

Abstract
Biomolecular NMR now contributes routinely to every step in the development of new chemical entities ahead of clinical trials. The versatility of NMR — from detection of ligand binding over a wide range of affinities and a wide range of drug targets with its wealth of molecular information, to metabolomic profiling, both ex vivo and in vivo — has paved the way for broadly distributed applications in academia and the pharmaceutical industry. Proteomics and initial target selection both benefit from NMR: screenings by NMR identify lead compounds capable of inhibiting protein–protein interactions, still one of the most difficult development tasks in drug discovery. NMR hardware improvements have given access to the microgram domain of phytochemistry, which should lead to the discovery of novel bioactive natural compounds. Steering medicinal chemists through the lead optimisation process by providing detailed information about protein–ligand interactions has led to impressive success in the development of novel drugs. The study of biofluid composition — metabonomics — provides information about pharmacokinetics and helps toxicological safety assessment in animal model systems. In vivo, magnetic resonance spectroscopy interrogates metabolite distributions in living cells and tissues with increasing precision, which significantly impacts the development of anticancer or neurological disorder therapeutics. An overview of different steps in recent drug discovery is presented to illuminate the links with the most recent advances in NMR methodology.
Introduction
The tedious process of drug discovery requires interdisciplinary team play. Marketable, patented medicine is cultivated by different specialists contributing to each step of the value chain: from the first stage of target selection, through its identification and optimisation to a validated drug that can be administered safely. The process relies on continuous innovation and the improvement of their underlying methodologies. This article briefly points out the consecutive steps, as shown in Figure 1 , where recent advances in NMR spectroscopy are increasingly contributing to pharmaceutical drug discovery.
Figure 1.
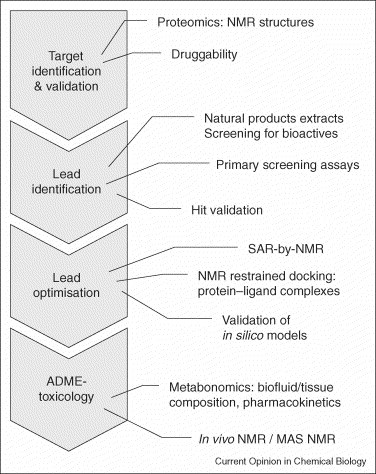
Flow scheme of the contributions of biomolecular NMR to the drug discovery phases.
Proteomics and target selection
Proteomics and structural genomics initiatives often implement strategies for high-throughput cloning, expression (for a comparison of cell-based and cell-free expression protocols see [1]), purification and structure determination to feed the demand for three-dimensional structures of gene products. X-ray crystallography and NMR spectroscopy provide the only sources of experimental data at high, often atomic, resolution. Whether there is a correlation between NMR spectral quality and the success rate of crystallisation is a traditional matter of debate. In one initiative, X-ray crystallography is used as the sole method for structure determination; here, protein samples are subjected to fine-screen or coarse-screen crystallisation trials on the prediction basis of 1D 1H NMR spectra [2].
In a different initiative, a statistical analysis failed to distinguish the HSQC (heteronuclear single quantum correlation) spectra quality distributions of proteins that did and did not successfully yield crystal structures [3•]. Protein samples whose HSQC spectra qualified them as poor or unfolded, crystallised with good diffraction properties and vice versa. The authors indicated that unstructured or molten-globule-like proteins with poor HSQC quality should be subjected to HetNOE data and/or 15N transverse or longitudinal relaxation analysis. This procedure provides more quantitative data and may allow detection of equilibria between folded and unfolded states or some partially folded character in the solution state. The crystallisation trials may drive some samples into the folded conformation by mass action effects. Another large-scale analysis of the results of structural genomics initiatives recommends that both methodologies should be used in parallel because of the complementary success rates [4•]. This approach seems justified given the dramatic improvements in the speed of NMR structure determination [5].
