH3T3 cells were pretreated with poly(I:C) or reovirus at an MOI of 20, 6 hours before reovirus/ISVP infection at an MOI of 20, and then processed as in b.
Cathepsin B and L inhibitors prevent reovirus oncolysis of susceptible cancer cells
To characterize the proteolytic requirement of susceptible cancer cells to reovirus, we first investigated the proteases cathepsin B and L. Both have been reported to mediate reovirus disassembly and permit reovirus infection.14 , 17 In addition, these proteases have been implicated in the progression of various cancers, including gliomas,38 , 39 and are elevated by Ras activation in NIH3T3 cells.40 We determined whether cathepsin B and L are critical in promoting permissiveness to reovirus in susceptible cell lines. Reovirus-susceptible cells treated or untreated for 6 hours with 10 μmol/l of the cathepsin B (CA-074) and L (Inhibitor III) (Invitrogen) were lysed, and equal amounts of protein from each sample were assayed for cathepsin B or L protease activity, respectively. Both inhibitors strongly impeded the activity of their respective protease and also had some inhibiting effect on the activity of the other cathepsin (Figure 5a ). We then found that inhibition of cathepsin B and L 1 hour before reovirus infection significantly protected susceptible cells against reovirus oncolysis 48 h.p.i. (Figure 5b). The combination of both inhibitors had the strongest effect in blocking reovirus oncolysis in the susceptible cell lines U87, Ras-transformed NIH3T3, and L929. As anticipated, ISVP infection was unaffected by these inhibitors. Furthermore, this result was specific to cathepsins because we did not find protection from infection when these cell lines were treated with the broad-spectrum synthetic matrix metalloproteinase inhibitors AG3340 or BB-94 (data not shown).
Figure 5.

Activity and involvement of the proteases cathepsin B and L for reovirus oncolysis. (a) The specific inhibitors CA-074 (Cathepsin B) and Inhibitor III (Cathepsin L) (Invitrogen) were added to cells at a concentration of 10 μmol/l for 6 hours before lysis. An equal amount of protein was assessed for cathepsin L or B activity as measured by fluorescence, following the procedure recommended by the manufacturer (Invitrogen). Results are presented as percentages of the total activity of untreated cells measured in relative fluorescence units. (b) Susceptible cells were treated with 10 μmol/l of either or both cathepsin B and L inhibitors for 1 hour before infection with reovirus or ISVP with an MOI of 20. Reovirus oncolysis was assessed by MTT 48 hours after infection; the remaining viability is presented.
Productive reovirus infectionin tumors of the in vitro disassembly-restrictive U118 glioma cell line
Since the microenvironment of tumors in vivo contains a number of proteases that may help promote reovirus disassembly, we decided to challenge the reovirus-restrictive U118 cell line grown as a tumor in mice with a single intratumoral injection of reovirus. U118 tumors were allowed to grow until the establishment of a palpable mass and were then challenged with 1 × 107 plaque forming units (pfu) of live reovirus or saline intratumorally. We observed a lack of tumor growth in animals treated with reovirus (Figure 6a ). Hematoxylin and eosin stained sections showed necrosis of tumor cells after live reovirus treatment, and immunohistochemistry with a polyclonal anti-reovirus antibody showed the presence of reovirus (brown) in the U118 tumor; uninfected tumors showed no staining (Figure 6b). We then determined the levels of cathepsin B and L activity from in vivoglioma tumor samples grown in severe combined immunodeficiency mice compared to their respective glioma cells grown in culture, finding elevated cathepsin activity for both U87 and U118 tumors in vivo (Figure 6c). These results provide additional evidence that cell lines resistant to reovirus oncolysis in vitro can be rendered permissive once grown in animals and that this may be due to the proteolytic microenvironment of the tumor. The permissivenessof the U118 cells in vivo (in contrast to their resistance in vitro) was not a consequence of upregulated Ras in the in vivosetting, since Ras–GTP levels from U118 tissue lysates remained low—as in U118 cells grown in culture (Figure 6d).
Figure 6.
