

The failure of remyelination in multiple sclerosis is largely unexplained. Lindner et al. report that glial cells in demyelinating lesions show increased expression of fibroblast growth factor 9 (FGF9). This induces astrocyte-dependent responses that inhibit remyelination and stimulate expression of pro-inflammatory chemokines, supporting a feedback loop that amplifies disease activity.
Keywords: multiple sclerosis (MS), FGF, chemokines, remyelination, inflammation

The failure of remyelination in multiple sclerosis is largely unexplained. Lindner et al. report that glial cells in demyelinating lesions show increased expression of fibroblast growth factor 9 (FGF9). This induces astrocyte-dependent responses that inhibit remyelination and stimulate expression of pro-inflammatory chemokines, supporting a feedback loop that amplifies disease activity
Abstract
Remyelination failure plays an important role in the pathophysiology of multiple sclerosis, but the underlying cellular and molecular mechanisms remain poorly understood. We now report actively demyelinating lesions in patients with multiple sclerosis are associated with increased glial expression of fibroblast growth factor 9 (FGF9), which we demonstrate inhibits myelination and remyelination in vitro. This inhibitory activity is associated with the appearance of multi-branched ‘pre-myelinating’ MBP+/PLP+ oligodendrocytes that interact with axons but fail to assemble myelin sheaths; an oligodendrocyte phenotype described previously in chronically demyelinated multiple sclerosis lesions. This inhibitory activity is not due to a direct effect of FGF9 on cells of the oligodendrocyte lineage but is mediated by factors secreted by astrocytes. Transcriptional profiling and functional validation studies demonstrate that these include effects dependent on increased expression of tissue inhibitor of metalloproteinase-sensitive proteases, enzymes more commonly associated with extracellular matrix remodelling. Further, we found that FGF9 induces expression of Ccl2 and Ccl7, two pro-inflammatory chemokines that contribute to recruitment of microglia and macrophages into multiple sclerosis lesions. These data indicate glial expression of FGF9 can initiate a complex astrocyte-dependent response that contributes to two distinct pathogenic pathways involved in the development of multiple sclerosis lesions. Namely, induction of a pro-inflammatory environment and failure of remyelination; a combination of effects predicted to exacerbate axonal injury and loss in patients.
Introduction
Experimental studies demonstrate the default response to immune-mediated demyelination in the CNS is rapid remyelination by oligodendrocytes derived from an endogenous pool of mitotic CSPG4+ (also known as NG2+) oligodendrocyte progenitor cells (OPCs; Tripathi et al., 2010). However, in multiple sclerosis this repair mechanism frequently fails resulting in chronically demyelinated plaques of gliotic scar tissue (Franklin and Ffrench-Constant, 2008). This has profound pathophysiological consequences as loss of myelin not only disrupts axonal function per se, but also compromises the physical integrity of affected axons by increasing their susceptibility to inflammatory mediators (Redford et al., 1997) and disrupting trophic support provided by myelinating oligodendrocytes (Fünfschilling et al., 2012). This combination of effects will exacerbate axonal loss in patients (Nave, 2010; Franklin et al., 2012) and restoring endogenous remyelination is now considered an attractive strategy to prevent further axonal damage and restore function (Irvine and Blakemore, 2008; Duncan et al., 2009; Deshmukh et al., 2013). However achieving this goal in multiple sclerosis requires a far better understanding of why remyelination fails and how this is related to the inflammatory component of the disease.
Failure of remyelination is not due to an intrinsic defect in cells of the oligodendrocyte lineage as demonstrated by the presence of remyelinated ‘shadow plaques’ in many patients at autopsy (Prineas and Connell, 1979; Prineas et al., 1993; Raine and Wu, 1993; Patrikios et al., 2006; Patani et al., 2007). However, the efficacy of this endogenous repair mechanism declines with disease progression (Patani et al., 2007; Goldschmidt et al., 2009). Studies in rodents suggest this may be due to the cumulative effects of repeated or sustained episodes of inflammatory demyelination on OPC recruitment or survival (Linington et al., 1992), which will be exacerbated by effects related to normal ageing (Shen et al., 2008). Nonetheless in patients the mechanisms responsible for failure of remyelination remain obscure. The continued presence of significant numbers of OPCs in many lesions suggests this involves factors that inhibit OPC differentiation (Scolding et al., 1999; Chang et al., 2002; Wolswijk, 2002; Kuhlmann et al., 2008), although effects reducing OPC recruitment and/or survival will also impact on remyelination efficacy (Piaton et al., 2009).
Experimental studies suggest a large number of different mechanisms are available that might inhibit OPC differentiation and remyelination in multiple sclerosis. These include factors affecting interactions with extracellular matrix components, altered availability of growth factors and/or OPC guidance cues or which trigger inappropriate re-expression of developmental signalling pathways (reviewed in Kotter et al., 2011). Convincing arguments are available to support a role for each of these in lesion development, but their relative importance with respect to failure of remyelination remains unclear. Resolving this question is important as it will determine which effector pathway(s) provide the most appropriate target for treatment strategies designed to restore endogenous remyelination.
One potential target is signal transduction by members of the fibroblast growth factor (FGF) family (Bansal, 2002; Mohan et al., 2014), which play important roles in coordinating neuronal and glial differentiation and survival during CNS development (Guillemot and Zimmer, 2011). In the context of multiple sclerosis, previous studies focused on the possible involvement of FGF2 (Sarchielli et al., 2008), a pleiotrophic growth factor expressed by macrophages and microglia in active white matter lesions (Clemente et al., 2011). However, it is difficult to deduce the functional significance of increased expression of FGF2, as it can exert both potentially beneficial as well as detrimental effects in the adult CNS. On one hand it may limit axonal loss by virtue of its neuroprotective properties (Ruffini et al., 2001; Rottlaender et al., 2011) or enhance remyelination by supporting the expansion and specification of OPCs derived from neural stem cells (McKinnon et al., 1990; Azim et al., 2012). However, any benefit provided by these effects are thought to be negated by other effects that may inhibit OPC differentiation and compromise remyelination (Bansal, 2002; Butt and Dinsdale, 2005; Tobin et al., 2011; Azim et al., 2012; Mierzwa et al., 2013). Unravelling the pathophysiological significance of FGF2 is complicated further by expression of other FGF family members in the CNS that signal using the same FGF receptor (FGFR) isoforms (Tacer et al., 2010; Mohan et al., 2014). These include FGF9 that is expressed constitutively by many adult neurons, and mimics the ability of FGF2 to suppress myelin protein expression by differentiating OPCs (Cohen and Chandross, 2000); an observation that led us to investigate whether it might also contribute to lesion formation in multiple sclerosis.
We now report lesion formation is associated with increased expression of fibroblast growth factor 9 (FGF9) by astrocytes and oligodendrocytes and demonstrate this pleiotropic growth factor inhibits myelination and remyelination in vitro. This is not due to a direct effect of FGF9 on cells of the oligodendrocyte lineage, but is an off target effect mediated by soluble astrocyte-derived factors that inhibit the terminal differentiation of myelinating oligodendrocytes. In tissue culture this is associated with the appearance of multi-branched ‘pre-myelinating’ oligodendrocytes that extend processes that interact with axons, but fail to form myelin sheaths; an oligodendrocyte phenotype reminiscent of that seen in chronically demyelinated multiple sclerosis lesions (Chang et al., 2002).
Transcriptional profiling of myelinating cultures revealed FGF9 modulated expression of genes involved in several pathways identified previously as contributing to failure of remyelination in multiple sclerosis, in particular several associated with components of the extracellular matrix (Kotter et al., 2011). These included effects predicted to impact on remyelination due to altered CD44/hyaluronan signalling (Tuohy et al., 2004; Back et al., 2005; Chang et al., 2012; Bugiani et al., 2013), as well as increased expression of chondroitin sulphate proteoglycans (Siebert et al., 2011) and fibronectin (Maier et al., 2005; Sisková et al., 2006, 2009; Stoffels et al., 2013). These changes were associated with significantly increased expression of mRNAs encoding ADAMTS1 and other metalloproteases associated with extracellular matrix remodelling. Subsequent experiments confirmed increased expression/activity of these tissue inhibitors of metalloproteinases (TIMP)-sensitive proteases contribute significantly to the mechanisms by which FGF9 inhibits myelination. In addition FGF9 also induced an innate immune signature characterized by increased astrocytic expression of Ccl2 and Ccl7; pro-inflammatory chemokines that contribute to the development of inflammatory responses in the CNS (Fuentes et al., 1995; Mahad and Ransohoff, 2003; Thompson and Van Eldik, 2009; Renner et al., 2011; Trujillo et al., 2013).
In summary our data demonstrate that FGF9 can initiate a complex astrocytic response predicted to compromise remyelination, while at the same time stimulating microglial/macrophage recruitment in multiple sclerosis lesions; a combination of effects predicted to exacerbate axonal loss and accumulation of disability in patients with progressive multiple sclerosis (Franklin et al., 2012). We suggest inhibition of FGF-mediated signal transduction in astrocytes may provide an effective target for treatment strategies that simultaneously enhance endogenous remyelination and suppress intrinsic pro-inflammatory responses in the CNS.
Materials and methods
Immunohistochemical analysis of human tissues
Immunohistochemical studies were performed on archived formalin-fixed, paraffin-embedded brain material from 14 patients with multiple sclerosis and six controls without neurological disease or evidence of brain lesions (Table 1). Immunohistochemical studies were performed on consecutive deparaffinized 5 µm sections after endogenous peroxidase was blocked with H2O2/methanol. Antigen retrieval was performed using citrate buffer (pH 6.0) and non-specific antibody binding was blocked by incubating sections in 10% foetal calf serum. Primary antibodies to FGF9 (rabbit polyclonal, Abcam, 1:1000), carbonic anhydrase II (CAII; sheep polyclonal, The Binding Site, 1:1000), glial fibrillary acidic protein (GFAP; mouse monoclonal; Neomarkers; 1:200) and amyloid precursor protein (APP; mouse monoclonal; Chemicon, 1:1000) were applied overnight at 4°C. Antibody binding was routinely visualized using appropriate species-specific horse radish peroxidase conjugated secondary antibodies and 3,3’-diaminobenzidine (DAB). Cell nuclei were counterstained with haematoxylin before mounting.
Table 1.
