

Abstract
The aldehyde inhibitor Z-Ala-Ala-Phe-CHO has been synthesized and shown by 13C-NMR to react with the active site serine hydroxyl group of alpha-chymotrypsin to form two diastereomeric hemiacetals. For both hemiacetals oxyanion formation occurs with a pKa value of ~ 7 showing that chymotrypsin reduces the oxyanion pKa values by ~ 5.6 pKa units and stabilizes the oxyanions of both diastereoisomers by ~ 32 kJ mol− 1. As pH has only a small effect on binding we conclude that oxyanion formation does not have a significant effect on binding the aldehyde inhibitor. By comparing the binding of Z-Ala-Ala-Phe-CHO with that of Z-Ala-Ala-Phe-H we estimate that the aldehyde group increases binding ~ 100 fold. At pH 7.2 the effective molarity of the active site serine hydroxy group is ~ 6000 which is ~ 7 × less effective than with the corresponding glyoxal inhibitor. Using 1H-NMR we have shown that at both 4 and 25 °C the histidine pKa is ~ 7.3 in free chymotrypsin and it is raised to ~ 8 when Z-Ala-Ala-Phe-CHO is bound. We conclude that oxyanion formation only has a minor role in raising the histidine pKa and that the aldehyde hydrogen must be replaced by a larger group to raise the histidine pKa > 10 and give stereospecific formation of tetrahedral intermediates. The results show that a large increase in the pKa of the active site histidine is not needed for the active site serine hydroxyl group to have an effective molarity of 6000.
Abbreviations: Z, benzyloxycarbonyl
Keywords: Chymotrypsin, Aldehyde inhibitor, Hemiacetal, Oxyanion, Effective molarity
Highlights
-
•
Z-Ala-Ala-Phe-CHO forms two diastereomeric hemiacetals with chymotrypsin.
-
•
The oxyanions of both hemiacetals have pKa values of ~ 7.
-
•
The oxyanion has only a small effect on inhibitor binding and on the pKa of his-57.
-
•
The aldehyde group of the inhibitor increases inhibitor binding 100-fold.
-
•
The effective molarity of the active site serine hydroxyl is ~ 6000 M.
1. Introduction
Specific peptide aldehyde inhibitors are usually formed by replacing the carboxy terminal carboxylate group with an aldehyde group. Such substrate derived aldehyde inhibitors are often highly specific and potent inhibitors of the serine proteases. Consequently they have been used to target specific serine proteases such as prostate specific antigen [1], the proteasome [2] and the SARS 3Cl protease [3] in the hope that they can be used to help in the treatment of conditions such as prostate cancer and severe acute respiratory syndrome.
We have recently developed an approach which enables us to quantify how much a peptide warhead contributes to inhibitor potency [4]. This has been used to determine how the glyoxylate group and the carboxylate group contribute to inhibitor potency with the serine protease chymotrypsin. In this paper we use the same approach to determine how the aldehyde group contributes to inhibitor potency with chymotrypsin.
The rate-limiting step in peptide catalysis by the serine proteases is thought to be either formation or breakdown of a tetrahedral intermediate formed by the addition of the hydroxyl group of the catalytic serine to the carbonyl carbon of the peptide bond being hydrolysed. The tight binding of aldehydes to the cysteine protease papain was attributed to the formation of a thiohemiacetal by the addition of the thiolate ion of the catalytic cysteine to the carbonyl of the peptide bond being hydrolysed analogous to the tetrahedral intermediate formed during catalysis [5]. Similar proposals were made when aldehydes were found to be potent inhibitors of the serine protease elastase [6]. Using 1H-NMR indirect evidence for hemiacetal formation with the serine protease chymotrypsin was obtained using saturation transfer [7], [8], [9]. Direct observation of a hemiacetal signal was reported using 19F-NMR with N-Acetyl-p-fluorophenylalinals and α-chymotrypsin [10], [11]. 13C-NMR studies of the aldehyde inhibitor Acetyl-Phe-CHO binding to α-chymotrypsin have shown two hemiacetals are formed [12]. 13C-NMR has shown that the aldehyde inhibitor leupeptin (N-Acetyl-Leu-Leu-Leu-Arg-CHO) also forms two diastereomeric hemiacetals with trypsin [13]. However, with Acetyl-Leu-Phe-CHO it was reported that only one hemiacetal signal was observed [14]. The fact that only small changes (≤ 4 fold) in Ki values have been observed from pH 3–8 when aldehyde inhibitors bind to chymotrypsin, has led to the suggestion that the hemiacetal formed with chymotrypsin remains neutral from pH 3–8 [11], [15]. Likewise, the fact that the chemical shifts of the hemiacetals observed with Acetyl-Phe-CHO [12] or Acetyl-Leu-Phe-CHO [14] did not titrate significantly at pHs > 7 has been used to support the suggestion that the hemiacetals are neutral from pH 3–8. However, with trypsin the hydroxy groups of both the diastereomeric hemiacetals formed with leupeptin and Acetyl-Leu-Phe-CHO ionized showing oxyanion formation with pKa values of 4.69 and 5.67 [13]. We have also shown that there is oxyanion formation (pKa ~ 5) in chymotrypsin glyoxal inhibitor complexes formed with Z-Ala-Ala-Phe-COCHO (Scheme 1D) [16]. Therefore in this study we use 13C-NMR to determine whether there is oxyanion formation in the corresponding chymotrypsin-aldehyde inhibitor complexes formed with Z-Ala-Ala-Phe-CHO (Scheme 1C) from pH 3 to 11. This allows us to compare how chymotrypsin stabilizes oxyanion formation in hemiacetals and hemiketals [16], [17].
Scheme 1.

Structure of inhibitors.
R is ZAA (Z-Ala-Ala).
Recently we have developed a procedure that allows us to quantify hemiketal formation in chymotrypsin–glyoxal inhibitor complexes and to determine the effective molarity of the hydroxyl group of the catalytic serine residue in these complexes [4]. In this study we use a similar approach to quantify hemiacetal formation in a chymotrypsin–aldehyde inhibitor complex and to determine the effective molarity of the hydroxyl group of the catalytic serine residue in this complex. We also undertake an extensive 1H-NMR study of how the ionization state of the active site histidine residue changes when aldehyde inhibitors are bound. This allows us to determine how oxyanion formation affects the histidine pKa and also whether a high histidine pKa is required for the active site serine hydroxyl group to have a high effective molarity. Finally we discuss the mechanistic significance of these results.
2. Materials and methods
2.1. Materials
l-[1-13C]Phenylalanine (99 at.%) was obtained from Cambridge Isotope laboratories, Inc. (50 Frontage Road, Andover, MA 01810-5413 USA). All other chemicals were obtained from Sigma-Aldrich Chemical co., Gillingham, Dorset, U.K.
2.2. Synthesis of Z-Ala-Ala-Phe-H, Z-Ala-Ala-Phe-COOCH3 and Z-Ala-Ala-Phe-COCHO
Z-Ala-Ala-Phe-H (Scheme 1A) was synthesized as described by Petrillo et al. [4]. Z-Ala-Ala-Phe-13COOCH3 and Z-Ala-Ala-Phe-13COCHO (Scheme 1D) were synthesized as described by Cosgrove et al.,[18].