For target selection, the therapeutically relevant targets should be both ‘disease-modifying’ and ‘druggable’ [6•]. Therefore, the three-dimensional structure of a new protein target is inspected for energetic focal points, often called ‘hot spots’, on its surface (Figure 2 ), which are the major contributors to the binding energies of ligands. Geometry-based algorithms (e.g. the flood-fill algorithm implemented within Insight (Accelrys)) heuristically search for concave invaginations. A simple model was obtained by a statistical regression analysis of successful and fruitless NMR screening approaches. The polarity, the surface complexity, the ‘roughness’ and the ‘compactness’ of the corresponding protein pockets are combined to predict the success rate — the ‘druggability’ — of the potential target [7••]. The linear combination of the mentioned properties is used to predict the ‘druggability’ of new target proteins. To delimitate the costs of expensive high-throughput screening (HTS) approaches, the authors put forward the use of NMR-based pre-screening with a diverse fragment library to experimentally assess and validate the general druggability of the protein target.
Figure 2.
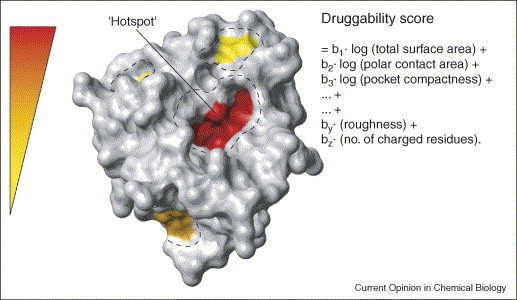
To predict potential druggability, the first step is to identify possible binding sites on the protein surface. Next, characteristics of the putative sites such as hydrophobicity, shape and charge are calculated and submitted to a numerical equation. The weight coefficients bi are known from a statistical analysis of previous screening trials.
This tool for risk assessment might prove handy to spur intensive research on those proteins (and their large molecular weight complexes) that could be targeted with orally bioavailable drugs, especially given recent breakthroughs in small-molecule disruption of protein–protein interactions [8••] (a selection of recent examples is given in Table 1 ). In some cases, inhibitors selective in targeting a single downstream signalling pathway will have to be designed. Blocking enzyme activity per se and therefore indiscriminately disrupting all downstream signaling might not be reasonable [9].
Figure 3.
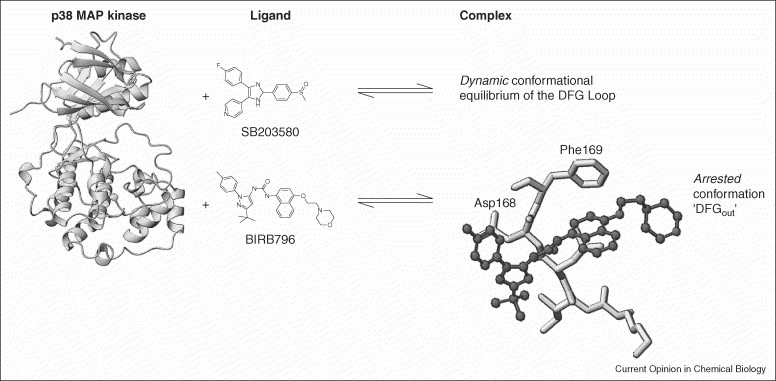
The effects of p38 MAP kinase inhibitors are characterized by the analysis of 2D TROSY spectra of selectively labeled 15N-Phe-p38. The binding of diarylurea compounds such as BIRB796 locks the dynamic in/out exchange into the out-conformation of the highly conserved Asp-Phe-Gly loop (DFG). The unique selection of the conformational space of the involved protein loop enhances the selectivity of the inhibitor.
Table 1.
Some examples: recent NMR-based approaches to pharmaceutically relevant targets
| Protein–protein interaction | NMR applications | Refs |
|---|---|---|
| Hydrophobic BH3-binding groove of Bcl-XL | Fragment screening, SAR-by-NMR, fragment linkage led to small molecule ABT-737 | [10] |
| HIV-1 envelope protein: gp41 | Solution structure, mapping interaction of human antibody epitope D5 | [11, 12] |
| HIV-1: capsid domain | Mapping interactions with peptide inhibitors | [13] |
| Bid (Bcl-2 family member) | Fragment screening, SAR by ILOE (in detail see below) | [14•] |
| P53–MDM2 | Binding studies with small-molecule inhibitor RITA | [15] |
| Malaria surface protein-1 with monoclonal antibody | NMR cross saturation | [16] |
| SARS coronavirus nucleocapsid | Solution structure, characterisation of the dimer interface | [17] |
| Ubiquitin-related modifier SUMO with E2 | NMR restrained docking complex by HADDOCK approach | [18] |
| Protein-cosubstrate interaction | ||
| P38 MAP kinase | Binding studies and protein dynamics in the presence of small-molecule inhibitors (Figure 3) | [19] |
| Protein–polypeptide interaction | ||
| HCV NS3 protease with substrate-based hexapeptide | SAR optimisation guided by molecular modelling and NMR led to a clinical phase macrocyclic inhibitor | [20•] |
| RNA–protein interaction | ||
| HIV-1: TAR RNA with a cyclic peptidomimetic | Solution structure | [21] |
| VEGF165 with aptamer | NMR binding studies with the nucleic acid-based inhibitor Macugen | [22] |
| DNA–protein interaction | ||
| HIV-1: integrase with DNA quadruplex | Solution structure of DNA quadruplex, Docking to tetrameric model of HIV-1 integrase | [23] |
Lead identification
General aspects of NMR screening techniques for drug discovery have been the subject of several recently published reviews [24, 25, 26, 27]. Here, we focus on examples published in 2005.