Summary of FGF9 expression in healthy controls and patients with multiple sclerosis
| FGF9 expression |
||||||||
|---|---|---|---|---|---|---|---|---|
| Patient ID | Disease type | Age | Gender | GM | WM | NAWM | Lesion rim | Lesion core |
| 8-04-1 | Control | 36 | F | +++ | + | |||
| 579-91-3 | Control | 46 | M | +++ | + | |||
| 28-03-2 | Control | 42 | F | +++ | + | |||
| 132-92-4 | Control | 65 | M | +++ | + | |||
| 421-91-5 | Control | 80 | F | ++ | + | |||
| 68-93-4 | Control | 83 | M | +++ | + | |||
| 581-96-10D | Acute MS | 35 | M | ++ | + | +++ | ++ | |
| 90-09-6 | Acute MS | 69 | F | +++ | + | +++ | ++ | |
| A01-144 | Acute MS | 34 | F | +++ | + | +++ | ++ | |
| S403-97 | Acute MS | 45 | M | +++ | + | ++ | ++ | |
| 270-99-2 | Acute MS | 45 | M | +++ | + | ++ | ++ | |
| 70-93-6 | Acute MS | 78 | M | +++ | + | +++ | +++ | |
| Spanien C2 | RRMS | 40 | F | +++ | + | ++ | ++ | |
| 39-03-15 | Chronic active | 67 | M | +++ | - | + | - | |
| 146-01-8 | Chronic active | 41 | M | +++ | - | + | - | |
| 144-90-3 | Chronic active | 77 | F | +++ | - | + | - | |
| 67-05-9 | Chronic active | 34 | M | +++ | - | + | - | |
| 244-94-7 | Chronic inactive | 81 | F | +++ | - | - | - | |
| 285-81-1 | Chronic inactive | 78 | F | +++ | - | - | - | |
| 72-83-6 | Chronic inactive | 64 | F | +++ | - | - | - | |
GM = grey matter; WM = white matter; MS = multiple sclerosis; NAWM = normal appearing white matter; RRMS = relapsing remitting multiple sclerosis.
Fluorescence double immunostainings were successfully performed on biopsy tissue from five patients with early active demyelinating lesions using anti-FGF9 (Abcam, 1:400), anti-NogoA (11C7, 1:4000, kindly provided by Martin Schwab, University and ETH Zurich, Switzerland) and anti-GFAP (Dako, mouse monoclonal; 1:300) antibodies, respectively. Anti-rabbit-Cy3 (1:200) and anti-mouse-Alexa Fluor® 488 (1:500) secondary antibodies were used to detect bound primary antibodies. Sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole). The density of FGF9 immunoreactive astrocytes was determined using an ocular morphometric grid at a magnification of ×400. Demyelinating lesion areas as well as periplaque tissue (3/5) and normal-appearing white or grey matter (3/5) were available for analysis. Astrocytes were identified by virtue of their nuclear and cytoplasmic morphology. The percentage of FGF9-immunoreactive astrocytes was determined for lesion and periplaque tissue.
Cell culture
Dissociated myelinating cultures were generated as described previously (Elliott et al., 2012). Briefly, neurospheres were generated from striata of post-natal Day 1 Sprague-Dawley rats by resuspending dissociated cells in 20 ml neurosphere media [Dulbecco’s modified Eagle medium (DMEM)/F12 (1:1, DMEM containing 4500 mg/l glucose), supplemented with 0.105% NaHCO3, 2 mM glutamine, 5000 IU/ml penicillin, 5 µg/ml streptomycin, 5.0 mM HEPES, 0.0001% bovine serum albumin, (all from Invitrogen), 25 µg/ml insulin, 100 µg/ml apotransferrin, 60 µM putrescine, 20 nM progesterone, and 30 nM sodium selenite (all from Sigma)] and plating them in a 75 cm3 tissue culture flask. The culture was supplemented with 20 ng/ml mouse submaxillary gland epidermal growth factor (R&D Systems), and maintained at 37°C in a humidified atmosphere of 7% CO2/93% air. Every 2–3 days, 5 ml neurosphere medium and 4 µl EGF were added to the flask. Neurosphere-derived astrocytes were obtained by triturating neurospheres and plating them onto poly-L-lysine (13 µg/ml) coated cover slips (13-mm diameter, VWR International) in low glucose DMEM supplemented with 10% foetal bovine serum and 2 mM L-glutamine (Sigma) until they formed a confluent monolayer.
Neurosphere-derived astrocytes conditioned media was obtained from confluent neurosphere-derived astrocyte monolayers grown in differentiation medium without insulin (see below) in the presence or absence of 100 ng/ml FGF9 for 3 days. These supernatants were immediately used to treat myelinating cultures (3:1 vol/vol supernatant/fresh differentiation medium without insulin) from 18 to 28 days in vitro (DIV) in the presence or absence of a neutralizing antibody specific for human FGF9 (R&D Systems, 10 μg/ml).
Myelinating spinal cord cultures were derived from embryonic Day 15.5 Sprague-Dawley embryos. Spinal cords were dissected out, meninges removed, minced with a scalpel blade and enzymatically dissociated with 2.5% trypsin (Invitrogen) and 1.33% collagenase (ICN Pharmaceuticals). Enzymatic activity was stopped by adding 1 ml of a solution containing 0.52 mg/ml soybean trypsin inhibitor, 3.0 mg/ml bovine serum albumin, and 0.04 mg/ml DNase (Sigma). The tissue was then triturated, centrifuged and resuspended in plating medium [50% DMEM, 25% horse serum, 25% Hank’s Balanced Salt Solution (HBSS)], and thereafter 150 000 cells in 50 µl were plated on cover slips coated with a confluent monolayer of neurosphere-derived astrocytes. Coverslips were placed in 35 mm Petri dishes (three per dish) and left in the incubator to attach for 2 h, and subsequently 450 µl of plating medium and 600 µl of differentiation medium [DMEM (4500 mg/ml glucose), 10 ng/ml biotin, 0.5% hormone mixture (1 mg/ml apotransferrin, 20 mM putrescine, 4 μM progesterone, and 6 μM selenium; formulation based on Bottenstein and Sato, 1979), 50 nM hydrocortisone, and 0.1 μg/ml insulin (all reagents from Sigma)] was added. Every 2 days 500 µl of media was withdrawn and replaced by fresh differentiation media. From 12 DIV onwards cultures were fed with insulin free differentiation medium. The effect of recombinant human FGF9 and other soluble factors (all obtained from R&D Systems) on myelinating cultures was investigated between 18 and 28 DIV. To block proliferation, cultures were treated with 20 µM cytosine β-D-arabinofuranoside (AraC, Sigma). ADAMTS and matrix metalloproteinase (MMP) activity was inhibited by adding a cocktail containing 3 µg/ml recombinant human tissue inhibitor of MMP1, MMP2 and MMP3 (TIMP1, TIMP2 and TIMP3; all from R&D Systems).
A2B5+ OPCs were isolated from post-natal Day 1 rat cortex. Briefly, meninges were removed and tissue digested with a neural tissue dissociation kit containing Papain (Miltenyi Biotec) according to the manufacturer’s instructions. Tissue was digested for 25 min at 37°C and then dissociated manually to give a single cell suspension. Cells were centrifuged at 300 g for 10 min, after which OPCs were purified by MACS magnetic bead separation using anti-A2B5 MicroBeads (Miltenyi Biotec). A2B5+ cells were resuspended in Basal Chemically Defined medium [DMEM, 4 mM L-glutamine, 1 mM sodium pyruvate, 0.1% bovine serum albumin (BSA), 50 µg/ml apo-transferrin, 5 µg/ml insulin, 30 nM sodium selenite, 10 nM D-biotin and 10 nM hydrocortisone] containing 20 ng/ml PDGF-AA and 20 ng/ml FGF2 and plated on poly-L-lysine (13 µg/ml) coated coverslips at a density of 1–2 × 104 cells/cm2. After 3 DIV PDGF/FGF2 was withdrawn and cells were allowed to differentiate for up to 7 days in modified Sato’s medium (Bottenstein and Sato, 1979; DMEM containing 4500 mg/l glucose, 2 mM glutamine, 5000 U/ml penicillin, 5 µg/ml streptomycin, 10 µg/ml insulin, 100 µg/ml apo-transferin, 16.1 µg/ml putrescine, 60 ng/ml progesterone, 30 nM sodium selenite, 0.4 µg/ml triiodo-L-thyronine, 0.4 µg/ml L-thyroxine T4, and 0.1 mg/ml BSA) in the absence or presence of FGF9. Cells were fed twice a week by replacing half the culture supernatant with fresh media.
Organotypic slice cultures
Organotypic cultures were established and analysed as described previously (Zhang et al., 2011). Briefly, postnatal Day 1–2 mouse pups were decapitated and their brains were placed into ice-cold HBSS. Cerebellum and brainstem were cut into 200–300 μm sagittal slices using a McIlwain tissue chopper. The slices were placed onto Millicell®-CM organotypic culture inserts (Millipore) in medium containing 50% minimal essential medium with Earle’s salts, 25% Earle’s Balanced Salt Solution, 25% heat-inactivated horse serum, GlutaMAX-II supplemented with penicillin-streptomycin, amphotericin B (Invitrogen) and 6.5 mg/ml glucose (Sigma). Medium was changed every 2 days. After 10 days in culture, demyelination was induced by addition of 0.5 mg/ml lysophosphatidyl lysolecithin (LPC; Sigma) to the medium for ∼16 h, after which the slices were transferred back into normal medium ±100 ng/ml FGF9. Cultures were maintained for a further 14 days and then fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 1 h while still attached to the membrane inserts. They were then rinsed in PBS for 10 mins (three times) and blocked with 3% heat inactivated horse serum, 2% BSA, and 0.5% Triton™ X-100 in PBS for 1 h. Slices were incubated with primary antibodies overnight, washed with PBS once for 10 min and then three times for 1 h. Appropriate secondary antibodies (Alexa Fluor®, Invitrogen) were then applied overnight after which the slices were washed twice for 1 h in PBS and then mounted.
Primary antibodies used for these studies were specific for: myelin oligodendrocyte glycoprotein (MOG, 1:500, Abcam), myelin basic protein (MBP, 1:500, Serotec), proteolipid protein (PLP, 1:100, AA3 hybridoma supernatant supplied by S. Barnett), and neurofilament (NFH, 1:50000, EnCor). Myelination was quantified by confocal microscopy as described previously (Zhang et al., 2011) using stacks of images of myelin (MOG/MBP) and neurofilament immune labelling obtained at 1 -μm intervals in white matter areas at ×40 magnification. Four slices (four image stacks) were quantified per treatment/control and the experiment repeated three times using cultures from different litters of postnatal Day 1 mice.