2.3. Synthesis of Z-Ala-Ala-Phe-13CH2OH
Z-Ala-Ala-Phe-13COOCH3 (220 mg, 0.48 mmol, 1 equiv.) was suspended in anhydrous tetrahydrofuran (2 mL) and added to a stirred suspension of LiAlH4 (125 mg, 3.29 mmol, 7 equiv.) in anhydrous THF (3.5 mL) under nitrogen at 0 °C. The reaction mixture was left to stir for 1 h at 0 °C followed by a further 30 min at room temperature. The reaction mixture was cooled to 0 °C and ethyl acetate (5 mL) was cautiously added dropwise to the mixture, followed by dropwise addition of ice cold water (5 mL). The resulting mixture was stirred for a further 30 min and the layers partitioned. The aqueous layer was extracted with ethyl acetate (2 × 10 mL) and the combined organic layers were washed with brine (10 mL), dried over MgSO4, filtered and solvent was removed in vacuo to give the crude product. Purification by silica chromatography using CH2Cl2-MeOH; 15:1 as an eluant gave the product as a white solid. M.p. 170–175 °C.
The yield was 0.112 g (0.248 mmol 54%). 13C-NMR analysis gave the following data. δC (100 MHz, d6-DMSO) 18.1 (C, CHCH3), 18.5 (C, CHCH3), 36.3 (1C, C6H5 CH2), 48.2 (1C, CHCH3), 50.0 (1C, CHCH3), 52.4 (1C, C6H5CH2 CH), 62.1 (13 CH2OH), 65.4 (1C, O— CH2Ph), 125.9–129.1 (10C, CH CH), 137.0 (1C, CH C ), 137.5(1C, CH C ), 155.7 (1C, O— CO—NH), 171.6 (1C, CONH), 172.0 (1C, CONH).
2.4. Synthesis of Z-Ala-Ala-Phe-13CHO
A sonicated suspension of Z-Ala-Ala-Phe-13CH2OH (94 mg, 0.22 mmol, 1 equiv.) in anhydrous CH2Cl2 (3 mL) was added to a stirred solution of pyridinium chlorochromate (315 mg, 1.46 mmol, 6.5 equiv.) and Celite (280 mg) in anhydrous CH2Cl2 (2 mL). The reaction mixture was left to stir for 6 h after which time silica (ca. 2 g) was added and solvent was removed in vacuo. The residue was purified by silica chromatography using ethylacetate as an eluant. This gave the product as a white solid.
The yield was 0.038 g (0.085 mmol, 41%). 13C-NMR analysis gave the following data. δC (100 MHz, d6-DMSO) 18.1 (C, CHCH3), 18.4 (C, CHCH3), 33.2 (1C, C6H5 CH2) 47.9 (1C, CHCH3), 49.9 (1C, CHCH3), 59.5 (1C, C6H5CH2 CH), 65.3 (1C, O— CH2Ph), 89.8 (1C, — 13 CH(OH)2), 126.3–129.2 (10C, CH CH), 137.0 (1C, CH C ), 137.5(1C, CH C ), 155.7 (1C, O— CO—NH), 171.0 (1C, CONH), 172.7 (1C, CONH), 200.2 (1C, — 13 CHO)
2.5. Chymotrypsin solutions
α-Chymotrypsin (crystallized and lyophilized) was obtained from the Sigma Chemical Co. The amount of fully active enzyme (~ 77%) was determined as described by Finucane et al. [19].
2.6. Determination of Ki values
The Ki value for inhibition of the chymotrypsin-catalyzed hydrolysis of succinyl-Ala-Ala-Pro-Phe-pna at pH 7.2 in 0.1 M potassium phosphate buffer was determined by determining KM(obs) values at different inhibitor concentrations and then plotting KM(obs) versus the inhibitor concentration. Ki was determined from the intercept on the x-axis. All other Ki values were determined when [S0] ≪ KM by as described by [4].
2.7. NMR spectroscopy
NMR spectra at 11.75 T were recorded with a Bruker Avance DRX 500 standard-bore spectrometer operating at 125.7716 MHz for 13C-nuclei. 10 mm-diameter NMR tubes containing ~ 3 mL samples were used for 13C-NMR spectroscopy. The 13C-NMR spectral conditions for the samples were: 8192 time domain data points; spectral width 236.6 ppm; acquisition time 0.138 s; 0.6 s relaxation delay time; 90 °C pulse angle; 2048 transients recorded per spectrum. Waltz-16 composite pulse 1H decoupling of 1.0 W was used which was reduced to 0.026 W during the relaxation delay to minimize dielectric heating but maintain the Nuclear Overhauser Effect.
1H-NMR spectra were recorded at 500 MHz using 5-mm-diameter NMR tubes. Spectral conditions used for observing H-bonded protons at 12–18 ppm were: 32768 time domain data points, spectral width 40 ppm, acquisition time 0.819 s, 1.0 s relaxation delay, 90° pulse angle, 4 dummy scans and 512 transients were recorded per spectrum. Water suppression was carried out with the Watergate W5 pulse sequence [20] and the 1331 pulse sequence [21].
All spectra were transformed using an exponential weight factor of 50 Hz. Both 1H and 13C chemical shifts are quoted relative to tetramethylsilane at 0.00 ppm. The chemical shift of d6-dimethyl sulfoxide at 38.7 ppm was used as a secondary reference for 13C-NMR spectra obtained in aqueous solutions. Aqueous samples contained 10% (v/v) 2H2O to obtain a deuterium lock signal.
3. Results
3.1. 13C-NMR spectra of Z-Ala-Ala-Phe-13CHO in an aqueous solution
In an aqueous solvent at pH 7.2 containing 10 mM sodium phosphate buffer, 10% d6-DMSO and 10% 2H2O Z-Ala-Ala-Phe-13CHO (Scheme 1C) gave signals at 202.7 and 91.0 ppm with JCH values of 183.0 ± 0.3 Hz and 163.4 ± 0.1 Hz respectively. T1 (spin lattice relaxation times) were determined by the inversion recovery method. The signal at 91 ppm had a spin lattice relaxation time of 0.61 ± 0.10 s. Using quantitative 13C-NMR it was determined that the hydration constant (KH1obs) was 13.9 ± 0.7 (Mean of 13 determinations.).
3.2. 13C-NMR of α-chymotrypsin inhibited by Z-Ala-Ala-Phe-13CHO
On adding Z-Ala-Ala-Phe-13CHO (Fig. 1a) to α-chymotrypsin (Fig. 1b) new signals were observed at 99.2 ppm (JCH 158.8 Hz) and 95.5 ppm (JCH 169.3 Hz) at pH 2.93 (Fig. 1c). The signal at 38.7 ppm is due the d6 = DMSO (1.8% (v/v)) used to make the inhibitor stock solution and the signal at 91.2 ppm (JCH 163.4 Hz) (Fig. 1c) is due to a small excess of the inhibitor (Fig. 1a). The signals at 99.2 and 95.5 had T1 values of 0.64 ± 0.07 s and 0.69 ± 0.08 s respectively and are assigned to diastereomeric hemiacetals formed when the active site serine hydroxyl group reacts with the 13C-enriched inhibitor aldehyde group. A hydrate cannot form diastereoisomers and would only give one NMR signal. Therefore the fact that we observe two 13C-NMR signals confirms that two diastereomeric hemiacetals have been formed and that the inhibitor is not bound as a hydrate. The inhibitor was tightly bound at both acid and alkaline pHs (Table 5) and we have calculated that more than 98% of the enzyme is saturated with inhibitor from pH 3.2 to 10.6.
Fig. 1.