The fragment-based approach for primary screening has proved to be viable for the identification of lead molecules [28]. The probability of detecting the binding of a low molecular weight (MW ∼200–300 Da) fragment with high sensitivity exceeds that of ligands with MW of ∼500 Da. The functional groups of fragment-based libraries should already include synthetically accessible starting points for chemical linkage. In a follow-up screen, chemical building-block fragments with masked linker groups can be used, an optimisation step in library design called the ‘fragment pair concept’. One can consider fragment pairs of a synthesis and a screening fragment [29••]. Another major issue for any library design is the identification of reactive false positives that oxidize or alkylate a protein target. An assay called ALARM-NMR has developed to rule out these compounds. It monitors 13C chemical shift changes of the human La antigen caused by the test compound or mixture. To validate the unwanted reactivity, the same experiment is repeated in the presence of DTT [30].
Several NMR strategies, which follow on from the initial screening trials, have been proposed. Spin-labeled adenine analogues can be used to detect allosteric ligands at ATP-binding drug targets by paramagnetic relaxation enhancement [31]. The same effect can be utilised when the ATP-complexed magnesium ion is replaced by the paramagnetic manganese ion [32]. By these methods, the proximity of potential allosteric inhibitors can be estimated. The effects of antagonists on protein–protein interactions can elegantly be monitored by NMR spectroscopy. When a small protein (e.g. ∼20 kDa) binds to larger one, the NMR resonances vanish because of excessive line broadening of enhanced relaxation effects in the large macromolecular complex. But the initial NMR spectrum of the small protein can be restored upon addition of an antagonist, which disrupts the protein–protein complex [33].
Improvements stemming from NMR hardware development have also been reported. Better shielding of NMR magnets in combination with cryo-probe technology enables the combination of liquid chromatography with NMR (LC/NMR). Plant extracts can be investigated in the microgram domain to explore an extraordinary reservoir of novel molecules with potential medical use [34]. Binding to protein targets can be detected by intermolecular magnetisation transfer (e.g. the saturation transfer difference (STD) NMR technique). During the search for novel antibiotics, on-flow LC/1H-NMR can also be combined with bioassays of the sampled fractions. The 1D NMR spectra can be used as ‘barcodes’ to guide the fractionation of crude natural mixtures, thus avoiding the repetition of biological testing [35]. For the same reason, multivariate pattern recognition is applied to the complex NMR data, which is obtained from HTS extracts from a diverse collection of plants and marine organisms. Different samples that may contain the same bioactive compounds can be identified or clustered together [36]. A further step towards automated analysis of screening spectra was published recently. A wavelet de-noising step can be applied to 1D/2D screening data prior to common algorithms of data reduction and clustering, which improves the separation of outliers (‘hits’) from the cluster representing spectra of protein with non-binding ligands [37].
Structure-guided lead optimisation
Advances in modelling and NMR data-driven docking procedures allow the determination of approximate structures of biomolecular complexes with rising precision. 15N chemical shift perturbations of amide resonances upon addition of a ligand (if the binding partner is another protein or biomolecule; see review [38]) can routinely be detected and used to principally map the binding interface. This data is subjected as restraints to the docking methods, which model the complex from the individual atomic coordinates. The current algorithms allow the incorporation of other NMR-derived data such as cross-saturation transfer experiments, nuclear Overhauser effects (NOEs) [39], residual dipolar couplings (RDCs) [40] and even data derived from mutational analysis or amino-acid-specific labeling. The recent success in disrupting protein–protein interactions for drug discovery surely fuels the interest in docking protein–protein complexes to guide subsequent research. Applicable tools to model docking complexes with small molecules, which help medicinal chemists to judge structure–activity relationships, have recently been published. One approach can be used as an approximation even in the weak-binding regime of small fragment molecules [41]. Another approach incorporates NMR-NOE data, which can be measured in the tight-binding regime of drug candidates at an advanced optimisation stage [42]. A brief overview of NMR methods, which detect ligand interaction with the target, is given in Table 2 .