Immunofluorescence microscopy
Cultures were fixed with 4% PFA for 20 min at room temperature, washed in PBS, permeabilized in 0.5% Triton™ X-100/PBS for 15 min at room temperature, washed with PBS, and blocked with 1% BSA/10% horse serum/PBS for 1 h at room temperature. Primary antibodies used were: SMI-31 (1:1500, Abcam), Z2 (anti-MOG, 1:200, clone Z2), PLP/DM20 (1:100, hybridoma supernatant supplied by S. Barnett), MBP (1:100, Millipore), O4 (IGKV1D-37, 1:500, R&D Systems), NG2 (1:200, Millipore), OLIG2 (1:1000, Millipore), Caspr (1:500, Abcam) and GFAP (1:500, Dako). These were diluted in blocking buffer and incubations performed either at room temperature for 1 h or overnight at 4°C. After gentle washing bound antibodies were visualized using appropriate combinations of species/isotype specific fluorochrome-conjugated secondary antibodies (1:400, Invitrogen) after incubation at room temperature for 15 min. Cover slips were then washed with PBS followed by distilled water and mounted with Mowiol® 4-88 (Calbiochem) containing the nuclear stain DAPI. Cell proliferation was analysed using the Click-iT® EdU Alexa Fluor® 594 Kit (Invitrogen) following manufacturer’s instructions, co-stained with DAPI and other markers and imaged. Images were captured using an Olympus BX51 fluorescent microscope and Image-Pro software. For quantitative analysis of neurite density and myelination, 10 random images from each of three coverslips were taken at ×10 magnification. All experiments were performed in triplicate providing a total of 270 images per condition for quantification and analysis. Neurite density was quantified as described previously (Elliott et al., 2012), myelination and cell counts were quantified using CellProfiler cell image analysis software (Carpenter et al., 2006). The pipe lines developed for this study are available at https://github.com/muecs/cp.
In situ hybridization analysis
In situ hybridization studies were carried out using fresh frozen tissue samples provided by the UK Multiple Sclerosis Tissue Bank (UK Multicentre Research Ethics Committee, MREC/02/2/39). Synthetic digoxigenin-labelled riboprobes (cRNA) were generated from recombinant pCRTMII-Topo® plasmid containing a 606 bp cDNA insert of human FGF9 sequence (5’-2809-3414-3’). Transcription was done from both sides with either SP6 or T7 RNA polymerase, generating antisense or sense (control) cRNA probes. In situ hybridization was performed on cryosections of freshly frozen tissues as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993; Graumann et al., 2003). In situ hybridization signal was revealed by alkaline phosphatase with BCIP (5-bromo-4-chloro-3′-indolyphosphate) and NBP (nitro-blue tetrazolium) as substrate. For immunohistochemistry tissue sections were washed twice with PBS, thereafter sections were treated with 0.6% hydrogen peroxide in methanol for 30 min and with blocking buffer (1% normal donkey serum, 0.1% Triton™ X-100, 0.05% Tween) for 1 h. Sections were incubated with the following primary antibodies overnight at 4°C: rabbit anti-OLIG2 (1:500, Millipore), and rabbit anti-GFAP (1:2000, DakoCytomation). Secondary biotinylated antibodies (Vector Laboratories, 1:500) were applied for 2 h at room temperature, followed by ABC complex reagent (Vector Labs) for 30 min. Colour reaction was performed with 3-amino-9-ethylcarbazole. Luxol Fast blue and haematoxylin and eosin staining were performed according to standard protocols.
RNA extraction and microarray analysis
RNA was extracted from myelinating cultures grown in the presence or absence of FGF9 (100 ng/ml) for 24 h using the Qiagen RNeasy® Micro kit according to manufacturer’s instructions. RNA quality and integrity was checked using the Agilent Bioanalyzer 6000 Nano LabChip platform. RNA was then used for microarray expression analysis and quantitative reverse transcription (qRT)-PCR. The total RNAs were processed and labelled with biotin using Ambion® WT Expression Kit following the Affymetrix GeneChip® WT Terminal Labeling and Hybridization protocol. The processed RNAs were hybridized to Affymetrix GeneChip® Rat Gene 1.0 ST Arrays using manufacturer's protocols for using the Fluidics Station 450. The hybridized arrays were scanned on the Gene Array Scanner 3000-7G. The data analysis was carried out in Partek Genomics Suite (version 6.6, Partek) software. Control and FGF treatment groups were generated with three replicates per group. This data set has been deposited in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/ accession number GSE52753). Probe set level data were normalized using the GCRMA normalization method and summarized to transcript cluster level using One-Step Tukey's Biweight method. Differential expression analysis was carried out by performing a two-way ANOVA test on the normalized expression values. Differentially expressed gene lists were generated based on the ANOVA linear fold-change > ±1.4 (±2.639-fold change in linear scale) at a FDR adjusted P-value of <0.01. The differentially expressed genes were analysed in Partek Pathway for enriched pathways using the Kegg database for rat.
Quantitative real-time PCR
Changes in expression of selected genes of interest was verified using RNA obtained from additional myelinating cultures and monolayers of neurosphere-derived astrocytes grown in the presence or absence of FGF9. Following RNA extraction, cDNA was synthesized using the Qiagen QuantiTect® Reverse Transcription Kit following the manufacturer’s instructions. Cycling parameters were as follows: first cycle (DNA wipeout step) 42°C for 2 min, after adding reverse transcriptase, reaction buffer and primer mix second cycle: 42°C for 20 min, then 3 min for 95°C. Real-time PCR was performed using SYBR® Green master mix (Applied Biosystems), 10 ng cDNA template, 50 pmol/µl of each primer and distilled water. Primers were designed using the NCBI nucleotide data base and Primer 3 software (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) (Rozen and Skaletsky, 2000). Primer sequences were checked with BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and were purchased from Sigma or IDT. The reaction was amplified in an Applied Biosystems Fast Real-Time PCR System (ABI 7500) using the following cycle settings: 50°C for 5 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 1 min, and a final dissociation step at 95°C for 15 s. Melt curve analysis was then performed between 75–99°C in 1°C increments. The comparative CT method (or the 2−ΔCT method) (Livak and Schmittgen, 2001; Schmittgen and Livak, 2008) was used to determine differences in gene expression between FGF9 and control samples. For statistical analysis, a paired two-sample t-test was used on the mean ΔCT for each experimental repeat.
Enzyme-linked immunosorbent assay
CCL2 and Hyaluronan concentrations were measured in supernatants harvested from myelinating cultures (DIV18) or astrocytes monolayers grown in the presence or absence of FGF9 for 24 h using mouse/rat CCL2 or Hyaluronan Quantikine ELISA kits (R&D Systems) according to the manufacturer’s instructions. Aggrecanase activity was assayed in supernatants from myelinating cultures (DIV18) treated with or without FGF9 for 72 h using an ELISA-based Aggrecanase Activity Assay Kit (Abnova) following manufacturer’s instructions. In brief, enzyme activity was assessed by the capacity of test samples to digest a recombinant fragment of human aggrecan. This proteolytic cleavage releases an ARGSVIL-peptide, which is then quantified by ELISA using appropriate specific antibodies.
Results
Tissue damage in multiple sclerosis is associated with increased expression of FGF9
We investigated FGF9 expression in post-mortem CNS tissue from seven control donors and in white matter lesions from 14 cases of multiple sclerosis (six acute, one relapsing remitting, seven chronic; Table 1). In agreement with previous studies (Todo et al., 1998) FGF9 was expressed constitutively by neurons and occasionally by glia and cells associated with the vasculature (Fig. 1A). Neurons exhibited a punctate pattern of cytoplasmic staining that was superimposed on a background of extracellular immunoreactivity, which may be due to FGF9 in axons and/or dendrites, or bound to components of the extracellular matrix. FGF9 immunoreactivity was far less pronounced in subcortical white matter in which weak staining was observed in scattered neurons, and less frequently in astrocytes and oligodendrocytes (Fig. 1B). This contrasted to the situation in multiple sclerosis which was associated with a generalized increase of FGF9 immunoreactivity in white matter, in particular in active lesions, defined by the abundance of macrophages, containing early myelin degradation products (Fig. 1C and D). Staining was most pronounced in oligodendrocytes, although astrocytes and other cells, putatively identified as OPCs were also immunoreactive (Fig. 1F). This was not only seen in acute active lesions, but was also observed, albeit to a lesser extent, at the rim of slowly expanding white matter lesions from patients with longstanding progressive disease (Fig. 1 G–I). Normal-appearing white matter exhibited variable immunoreactivity in oligodendrocyte-like cells, astrocytes and vessel associated cells that were intermediate between that in control white matter and active lesion areas (Fig. 1E). However, FGF9 immunoreactivity was minimal in the centre of slow expanding white matter lesions apart from occasional weakly staining astrocytes (Fig. 1J). A similar lack of staining for FGF9 was apparent in chronic inactive lesions (Fig 1K and L) where immunoreactivity for FGF9 was restricted to occasional axonal spheroids, and cells morphologically consistent with being astrocytes and endothelial cells (Fig. 1M and N).
Figure 1.