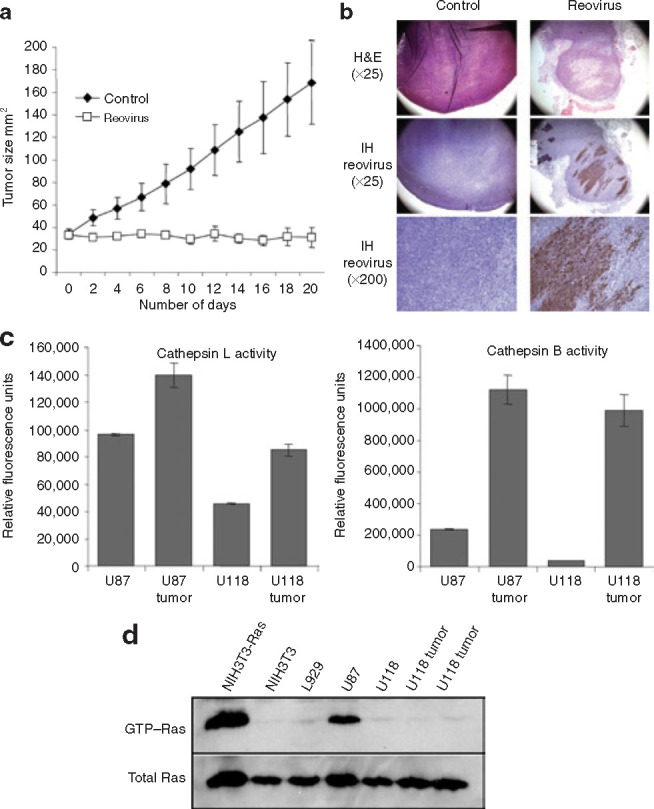
Reovirus infection of tumors derived from the in vitro reovirus-resistant glioma U118 cells. (a) Approximately 1 × 107 U118 cells were injected subcutaneously in severe combined immunodeficiency/non-obese diabetic mice and, once the tumor reached palpable size (usually ∼5 × 5 mm2), a single injection of 1 × 107pfu of reovirus (n = 5) or saline (n= 5) was performed intratumorally. Tumor size was measured every second day following reovirus injection. (b) Hematoxylin and eosin (H&E) and immunohistochemistry (IH) of control and reovirus treated U118 tumors. H&E stained section (×400 magnification) shows necrosis of tumor cells 20 days after live reovirus treatment; IH stained section (×400 magnification) of remaining tumor cells reveals reovirus proteins (brown) whereas control tumor shows no staining. (c) Cells and tumor tissues were lysed; an equal amount of protein was assessed for cathepsin B or L activity, as in Figure 5, and measured as relative fluorescence units. (d) Equal amount of protein from cells or tissue samples were subjected to a Ras activation assay kit, as described in Materials and Methods; Ras–GTP levels were measured using a RAS10 monoclonal antibody followed by incubation in goat anti-mouse-horseradish peroxidase antibody and were visualized using enhanced chemiluminescence (Amersham Biosciences).
Discussion
Reovirus is currently being evaluated in clinical trials as a novel anti-cancer therapeutic23 , 24 (http://www.oncolyticsbiotech.com). The majority of cancer cell lines are susceptible to infection and killing by reovirus, and only a small number are resistant.26 , 32 , 33 , 34 , 41 In spite of intensive preclinical studies, the mechanisms underlying the susceptibility of cancer cells are not fully understood. In this study, we focused on host-mediated proteolytic reovirus disassembly as an important mediator of susceptibility. Using radiolabeled, fluorescently labeled, and reovirus virions and infectious subvirion particles, we found that reovirus-resistant cancer cells and non-transformed cells restrict infection in vitro because they fail to mediate viral disassembly and penetration. Our data show that proteolysis of the reovirus outer-capsid protein is necessary for the selective infection and killing of cancer cells.
The demonstration that proteolytic disassembly of reovirus can be a primary determinant of susceptibility requires a revision of the proposed model,25 which we call the “reovirus-ras” model, for the molecular basis of reovirus oncolysis. According to the reovirus-ras model, ras signaling (through an unknown mechanism perhaps through p38 signaling27) releases a block in translation of viral transcripts by the dsRNA-activated protein kinase (PKR) found in resistant cells. In support of the model, viral transcripts were found to be equivalent with respect to both susceptible and resistant cells. However, we have four observations that are inconsistent with the model. First, we found a restriction of reovirus uncoating in reovirus-resistant cancer cell lines and non-transformed cells. Infection by reovirus requires the disassembly of its outermost capsid in order to penetrate the host and initiate viral gene transcription.9 , 10 , 11 Second, we found that ISVPs efficiently infected “reovirus-resistant” cells. Golden et al. also found that the addition of proteases to the media of resistant cells rendered them susceptible to infection.31 Third, the infection of reovirus-resistant cells by ISVPs was not prevented in cells transformed by pretreatment with synthetic dsRNA. The reovirus-ras model predicts that this treatment would activate PKR, resulting in a translation block and an unproductive infection. Finally, consistent with the ineffective penetration of the virus in resistant cells, we found a lack of reovirus S1 transcripts in resistant cells rather than the equivalent levels of transcripts predicted by that model. Taken together, these results suggest that reovirus disassembly is a critical determinant for oncolysis of cancer cells and a primary factor in resistance to reovirus oncolysis.