Table 2.
NMR methods for the detection of ligand binding and its derived informationa
| Effect | Observation | Used for | Information |
|---|---|---|---|
| Chemical shift perturbation | Ligand/target | Structural information | Identifies binding epitope, delivers restraints for 3D structure calculation |
| Screening/hit validation | Identifies binders, SAR-by-NMR | ||
| Intermolecular magnetisation transfer | |||
| Saturation transfer difference (STD) NMR | Ligand | Primary screening | Identifies weak binders, build-up curve identifies interacting functional groups |
| (Reverse) NOE pumping | Ligand | Characterisation | Identifies binders, alternative to the more robust STD method |
| WaterLOGSY | Ligand | Primary screening | Identifies weak binders |
| SLAPSTICK (requires spin-labeled protein) | Ligand | Primary screening | Highly sensitive detection of binders |
| SAR by ILOE [14•] | Ligand-to-ligand | Compound optimisation | Detects protein-mediated ligand–ligand interactions (competition for the same binding site) |
| INPHARMA method [44] | Ligand-to-ligand | Compound optimisation | Detects protein mediated ligand–ligand interactions (competition for the same binding site) |
| Rotational dynamics | |||
| T2 relaxation, T1ρ, Line broadening | Ligand | Characterisation, primary screening | Binding enhances relaxation, affinity estimation, build-up curve identifies interacting functional groups |
| Sign of transferred intramol. NOE | Ligand | Characterisation | Interaction of tight binders with the target |
| Surface protection | |||
| H2O/D2O exchange rates | Target | Characterisation | Identifies binding epitope |
| Translational dynamics | |||
| DOSY | Ligand | Characterisation | Binding slows diffusion rates |
| Molecular orientation | |||
| Residual dipolar couplings | Ligand/target | Structure determination | Delivers restraints for 3D structure calculation |
The relative orientation of two competitive ligands is important in the design of high-affinity drug candidates from weakly bound fragments. The nuclear Overhauser effect can be transferred from one ligand to the competitive ligand if the two are undergoing rapid exchange. The magnetisation of the first ligand is transferred to the protons of the protein and there it spreads over the interaction surface by spin-diffusion. After replacement, the second ligand picks it up and a correlation peak between both ligand resonances evolves. This information is used for subsequent linker design. The method is described as INPHARMA (protein-mediated interligand NOEs for pharmacophore mapping [44•]) or SAR by ILOE (structure–activity relationship by interligand nuclear Overhauser effect [14•]), which seems to be applicable to any combination of ligands weakly bound to a common receptor.
Solid-state NMR is catching up with the achievements of solution-state NMR. Membrane proteins such as neurologically important GPCRs, transporters or ion pumps can be targeted for pharmaceutical drug discovery. Solid-state NMR in drug design still strongly relies on bioinformatic analysis, and the proposed models are refined by the input data of direct experimentation. NMR signals from the ligand provide useful information about its location within the membrane target, its own structure, the dynamic situation and its putative binding site (for review see [45]). The application has been demonstrated with ligand docking to the gastric H+/K+-ATPase, which has already been successfully targeted by omeprazole, lanzoprazole and pantoprazole [46•]. Homology models of the target protein were generated from the templates of the E1·Ca2+-ATPases and E2·(TG)-conformer, which are adopted during their catalytic cycle. Several analogues to known inhibitors were synthesised and the conformation of a representative was determined in the presence of gastric H+/K+-ATPase by solid-state NMR. The ligand structure was subsequently modelled into the putative binding volumes in the E1/E2 models. The obtained model is consistent with existing site-directed mutagenesis data.
ADME-toxicology
Assessing the pharmacokinetics, a common problem may occur: the potential drug candidate binds tightly to serum albumin and/or is inhibited and modified by cytochrome P450 enzymes. The NMR-feasible domains of these two proteins can be used to investigate possible interactions. Similar to the NMR methods mentioned for lead optimisation, the obtained information guides the medicinal chemist to design out these unacceptable in vivo properties [47].