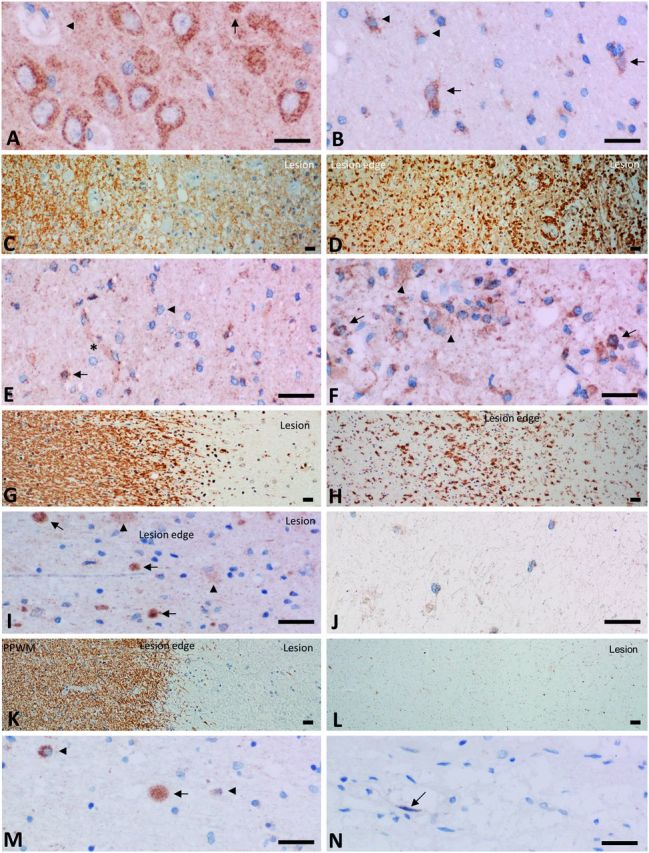
FGF9 is upregulated in multiple sclerosis lesions. (A) In control grey matter we detected intense extensive granular FGF9 immunoreactivity in the cytoplasm of cortical neurons superimposed on weaker diffuse granular immunoreactivity in the parenchyma. We also observed weak granular reactivity in some glia cells (arrows) and in association with the vessel wall (arrowhead). (B) Immunoreactivity was less pronounced in subcortical white matter although we still detected immunoreactivity in neurons (arrows), as well as weak reactivity in occasional cells with astrocyte morphology (arrowheads). (C and D) Active multiple sclerosis lesion of a patient with acute multiple sclerosis. (C) Myelin is lost in the plaque (right) in comparison to the periplaque white matter and is taken up by macrophages in the lesions. Immunocytochemical staining for MOG. (D) Immunocytochemistry for macrophages shows profound infiltration of the lesion (right) and massive microglia activation at the lesion edge (left). (E) Expression in normal appearing white matter is intermediate between that seen in active lesions and control white matter and is characterized by variable staining of oligodendrocyte-like cells (arrow), astrocytes (arrowhead) and vessel associated cells (asterisk). (F) Immunoreactivity for FGF9 was far more pronounced in active lesions compared to control white matter. Strong expression was observed in cells with oligodendrocyte morphology (arrows), weaker expression in astrocytes (arrowheads) as well as other cell types, possibly OPCs. Rim of an early active lesion from case of acute multiple sclerosis, disease duration, 2 months. (G and H) Slowly expanding lesions in progressive multiple sclerosis are defined by myelin loss and the presence of some macrophages at the lesion border with myelin degradation products (G); there is massive microglia activation at the lesions edge and some immunoreactive cells have macrophage morphology (H). (I) FGF9 immunoreactivity is increased at the edge of slowly expanding white matter lesions from cases of progressive disease. In this case immunoreactivity is associated with axonal spheroids (arrows) and glia (arrowhead). Rim of a slowly expanding lesion from a case of primary progressive multiple sclerosis. (J) The centre of chronic active lesions show very limited astrocytic immunoreactivity for FGF9. (K and L) Inactive lesions show myelin loss with a sharp border towards the periplaque white matter (PPWM) (K) and no immunoreactivity for macrophage antigens (e.g. CD68) within or around the lesion (L). Immunoreactivity for FGF9 across inactive chronic lesions is far lower than in control white matter as seen at the lesion rim (M) where immunoreactivity is associated with an axonal spheroid (arrow) as well as weaker and variable staining of glia (arrowheads). In the centre of the same lesion (N) weak immunoreactivity is seen associated with a single cell, morphologically consistent with an endothelial cell. (K–N) Inactive lesion from a case of secondary progressive multiple sclerosis with a duration of >35 years. Scale bar in all panels corresponds to 20 µm.
The cellular distribution of FGF9 immunoreactivity in active lesions was then examined in more detail using biopsies of early active demyelinating lesions. This confirmed neurons, GFAP+ astrocytes and occasional NogoA+ oligodendrocytes can all express FGF9 in these lesions, but also revealed there was substantial variation in the number of FGF9 immunoreactive glia between cases (Supplementary Fig. 1). Approximately half of the astrocytes in these lesions (50.94 ± 0.08 per cent) and surrounding periplaque grey matter (50.67 ± 0.12 per cent) were immunoreactive for FGF9. In contrast no FGF9+ reactive astrocytes were detected in normal-appearing grey matter in the three cases where this was available. In addition to this pronounced astrocytic response a small number of NogoA+ cells expressing FGF9 were also detected in and around these early demyelinating lesions (Supplementary Fig. 1).
To determine whether glial immunoreactivity for FGF9 was due to de novo synthesis, or uptake from the extracellular milieu we performed in situ hybridization studies in combination with immunohistochemistry for OLIG2 and GFAP on fresh frozen tissue sections from active inflammatory lesions (Fig. 2A–D, K and L) and chronic inactive lesions (Fig. 2E–H). This study detected variable, but high expression of FGF9 in active lesions (Fig. 2C and D), as well as increased expression associated with the edge of chronic inactive lesions (Fig. 2G and H) compared to normal-appearing white matter (Fig. 2I). Immunohistochemical co-localization studies identified Fgf9 mRNA transcripts in some but not all OLIG2+ cells associated with these lesions (Fig. 2K, arrows), as well as in a small numbers of GFAP+ astrocytes (Fig. 2L, arrowheads). These findings confirm glial expression of FGF9 is upregulated at sites of ongoing lesion formation in multiple sclerosis.
Figure 2.
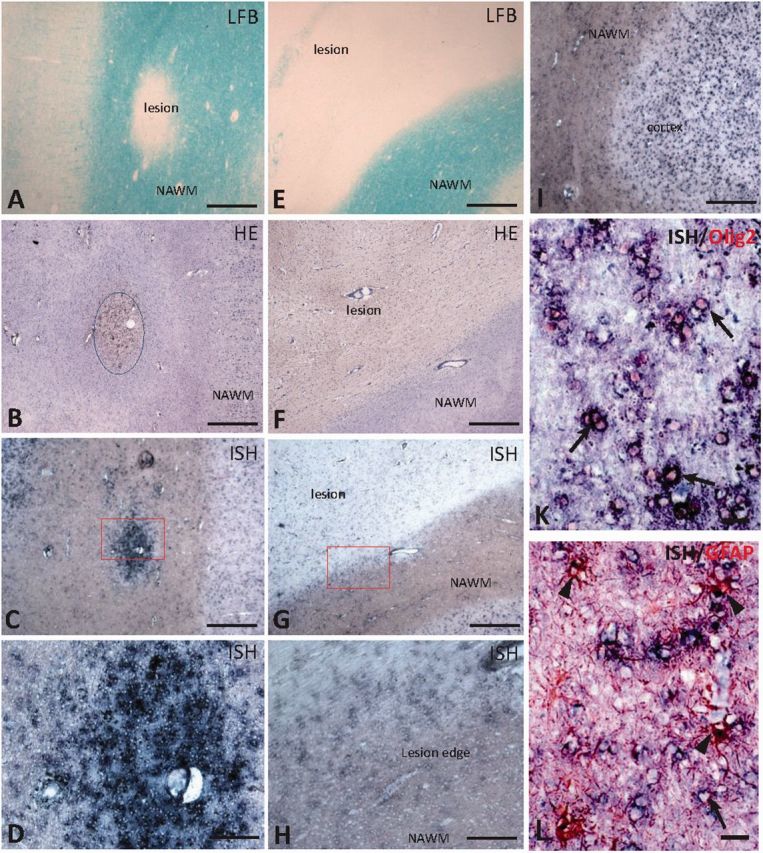
In active lesions FGF9 is expressed by cells of the oligodendrocyte lineage. In situ hybridization (ISH) was performed on fresh frozen post-mortem multiple sclerosis brain tissue with active and chronic active white matter lesions identified by Luxol Fast blue (LFB) (A and E, respectively). B and F show the corresponding haematoxylin and eosin (HE) staining of consecutive tissue sections. In situ hybridization for FGF9 transcripts show high level of expression within active lesion (C and D, higher magnification), increased expression in the edge of chronic active lesions (G and H, higher magnification), and high expression in cerebral cortex (I), whereas in normal-appearing white matter (NAWM) FGF9 is expressed at low levels (H and I). Immunohistochemical colocalization detects FGF9 transcripts mainly in OLIG2-positive cells (K, arrows), but also in a small number of GFAP-positive cells (L, arrows) although most GFAP-positive cells were negative (arrowhead). Scale bar = 500 µm in A–C, E–G and I; 100 µm in D and H; and 25 µm in K and L.
Increased availability of FGF9 compromises (re)myelination in vitro
FGF9 plays important roles in co-ordinating cell proliferation, differentiation and survival during development, but its expression is strongly downregulated before birth in the CNS. Re-expression of FGF9 in the adult CNS might therefore be expected to have a significant impact on lesion development in multiple sclerosis; a concept supported by the ability of FGF9 to suppress myelin protein expression by OPC (Cohen and Chandross, 2000). We therefore investigated the effects of adding FGF9 to myelinating cultures derived from embryonic rat spinal cord by immunofluorescence microscopy, a model system that allows us to quantify effects on all stages of the oligodendroglial cell lineage, as well as myelination and neurite outgrowth. FGF9 inhibits myelination in a dose dependent manner (Fig. 3A and B), >90% inhibition being obtained when FGF9 was present at a concentration of 100 ng/ml (18–28 DIV) (Fig. 3B and Supplementary Fig. 2). Failure of myelination was associated with the appearance of a population of ‘pre-myelinating’ oligodendrocytes that exhibited a granular pattern of PLP and MBP immune reactivity in their cytosol. These cells were observed to extend processes that ended in large membrane expansions or alternatively, aligned along and interacted with SMI31+ neurites (Fig. 3A and Supplementary Fig. 2); an oligodendrocyte phenotype associated with chronically demyelinated multiple sclerosis lesions (Chang et al., 2002).
Figure 3.
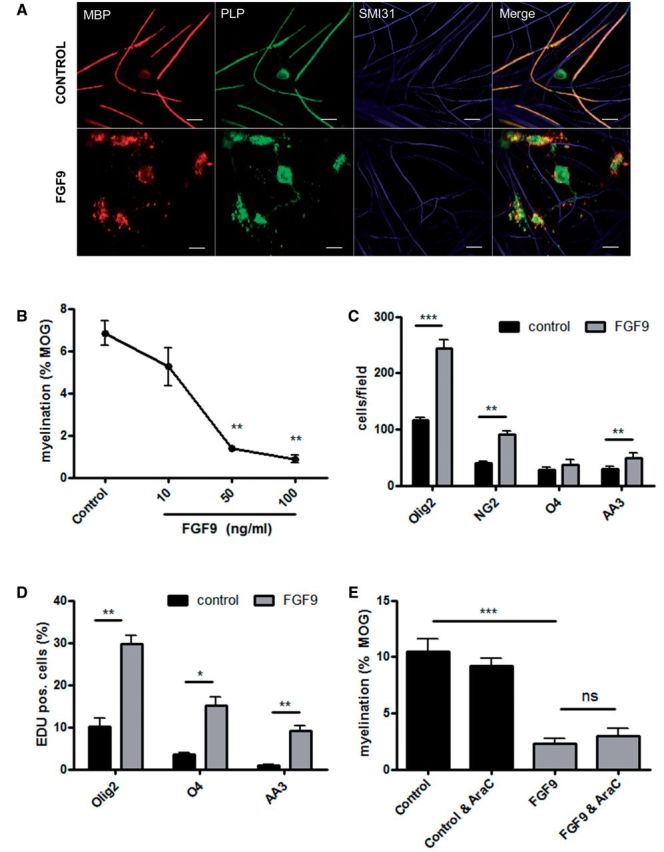
FGF9 inhibits myelination and expands OPC numbers in vitro. Oligodendrocytes in untreated cultures myelinate multiple axons (A); MBP (red) reactivity is predominantly associated with myelin sheaths, whereas PLP (green) immunoreactivity is also present in the oligodendrocyte cell body. Treatment with 100 ng/ml FGF9 from 18–28 DIV is associated with the appearance of multi-branched oligodendrocytes that exhibit a granular staining pattern for MBP and PLP in the cell body and fail to generate myelin sheaths. MBP, red; PLP, green; SMI31, blue. Scale bar = 20 µm. Inhibition of myelination by FGF9 in these cultures is dose-dependent (B); myelination was quantified using a MOG-specific antibody as described in the text. This treatment is associated with increased numbers of OLIG2+ (P < 0.001), NG2+ (P < 0.01) and AA3+ (P < 0.01) cells. There was also a trend for O4+ cell numbers to increase, but this did not reach significance (P = 0.055) (C). This increase in OPC numbers was due to proliferation as demonstrated by the presence of increased numbers of OLIG2, O4 and AA3 (PLP/DM20) immunoreactive cells labelled with EdU (DIV 18–22) (D). However, the ability of FGF9 to inhibit myelination is independent of this proliferative response as inhibition of proliferation by cytosine arabinoside (AraC, 20 µM) was unable to abrogate inhibition of myelination by FGF9 (E). Data are presented as mean ± SEM obtained from a minimum of three experiments; *P < 0.05; **P < 0.01; ***P < 0.001.