We have no data that explain the discordance between our observations and those that led to the development of the reovirus-ras model. That model, which was largely based on studies of murine fibroblasts transfected with high levels of oncogenic ras, was never proposed to account for all cases of susceptibility. Our studies demonstrate that NIH3T3 cells have very low levels of binding and entry of reovirus, so the reovirus-ras model may apply most directly to cell lines with inefficient viral entry; its generalization to the signaling milieu of human cancers remains uncertain. Since reovirus infects a vast majority of cancer cell lines and since most of these do have ras activation, it follows that mutations leading to activated ras may contribute to reovirus susceptibility by increasing protease activity40 (e.g., by increasing expression or reducing expression of an inhibitor) or otherwise facilitating viral entry and disassembly. Studies that address these questions are currently underway in our laboratories.
Many other viruses (adenovirus, Newcastle disease virus, measles, severe acute respiratory syndrome, Ebola, etc.) also rely on proteolytic processing of a capsid or envelope protein to permit cell entry and replication.42 , 43 , 44 , 45 , 46 In this study, we found that specific inhibitors of cathepsin B and L block reovirus oncolysis but that broad-spectrum matrix metalloproteinase inhibitors did not. Others have also reported that a number of different proteases (cathepsin S, neutrophil elastase, typsin, and others) are capable of converting intact reovirus virions into ISVPs that can enter cells without additional proteolysis.8 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Elevated protease expression and/or activity are common consequences of tumorigenesis (e.g., invasion, angiogenesis, and metastasis).38 , 39 Consistent with this, we found that tumors expressed more cathepsin B and L activity than their in vitro counterparts. The in vivoproteolytic microenvironment may explain why a highly resistant cell line became susceptible to reovirus-induced oncolysis when established as a tumor in vivo.
There are three broad implications of our study for the use of oncolytic viruses in the clinic. First, it is encouraging that the efficiency of some viruses might be enhanced in vivoby the protease-rich environment of the tumor. However, this complicates predictions of efficacy based on in vitro assays because cells that are resistant in vitro may be susceptible in patients. Second, improving disassembly and viral entry may be important strategies to employ in the design and use of oncolytic viruses for clinical use. Isolation of reovirus variants that disassemble more efficiently or require cancer-type specific proteases for uncoating may increase the efficacy of reovirus in vivo and provide further specificity. A related strategy has been used with a measles virus variant specifically engineered to selectively fuse to and infect cancer cells that overexpress metalloproteinases.44 Finally, a better understanding of the molecular basis of reovirus oncolysis will allow us to properly select the patients who are most likely to benefit from this treatment and optimize its use.
Materials and Methods
Cells and reovirus. Human malignant glioma cell lines U87, U118, U251, and U343, Burkitt lymphoma cell lines Raji and Daudi, the mouse fibroblasts NIH3T3 cell line, and L929 cells were obtained from American Type Culture Collection and were maintained as described.33 , 34 , 41 Activated Ras constructs in the pBABE retroviral vector were generously provided by P. W. Lee (Dalhousie University, Halifax, Canada), and transfection and selection of NIH3T3 cells were as described.27 The Dearing strain of reovirus serotype 3 and intermediate subviral particles were obtained as described.32 Purified virions containing [35S]-labeled proteins were obtained by culturing the cells with [35S]-methionine (Amersham Biosciences, Arlington Heights, IL) into a medium of plated cells (∼50 μCi/ml) for 12 hours at 12 h.p.i. Cells were collected and freeze-thawed three times; [35S]-radiolabeled reovirus was purified as described. For Cy3-labeled reovirus, Cy3 monofunctional reactive dye (Amersham Biosciences, Arlington Heights, IL) was added to purified reovirus virions (5 × 1010particles per ml) and incubated at 25°C for 45 minutes. Conjugated virus was dialyzed at 4°C against phosphate-buffered saline (PBS) (pH 7.0) overnight to remove free dye. Conjugation of reovirus with fluorescent dyes has been shown to yield labeling of viral outer-capsid proteins σ1, σ3, μ1, and λ2 as well as a fivefold decrease in viral infectivity.19 However, we did not detect a significant decrease in infectivity of our conjugated reovirus.