To investigate the pharmacodynamics, high-throughput NMR can be used to screen biofluid composition for metabolic evidence of drug toxicity or therapeutic progress. Analogous methods for plant extracts such as coupled LC/NMR techniques can be applied. In vivo magnetic resonance spectroscopy (MRS) with the latest high-field instruments enables the analysis of metabolic composition with great spectral dispersion in living tissues [48]. Complementary, ex vivo spectra can be recorded by solid-state NMR from tissue samples that are directly placed into the magic-angle-spinning rotor [49]. Besides the benefits from cryo-probe technology, steady progress has been made because of growing databases, which store the information obtained from animal models and patients. Physiological variations due to species, age, gender, nutrition state etc. are automatically analysed by pattern recognition, and subjected to expert systems [50]. Thus, metabonomic approaches based on information-rich spectroscopic data sets can be used to evaluate normal physiological variation and to investigate drug safety issues.
Conclusion
Truly, NMR is a versatile technique in combination with its unprecedented sensitivity and atomic resolution. Emancipated from academia, NMR routinely contributes to different aspects of drug discovery in the pharmaceutical industry. In this review, readily available NMR methods have been outlined, which most recently contributed to the concatenated steps of target validation and selection, lead discovery and ADME-toxicology. Improvements in hardware and instrumentation, together with the fast-growing field of bioinformatics push the frontier of amendable targets in modern drug discovery.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgement
Work supported by the Center for Biomolecular Magnetic Resonance through a grant of the state of Hesse.
References
- 1.Tyler R.C., Aceti D.J., Bingman C.A., Cornilescu C.C., Fox B.G., Frederick R.O., Jeon W.B., Lee M.S., Newman C.S., Peterson F.C. Comparison of cell-based and cell-free protocols for producing target proteins from the Arabidopsis thaliana genome for structural studies. Proteins. 2005;59:633–643. doi: 10.1002/prot.20436. [DOI] [PubMed] [Google Scholar]
- 2.Page R., Peti W., Wilson I.A., Stevens R.C., Wuthrich K. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc Natl Acad Sci USA. 2005;102:1901–1905. doi: 10.1073/pnas.0408490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Snyder D.A., Chen Y., Denissova N.G., Acton T., Aramini J.M., Ciano M., Karlin R., Liu J., Manor P., Rajan P.A. Comparisons of NMR spectral quality and success in crystallization demonstrate that NMR and X-ray crystallography are complementary methods for small protein structure determination. J Am Chem Soc. 2005;127:16505–16511. doi: 10.1021/ja053564h. [DOI] [PubMed] [Google Scholar]; See annotation to [4•].
- 4•.Yee A.A., Savchenko A., Ignachenko A., Lukin J., Xu X., Skarina T., Evdokimova E., Liu C.S., Semesi A., Guido V. NMR and X-ray crystallography, complementary tools in structural proteomics of small proteins. J Am Chem Soc. 2005;127:16512–16517. doi: 10.1021/ja053565+. [DOI] [PubMed] [Google Scholar]; Both articles present [3•, 4•] a profound analysis of an unbiased approach to produce high-quality protein structures. The correlation between NMR spectral quality and the success in structure determination by X-ray crystallography are discussed in context to structural proteomics pipelines.
- 5.Liu G., Shen Y., Atreya H.S., Parish D., Shao Y., Sukumaran D.K., Xiao R., Yee A., Lemak A., Bhattacharya A. NMR data collection and analysis protocol for high-throughput protein structure determination. Proc Natl Acad Sci USA. 2005;102:10487–10492. doi: 10.1073/pnas.0504338102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6•.Hajduk P.J., Huth J.R., Tse C. Predicting protein druggability. Drug Discov Today. 2005;10:1675–1682. doi: 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]; This review provides a valuable introduction to the different strategies of integrating the protein druggability data with bioinformatic approaches of target selection.
- 7••.Hajduk P.J., Huth J.R., Fesik S.W. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]; This article presents a detailed examination of NMR-based screening data obtained from numerous protein targets. Targets, binding sites and hit rates are analysed and a simple model of scoring protein druggability is derived.