Inhibition of myelination by FGF9 in these cultures was accompanied by a significant increase in the number of OLIG2+, NG2+ and PLP+ cells (Fig. 3C and Supplementary Fig. 3); an effect associated with proliferation as demonstrated by incorporation of EdU (5-ethynyl-2′-deoxyuridine) into cells expressing oligodendrocyte lineage-specific markers (Fig. 3D). Incorporation of EdU was increased 3-fold in OLIG2+ cells, 4–5-fold in O4+ cells and almost 10-fold in PLP+ cells. This effect of FGF9 on the oligodendroglial lineage was reminiscent of that reported for FGF2, a well-characterized OPC mitogen that inhibits OPC differentiation by limiting their ability to drop out of cell cycle. We therefore performed experiments to determine whether this represents a generalized response to FGF signal transduction in this culture system. However screening nine family members revealed this is not the case. Only FGF9 and to a lesser extent FGF2 stimulated an increase in OLIG2+ cell numbers while at the same time inhibiting myelination (Supplementary Fig. 3C and Supplementary Data).
We then went on to explore whether inhibition of myelination by FGF9 in this culture system was dependent on maintaining OPCs in cell cycle using AraC to eliminate proliferating cells (Fig. 3E and Supplementary Fig. 3). Control experiments revealed that although treatment with AraC from 18 to 28 DIV significantly reduced cell proliferation, this had no effect on myelination per se; an observation indicating myelination during this period is performed by a pre-existing pool of non-proliferating OPCs. AraC also eliminated the proliferative response induced by FGF9; however, this failed to abrogate the effect of FGF9 on myelination demonstrating this is independent of its ability to act as a glial mitogen (Fig. 3E). In contrast to these marked effects on cells of the oligodendrocyte lineage and myelination FGF9 had no effect on neurite density as determined by SMI31 immune reactivity (100 ng/ml; DIV 18–28; Control 77.87 ± 3.11; FGF9 treated 77.75 ± 5.09; percent SMI31+ pixels per field).
These experiments demonstrate FGF9 inhibits myelination in a developmental setting, but it remained unclear whether it would also compromise remyelination. To explore this further we addressed this question by monitoring the effect of FGF9 on remyelination in cerebellar slice cultures demyelinated by treatment with LPC (Fig. 4). Analysis of cultures 14 days after exposure to LPC revealed remyelination was significantly reduced in the presence of FGF9 compared to demyelinated cultures that were not exposed to this factor (P < 0.01) (Fig. 4).
Figure 4.

FGF9 compromises remyelination in organotypic slices. Cerebellar slices from post-natal Day 2 mouse pups were cultured for 10 days and then demyelinated using LPC for 16 h. Cultures were then allowed to remyelinate over the next 14 days in the presence or absence of FGF9 (100 ng/ml) after which remyelination was assessed using antibodies specific for MOG and PLP to label myelin, and neurofilament (NFH) to label axons. Confocal immunofluorescence microscopy shows treatment with FGF9 leads to less remyelination (A) as demonstrated by the reduction in MOG+ internodes (red) and PLP+ internodes (green) encasing NFH+ axons (blue). Scale bar = 10 µm. Remyelination was quantified using image analysis to define co-localization of staining for myelin (PLP+ or MOG+) and axons (NFH+) (B)—a quantification of myelinated internodes we term the remyelination index (amount of myelin/axon). Irrespective of the antibody used to identify myelinated internodes, treatment with FGF9 (F) reduced remyelination compared to untreated cultures (C). Mean ± SEM (n = 3, at least six slices per experiment; ***P < 0.001; **P < 0.01.
FGF9 is a stage dependent mitogen for OPCs
The observation that FGF9 induced OPC proliferation in myelinating cultures was unexpected as previous studies reported FGF9 is not a mitogen for purified OPCs (Fortin et al., 2005). However, as OPCs differentiate into mature oligodendrocytes they sequentially express different high affinity receptors for FGF9. Receptor expression changes from FGFR3 in OPCs to FGFR2 in mature myelinating oligodendrocytes; activation of the latter by FGF9 triggering changes in process outgrowth without affecting expression of myelin proteins (Fortin et al., 2005). These results were however obtained after short term (48 h) exposure of either early or highly differentiated progenitors to FGF9. This prompted us to investigate the effect of continuous exposure to FGF9 on OPC differentiation. Using A2B5+ progenitor cells immunopurified from the neonatal CNS we confirmed FGF9 is not a mitogen for OPCs during their initial stages of differentiation in Sato’s medium (DIV 1–3; Fig. 5A). However, they subsequently become responsive to its mitogenic potential as later exposure (DIV 4–7) doubles incorporation of EdU (Fig. 5A). This proliferative response was associated with inhibition of differentiation as demonstrated by analysis of cultures continuously exposed to FGF9 for 7 days; the number of OPCs expressing O4, PLP and MOG being reduced by 30%, 86% and >90%, respectively (Fig. 5B and C).
Figure 5.

FGF9 inhibits OPC differentiation directly and induces secretion of myelination-inhibiting factors by astrocytes. Proliferation of immunopurified A2B5+ OPCs cultured in the presence or absence of FGF9. FGF9 is not initially an OPC mitogen (DIV 1–3), but progenitors become responsive as they differentiate (DIV 4–7) (A). This proliferative response is associated with inhibition of OPC differentiation assessed using three stage specific markers (O4, PLP and MOG) (B and C). B provides representative images demonstrating the effect on expression of PLP and MOG. Scale bar = 100 µm. Supernatants from FGF9 treated and untreated neurosphere-derived astrocytes were harvested and added to myelinating cultures in the presence or absence of a neutralizing anti-FGF9 antibody (10 μg/ml) from 18 to 28 DIV (D). Quantifying PLP+ myelination reveals the FGF9-specific antibody neutralized the ability of FGF9 to inhibit myelination, but failed to block inhibitory activity present in supernatants harvested from FGF9-treated astrocytes. (A, C and D) Data represent means ± SEM from at least three independent experiments; ***P < 0.001; **P < 0.01; *P < 0.05, n.s. = not significant.
These results suggest a direct effect of FGF9 on cells of the oligodendrocyte lineage would account for its ability to inhibit myelination. However, our data question the validity of this supposition. Specifically, inhibition of myelination by FGF9 in the far more complex cellular environment of our myelinating cultures was neither dependent on its mitogenic potential (Fig. 3E), nor was it associated with a net decrease in O4+ or PLP+ cell numbers (Fig. 3C). In fact the converse was true, FGF9 increased the absolute number of O4+ and PLP+ cells in these cultures by 37% and 68%, respectively (Fig. 3C). We reasoned this dichotomy could be explained by an off target effect of FGF9 that compensated for any direct effect of FGF9 on OPC differentiation, but which at the same time might also adversely affect myelination. The most likely candidates being astrocytes as they constitutively express FGFR3 (Young et al., 2010), a preferred high affinity receptor for FGF9 (Goetz and Mohammadi, 2013).
To test this hypothesis we grew myelinating cultures (18–28 DIV) in the presence of supernatants harvested from naïve or FGF9 treated neurosphere-derived astrocytes in the presence or absence of a neutralizing antibody to eliminate any direct effect of residual FGF9. Control experiments established this antibody had no deleterious effect on myelination per se, but completely neutralized the ability of FGF9 to inhibit myelination (Fig. 5D). However, the inhibitory activity of supernatants harvested from FGF9-treated astrocytes could not be abrogated by this FGF9-specific antibody demonstrating inhibition of myelination is dominated by mechanisms involving one or more astrocyte-derived factors (Fig. 5D).
FGF9 activates immune and neurobiological pathways relevant for lesion formation
To identify mechanisms by which FGF9 inhibits myelination we performed a microarray analysis on myelinating cultures (DIV 18) grown in the presence or absence of FGF9 for 24 h. This identified 3262 transcripts differentially regulated by FGF9 (1752 upregulated, 1510 downregulated; fold-change > ±1.4; P < 0.01; Fig. 6 and Table 2). Pathway enrichment analysis using the 731 most differentially regulated transcripts (fold change > ±2.2; P < 0.01) identified 17 enriched pathways (P < 0.05; Fisher’s Exact Test; Supplementary Table 1). The most highly enriched was the cell cycle pathway, an observation that reflects the proliferative response to FGF9. However three of the four next most enriched pathways are associated with both neurobiological and immune functions (transforming growth factor beta signalling, P = 0.00025; cytokine-cytokine receptor interaction, P = 0.00098; Jak-STAT signalling, P = 0.0048); an observation suggesting that in addition to any effect on remyelination increased availability of FGF9 may also modulate immune function within the CNS. This hypothesis is supported by the observation that FGF9 increased expression of a cluster of ‘pro-inflammatory’ genes (Ccl7, Cd93, Cd63, Ccl2, Il1rap, Tnfrsf10b and Il1r1) more commonly discussed in the context of innate immunity rather than their neurobiological effects. The relative magnitude of this effect is apparent when significance (log10 P) is plotted against fold-change in expression which highlights the profound effects of FGF9 on expression of Ccl7 (ranking 1; 31-fold increase), Cd93 (ranking 4; 17-fold increase), Il1rap (ranking 55; 5-fold increase) and Ccl2 (ranking 56; 5-fold increase) (Fig. 6 and Table 2). This raises the possibility that in addition to its ability to inhibit (re)myelination, FGF9 may also promote a pro-inflammatory environment in the CNS, a combination of effects predicted to exacerbate axonal injury and loss in multiple sclerosis lesions (Franklin et al., 2012).