Effect of pH on the 13C-NMR spectra from the Z-Ala-Ala-Phe-13CHO in the presence of α-chymotrypsin. 13C-NMR acquisition and processing parameters were as described in the Materials and methods section. Sample conditions were (a) 3.0 ml, 0.2 mM Z-Ala-Ala-Phe-13CHO, 1.7% (v/v) d6-DMSO, 1 mM HCl, (b) 3.0 ml, 1.7 mM α-chymotrypsin. Samples (c)–(h) were 3.0 ml and contained 1.7 mM α-chymotrypsin, 2.1 mM Z-Ala-Ala-Phe-13CHO, 1.8% (v/v) d6-DMSO. Samples (i) and (j) were 3.1 ml and contained 1.7 mM α-chymotrypsin, 2.0 mM Z-Ala-Ala-Phe-13CHO, 1.8% (v/v) d6-DMSO. All samples contained 10% 2H2O which was used as a deuterium lock signal and samples (b)–(h) also contained ~ 10 mM potassium phosphate to help maintain stable pH values.
Table 5.
The effect of pH on the formation of hemiacetals or hemiketals in chymotrypsin inhibitor complexes.
| pH | Ki(obs)a (μM) |
KHA(obs) |
KHK(obs) |
||
|---|---|---|---|---|---|
| ZAAF-H | ZAAF-CHO | ZAAF-COCHO | ZAAF-CHO | ZAAF-COCHO | |
| 3.2 | 1100 ± 60 | 5.4 ± 0.4 | 15.0b | 3100 | 1200 |
| 7.2 | 170 ± 10 | 1.7 ± 0.4 | 0.37b | 1500 | 1200 |
| 10.6 | 1100 ± 110 | 5.5 ± 0.8 | 0.967b | 2900 | 2800 |
Present work, errors are the standard deviations of 3 determinations.
[16].
Hemiacetals like hemiketals are expected to have chemical shifts ~ 4 ppm larger than their hydrates [16]. Therefore the signal at 95.5 ppm has a chemical shift close to that expected if the hydroxyl group of the active site serine of chymotrypsin forms a hemiacetal with the inhibitor aldehyde group. If this process is stereospecific then only one hemiacetal signal will be observed. However, as two hemiacetal signals are observed then we can conclude that hemiacetal formation is not stereospecific.
From the areas of signals at 99.2 ppm and 95.5 ppm (Fig. 1c) the proportions of the corresponding hemiacetals at pH 2.93 were 22.3 ± 0.6% and 77.7 ± 0.6% respectively. Similar proportions (22.6 ± 2.1% and 77.4 ± 2.1%) were obtained at pH 5.85 (Fig. 1e). Due to line broadening accurate measurements could not be made at intermediate pHs. However, similar proportions were also obtained at pHs 10.26 (Fig. 1i, 20.8 ± 1.0% and 79.2 ± 1.0%) and pH 11.10 (Fig. 1j, 26.5 ± 3.0% and 73.5 ± 3.0%). The mean values are 23 ± 2.4% for the diastereoisomer with a signal at 99.2 ppm and 77 ± 2.4% for the diastereoisomer with a signal at 95.5 ppm.
Using 13C-NMR two diastereomeric thiohemiacetals were first observed when the cysteine protease papain was incubated with an aldehyde inhibitor [23]. Subsequently two hemiacetals were observed by 13C-NMR when aldehyde inhibitors reacted with the serine proteases chymotrypsin [12] and trypsin [13]. The hemiacetals formed when leupeptin (N-acetyl-Leu-Leu-Arg-CHO) reacted with trypsin [13] had similar chemical shifts and proportions except the signal at higher chemical shift predominated (86%) whereas in the present work the signal at lower chemical shift predominated (77.4–79.2%).
3.3. Quantifying the role of the aldehyde group in inhibitor binding
The Ki value for the inhibition of the chymotrypsin catalyzed hydrolysis of succinyl-Ala-Ala-Pro-Phe-pna by the aldehyde inhibitor Z-Ala-Ala-Phe-CHO at pH 7.2 was determined from the dependence of the observed KM value for succinyl-Ala-Ala-Pro-Phe-pna on the concentration of the inhibitor present. From the Ki values of ZAAF-H and ZAAF-CHO we estimate that the aldehyde group increases binding ~ 100 fold (Table 1 ).
Table 1.
Inhibition of chymotrypsin by peptide aldehyde and peptide glyoxal inhibitors at pH 7.2: Contribution of the aldehyde and glyoxal groups to inhibition.
| Inhibitor | Ki(obs) (μM) | KiZAAF-H/KiZAAF-CHO | KiZAAF-H/KiZAAF-COCHO |
|---|---|---|---|
| ZAAF-H | 170 ± 10a | ||
| ZAAF-CHO | 1.7 ± 0.4a | 100 | |
| ZAAF-COCHO | 0.37b | 460 |
Present work, errors are the standard deviations of 3 or more determinations.
The glyoxal group of ZAAF-COCHO is bound 4.6 times more tightly than the aldehyde group of ZAAF-CHO (Table 1). 13C-NMR has shown that ZAAF-CHO reacts with chymotrypsin to form two hemiacetals, one smaller signal with a chemical shift of 99.2 ppm and the other larger signal with a chemical shift of 95.5 ppm at pH 2.93 (Fig. 1c). Scheme 2 is the minimal scheme we have used to describe hemiacetal formation. KH1obs is the hydration constant for aldehyde inhibitor (KH1(obs) = hydrate/aldehyde = [Ahyd] / [Aald]). Ks is the dissociation constant (Ks = ([E][Aald]) / [EAald]) of the non-covalent chymotrypsin–aldehyde inhibitor complex (EAald). KHA1 and KHA2 are the equilibrium constants for the formation of hemiacetal-1 and -2 respectively (Scheme 2). The experimentally determined disassociation constant Ki(obs) = [E]([Aald] + [Ahyd]) / ([EAald] + [Hemiacetal-1] + [Hemiacetal-2]) and so
| (1) |
| (2) |
| (3) |
| (4) |
Scheme 2.
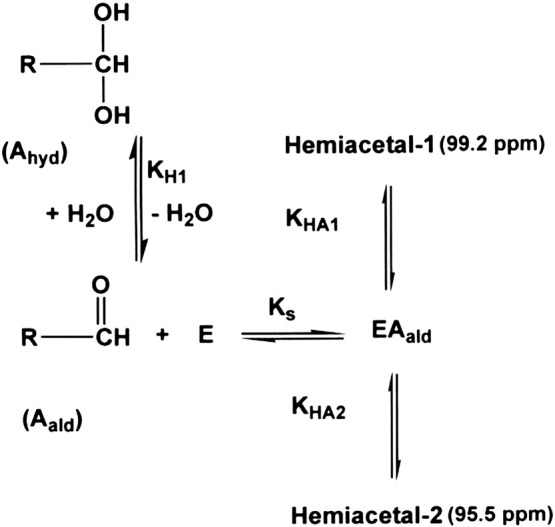
Minimal scheme for hemiacetal formation by chymotrypsin.
Assuming Ki(obs) for ZAAF-H (Scheme 1A) was a good approximation for the disassociation constant (Ks) of the corresponding chymotrypsin–ZAAF-CHO complex (EAald in Scheme 2) KHA(obs) (Eq. (4)) was calculated. The KHA(obs) of 1500 for hemiacetal formation with ZAAF-CHO was similar in magnitude to KHK(obs) for hemiketal formation with ZAAF-COCHO (Table 2 ). Using Eq. (3), the value of 1500 for KHK(obs) and the mean proportions of 77% and 23% for the diastereoisomers at 95.5 ppm and 99.2 ppm respectively KHA1 and KHA2 were determined for the two diastereoisomers (Table 2).
Table 2.