- 8••.Fry D.C., Vassilev L.T. Targeting protein-protein interactions for cancer therapy. J Mol Med. 2005;83:955–963. doi: 10.1007/s00109-005-0705-x. [DOI] [PubMed] [Google Scholar]; This excellent article reexamines the progress that has been made in disrupting protein–protein interactions by small molecules. The analysis focuses primarily on the structural characteristics of the involved binding sites with regard to known ligands and their drug-likeness.
- 9.Roehrl M.H., Kang S., Aramburu J., Wagner G., Rao A., Hogan P.G. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci USA. 2004;101:7554–7559. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A., Bruncko M., Deckwerth T.L., Dinges J., Hajduk P.J. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 11.Jaroniec C.P., Kaufman J.D., Stahl S.J., Viard M., Blumenthal R., Wingfield P.T., Bax A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44:16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 12.Miller M.D., Geleziunas R., Bianchi E., Lennard S., Hrin R., Zhang H., Lu M., An Z., Ingallinella P., Finotto M. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc Natl Acad Sci USA. 2005;102:14759–14764. doi: 10.1073/pnas.0506927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sticht J., Humbert M., Findlow S., Bodem J., Muller B., Dietrich U., Werner J., Krausslich H.G. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12:671–677. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- 14•.Becattini B., Pellecchia M. SAR by ILOEs: an NMR-based approach to reverse chemical genetics. Chemistry. 2006;12:2658–2662. doi: 10.1002/chem.200500636. [DOI] [PubMed] [Google Scholar]; This publication describes an application to identify fragments during lead optimisation that bind in spatial proximity on the surface of the target protein. Covalent linkage could lead to higher affinity bidentate compounds. A similar approach is described in [43].
- 15.Krajewski M., Ozdowy P., D'Silva L., Rothweiler U., Holak T.A. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat Med. 2005;11:1135–1136. doi: 10.1038/nm1105-1135. [DOI] [PubMed] [Google Scholar]
- 16.Morgan W.D., Frenkiel T.A., Lock M.J., Grainger M., Holder A.A. Precise epitope mapping of malaria parasite inhibitory antibodies by TROSY NMR cross-saturation. Biochemistry. 2005;44:518–523. doi: 10.1021/bi0482957. [DOI] [PubMed] [Google Scholar]
- 17.Chang C.K., Sue S.C., Yu T.H., Hsieh C.M., Tsai C.K., Chiang Y.C., Lee S.J., Hsiao H.H., Wu W.J., Chang C.F. The dimer interface of the SARS coronavirus nucleocapsid protein adapts a porcine respiratory and reproductive syndrome virus-like structure. FEBS Lett. 2005;579:5663–5668. doi: 10.1016/j.febslet.2005.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding H., Yang Y., Zhang J., Wu J., Liu H., Shi Y. Structural basis for SUMO-E2 interaction revealed by a complex model using docking approach in combination with NMR data. Proteins. 2005;61:1050–1058. doi: 10.1002/prot.20695. [DOI] [PubMed] [Google Scholar]
- 19.Vogtherr M., Saxena K., Hoelder S., Grimme S., Betz M., Schieborr U., Pescatore B., Robin M., Delarbre L., Langer T. NMR Characterization of kinase p38 dynamics in free and ligand-bound forms. Angew Chem Int Ed Engl. 2006;45:993–997. doi: 10.1002/anie.200502770. [DOI] [PubMed] [Google Scholar]
- 20•.Tsantrizos Y.S. The design of a potent inhibitor of the hepatitis C virus NS3 protease: BILN 2061 – from the NMR tube to the clinic. Biopolymers. 2004;76:309–323. doi: 10.1002/bip.20127. [DOI] [PubMed] [Google Scholar]; This paper details how an oligopeptide was optimised to a macrocyclic β-strand mimic by the combination of molecular modeling and NMR studies.