Reovirus binding analysis. Cells were plated in triplicates at a density of 105 per well in a 6-well culture dish and incubated for 24 hours before treatment. [35S]-methionine-labeled reovirions (2.5 × 104 counts per minute/sample corresponding to about 1,000 MOI) were added to cells in minimal medium and allowed to bind to cells for 1 hour at 4°C. Cells were subsequently washed twice with PBS to remove unbound virions, and samples were collected by lysis of the cells (PBS containing 1% Triton X-100, 0.5% sodium deoxycholate, and 1 mmol/l EDTA); samples were then exposed to scintillation counting. For negative binding control, rabbit polyclonal anti-reovirus type-3 serum was added together with labeled virus.
Reovirus disassembly analysis. Cells were treated as described previously. After binding, fresh medium was added to cells, which were then returned to a 37 °C incubator. At the indicated times p.i., cells were washed in PBS and lysed. Lysates were cleared of debris by centrifugation, and supernatants were submitted to SDS-PAGE followed by autoradiography. For E64 protease inhibitor (Sigma-Aldrich, St. Louis, MO), cells were exposed to 100 μmol/l of E64 in culture medium 1 hour before reovirus infection. After binding, E64 was again added to the cells to yield a final concentration of 100 μmol/l. Western blotting (on equal amounts of protein from lysates) with a specific anti-μ1 reovirus antibody (10H2) (1/1,000)47 was performed for the same experiment but using unlabeled reovirions.
Reovirus uptake and trafficking. Cells were grown on slides and infected with Cy3-labeled reovirus at an MOI of 5,000 pfu/cells (the minimum amount of virus to detect fluorescence) for 1 hour at 4°C. Cells were subsequently washed in PBS and returned to 37°C in fresh medium for 24 hours. Cells were fixed in 4% paraformaldehyde for 20 minutes at room temperature, followed by four washes with PBS. The fixed cells were treated with 10% goat serum in PBS and then incubated with mouse monoclonal lysosomal associated membrane protein 2 lysosomal marker (1/250) (Abcam, Cambridge, MA) followed with secondary FITC–conjugated rabbit anti-mouse IgG (1/250) (Sigma-Aldrich, St. Louis, MO) for 1 hour at room temperature; cells were then photographed with a fluorescent Zeiss microscope using the multiple acquisition software provided by Zeiss (magnification ×400).
Reovirus infection. Approximately 105 cells of each cell line were dispensed into 12-well plates or 2-well glass slides and infected with reovirus or ISVPs at an MOI of 20 pfu/cell. For E64 protease inhibitor (Sigma-Aldrich, St. Louis, MO) treatment, cells were exposed to 100 μmol/l of E64 in culture medium 1 hour before reovirus infection. For synthetic dsRNA (poly(I:C), Amersham Biosciences, Arlington Heights, IL) treatment, cells were exposed to 20 μg/ml of the synthetic dsRNA in the medium for a period of 6 hours before reovirus or ISVP infection. Virus was allowed to bind for 45 minutes at 4°C, after which E64 was again added to the cells to a final concentration of 100 μmol/l. For metabolic labeling, [35S]-methionine was added to the culture medium at 18 h.p.i. for a period of 6 hours; then viral protein synthesis was assessed as previously described.32 , 41 For immunofluorescent studies, cells were grown on slides and processed as described but then incubated with rabbit polyclonal anti-reovirus type-3 serum (diluted 1/5,000 in PBS) for 1 hour at room temperature, washed, and incubated with secondary antibody (FITC–conjugated goat anti-rabbit IgG diluted 1/250 in PBS) (Cedarlane, Hornby, Canada) for 1 hour at room temperature.