The effective molarity of the catalytic serine of the α-chymotrypsin when forming hemiketals with glyoxal inhibitors or hemiacetals with aldehyde inhibitors at pH 7.2.
| Inhibitor | Ki(obs)a (μM) |
KHK(obs) | KHA(obs) | KH1 (M− 1) |
Effective molarity (M) |
ΔG at 25 °C (kJ/mol) |
|---|---|---|---|---|---|---|
| ZAAF-COCHO | 0.37b | 1200 | 0.028 | 41,000 | − 26.3 | |
| ZAAF-CHO | 1.7 ± 0.39 | 1500 | 0.25 | 6000 | − 21.5 | |
| Diastereoisomer | KHA1 | KHA2 | ||||
| ZAAF-CHO | Hemiacetal-1 | 340 | 0.25 | 1400 | − 17.9 | |
| ZAAF-CHO | Hemiacetal-2 | 1100 | 0.26 | 4600 | − 20.9 | |
Present work, errors are the standard deviations of 3 determinations.
[16].
3.4. Effective molarity of the hydroxyl group of the catalytic serine in α-chymotrypsin
Hemiacetal formation in the chymotrypsin–aldehyde inhibitor complex is a first order process (KHAobs = [hemiacetal] / [EA]) whereas the hydration of the free aldehyde inhibitor in water is a second order process (KH1 = [hemiacetal] / [aldehyde][H2O]). KH1 is calculated by dividing KH1(obs) (KH1(obs) = [hemiacetal] / [aldehyde]) by the molarity of water. The ratio KHAobs/KH1 is therefore the effective molarity of the serine hydroxyl group in hemiacetal formation. This effective molarity is the molarity of water required to be as effective as the catalytic serine hydroxyl group in forming a hemiacetal with the inhibitor aldehyde group. The effective molarity of the active site serine hydroxyl group is ~ 7 times lower for hemiacetal formation with ZAAF-CHO (Scheme 1C) compared to hemiketal formation with the keto carbon of the glyoxal group of ZAAF-COCHO (Scheme 1D).
3.5. Effect of pH on the 13C-NMR signals from Z-Ala-Ala-Phe-CHO in the presence and absence of α-chymotrypsin
On increasing the pH the chemical shift of both the large signal at 95.5 and the small signal at 99.2 ppm increased (Fig. 1a–j). The chemical shift of the large signal (Fig. 1c) increased from at 95.55 ± 0.02 ppm to 97.66 ± 0.02 ppm with a pKa of 6.86 ± 0.05 (Fig. 2B). There was also a further small increase in chemical shift to 98.99 ± 0.04 ppm with a pKa of 10.65 ± 0.12 (Fig. 2B). The total titration shift of 3.44 ppm is similar to that of 3.53 ppm observed for formation of the hemiketal oxyanion with the corresponding hemiketal formed by the glyoxal inhibitor Z-Ala-Ala-Phe-13COCHO reacting with chymotrypsin(Table 3 ). Therefore the titration shift is assigned to formation of the hemiacetal oxyanion when Z-Ala-Ala-Phe-CHO forms a hemiacetal with chymotrypsin. Ionization of the isoleucine-16/aspartate-194 ion pair causes a conformational change which inactivates chymotrypsin [19], [24]. The binding of ligands and or chemical modification of the active site serine hydroxyl is known to raise the pKa of the isoleucine-16/aspartate-194 ion pair to a value of 10.5–11.0 [16], [19] and so we assign the pKa of 10.65 to ionization of the isoleucine-16/aspartate-194 ion pair. The smaller signal at 99.2 ppm(Fig. 1c) titrated with a similar pKa of 7.0(Fig. 2C) but the titration shift (δ2-δ1) was smaller (Table 3).
Fig. 2.

Effect of pH on the chemical shift of the hydrate and the hemiacetal 13C-NMR signals of Z-Ala-Ala-Phe-13CHO observed in the presence of α-chymotrypsin. In (A) and (C) the continuous lines were calculated using the equation δobs = δ1 / (1 + Ka/[H]) + δ2 / (1 + [H]/Ka) and for the hydrate signal in (A) the fitted parameters were pKa = 13.23 ± 0.01, δ1 = 91.04 ± 0.01 ppm and δ2 = 95.28 ± 0.24 ppm. For the hemiacetal signal in (C) the fitted parameters were pKa = 7.00 ± 0.16, δ1 = 99.25 ± 0.02 ppm and δ2 = 100.14 ± 0.02. ppm. For the hemiacetal signal in (B) the continuous line was calculated using the equation δobs = (δ1[H]2 + δ2Ka1[H] + Ka1Ka2Ka3)/([H]2 + Ka1[H]) + Ka1Ka2) and the fitted parameters pKa1 = 6.86 ± 0.05, pKa2 = 10.65 ± 0.02, δ1 = 95.55 ± 0.02 ppm, δ2 = 97.66 ± 0.02 ppm and δ3 = 98.99 ± 0.04 ppm.
Table 3.
Titration constants from pH dependent changes in the chemical shifts of the 13C-enriched carbons of Z-Ala-Ala-Phe-13CHO and Z-Ala-Ala-Phe-13COCHO when they form complexes with α-chymotrypsin.
3.6. Oxyanion stabilization
The pKa values of ~ 7.0 for the hemiacetal oxyanions in the inhibitor complexes are much smaller than the oxyanion pKa of 13.23 ± 0.1 for the inhibitor hydrate (Fig. 2A). This shows that chymotrypsin is stabilizing the hemiacetal oxyanion of both diastereoisomers by similar amounts. In order to quantify this stabilization we need to determine the hemiacetal pKa in the absence of enzyme stabilization. This can be done using free energy relationships [25]. The pKa of ZAAF-13COOH (Scheme 1B) was determined by 13C-NMR to be 3.52 ± 0.03. Using the relationship σ* = (4.644-pKa)/1.7 [26] for ZAAF-COOH the substituent constant, σ* = 0.66 for the substituent ZAAF—. The substituent constant for EO— is 1.68 [27] and using these σ* values and the relationship pKa = 15.9–1.42 Σσ* [28] we estimate that the pKa of the oxyanion of the hemiacetal formed when chymotrypsin and Z-Ala-Ala-Phe-CHO react will be 12.6. Therefore chymotrypsin has lowered the pKa of the larger hemiacetal-1 (Fig. 1c) by 12.6–7.0 = 5.6 pKa units and the pKa of the smaller hemiacetal-2 (Fig. 1c) by a similar amount (Table 4 ). The oxyanions of both hemiacetals are therefore both stabilized by ~ 32 kJ mol− 1 (Table 4).
Table 4.
Hemiacetal and hemiketal oxyanion stabilization in aldehyde (ZAAF-CHO) and glyoxal (ZAAF-COCHO) inhibitor complexes formed with α-chymotrypsin.
| Complex formed | Inhibitor complexed with chymotrypsin | Oxyanion pKa |
pKa decrease |
Oxyanion stabilization |
|
|---|---|---|---|---|---|
| pKaw (water) |
pKae (enzyme) |
pKaw–pKae | ΔG (kJ mol− 1) |
||
| Hemiacetal-1 | ZAAF-13CHO | 12.6a | 7.0b | 5.6 | 32.0 |
| Hemiacetal-2 | ZAAF-13CHO | 12.6a | 6.9c | 5.7 | 32.5 |
| Hemiketal | ZAAF13COCHO | 9.5d | 4.6e | 4.9 | 28.0 |
pKa of hemiacetal in water calculated as described in the text.
pKa of hemiacetal-1 formed with α-chymotrypsin as in Scheme 1.
pKa of hemiacetal-2 formed with α-chymotrypsin as in Scheme 1.
pKa of the hemiketal in water calculated as described for the hemiacetal except that a substituent constant, σ* = 2.15 was used for the aldehyde group of the glyoxal inhibitor [25].
pKa of hemiketal formed with chymotrypsin [16].