- 21.Leeper T.C., Athanassiou Z., Dias R.L., Robinson J.A., Varani G. TAR RNA recognition by a cyclic peptidomimetic of Tat protein. Biochemistry. 2005;44:12362–12372. doi: 10.1021/bi0510532. [DOI] [PubMed] [Google Scholar]
- 22.Lee J.H., Canny M.D., De Erkenez A., Krilleke D., Ng Y.S., Shima D.T., Pardi A., Jucker F. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc Natl Acad Sci USA. 2005;102:18902–18907. doi: 10.1073/pnas.0509069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phan A.T., Kuryavyi V., Ma J.B., Faure A., Andreola M.L., Patel D.J. An interlocked dimeric parallel-stranded DNA quadruplex: a potent inhibitor of HIV-1 integrase. Proc Natl Acad Sci USA. 2005;102:634–639. doi: 10.1073/pnas.0406278102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlomagno T. Ligand-target interactions: what can we learn from NMR? Annu Rev Biophys Biomol Struct. 2005;34:245–266. doi: 10.1146/annurev.biophys.34.040204.144419. [DOI] [PubMed] [Google Scholar]
- 25.Shimotakahara S., Furihata K., Tashiro M. Application of NMR screening techniques for observing ligand binding with a protein receptor. Magn Reson Chem. 2005;43:69–72. doi: 10.1002/mrc.1492. [DOI] [PubMed] [Google Scholar]
- 26.Schade M., Oschkinat H. NMR fragment screening: tackling protein-protein interaction targets. Curr Opin Drug Discov Devel. 2005;8:365–373. [PubMed] [Google Scholar]
- 27.Wishart D. NMR spectroscopy and protein structure determination: applications to drug discovery and development. Curr Pharm Biotechnol. 2005;6:105–120. doi: 10.2174/1389201053642367. [DOI] [PubMed] [Google Scholar]
- 28.Zartler E.R., Shapiro M.J. Fragonomics: fragment-based drug discovery. Curr Opin Chem Biol. 2005;9:366–370. doi: 10.1016/j.cbpa.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29••.Schuffenhauer A., Ruedisser S., Marzinzik A.L., Jahnke W., Blommers M., Selzer P., Jacoby E. Library design for fragment based screening. Curr Top Med Chem. 2005;5:751–762. doi: 10.2174/1568026054637700. [DOI] [PubMed] [Google Scholar]; This innovative reference gives valuable practical advice to optimise a generic fragment screening library. The authors describe that functional groups preferred by chemists for modification and linking are also preferentially involved in binding to the protein target. They suggest that fragment pairs are to be screened, where the second compound contains a masked linker group.
- 30.Huth J.R., Mendoza R., Olejniczak E.T., Johnson R.W., Cothron D.A., Liu Y., Lerner C.G., Chen J., Hajduk P.J. ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J Am Chem Soc. 2005;127:217–224. doi: 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- 31.Jahnke W., Blommers M.J., Fernandez C., Zwingelstein C., Amstutz R. Strategies for the NMR-based identification and optimization of allosteric protein kinase inhibitors. Chem Bio Chem. 2005;6:1607–1610. doi: 10.1002/cbic.200500100. [DOI] [PubMed] [Google Scholar]
- 32.McCoy M.A., Senior M.M., Wyss D.F. Screening of protein kinases by ATP-STD NMR spectroscopy. J Am Chem Soc. 2005;127:7978–7979. doi: 10.1021/ja0425942. [DOI] [PubMed] [Google Scholar]
- 33.D'Silva L., Ozdowy P., Krajewski M., Rothweiler U., Singh M., Holak T.A. Monitoring the effects of antagonists on protein-protein interactions with NMR spectroscopy. J Am Chem Soc. 2005;127:13220–13226. doi: 10.1021/ja052143x. [DOI] [PubMed] [Google Scholar]
- 34.Wolfender J.L., Queiroz E.F., Hostettmann K. Phytochemistry in the microgram domain - a LC-NMR perspective. Magn Reson Chem. 2005;43:697–709. doi: 10.1002/mrc.1631. [DOI] [PubMed] [Google Scholar]
- 35.Politi M., Chávez M.I., Cañada F.J., Jiménez-Barbero J. Screening by NMR: a new approach for the study of bioactive natural products? The example of Pleurotus ostreatus hot water extract. Eur J Org Chem. 2005;7:1392–1396. [Google Scholar]