Progeny virus production. Approximately 2 × 105 cells grown in 6-well plates were infected with reovirus at an MOI of 20. At 48 h.p.i., the plates were frozen and stored at –70°C until use. To assay for progeny virus production, the plates were subjected to three rounds of freeze-thaw, and serial dilution of the supernatants were used for plaque titration on L929 cells. All titration experiments were repeated in triplicate.
Northern blot analysis. Digoxygenin-labeled riboprobes were generated from the plasmid pGEM-4Z-S1, which contained the open reading frame of the S1 complementary DNA. The plasmid was linearized by restriction enzyme SsaI. Antisense probes to detect positive strand transcript were synthesized using the T7 polymerases and DIG-RNA labeling reagents (Roche Diagnostics, Laval, Canada) according to the manufacturer's recommendation. Probes were precipitated with ethanol and then assessed for quality by agarose gel electrophoresis. Cells were infected with reovirus at an MOI of 20 pfu and the virus was allowed to bind for 1 hour at 4°C; inoculum was then replaced by fresh medium and put at 37°C before total cellular RNA was extracted 24 h.p.i. using RNeasy according to the manufacturer's protocol (Qiagen Inc, Mississauga, Canada). Five micrograms of total RNA were resolved on 1.2% agarose-formaldehyde gel and transferred electrophoretically (0.5 A overnight at 4°C) to Hybond-NTM nylon membranes (Amersham Biosciences, Arlington Heights, IL) in 0.5× Tris/acetate/EDTA buffer (pH 7). After prehybridization for 5 hours, the hybridizations were carried out at 50–55°C for 24–36 hours, followed by high-stringency washing at 68°C in 0.1× SSC, 0.1% sodium dodecyl sulfate. Anti-DIG-HRP (Roche) was used for detection of the probe using enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL) on film (Kodak, Chalon-sur-Chaone, France).
Viability assay. Cells grown to 50% confluence were infected with reovirus or ISVP at an MOI of 20. Cell viability was measured at 48 h.p.i. by MTT assay (Sigma-Aldrich, St. Louis, MO) and by WST-1 assay (Roche Diagnostic, Laval, Canada) to measure cell viability of the Burkitt lymphoma Daudi cells as previously described.32 , 48
Cathepsin B and L inhibition and activity. The specific inhibitors CA-074 (Cathepsin B) and Inhibitor III (Cathepsin L) (Invitrogen, Burlington, Canada) were added to cells at a concentration of 10 μmol/l 1 hour before reovirus infection. Cathepsin B or L activity in susceptible and resistant cells, as well as in tumor tissues, was quantified using a proteolytic activity kit (Invitrogen, Burlington, Canada) according to the manufacturer's protocol.
Severe combined immunodeficiency mice studies. Six to eight-week old Fox-Chase severe combined immunodeficiency mice were obtained from the Jackson Laboratory (Bar Harbor, ME). The animals were maintained under specific pathogen-free conditions and according to a protocol approved by the University of Calgary Animal Care Committee. As a xenograft model, 1.0 × 107 U118 glioma cells were injected subcutaneously in the hind flank of the mice. Once palpable tumors were established (day 0), 1.0 × 107pfu of live reovirus in PBS was administered intratumorally (experimental group) or PBS was administered alone (control group). Two-dimensional tumor measurements were performed with calipers every other day for 25 days or until the animals showed severe morbidity due to excess tumor burden or due to complications arising from viral infection. For histology and immunohistochemistry studies, tumors (or remaining masses) taken from animals on day 20 after intratumoral reovirus (or saline) injection were fixed in 10% neutral buffered formalin, embedded in paraffin for histological analysis, and then processed as described.32 , 41
Ras activity. Ras–GTP levels from established human glioma cells lines, parental NIH3T3 and NIH3T3 transformed with Ras, L929 cells, and tumor tissues derived subcutaneously from U118 cells grown in severe combined immunodeficiency/non-obese diabetic mice were measured using a Ras activation assay kit (Upstate Biotechnology, Lake Placid, NY) according to manufacturer protocol and as described previously.32
Acknowledgments
This work was supported by grants to P.A.F. and D.L.S. from the National Cancer Institute of Canada with funds from the Canadian Cancer Society. L.A.S was supported by grant AI045990 from the National Institutes of Health. T.A. is supported by the Canadian Institute for Health Research and by the Alberta Heritage Foundation for Medical Research. A.L. holds a postdoctoral fellowship from the Alberta Heritage Foundation for Medical Research. We thank Clark H. Smith and his family for their generous support.