3.7. Effect of pH on hemiacetal and hemiketal formation
With the glyoxal inhibitor (ZAAF-COCHO) there is a ~ 40 fold decrease in binding when the pH is decreased from 7.2 to 3.2 and there is a smaller ~ 2.6 fold decrease in binding on increasing the pH from 7.2 to 10.6 (Table 5 ). However, with the aldehyde inhibitor (ZAAF-CHO) there is a much smaller decrease in binding of ~ 3 when the pH is decreased to from 7.2 to 3.2 (Table 5). However, from pH 7.2 to 10.6 there is an ~ 3 fold decrease in binding for both aldehydes and glyoxals (Table 5). A similar small 4 fold increase in Ki from pH 8 to 3 with Bz-Phe-CHO and chymotrypsin has led to the suggestion that the hemiacetal is neutral throughout the pH range 3–8 [11], [15]. As our results show that oxyanion formation occurs according to a pKa of ~ 7 then we conclude that this suggestion is incorrect and the hemiacetal is not neutral at pH 7 or higher.
With ZAAF-COCHO hemiketal formation (KHKobs) is unchanged from pH 7.2 to 3.2 (Table 5)
However, as a result of the better binding of the aldehyde compared to the glyoxal at pH 3.2 there is a ~ 2 fold increase in hemiacetal formation (KHAobs) from pH 7.2 to 3.2 with ZAAF-CHO (Table 5). In contrast with ZAPF-COCHO hemiketal formation decreases from pH 7.2 to 3.2 or 10.2 [4].
The binding of ZAAF-H decreased 6 fold from pH 7.2 to 10.2 and both aldehyde and glyoxal binding decreased ~ 3 fold from pH 7.2 to 10.2 (Table 5). Similar decreases were observed with ZAPF-H and ZAPF-COCHO [4] supporting the suggestion [16] that this increase is due to a conformational change resulting from the ionization of the isoleucine-16/aspartate-194 ion pair in chymotrypsin.
3.8. 1H-NMR of the hydrogen bonded protons of α-chymotrypsin and of its complex with Z-Ala-Ala-Phe-CHO
In chymotrypsin signals at ~ 18 and ~ 13 ppm have been assigned to the Nδ1 and Nε2 protons of the positively charged imidazolium ring of histidine-57 and the signal at ~ 15 ppm has been assigned to the Nδ1 proton of the neutral imidazole ring [16], [29], [30], [31], [32]. In the glyoxal complex of α-chymotrypsin with Z-Ala-Ala-Phe-COCHO the signals at ~ 18 ppm and ~ 13 ppm are present at both low and high pHs at 25 °C demonstrating that the binding of the glyoxal inhibitor has raised the pKa of the active site histidine to a value > 11 [16]. In contrast, in the aldehyde complex of α-chymotrypsin and Z-Ala-Ala-Phe-CHO the signals were barely discernable at 25 °C (Fig. 3A) and they were similar to those observed from free α-chymotrypsin under the same conditions (Fig. 3B). However, at 4 °C the signals at 17.9 ppm and ~ 13.0 ppm were clearly visible at low pH, both in the presence (Fig. 3C1) and absence (Fig. 3D1) of Z-Ala-Ala-Phe-CHO. Unlike with the analogous glyoxal inhibitor [16] these signals disappeared on increasing the pH and were replaced by a single large signal at 14.7 ppm at pHs > 9.4 and 4 °C (Fig. 3C8). This signal (Fig. 3C8) was similar to those observed with free α-chymotrypsin at 4 °C (Fig. 3D8). These signals were barely visible at 25 °C (Fig. 3A8,B8). This shows that there is more exchange broadening with the aldehyde inhibitor compared to its glyoxal analogue. Therefore the exchange rates are faster with the aldehyde inhibitor than with the glyoxal inhibitor. It also shows that with the aldehyde inhibitor the histidine pKa must be in the range 5–9. These spectra and the glyoxal inhibitor spectra were obtained using the Watergate W5 pulse sequence [20]. The 1331 water suppression pulse sequence [21] is effective over a smaller chemical shift range of 14–20 ppm, but it is more effective at detecting exchange broadened signals [14]. Therefore we have used the 1331 pulse sequence to follow the ionization of the imidazolium ion of histidine-57 by the pH titration of the Nδ1 proton from ~ 18 to ~ 15 ppm in the presence and absence of Z-Ala-Ala-Phe-CHO at 25 and 4 °C (Fig. 4 ). From the pH dependence of the chemical shifts of the H-bonded protons (Fig. 5 ) the histidine-57 pKa values in free chymotrypsin and in the Z-Ala-Ala-Phe-CHO-chymotrypsin complex were 7.32 ± 0.18 (Fig. 5D) and 7.97 ± 0.20 (Fig. 5C) respectively at 4 °C. Similar values of 7.35 ± 0.09 and 7.87 ± 0.18 were obtained for the free chymotrypsin (Fig. 5B) and the Z-Ala-Ala-Phe-CHO–chymotrypsin complex (Fig. 5A) respectively at 25 °C. The histidine pKa value for the free enzyme is similar to the values of 6.7 and 7.5 reported for α- and δ-chymotrypsin respectively [16], [32]. The increase in the histidine pKa from 7.3 to 7.9 on adding Z-Ala-Ala-Phe-CHO to α-chymotrypsin contrasts with the observed decrease in the histidine pKa to 6.5 when Acetyl-Leu-Phe-CHO binds to α-chymotrypsin [14].
Fig. 3.

pH dependence of the 1H-NMR spectra of α-chymotrypsin and its complex with Z-Ala-Ala-Phe-13CHO at 4 and 25 °C obtained using the Watergate-5 pulse sequence. 1H-NMR acquisition and processing parameters were as described in the Materials and methods section. All samples contained 0.6-0.7% (v/v) d6-dimethyl sulphoxide, 9.4–10.0 mM potassium phosphate and 10% (v/v) 2H2O which was used as a deuterium lock signal. Sample conditions were; (A) volumes 0.75–0.80 mL, 1.71–1.82 mM α-chymotrypsin and 1.76–1.88 mM Z-Ala-Ala-Phe-CHO; (B) volumes 0.75–0.82 mL, 1.69–1.84 mM α-chymotrypsin and 1.72–1.88 mM Z-Ala-Ala-Phe-CHO; (C) volumes 0.75–0.78 mL, 1.76–1.83 mM α-chymotrypsin and 1.81–1.88 mM Z-Ala-Ala-Phe-CHO;(D) volumes 0.75–0.78 mL, 1.76–1.83 mM α-chymotrypsin and 1.81–1.88 mM Z-Ala-Ala-Phe-CHO.
Fig. 4.

pH dependence of the 1H-NMR spectra of α-chymotrypsin and its complex with Z-Ala-Ala-Phe-13CHO at 4 and 25 °C obtained using the 1331-pulse sequence. 1H-NMR acquisition and processing parameters were as described in the Materials and methods section. All samples contained 0.6–0.7% (v/v) d6-dimethyl sulphoxide and 10% (v/v) 2H2O which was used as a deuterium lock signal. Sample conditions were; (A) volumes 0.75–0.79 mL, 1.76–1.84 mM α-chymotrypsin, 1.80–1.88 mM Z-Ala-Ala-Phe-CHO and 9.5–10.0 mM potassium phosphate; (B) volumes 0.75–0.82 mL, 1.72–1.84 mM α-chymotrypsin, 1.74–1.88 mM Z-Ala-Ala-Phe-CHO and 9.3–10.0 mM potassium phosphate; (C) and (D) volumes 0.75–0.80 mL, 1.72–1.83 mM α-chymotrypsin, 1.81–1.88 mM Z-Ala-Ala-Phe-CHO and 9.4–10.0 mM potassium phosphate.