Figure 6.

Transcriptional profiling of the effects of FGF9 in myelinating cultures. (A) Volcano plot of fold-change versus log10 significance reveals FGF9 upregulated a cluster of pro-inflammatory genes (dark blue) including Ccl7 and Ccl2; induced expression changes indicative of extracellular matrix remodelling (red); modulated the relative expression of IL6/gp130 gene family members (light blue) and uniformly reduced expression of myelin associated genes. (B) Aggrecanase activity was assayed in supernatants from myelinating cultures grown in the presence or absence of FGF9 for 72 h. Cell culture supernatants protolytically cleave a recombinant aggrecan substrate into ARGSVIL-peptide which is then measured by ELISA. Data represent the mean ± SEM of seven independent experiments, *P < 0.05. (C) Myelinating cultures were treated with FGF9 for 10 days in the absence or presence of tissue inhibitors of metalloproteinases (TIMP1, TIMP2 and TIMP3; each at 1 µg/ml) after which myelination was quantified, as described in the text. This cocktail of TIMPS had no effect on basal levels of myelination, but abrogated inhibition of myelination by FGF9. Representative data (mean ± SEM) from one of three independent experiments.
Table 2.
Genes shown in the volcano plot
| # | Gene | Fold-change | P-value | Rank |
|---|---|---|---|---|
| Pro-inflammatory genes | ||||
| 1 | * Ccl7 | 31.03 | 7.28 × 1020 | 1 |
| 2 | Cd93 | 17.48 | 9.60 × 1017 | 4 |
| 3 | Cd63 | 8.89 | 7.42 × 1017 | 21 |
| 4 | Il1rap | 4.96 | 1.32 × 1018 | 55 |
| 5 | * Ccl2 | 4.96 | 6.46 × 1017 | 56 |
| 6 | Tnfrsf10b | 3.19 | 2.50 × 1011 | 110 |
| 7 | Il1r1 | 2.04 | 1.88 × 1007 | 426 |
| 8 | * Cxcl12 | −1.59 | 4.40 × 1003 | 28 267 |
| Extracellular matrix genes | ||||
| 9 | * Adamts1 | 19.83 | 1.60 × 1016 | 2 |
| 10 | Itga5 | 7.04 | 1.64 × 1015 | 29 |
| 11 | Igfbp3 | 6.67 | 4.37 × 1017 | 32 |
| 12 | Adam12 | 5.33 | 2.30 × 1010 | 49 |
| 13 | * Has2 | 5.09 | 2.89 × 1011 | 51 |
| 14 | * Adamts7 | 4.74 | 2.50 × 1014 | 60 |
| 15 | Chst3 | 3.59 | 4.96 × 1016 | 88 |
| 16 | Adamts9 | 2.93 | 4.20 × 1010 | 131 |
| 17 | Adamts14 | 2.84 | 5.26 × 1018 | 143 |
| 18 | * Cd44 | 2.65 | 2.61 × 1016 | 179 |
| 19 | Fn1 | 2.13 | 4.42 × 1006 | 371 |
| 20 | Adamts4 | 2.08 | 2.47 × 1009 | 397 |
| 21 | Lama5 | 1.71 | 1.63 × 1007 | 778 |
| 22 | Mmp15 | 1.67 | 4.30 × 1012 | 833 |
| 23 | * Mmp3 | 1.65 | 4.00 × 1005 | 860 |
| 24 | Itgb1 | 1.58 | 6.28 × 1011 | 1062 |
| 25 | Itgb8 | 1.54 | 1.47 × 1013 | 1184 |
| 26 | Icam1 | 1.44 | 6.36 × 1006 | 1554 |
| 27 | Mmp11 | 1.43 | 1.11 × 1003 | 1627 |
| IL6/Gp130 family genes | ||||
| 28 | * Clcf1 | 14.19 | 5.92 × 1020 | 6 |
| 29 | * Il11 | 9.26 | 1.47 × 1015 | 19 |
| 30 | * Lif | 5.70 | 3.42 × 1011 | 42 |
| 31 | Osmr | 2.26 | 5.20 × 1013 | 302 |
| 32 | Il6 | 1.26 | 7.39 × 1003 | 3104 |
| 33 | Stat2 | −2.11 | 5.91 × 1010 | 28 775 |
| 34 | * Cntf | −2.42 | 4.04 × 1011 | 28 895 |
| Myelin genes | ||||
| 35 | Plp1 | −1.39 | 1.18 × 1011 | 27 534 |
| 36 | Cnp | −1.51 | 3.51 × 1010 | 28 057 |
| 37 | Opalin | −1.60 | 2.61 × 1005 | 28 296 |
| 38 | * Mog | −1.69 | 3.45 × 1008 | 28 414 |
| 39 | Omg | −1.70 | 1.14 × 1012 | 28 443 |
| 40 | * Mbp | −1.72 | 6.02 × 1009 | 28 479 |
| 41 | * Mag | −1.74 | 8.45 × 1009 | 28 497 |
| 42 | Mobp | −2.93 | 4.52 × 1012 | 29 021 |
| Other genes | ||||
| 43 | * Hbegf | 10.86 | 7.99 × 1017 | 11 |
| 44 | * Vgf | 4.56 | 2.58 × 1012 | 65 |
| 45 | * Fgf2 | 1.55 | 7.14 × 1009 | 1140 |
| 46 | * Hgf | −8.02 | 1.39 × 1020 | 19 788 |
Highest rank corresponds to highest fold-change.
*Genes validated by quantitative PCR.
In addition to this ‘pro-inflammatory’ effect FGF9 modulated expression of multiple genes associated with pathways implicated previously in failure of remyelination. These include changes indicative of altered hyaluronan/CD44 signalling (Has2, Cd44; Tuohy et al., 2004; Back et al., 2005; Sloane et al., 2010; Bugiani et al., 2013), integrin/fibronectin associated effects (Itga5, Fn1; Stoffels et al., 2013), as well as changes predicted to affect availability of soluble factors known to support OPC migration, differentiation and/or survival (Stark and Cross, 2006) such as Cntf (Stankoff et al., 2002), Cxcl12 (Patel et al., 2010) and Fgf2 (Clemente et al., 2011).
To identify which of these pathways might contribute to inhibition of myelination we first validated selected candidates by quantitative PCR using reverse-transcribed mRNA isolated from myelinating cultures and astrocyte monolayers cultured for 24 h in the presence or absence of FGF9. This strategy revealed FGF9 upregulated astrocytic expression of a number of proteases in particular ADAMTS1, ADAMTS7 and MMP3 (Table 2 and Supplementary Table 2). These mediate a wide range of biological processes including extracellular matrix remodelling which led us to speculate they may adversely affect (re)myelination. This hypothesis received further support when assays using aggrecan as a ‘global’ substrate for ADAMTS and MMP family members revealed FGF9 significantly increased aggrecanase activity in supernatants harvested from myelinating cultures (Fig. 6B). To determine whether this protease activity was actively involved in mediating inhibition of myelination we used a cocktail of physiological protease inhibitors (TIMP1, TIMP2 and TIMP3) known to inhibit a wide range of ADAMTS and MMP family members. The inhibitors had no effect on myelination per se but abrogated the inhibitory effect of FGF9 which was reduced by ∼37% (n = 3; FGF9 versus untreated control, 78.2 ± 5.8% inhibition, P < 0.01; FGF9 + TIMP versus untreated control, 49.1 ± 11.8 inhibition, not significant) (Fig. 6C). Induction of TIMP-sensitive proteases therefore makes a significant contribution to the mechanisms by which FGF9 inhibits myelination in this culture system.
Why this cocktail of inhibitors fails to completely abrogate the effect of FGF9 remains unclear. This may be due to incomplete inhibition of the relevant proteases, or alternatively involvement of other factors such as chemokines (Ccl2, Ccl7), IL6/gp130 cytokine family members (Il11, Lif, Cntf) and growth factors (Hbegf, Vgf, Hgf). To explore this possibility we screened a number of soluble factors that our data suggest are expressed by astrocytes. For example our validation strategy indicates CCL2 is derived primarily from astrocytes, whereas Has2 is upregulated in some other cell type (Supplementary Table 2). To validate this interpretation we determined the concentrations of CCL2 and hyaluronic acid (a surrogate marker for increased HAS2 activity) in supernatants harvested from myelinating cultures and astrocyte monolayers grown in the presence or absence of FGF9 (Supplementary Fig. 4). Basal concentrations of CCL2 were similar in myelinating cultures and astrocyte monolayers and upregulated to similar extents, 16 - and 14-fold, respectively, confirming astrocytes were a major source of FGF9-induced CCL2 production. In contrast basal and induced levels of hyaluronic acid were far higher in supernatants harvested from myelinating cultures than astrocyte monolayers indicating astrocytes are not the major cellular source of hyaluronic acid in myelinating cultures. Screening selected candidates such as CCL2, CCL7 and IL11 has, however, as yet failed to identify any that replicate the effects of FGF9. Similarly CNTF, which is downregulated by FGF9, was unable to abrogate the effects of FGF9 (Supplementary Fig. 4).
Discussion
Signal transduction by members of the FGF family influences OPC proliferation, survival and differentiation in the rodent CNS, observations that stimulated speculation aberrant FGF expression may play a role in the pathogenesis of multiple sclerosis (McKinnon et al., 1990; Bansal and Pfeiffer, 1994; Goddard et al., 2001; Oh et al., 2003; Fortin et al., 2005; Tobin et al., 2011; Furusho et al., 2011, 2012; Mohan et al., 2014). This concept has until now focused on FGF2, a well-characterized OPC mitogen (McKinnon et al., 1990) that is over expressed in multiple sclerosis tissues (Sarchielli et al., 2008; Clemente et al., 2011) and which acts as a negative regulator of remyelination in the adult CNS (Tobin et al., 2011). We now report expression of FGF9 is also upregulated in multiple sclerosis lesions and provide evidence this will result in a complex cellular response predicted to exacerbate disease activity in patients.
Our data demonstrate expression of FGF9 is not only upregulated in early active lesions, but remains elevated at sites of ongoing tissue damage in patients with longstanding progressive disease. At these sites induction of FGF9 was observed in GFAP+ astrocytes, OLIG2+ and occasionally more mature NOGO-A+ oligodendrocytes, a complex cellular response consistently associated with a background of diffuse extracellular immunoreactivity. The latter is attributed to FGF9 binding to heparin sulphate proteoglycans (HSPG) in the extracellular matrix, a property that sequesters its direct biological effects to the immediate vicinity of its site of synthesis (Harada et al., 2009; Goetz and Mohammadi, 2013). FGF9 expression was far lower in adjacent normal-appearing white matter and largely absent in chronically demyelinated inactive lesions, a pattern that suggests induction represents a localised glial response to ongoing tissue damage. More detailed analysis of lesion biopsies revealed considerable variation in the number of FGF9 immunoreactive glia associated with early active lesions.