Footnotes
published online 22 May 2007
References
- 1.Tyler KL, Fields B. Reoviruses. Lippincott-Raven; Philadelphia, PA: 1996. [Google Scholar]
- 2.Ridinger DN, Spendlove RS, Barnett BB, George DB, Roth JC. Evaluation of cell lines and immunofluorescence and plaque assay procedures for quantifying reoviruses in sewage. Appl Environ Microbiol. 1982;43:740–746. doi: 10.1128/aem.43.4.740-746.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson GG, Muldoon RL. Viruses causing common respiratory infection in man. IV. Reoviruses and adenoviruses. J Infect Dis. 1973;128:811–866. doi: 10.1093/infdis/128.6.811. [DOI] [PubMed] [Google Scholar]
- 4.Sabin AB. Reoviruses. A new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described. Science. 1959;130:1387–1389. doi: 10.1126/science.130.3386.1387. [DOI] [PubMed] [Google Scholar]
- 5.Rosen L, Evans HE, Spickard A. Reovirus infections in human volunteers. Am J Hyg. 1963;77:29–37. doi: 10.1093/oxfordjournals.aje.a120293. [DOI] [PubMed] [Google Scholar]
- 6.Rosen L. [Respiratory enterovirus and reovirus infections.] Arch Gesamte Virusforsch. 1963;13:272–280. doi: 10.1007/BF01243853. [DOI] [PubMed] [Google Scholar]
- 7.Jane-Valbuena J, Breun LA, Schiff LA, Nibert ML. Sites and determinants of early cleavages in the proteolytic processing pathway of reovirus surface protein sigma3. J Virol. 2002;76:5184–5197. doi: 10.1128/JVI.76.10.5184-5197.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandran K, Nibert ML. Protease cleavage of reovirus capsid protein mu1/mu1C is blocked by alkyl sulfate detergents, yielding a new type of infectious subvirion particle. J Virol. 1998;72:467–475. doi: 10.1128/jvi.72.1.467-475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. Two modes of entry of reovirus particles into L cells. J Gen Virol. 1979;45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- 10.Chandran K, Farsetta DL, Nibert ML. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturzenbecker LJ, Nibert M, Furlong D, Fields BN. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bodkin DK, Nibert ML, Fields BN. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J Virol. 1989;63:4676–4681. doi: 10.1128/jvi.63.11.4676-4681.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J Virol. 1990;64:1830–1833. doi: 10.1128/jvi.64.4.1830-1833.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebert DH, Deussing J, Peters C, Dermody TS. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 15.Amerongen HM, Wilson GA, Fields BN, Neutra MR. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J Virol. 1994;68:8428–8432. doi: 10.1128/jvi.68.12.8428-8432.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dermody TS, Nibert ML, Wetzel JD, Tong X, Fields BN. Cells and viruses with mutations affecting viral entry are selected during persistent infections of L cells with mammalian reoviruses. J Virol. 1993;67:2055–2063. doi: 10.1128/jvi.67.4.2055-2063.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baer GS, Dermody TS. Mutations in reovirus outer-capsid protein sigma3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol. 1997;71:4921–4928. doi: 10.1128/jvi.71.7.4921-4928.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wetzel JD, Wilson GJ, Baer GS, Dunnigan LR, Wright JP, Tang DS. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J Virol. 1997;71:1362–1369. doi: 10.1128/jvi.71.2.1362-1369.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baer GS, Ebert DH, Chung CJ, Erickson AH, Dermody TS. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J Virol. 1999;73:9532–9543. doi: 10.1128/jvi.73.11.9532-9543.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golden JW, Schiff LA. Neutrophil elastase, an acid-independent serine protease, facilitates reovirus uncoating and infection in U937 promonocyte cells. Virol J. 2005;2:48. doi: 10.1186/1743-422X-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golden JW, Bahe JA, Lucas WT, Nibert ML, Schiff LA. Cathepsin S supports acid-independent infection by some reoviruses. J Biol Chem. 2004;279:8547–8557. doi: 10.1074/jbc.M309758200. [DOI] [PubMed] [Google Scholar]
- 22.Stoeckel J, Hay JG. Drug evaluation: Reolysin—wild-type reovirus as a cancer therapeutic. Curr Opin Mol Ther. 2006;8:249–260. [PubMed] [Google Scholar]