Fig. 5.

Effect of pH on the chemical shift of the Nδ1 hydrogen bonded proton of histidine-57 in α-chymotrypsin and its complex with Z-Ala-Ala-Phe-13CHO at 4 and 25 °C. In (A), (B), (C) and (D) the continuous lines were calculated using the equation δobs = δ1 / (1 + Ka / [H]) + δ2 / (1 + [H]/Ka) and the fitted parameters were: (A) pKa = 7.87 ± 0.18, δ1 = 17.85 ± 0.06 ppm and δ2 = 14.84 ± 0.06 ppm, (B) pKa = 7.35 ± 0.09, δ1 = 18.11 ± 0.05 ppm and δ2 = 14.73 ± 0.04. ppm, (C) pKa = 7.97 ± 0.20, δ1 = 17.79 ± 0.08 ppm and δ2 = 14.54 ± 0.08 ppm and (D) pKa = 7.32 ± 0.18, δ1 = 18.01 ± 0.12 ppm and δ2 = 14.56 ± 0.09 ppm.
4. Discussion
Aldehydes have been widely used as potent inhibitors of the serine proteases [1], [2], [3], [33] and our results show that the aldehyde group can increase inhibitor potency by a factor of 100 (Table 1). The potency of this inhibition is usually attributed to the ability of the aldehyde group to react with active site serine hydroxyl group to form a hemiacetal that mimics the tetrahedral intermediate formed during substrate catalysis [6], [15]. The fact that the aldehyde group of ZAAF-CHO is readily hydrated (KH1obs = 13.9) illustrates how readily hydroxyl groups react with aldehyde carbonyl carbon to give a tetrahedral species. However, solvent water hydroxyl groups compete with the enzymes catalytic serine hydroxyl decreasing the concentration of the non-hydrated aldehyde group which reacts with hydroxyl group of the catalytic serine residue. Therefore the observed Ki values are normally corrected to allow for this using Kiobs = Ki(1 + KHYD). In our case the Ki value for ZAAF-CHO would be decreased by a factor of 14.9 to 0.11 μM. For ZAAFCOCHO we have shown that the active site serine hydroxyl group of chymotrypsin reacts with the keto carbonyl of the glyoxal group which has a lower KH1obs = 1.58 therefore the observed Ki will only be reduced by a factor of 2.58 to 0.14 μM which is similar to the corrected Ki value of 0.11 μM obtained with the aldehyde inhibitor. Therefore the fact that glyoxal inhibitors are bound ~ 4 fold more tightly than the corresponding aldehyde inhibitor is due to the fact that with the glyoxal inhibitors hemiketal formation occurs at the less effectively hydrated keto carbon as opposed to the more heavily hydrated aldehyde carbon.
Inhibition of chymotrypsin by glyoxal inhibitors has been shown to be stereospecific with only one diastereomeric hemiketal being formed [16], [17]. However, with the corresponding peptide aldehyde inhibitor, inhibition is not fully stereospecific with two diastereoisomers being formed. The diastereoisomer at 95.5 ppm predominates (~ 75%) with the diastereoisomer at 99.2 ppm making up the remaining ~ 25% of the hemiacetal formed. Two diastereoisomers were also observed when N-Acetyl-Phe-CHO was incubated with chymotrypsin but the proportions and chemical shifts were not reported [12]. Likewise when Leupeptin was incubated with trypsin two similar diastereoisomers were detected with chemical shifts of 98.8 ppm (86%) and 97.2 ppm (14%) [13]. These titrated with pKa values of 4.69 and 5.67. In the present work both the chymotrypsin diastereoisomers formed with Z-Ala-Ala-Phe-CHO titrated but their pKa values were very similar at 6.9 and 7.0 (Table 4) suggesting that they are both in similar environments.
X-ray crystallographic studies of aldehyde inhibitors binding to serine proteases [14], [34], [35] has shown that hemiacetal formation is not stereospecific with aldehyde inhibitors with two diastereomeric hemiacetals being formed. One of these has the oxyanion in the oxyanion hole while in the other diastereoisomer the oxyanion points towards the active site histidine [14], [34], [35] and is hydrogen bonded to Nε2 of histidine-57. The oxyanion in the oxyanion hole is about 2.8 Å from the backbone amide nitrogens of serine-195 and glycine-193. However, in the other diastereoisomer the oxyanion is about 2.9 Å from the Nε2 nitrogen of the positively charged imidazolium ion of histidine-57. Therefore the oxyanions are in different electrostatic environments in the two diastereoisomers and so we would expect that they would have significantly different pKa values as is observed for the two diastereoisomeric hemiacetals observed in the Acetyl-Leu-Phe-CHO/trypsin complex [14]. The fact that in the Z-Ala-Ala-Phe-CHO/chymotrypsin complex the oxyanion pKa values were ~ 7 for both diastereoisomers suggests that the oxyanions in both diastereoisomers are in similar electrostatic environments.
Since the binding Z-Ala-Ala-Phe-CHO raised the pKa of histidine-57 to ~ 8 (Fig. 5) and the oxyanion pKa values of the diastereomeric hemiacetals were both ~ 7 (Fig. 2) it is expected that the positively charged histidine residue will play an important role in lowering the oxyanion pKa to ~ 7. The binding of Acetyl-Leu-Phe-CHO to α-chymotrypsin resulted in the formation of a hemiacetal signal at 102 ppm which did not titrate from pH 7 to 13 [14]. The chemical shift value of 102 ppm is larger than that observed (~ 100 ppm) when the oxyanion with Z-Ala-Ala-Phe-CHO is fully formed (Fig. 2B,C). This suggests that the oxyanion is fully formed at pH 7 with Acetyl-Leu-Phe-CHO and so should have a pKa < 6. The histidine pKa of 6.5 in the Acetyl-Leu-Phe-CHO complex [14] shows that it should be able to stabilize the oxyanion. However, with glyoxal inhibitors the histidine pKa is raised to > 11 therefore it is clear that while aldehyde inhibitors like glyoxal inhibitors are bound tightly to chymotrypsin, they are much less effective at raising the pKa of the active site histidine in chymotrypsin. Although oxyanion stabilization is more effective with the aldehyde hemiacetal oxyanion than with the glyoxal hemiketal oxyanion (Table 4) the pKa of the imidazolium ion of histidine-57 is only raised to ~ 8 with hemiacetal formation (Table 6 ) while it is raised to > 11 with hemiketal formation in glyoxal complexes [16] inhibitor and to > 10 when peptidyl trifluoromethyl ketones [36] or peptidyl boronic acid [37] inhibitors form negatively charged tetrahedral adducts with chymotrypsin. This shows that oxyanion formation only has a minor role in raising the pKa of the imidazolium ion of histidine-57 in aldehyde inhibitor complexes. Therefore replacing the aldehyde hydrogen with an aldehyde group [16], [17] or trifluoromethylene group [38] is the major factor in strengthening the hydrogen bond between histidine-57 and aspartate-102, raising the pKa of the imidazolium ion of histidine-57 and ensuring that tetrahedral intermediate formation is stereospecific. Similar effects are expected during catalysis with substrates.