Whether this variation in glial expression of FGF9 is due to differences in age or anatomical localization of lesions is currently under investigation, as are the molecular mechanisms responsible for its induction. Preliminary studies suggest this is not simply a glial response to T cell mediated inflammation, as increased expression of FGF9 is not observed in animals with experimental autoimmune encephalomyelitis. Moreover, FGF9 expression is increased at the active rim of slowly expanding white matter lesions in patients with long standing progressive disease, a site associated with minimal recruitment of lymphocytes from the periphery (Lassmann et al., 2012). One factor that might contribute to this response is decreased expression of Smad-interacting protein-1 (now known as ZEB2), which suppresses neuronal expression of FGF9 during cortical development (Seuntjens et al., 2009). Intriguingly, this transcription factor is expressed by oligodendrocytes and plays an essential role in CNS myelination (Weng et al., 2012), an observation that raises the possibility ZEB2 may also influence oligodendroglial expression of FGF9.
Analysis of the effects of FGF9 in vitro revealed it not only inhibited myelination/remyelination, but also induced expression of the pro-inflammatory chemokines Ccl2 and Ccl7. These pleiotropic effects are mediated by high affinity receptors for FGF9 (FGFR2, FGFR3; Hecht et al., 1995) that are expressed by many cells in the CNS including astrocytes (Reuss et al., 2000; Young et al., 2010), as well as OPCs and myelinating oligodendrocytes (Fortin et al., 2005). However, although FGF9 was found to inhibit the differentiation of isolated OPC into mature oligodendrocytes, this direct effect does not seem to be responsible for its ability to inhibit of myelination in the far more complex, multicellular environment of our myelinating cultures.
We were initially perplexed by this observation as previous studies failed to report any significant effect of FGF9 on OPC proliferation or differentiation (Fortin et al., 2005). However, this earlier study only examined short term responses of early OPCs or mature oligodendrocytes, whilst in the current study we examined the effect of continuous exposure to FGF9 on differentiating A2B5+ progenitors over a period of 7 days. This revealed that as they differentiate these cells enter a stage in which they become FGF9 sensitive; as demonstrated by their proliferative response that may reflect a developmental switch in expression of FGFR3 to FGFR2 (Fortin et al., 2005). Re-entry of these isolated progenitors into cell cycle was associated with inhibition of differentiation and a corresponding decrease in the appearance of PLP+ OPCs. However the reverse was observed when we explored the effects of FGF9 on the oligodendrocyte lineage in myelinating cultures. In this complex cellular environment FGF9 still acts as an OPC mitogen as demonstrated by EdU labelling studies, but in this case this results in a net increase in PLP+ cells. This dichotomy can be explained by off-target effects of FGF9 on other cells in the culture that override or compensate for its direct effect on the differentiation of early progenitors. Possible candidates are astrocyte-derived ‘pro-myelinating’ factors such as IL11 and LIF (Ishibashi et al., 2006; Zhang et al., 2006) that we found are upregulated by FGF9 in myelinating cultures. However despite any potential benefit this may provide, myelination was nonetheless inhibited by an astrocyte-dependent mechanism that inhibits differentiation MBP/PLP+ precursors into mature myelination-competent oligodendrocytes.
Transcriptional profiling revealed inhibition of myelination was associated with changes in gene expression affecting several pathways previously implicated as causes of remyelination failure in multiple sclerosis, in particular those involving interactions with components of the extracellular matrix (e.g. Adamts1, Has2, Chst3, Adamst9, Fn1, Cd44, Mmp3, Mmp11, Mmp15). These include CD44/hyaluronan signalling (Tuohy et al., 2004; Back et al., 2005; Chang et al., 2012; Bugiani et al., 2013), as well as effects associated with increased availability of chondroitin sulphate proteoglycans (Siebert et al., 2011) and fibronectin (Stoffels et al., 2013). The latter is of interest as fibronectin inhibits myelination via an integrin–dependent mechanism that disrupts vesicular traffic to sites of myelin assembly (Maier et al., 2005; Sisková et al., 2006, 2009); an effect that may contribute to the appearance of granular staining for MBP and PLP in the cytosol of oligodendrocytes in FGF9-treated myelinating cultures. It was also noted that processes extended by these cells appear to be able to interact with neurites but nonetheless fail to myelinate; a phenotype reminiscent of that seen in chronically demyelinated multiple sclerosis lesions (Chang et al., 2002).
These effects were accompanied by increased expression of protease families implicated in extracellular matrix remodelling (Howell and Gottschall, 2012; Lau et al., 2013), in particular a disintegrin and metalloproteinase with thrombospondin type 1 motif (ADAMTS) family members 1, 4, 7, 9 and 14; matrix metalloproteinase (MMP) family members 3, 11 and 15 and disintegrin and metalloproteinase domain (ADAM) 12. The activity of these metalloproteinases is regulated to differing extents by tissue inhibitors of metalloproteinases (TIMPs), a family of four physiological protease inhibitors. This was exploited to demonstrate increased expression/activity of TIMP-sensitive proteases contributes to the inhibitory effect of FGF9 on myelination. Further studies are now required to determine the relative contribution of specific proteases and their pathophysiological substrates. Our inability to completely abrogate the inhibitory effect of FGF9 on myelination using a combination of TIMPs may indicate additional pathways contribute to its downstream effects on myelination. However, it should be noted that the inhibitor cocktail used in this study only inhibited aggrecanase activity in cell culture supernatants by ∼50% raising the possibility more complete inhibition could fully abrogate the effect of FGF9. Moreover, screening other potential candidates has as yet failed to identify any that replicate the inhibitory activity of FGF9 (Supplementary Fig. 4).
In addition to transcriptional changes indicative of extracellular matrix remodelling, our micro array study revealed FGF9 enhanced expression of a cluster of ‘pro-inflammatory’ genes, in particular Ccl7 and Ccl2; the latter being validated at the protein level. These chemokines are expressed in multiple sclerosis lesions where they are thought to be involved in recruiting macrophages and microglia (McManus et al., 1998). This ability of FGF9 to induce this ‘pro-inflammatory’ response and at the same time inhibit remyelination has major implications with respect to its role in disease pathogenesis. Traditionally multiple sclerosis is viewed as an inflammatory disease driven by autoimmune response in the periphery, but immunomodulatory treatments that target this response and disease activity in relapsing-remitting multiple sclerosis generally fail to benefit patients with progressive forms of the disease. These immune modulatory treatments are thought to fail because accumulation of disability in these patients is driven by inflammatory mechanisms sequestered within the CNS that are largely independent of T cell recruitment from the periphery (Lassmann et al., 2012). Our data suggest glial expression of FGF9 will contribute to this scenario by virtue of its effects on astrocytes that inhibit remyelination and generate a pro-inflammatory chemokine response; a combination of effects predicted to exacerbate axonal loss while simultaneously recruiting more inflammatory cells into the developing lesion (Franklin et al., 2012). In summary our data identify FGF signalling in astrocytes as a novel target for both regenerative and anti-inflammatory therapies in multiple sclerosis.
Funding
This work was supported by the United Kingdom Multiple Sclerosis Society; The RS McDonald Charitable trust; Naomi Bramson Trust; Gemeinnützige Hertie Stiftung; Deutsche Forschungsgemeinschaft (TR128); Verein zur Therapieforschung für Multiple Sklerose-Kranke and BMBF (Clinical Competence Network Multiple Sclerosis), National Multiple Sclerosis Society of United States (Grant RG 4249A2), Swiss Multiple Sclerosis Society and the Austrian Science Fund (project W1205-B09, CCHD).