- 36.Pierens G.K., Palframan M.E., Tranter C.J., Carroll A.R., Quinn R.J. A robust clustering approach for NMR spectra of natural product extracts. Magn Reson Chem. 2005;43:359–365. doi: 10.1002/mrc.1562. [DOI] [PubMed] [Google Scholar]
- 37.Trbovic N., Dancea F., Langer T., Gunther U. Using wavelet de-noised spectra in NMR screening. J Magn Reson. 2005;173:280–287. doi: 10.1016/j.jmr.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 38.Bonvin A.M., Boelens R., Kaptein R. NMR analysis of protein interactions. Curr Opin Chem Biol. 2005;9:501–508. doi: 10.1016/j.cbpa.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Mallik B., Katragadda M., Spruce L.A., Carafides C., Tsokos C.G., Morikis D., Lambris J.D. Design and NMR characterization of active analogues of compstatin containing non-natural amino acids. J Med Chem. 2005;48:274–286. doi: 10.1021/jm0495531. [DOI] [PubMed] [Google Scholar]
- 40.van Dijk A.D., Fushman D., Bonvin A.M. Various strategies of using residual dipolar couplings in NMR-driven protein docking: application to Lys48-linked di-ubiquitin and validation against 15N-relaxation data. Proteins. 2005;60:367–381. doi: 10.1002/prot.20476. [DOI] [PubMed] [Google Scholar]
- 41.Schieborr U., Vogtherr M., Elshorst B., Betz M., Grimme S., Pescatore B., Langer T., Saxena K., Schwalbe H. How much NMR data is required to determine a protein-ligand complex structure? Chem Bio Chem. 2005;6:1891–1898. doi: 10.1002/cbic.200500092. [DOI] [PubMed] [Google Scholar]
- 42.Bertini I., Fragai M., Giachetti A., Luchinat C., Maletta M., Parigi G., Yeo K.J. Combining in silico tools and NMR data to validate protein-ligand structural models: application to matrix metalloproteinases. J Med Chem. 2005;48:7544–7559. doi: 10.1021/jm050574k. [DOI] [PubMed] [Google Scholar]
- 43.Lepre C.A., Moore J.M., Peng J.W. Theory and applications of NMR-based screening in pharmaceutical research. Chem Rev. 2004;104:3541–3675. doi: 10.1021/cr030409h. [DOI] [PubMed] [Google Scholar]
- 44•.Sanchez-Pedregal V.M., Reese M., Meiler J., Blommers M.J., Griesinger C., Carlomagno T. The INPHARMA method: protein-mediated interligand NOEs for pharmacophore mapping. Angew Chem Int Ed Engl. 2005;44:4172–4175. doi: 10.1002/anie.200500503. [DOI] [PubMed] [Google Scholar]; This paper details how the relative orientation of two competitive ligands weakly bound to a common protein target could be determined by NMR investigations.
- 45.Watts A. Solid-state NMR in drug design and discovery for membrane-embedded targets. Nat Rev Drug Discov. 2005;4:555–568. doi: 10.1038/nrd1773. [DOI] [PubMed] [Google Scholar]
- 46•.Kim C.G., Watts J.A., Watts A. Ligand docking in the gastric H+/K+-ATPase: homology modeling of reversible inhibitor binding sites. J Med Chem. 2005;48:7145–7152. doi: 10.1021/jm050326o. [DOI] [PubMed] [Google Scholar]; The authors demonstrate a major application of solid-state NMR in drug discovery. Structural NMR restraints of a bound inhibitor were used during a docking approach to an homology model.
- 47.Sun C., Huth J.R., Hajduk P.J. NMR in pharmacokinetic and pharmacodynamic profiling. Chem Bio Chem. 2005;6:1592–1600. doi: 10.1002/cbic.200500028. [DOI] [PubMed] [Google Scholar]
- 48.Gillies R.J., Morse D.L. In vivo magnetic resonance spectroscopy in cancer. Annu Rev Biomed Eng. 2005;7:287–326. doi: 10.1146/annurev.bioeng.7.060804.100411. [DOI] [PubMed] [Google Scholar]
- 49.Valonen P.K., Griffin J.L., Lehtimaki K.K., Liimatainen T., Nicholson J.K., Grohn O.H., Kauppinen R.A. High-resolution magic-angle-spinning 1H NMR spectroscopy reveals different responses in choline-containing metabolites upon gene therapy-induced programmed cell death in rat brain glioma. NMR Biomed. 2005;18:252–259. doi: 10.1002/nbm.955. [DOI] [PubMed] [Google Scholar]
- 50.Bollard M.E., Stanley E.G., Lindon J.C., Nicholson J.K., Holmes E. NMR-based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed. 2005;18:143–162. doi: 10.1002/nbm.935. [DOI] [PubMed] [Google Scholar]