Table 6.
Titration constants from pH dependent changes in the chemical shift of the Nδ1 proton of histidine-57 in α-chymotrypsin and in the Z-Ala-Ala-Phe-CHO/α-chymotrypsin complex at 4 and 25 °C.
| Inhibitor | °C | δ1 (ppm) |
δ2 (ppm) |
δ2–δ1 (ppm) |
pKa |
|---|---|---|---|---|---|
| ZAAF-13CHO | 25 | 17.85 | 14.84 | 3.01 | 7.87 |
| None | 25 | 18.11 | 14.73 | 3.38 | 7.35 |
| ZAAF-13CHO | 4 | 17.79 | 14.54 | 3.25 | 7.97 |
| None | 4 | 18.01 | 14.56 | 4.45 | 7.32 |
Therefore tetrahedral adduct formation only increases the strength of the hydrogen bond between the aspartate and histidine residues raising the pKa of the imidazolium ion of histidine-57 to a value > 10 if the leaving group peptide nitrogen is present or if an inhibitor carbon atom is present in an equivalent position. This suggests that the inhibitor aldehyde hydrogen atom is not large enough to compress histidine-57-aspartate-32 hydrogen bond and/or screen it from solvent so that the histidine pKa is raised to a value > 10. Likewise the aldehyde proton must be replaced by a larger group to ensure that the formation of the tetrahedral species is stereospecific.
The catalytic serine group is expected to have a pKa of ~ 15 and so for the imidazolium ion of histidine-57 to act as an effective base catalyst its pKa should be raised from its value of 7 in the free enzyme to a value similar to that of the active site serine hydroxyl group when a substrate binds [4], [22]. If the aldehyde inhibitor has only a small effect on the pKa of the active site histidine before tetrahedral adduct formation, then the fact that the effective molarity of the hydroxyl group of the catalytic serine group is only ~ 7 × lower with the aldehyde inhibitor compared with its equivalent glyoxal inhibitor (Table 2) shows that a large increase in the pKa of histidine-57 is not required to increase the reactivity of the serine hydroxyl group for formation of the tetrahedral hemiacetal. This suggests that the expected increase in the effective molarity of the active site serine hydroxyl required for tetrahedral intermediate formation during catalysis cannot be solely attributed to a large increase in the pKa of histidine-57. Therefore we propose that the primary reason for the increase in the effective molarity of the catalytic hydroxyl when it binds substrates will be the entropic advantage achieved by optimally aligning the catalytic serine's hydroxy group with the carbonyl carbon of the peptide being hydrolysed.
We conclude that a moiety larger than the aldehydic proton must be present if the pKa of the active site histidine is to be raised to a value > 10 and if there is to be stereospecific formation of a tetrahedral adduct. With aldehyde inhibitors there is stabilization of the hemiacetal oxyanion with its pKa being reduced by ~ 5.6 pKa units while the pKa of the active site histidine is only raised ~ 0.5 pKa units. Therefore we conclude that a large increase in the pKa of the active site histidine is not required for oxyanion stabilization or for the active site serine hydroxyl group to have an effective molarity of 6000. As the active site serine hydroxyl group is expected to have a pKa ~ 15 then we have argued [4], [16] that the large increase in the pKa of the active site histidine (i.e. pKa > 11) observed on binding glyoxal inhibitor is required for it to increase the reactivity of the serine hydroxyl group by general acid catalysis. But, with the aldehyde inhibitor there is only a small increase in the pKa of the active site histidine pKa to ~ 8. However, the effective molarity of the serine hydroxyl group with the aldehyde inhibitor is only ~ 7 fold smaller than that observed with the corresponding glyoxal inhibitor (Table 2). This clearly demonstrates that only a small part of the high reactivity of the serine hydroxyl group can be attributed to general base catalysis by the catalytic histidine residue of chymotrypsin.
Acknowledgements
This work was supported by the Irish Research Council and University College Dublin. Grant nos. 055637/Z/98 from the Wellcome Trust was used to purchase the NMR spectrometer used in these studies and Science Foundation Ireland funding was used to upgrade the NMR spectrometer.
References
- 1.LeBeau A.M., Singh P., Isaacs J.T., Denmeade S.R. Prostate-specific antigen is a “chymotrypsin-like” serine protease with unique P1 substrate specificity. Biochemistry. 2009;48:3490–3496. doi: 10.1021/bi9001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mroczkiewicz M., Winkler K., Nowis D., Placha G., Golab J., Ostaszewski R. Studies of the synthesis of all stereoisomers of MG-132 proteasome inhibitors in the tumor targeting approach. J. Med. Chem. 2010;53:1509–1518. doi: 10.1021/jm901619n. [DOI] [PubMed] [Google Scholar]
- 3.Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 4.Petrillo T., O'Donohoe C.A., Howe N., Malthouse J.P.G. Importance of tetrahedral intermediate formation in the catalytic mechanism of the serine proteases chymotrypsin and subtilisin. Biochemistry. 2012;51:6164–6170. doi: 10.1021/bi300688k. [DOI] [PubMed] [Google Scholar]
- 5.Westerik J.O., Wolfenden R. Aldehydes as inhibitors of papain. J. Biol. Chem. 1972;247:8195–8197. [PubMed] [Google Scholar]
- 6.Thompson R.C. Use of peptide aldehydes to generate transition-state analogs of elastase. Biochemistry. 1973;12:47–51. doi: 10.1021/bi00725a009. [DOI] [PubMed] [Google Scholar]
- 7.Chen R., Gorenstein D.G., Kennedy W.P., Lowe G., Nurse D., Schultz R.M. Evidence for hemiacetal formation between N-acyl-L-phenylalaninals and alpha-chymotrypsin by cross-saturation nuclear magnetic resonance spectroscopy. Biochemistry. 1979;18:921–926. doi: 10.1021/bi00572a030. [DOI] [PubMed] [Google Scholar]
- 8.Lowe G., Nurse D. Evidence for hemiacetal formation between alpha-chymotrypsin and hydrocinnamaldehyde by cross-saturation nuclear magnetic resonance spectroscopy. J. Chem. Soc. Chem. Commun. 1977:815–816. doi: 10.1021/bi00572a030. [DOI] [PubMed] [Google Scholar]
- 9.Wyeth P., Sharma R.P., Akhtar M. A proton-magnetic-resonance study of N-trifluoroacetyl-L-alanyl-L-phenylalaninal binding to alpha-chymotrypsin. Eur. J. Biochem. 1980;105:581–585. doi: 10.1111/j.1432-1033.1980.tb04535.x. [DOI] [PubMed] [Google Scholar]
- 10.Gorenstein D.G., Shah D.O. Proton and fluorine nuclear magnetic resonance spectroscopic observation of hemiacetal formation between N-acyl-p-fluorophenylalaninals and alpha-chymotrypsin. Biochemistry. 1982;21:4679–4686. doi: 10.1021/bi00262a025. [DOI] [PubMed] [Google Scholar]
- 11.Shah D.O., Gorenstein D.G. Fluorine nuclear magnetic resonance spectroscopy of “transition state analogue” complexes of the D and L enantiomers of N-Acetyl-p-fluorophenylalinal and alpha-chymotrypsin. Biochemistry. 1983;22:6096–6101. [Google Scholar]