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations:
- DIV
days in vitro
- OPC
oligodendrocyte progenitor cell
References
- Azim K, Raineteau O, Butt AM. Intraventricular injection of FGF-2 promotes generation of oligodendrocyte-lineage cells in the postnatal and adult forebrain. Glia. 2012;60:1977–90. doi: 10.1002/glia.22413. [DOI] [PubMed] [Google Scholar]
- Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11:966–72. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci. 2002;24:35–46. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- Bansal R, Pfeiffer SE. Inhibition of protein and lipid sulfation in oligodendrocytes blocks biological responses to FGF-2 and retards cytoarchitectural maturation, but not developmental lineage progression. Dev Biol. 1994;162:511–24. doi: 10.1006/dbio.1994.1105. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–17. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Postma N, Polder E, Dieleman N, Scheffer PG, Sim FJ, et al. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain. 2013;136:209–22. doi: 10.1093/brain/aws320. [DOI] [PubMed] [Google Scholar]
- Butt AM, Dinsdale J. Fibroblast growth factor 2 induces loss of adult oligodendrocytes and myelin in vivo. Exp Neurol. 2005;192:125–33. doi: 10.1016/j.expneurol.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, Chomyk AM, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol. 2012;72:918–26. doi: 10.1002/ana.23693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–73. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Clemente D, Ortega MC, Arenzana FJ, de Castro F. FGF-2 and Anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. Neurosci. 2011;31:14899–909. doi: 10.1523/JNEUROSCI.1158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R, Chandross K. Fibroblast growth factor-9 modulates the expression of myelin related proteins and multiple fibroblast growth factor receptors in developing oligodendrocytes. J Neurosci Res. 2000;61:273–87. doi: 10.1002/1097-4547(20000801)61:3<273::AID-JNR5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, et al. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502:327–32. doi: 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan ID, Brower A, Kondo Y, Curlee JF, Schultz RD. Extensive remyelination of the CNS leads to functional recovery. Proc Natl Acad Sci USA. 2009;106:6832–6. doi: 10.1073/pnas.0812500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott C, Lindner M, Arthur A, Brennan K, Jarius S, Hussey J, et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain. 2012;135:1819–33. doi: 10.1093/brain/aws105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–9. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–55. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol. 2012;8:624–34. doi: 10.1038/nrneurol.2012.200. [DOI] [PubMed] [Google Scholar]
- Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, et al. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol. 1995;155:5769–76. [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–21. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Nave KA, Bansal R. Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci. 2012;32:6631–41. doi: 10.1523/JNEUROSCI.6005-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Kaga Y, Ishii A, Hébert JM, Bansal R. Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J Neurosci. 2011;31:5055–66. doi: 10.1523/JNEUROSCI.4800-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard DR, Berry M, Kirvell SL, Butt AM. Fibroblast growth factor-2 inhibits myelin production by oligodendrocytes in vivo. Mol Cell Neurosci. 2001;18:557–69. doi: 10.1006/mcne.2001.1025. [DOI] [PubMed] [Google Scholar]
- Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166–80. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt T, Antel J, König FB, Brück W, Kuhlmann T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology. 2009;72:1914–21. doi: 10.1212/WNL.0b013e3181a8260a. [DOI] [PubMed] [Google Scholar]
- Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. 2003;13:554–73. doi: 10.1111/j.1750-3639.2003.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, Zimmer C. From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron. 2011;71:574–88. doi: 10.1016/j.neuron.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Harada M, Murakami H, Okawa A, Okimoto N, Hiraoka S, Nakahara T, et al. FGF9 monomer-dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet. 2009;41:289–98. doi: 10.1038/ng.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht D, Zimmerman N, Bedford M, Avivi A, Yayon A. Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors. 1995;12:223–33. doi: 10.3109/08977199509036882. [DOI] [PubMed] [Google Scholar]
- Howell MD, Gottschall PE. Lectican proteoglycans, their cleaving metalloproteinases, and plasticity in the central nervous system extracellular microenvironment. Neuroscience. 2012;217:6–18. doi: 10.1016/j.neuroscience.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain. 2008;131:1464–77. doi: 10.1093/brain/awn080. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, et al. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–32. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Stadelmann C, Hartung HP. Enhancing remyelination in disease–can we wrap it up? Brain. 2011;134:1882–900. doi: 10.1093/brain/awr014. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–58. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nature Rev Neurol. 2012;8:647–56. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- Lau LW, Cua R, Keough MB, Haylock-Jacobs S, Yong VW. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci. 2013;14:722–9. doi: 10.1038/nrn3550. [DOI] [PubMed] [Google Scholar]
- Linington C, Engelhardt B, Kapocs G, Lassman H. Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T cells and demyelinating antibody. J Neuroimmunol. 1992;40:219–24. doi: 10.1016/0165-5728(92)90136-9. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–14. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–9. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Maier O, van der Heide T, van Dam AM, Baron W, de Vries H, Hoekstra D. Alteration of the extracellular matrix interferes with raft association of neurofascin in oligodendrocytes. Potential significance for multiple sclerosis? Mol Cell Neurosci. 2005;28:390–401. doi: 10.1016/j.mcn.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mierzwa AJ, Zhou YX, Hibbits N, Vana AC, Armstrong RC. FGF2 and FGFR1 signaling regulate functional recovery following cuprizone demyelination. Neurosci Lett. 2013;548:280–5. doi: 10.1016/j.neulet.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan H, Friese A, Albrecht S, Krumbholz M, Elliott CL, Arthur A, et al. Transcript profiling of different types of multiple sclerosis lesions yields FGF1 as a promoter of remyelination. Acta Neuropathol Commun. 2014;2:178. doi: 10.1186/s40478-014-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010;468:244–52. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, et al. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci. 2003;23:883–94. doi: 10.1523/JNEUROSCI.23-03-00883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patani R, Balaratnam M, Vora A, Reynolds R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol. 2007;33:277–87. doi: 10.1111/j.1365-2990.2007.00805.x. [DOI] [PubMed] [Google Scholar]
- Patel JR, McCandless EE, Dorsey D, Klein RS. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci USA. 2010;107:11062–7. doi: 10.1073/pnas.1006301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129:3165–72. doi: 10.1093/brain/awl217. [DOI] [PubMed] [Google Scholar]
- Piaton G, Williams A, Seilhean D, Lubetzki C. Remyelination in multiple sclerosis. Prog Brain Res. 2009;175:453–64. doi: 10.1016/S0079-6123(09)17530-1. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Barnard RO, Kwon EE, Sharer LR, Cho ES. Multiple sclerosis: remyelination of nascent lesions. Ann Neurol. 1993;33:137–51. doi: 10.1002/ana.410330203. [DOI] [PubMed] [Google Scholar]
- Prineas JW, Connell F. Remyelination in multiple sclerosis. Ann Neurol. 1979;5:22–31. doi: 10.1002/ana.410050105. [DOI] [PubMed] [Google Scholar]
- Raine CS, Wu E. Multiple sclerosis: remyelination in acute lesions. J Neuropathol Exp Neurol. 1993;52:199–204. [PubMed] [Google Scholar]
- Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain. 1997;120:2149–57. doi: 10.1093/brain/120.12.2149. [DOI] [PubMed] [Google Scholar]
- Renner NA, Ivey NS, Redmann RK, Lackner AA, MacLean AG. MCP-3/CCL7 production by astrocytes: implications for SIV neuroinvasion and AIDS encephalitis. J Neurovirol. 2011;17:146–52. doi: 10.1007/s13365-010-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss B, Hertel M, Werner S, Unsicker K. Fibroblast growth factors-5 and -9 distinctly regulate expression and function of the gap junction protein connexin43 in cultured astroglial cells from different brain regions. Glia. 2000;30:231–41. [PubMed] [Google Scholar]
- Rottlaender A, Villwock H, Addicks K, Kuerten S. Neuroprotective role of fibroblast growth factor-2 in experimental autoimmune encephalomyelitis. Immunology. 2011;133:370–378. doi: 10.1111/j.1365-2567.2011.03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Ruffini F, Furlan R, Poliani PL, Brambilla E, Marconi PC, Bergami A, et al. Fibroblast growth factor-II gene therapy reverts the clinical course and the pathological signs of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther. 2001;8:1207–13. doi: 10.1038/sj.gt.3301523. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Di Filippo M, Ercolani MV, Chiasserini D, Mattioni A, Bonucci M, et al. Fibroblast growth factor-2 levels are elevated in the cerebrospinal fluid of multiple sclerosis patients. Neurosci Lett. 2008;435:223–8. doi: 10.1016/j.neulet.2008.02.040. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–40. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, Rayner PJ, Compston DA. Identification of A2B5-positive putative oligodendrocyte progenitor cells and A2B5-positive astrocytes in adult human white matter. Neuroscience. 1999;89:1–4. doi: 10.1016/s0306-4522(98)00548-x. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Seuntjens E, Nityanandam A, Miquelajauregui A, Debruyn J, Stryjewska A, Goebbels S, et al. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat Neurosci. 2009;12:1373–80. doi: 10.1038/nn.2409. [DOI] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–34. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert JR, Osterhout DJ. The inhibitory effects of chondroitin sulfate proteoglycans on oligodendrocytes. J Neurochem. 2011;119:176–88. doi: 10.1111/j.1471-4159.2011.07370.x. [DOI] [PubMed] [Google Scholar]
- Sisková Z, Baron W, de Vries H, Hoekstra D. Fibronectin impedes “myelin” sheet-directed flow in oligodendrocytes: a role for a beta 1 integrin-mediated PKC signaling pathway in vesicular trafficking. Mol Cell Neurosci. 2006;33:150–9. doi: 10.1016/j.mcn.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Sisková Z, Yong VW, Nomden A, van Strien M, Hoekstra D, Baron W. Fibronectin attenuates process outgrowth in oligodendrocytes by mislocalizing MMP-9 activity. Mol Cell Neurosci. 2009;42:234–42. doi: 10.1016/j.mcn.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Stark JL, Cross AH. Differential expression of suppressors of cytokine signaling-1 and -3 and related cytokines in central nervous system during remitting versus non-remitting forms of experimental autoimmune encephalomyelitis. Int Immunol. 2006;18:347–53. doi: 10.1093/intimm/dxh373. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci USA. 2010;107:11555–60. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffels JM, de Jonge JC, Stancic M, Nomden A, van Strien ME, Ma D, et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain. 2013;136:116–31. doi: 10.1093/brain/aws313. [DOI] [PubMed] [Google Scholar]
- Stankoff B, Aigrot MS, Noël F, Wattilliaux A, Zalc B, Lubetzki C. Ciliary neurotrophic factor (CNTF) enhances myelin formation: a novel role for CNTF and CNTF-related molecules. J Neurosci. 2002;22:9221–7. doi: 10.1523/JNEUROSCI.22-21-09221.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacer KF, Bookout AL, Ding X, Kurosu H, John GB, Wang L, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. 2010;24:2050–64. doi: 10.1210/me.2010-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes. Brain Res. 2009;1287:47–57. doi: 10.1016/j.brainres.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin JE, Xie M, Le TQ, Song SK, Armstrong RC. Reduced axonopathy and enhanced remyelination after chronic demyelination in fibroblast growth factor 2 (Fgf2)-null mice: differential detection with diffusion tensor imaging. J Neuropathol Exp Neurol. 2011;70:157–65. doi: 10.1097/NEN.0b013e31820937e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todo T, Kondo T, Nakamura S, Kirino T, Kurokawa T, Ikeda K. Neuronal localization of fibroblast growth factor-9 immunoreactivity in human and rat brain. Brain Res. 1998;783:179–87. doi: 10.1016/s0006-8993(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD. NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci. 2010;30:16383–90. doi: 10.1523/JNEUROSCI.3411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo JA, Fleming EL, Perlman S. Transgenic CCL2 expression in the central nervous system results in a dysregulated immune response and enhanced lethality after coronavirus infection. J Virol. 2013;87:2376–89. doi: 10.1128/JVI.03089-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuohy TM, Wallingford N, Liu Y, Chan FH, Rizvi T, Xing R, et al. CD44 overexpression by oligodendrocytes: a novel mouse model of inflammation-independent demyelination and dysmyelination. Glia. 2004;47:335–45. doi: 10.1002/glia.20042. [DOI] [PubMed] [Google Scholar]
- Weng Q, Chen Y, Wang H, Xu X, Yang B, He Q, et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron. 2012;73:713–28. doi: 10.1016/j.neuron.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolswijk G. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain. 2002;125:338–49. doi: 10.1093/brain/awf031. [DOI] [PubMed] [Google Scholar]
- Young KM, Mitsumori T, Pringle N, Grist M, Kessaris N, Richardson WD. An Fgfr3-iCreER(T2) transgenic mouse line for studies of neural stem cells and astrocytes. Glia. 2010;58:943–53. doi: 10.1002/glia.20976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Jarjour AA, Boyd A, Williams A. Central nervous system remyelination in culture–a tool for multiple sclerosis research. Exp Neurol. 2011;230:138–48. doi: 10.1016/j.expneurol.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Taveggia C, Melendez-Vasquez C, Einheber S, Raine CS, Salzer JL, et al. Interleukin-11 potentiates oligodendrocyte survival and maturation, and myelin formation. J Neurosci. 2006;26:12174–85. doi: 10.1523/JNEUROSCI.2289-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.