- 23.Morris DG, Forsyth PA, Paterson AH, Fonseca K, Difrancesco LM, Thompson BG. ASCO Annual Meeting. 2002. A phase I clinical trial evaluating intralesional Reolysin (reovirus) in histologically confirmed malignancies. Orlando, FL, USA. [Google Scholar]
- 24.Forsyth PA, Roldan G, George D, Wallace C, Morris DG, Cairncross J. A phase I trial of intratumoral (i.t.) administration of reovirus in patients with histologically confirmed recurrent malignant gliomas (MGs) Journal of Clinical Oncology, ASCO Proceedings. 2006 doi: 10.1038/sj.mt.6300403. [DOI] [PubMed] [Google Scholar]
- 25.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 27.Norman KL, Hirasawa K, Yang AD, Shields MA, Lee PW. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci USA. 2004;101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thirukkumaran CM, Luider JM, Stewart DA, Cheng T, Lupichuk SM, Nodwell MJ. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood. 2003;102:377–387. doi: 10.1182/blood-2002-08-2508. [DOI] [PubMed] [Google Scholar]
- 29.Smakman N, van den Wollenberg DJ, Borel Rinkes IH, Hoeben RC, Kranenburg O. Sensitization to apoptosis underlies KrasD12-dependent oncolysis of murine C26 colorectal carcinoma cells by reovirus T3D. J Virol. 2005;79:14981–14985. doi: 10.1128/JVI.79.23.14981-14985.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M, Egan C, Alain T, Urbanski SJ, Lee PW, Forsyth PA. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene. 2007 doi: 10.1038/sj.onc.1210189. (epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 31.Golden JW, Linke J, Schmechel S, Thoemke K, Schiff LA. Addition of exogenous protease facilitates reovirus infection in many restrictive cells. J Virol. 2002;76:7430–7443. doi: 10.1128/JVI.76.15.7430-7443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alain T, Kim M, Johnston RN, Urbanski S, Kossakowska AE, Forsyth PA. The oncolytic effect in vivo of reovirus on tumour cells that have survived reovirus cell killing in vitro. Br J Cancer. 2006;95:1020–1027. doi: 10.1038/sj.bjc.6603363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilcox ME, Yang W, Senger D, Rewcastle NB, Morris DG, Brasher PM. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93:903–912. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- 34.Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J Natl Cancer Inst. 2006;98:1546–1557. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 36.Farsetta DL, Chandran K, Nibert ML. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J Biol Chem. 2000;275:39693–39701. doi: 10.1074/jbc.M004562200. [DOI] [PubMed] [Google Scholar]
- 37.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 38.Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3:489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 39.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 40.Chambers AF, Colella R, Denhardt DT, Wilson SM. Increased expression of cathepsins L and B and decreased activity of their inhibitors in metastatic, ras-transformed NIH 3T3 cells. Mol Carcinog. 1992;5:238–245. doi: 10.1002/mc.2940050311. [DOI] [PubMed] [Google Scholar]
- 41.Alain T, Hirasawa K, Pon KJ, Nishikawa SG, Urbanski SJ, Auer Y. Reovirus therapy of lymphoid malignancies. Blood. 2002;100:4146–4153. doi: 10.1182/blood-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- 42.Medina-Kauwe LK. Endocytosis of adenovirus and adenovirus capsid proteins. Adv Drug Deliv Rev. 2003;55:1485–1496. doi: 10.1016/j.addr.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 43.Nagai Y, Klenk HD, Rott R. Proteolytic cleavage of the viral glycoproteins and its significance for the virulence of Newcastle disease virus. Virology. 1976;72:494–508. doi: 10.1016/0042-6822(76)90178-1. [DOI] [PubMed] [Google Scholar]
- 44.Springfeld C, von Messling V, Frenzke M, Ungerechts G, Buchholz CJ, Cattaneo R. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 2006;66:7694–7700. doi: 10.1158/0008-5472.CAN-06-0538. [DOI] [PubMed] [Google Scholar]
- 45.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Virgin HW, 4th, Mann MA, Fields BN, Tyler KL. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol. 1991;65:6772–6781. doi: 10.1128/jvi.65.12.6772-6781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang WQ, Senger D, Muzik H, Shi ZQ, Johnson D, Brasher PM. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003;63:3162–3172. [PubMed] [Google Scholar]