- 12.Shah D.O., Lai K., Gorenstein D.G. 13C NMR spectroscopy of “transition-state analogue” complexes of N-acetyl-L-phenylalaninal and alpha-chymotrypsin. J. Am. Chem. Soc. 1984;106:4272–4273. [Google Scholar]
- 13.Ortiz C., Tellier C., Williams H., Stolowich N.J., Scott A.I. Diastereotopic covalent binding of the natural inhibitor leupeptin to trypsin: detection of two interconverting hemiacetals by solution and solid-state NMR spectroscopy. Biochemistry. 1991;30:10026–10034. doi: 10.1021/bi00105a030. [DOI] [PubMed] [Google Scholar]
- 14.Neidhart D., Wei Y., Cassidy C., Lin J., Cleland W.W., Frey P.A. Correlation of low-barrier hydrogen bonding and oxyanion binding in transition state analogue complexes of chymotrypsin. Biochemistry. 2001;40:2439–2447. doi: 10.1021/bi002535a. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy W.P., Schultz R.M. Mechanism of association of a specific aldehyde “transition-state analogue” to the active site of alpha-chymotrypsin. Biochemistry. 1979;18:349–356. doi: 10.1021/bi00569a019. [DOI] [PubMed] [Google Scholar]
- 16.Spink E., Cosgrove S., Rogers L., Hewage C., Malthouse J.P.G. 13C and 1H NMR studies of ionizations and hydrogen bonding in chymotrypsin-glyoxal inhibitor complexes. J. Biol. Chem. 2007;282:7852–7861. doi: 10.1074/jbc.M611394200. [DOI] [PubMed] [Google Scholar]
- 17.Djurdjevic-Pahl A., Hewage C., Malthouse J.P.G. A 13C-NMR study of the inhibition of delta-chymotrypsin by a tripeptide-glyoxal inhibitor. Biochem. J. 2002;362:339–347. doi: 10.1042/0264-6021:3620339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cosgrove S., Rogers L., Hewage C., Malthouse J.P.G. An NMR study of the inhibition of pepsin by glyoxal inhibitors: mechanism of tetrahedral intermediate stabilization by the aspartyl proteinases. Biochemistry. 2007;46:11205–11215. doi: 10.1021/bi701000k. [DOI] [PubMed] [Google Scholar]
- 19.Finucane M.D., Hudson E.A., Malthouse J.P.G. A 13C-N.M.R. investigation of the ionizations within an inhibitor-alpha-chymotrypsin complex: evidence that both alpha-chymotrypsin and trypsin stabilize a hemiketal oxyanion by similar mechanisms. Biochem. J. 1989;258:853–859. doi: 10.1042/bj2580853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu M., Mao X., He C., Huang H., Nicholson J.K., Lindon J.C. Improved WATERGATE pulse sequences for solvent suppression in NMR spectroscopy. J. Magn. Reson. 1998;132:125–129. [Google Scholar]
- 21.Hore P.J. Solvent suppression in Fourier transform nuclear magnetic resonance. J. Magn. Reson. 1983;55:283–300. [Google Scholar]
- 22.Finucane M.D., Malthouse J.P.G. A study of the stabilization of tetrahedral adducts by trypsin and delta-chymotrypsin. Biochem. J. 1992;286:889–900. doi: 10.1042/bj2860889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamcsik M.P., Malthouse J.P.G., Primrose W.U., Mackenzie N.E., Boyd A.S.F., Russell R.A., Scott A.I. Structure and stereochemistry of tetrahedral inhibitor complexes of papain by direct NMR observation. J. Am. Chem. Soc. 1983;105:6324–6325. [Google Scholar]
- 24.Fersht A.R., Requena Y. Equilibrium and rate constants for the interconversion of two conformations of α-chymotrypsin. The existence of a catalytically inactive conformation at neutral pH. J. Mol. Biol. 1971;60:279–290. doi: 10.1016/0022-2836(71)90294-4. [DOI] [PubMed] [Google Scholar]
- 25.Perrin D.D., Dempsey B., Serjeant E.P. Chapman and Hall; London and New York: 1981. pKa Prediction for Organic Acids and Bases. [Google Scholar]
- 26.Takahashi S., Cohen L.A., Miller H.K., Peake E.G. Calculation of the pKa values of alcohols from σ* Constants and from the carbonyl frequencies of their esters. J. Org. Chem. 1971;36:1205–1209. [Google Scholar]
- 27.Howe N., Rogers L., Hewage C., Malthouse J.P.G. Oxyanion and tetrahedral intermediate stabilization by subtilisin: detection of a new tetrahedral adduct. Biochim. Biophys. Acta, Proteins Proteomics. 2009;1794:1251–1258. doi: 10.1016/j.bbapap.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Ballinger P., Long F.A. Acid ionization constants of alcohols. II. Acidities of some substituted methanols and related compounds. J. Am. Chem. Soc. 1960;82:795–798. [Google Scholar]
- 29.Bachovchin W.W. Confirmation of the assignment of the low-field proton resonance of serine proteases by using specifically nitrogen-15 labeled enzyme. Proc. Natl. Acad. Sci. U. S. A. 1985;82:7948–7951. doi: 10.1073/pnas.82.23.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markley J.L., Westler W.M. Protonation-state dependence of hydrogen bond strengths and exchange rates in a serine protease catalytic triad: bovine chymotrypsinogen A. Biochemistry. 1996;35:11092–11097. doi: 10.1021/bi961366k. [DOI] [PubMed] [Google Scholar]
- 31.Robillard G., Shulman R.G. High resolution nuclear magnetic resonance study of the histidine-aspartate hydrogen bond in chymotrypsin and chymotrypsinogen. J. Mol. Biol. 1972;71:507–511. doi: 10.1016/0022-2836(72)90366-x. [DOI] [PubMed] [Google Scholar]
- 32.Robillard G., Shulman R.G. High resolution nuclear magnetic resonance studies of the active site of chymotrypsin. J. Mol. Biol. 1974;86:519–540. doi: 10.1016/0022-2836(74)90178-8. [DOI] [PubMed] [Google Scholar]
- 33.Stein R.L., Strimpler A.M. Slow-binding inhibition of chymotrypsin and cathepsin G by the peptide aldehyde chymostatin. Biochemistry. 1987;26:2611–2615. doi: 10.1021/bi00383a030. [DOI] [PubMed] [Google Scholar]
- 34.Delbaere L.T.J., Brayer G.D. The 1.8 Å structure of the complex between chymostatin and Streptomyces griseus protease A. A model for serine protease catalytic tetrahedral intermediates. J. Mol. Biol. 1985;183:89–103. doi: 10.1016/0022-2836(85)90283-9. [DOI] [PubMed] [Google Scholar]
- 35.Kurinov I.V., Harrison R.W. Two crystal structures of the leupeptin–trypsin complex. Protein Sci. 1996;5:752–758. doi: 10.1002/pro.5560050420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang T.C., Abeles R.H. Inhibition of papain by nitriles: mechanistic studies using NMR and kinetic measurements. Arch. Biochem. Biophys. 1987;252:626–634. doi: 10.1016/0003-9861(87)90068-3. [DOI] [PubMed] [Google Scholar]
- 37.Zhong S., Haghoo K., Kettner C., Jordan F. Proton magnetic resonance studies of the active center histidine of chymotrypsin complexed to peptideboronic acids: solvent accessibility to the N-delta and N-epsilon sites can differentiate slow-binding and rapidly reversible inhibitors. J. Am. Chem. Soc. 1995;117:7048–7055. [Google Scholar]
- 38.Liang T.C., Abeles R.H. Complex of α-chymotrypsin and N-acetyl-L-leucyl-L-phenylalanyl trifluoromethyl ketone: structural studies with NMR spectroscopy. Biochemistry. 1987;26:7603–7608. doi: 10.1021/bi00398a011. [DOI] [PubMed] [Google Scholar]