

Abstract
Respiratory infections cause significant morbidity and mortality worldwide. Although an immune response is required to eliminate respiratory pathogens, if unchecked, it can damage surrounding tissues and block primary lung function. Based on our knowledge of immune T-cell activation, there are several pathways to which immune intervention could be applied. However, relatively few interventions target only those immune cells that are responding to antigens. OX40 and 4-1BB are members of the tumour necrosis factor receptor family and are expressed on the surface of T cells in several inflammatory conditions. Recently, the inhibition of OX40 has proved beneficial during influenza virus infection. This review highlights the recent advances in the manipulation of such molecules and how they have been applied to inflammatory conditions that are caused by viruses in the lung.
Respiratory infections account for more than four million deaths worldwide each year. Lower respiratory tract infections are the third leading cause of death worldwide [1] and are a WHO priority for vaccine development. The public recognition of the threat of respiratory infection has recently increased following the emergence of the severe acute respiratory syndrome-associated coronavirus 2, 3, 4, 5.
The elimination of respiratory viruses relies on the recruitment and activation of T lymphocytes. In the lung, this activation is often excessive, causing bystander tissue damage and airway occlusion. Therefore, a fine balance exists between the ability of the immune system to clear the virus and the damage that this response causes to the delicate architecture of the lung. Such immune-related problems are particularly evident in children less than one year of age, in whom the airways are small and not fully developed, as well as in the elderly.
The most unfortunate example of how exuberant immune responses cause severe complications was provided by the vaccine trials performed during the 1960s with a formalin-inactivated preparation of respiratory syncytial virus (RSV), which enhanced the disease severity, resulted in hospitalisation and caused vaccine-related deaths [6]. In mice, vaccination followed by an intranasal RSV challenge causes weight loss, cachexia and the occlusion of airways by inflammatory lymphocytes and eosinophils [7]. The depletion of immune-cell subsets or inflammatory cytokines indicates that disease is associated with an excessive immune response 8, 9. The detrimental outcome occurs as a result of one or more of the following: (i) the physical occlusion of the airways; (ii) the production of inflammatory mediators that enter the systemic circulation. Tumour necrosis factor (TNF), for example, causes cachexia, fever and appetite suppression if concentrations in the lung exceed a certain level and systemic spread occurs; (iii) damage caused by the biological action of immune cells. T lymphocytes are potent sources of cytokines. Helper and cytotoxic T cells produce IFN-γ and TNF during viral infection [10], which, in turn, recruit more immune cells to the lung and, in the case of TNF, cause the death of cells that express the appropriate receptors. Cytotoxic T cells and natural killer (NK) cells lyse virally infected targets by programmed cell death (apoptosis). The rate of viral infection and inflammatory cell death might exceed the ability of the lung microenvironment to remove apoptotic cells, which will contribute to occlusion and inflammation.
The development of anti-viral vaccines has been hampered by the detrimental effect of the early clinical trials, the age of the intended recipient and by strategies used by the virus to evade elimination by the immune system. A treatment that is effective during established viral infection and is not specific for particular viral strains would, therefore, be of great value. Because some of the debilitating effects of infection are caused by the occlusion of the airways by immune cells, strategies to inhibit inflammatory responses have been sought.
1. Coordinated activation of T lymphocytes
There are two main T-cell subsets: CD4+ T helper (Th) lymphocytes and CD8+ cytotoxic T lymphocytes (CTLs). CD4+ T cells can be further divided based upon the cytokines they secrete, into Th0, Th1, Th2 and Th3 cells (reviewed in [11]). CD4+ T cells help other cells by cognate interaction and/or the secretion of soluble mediators, such as cytokines. CD8+ T cells are also potent producers of cytokines and kill pathogen-infected cells or tumour cells directly [12]. The production of cytokines by T cells contributes significantly to the clinical symptoms that are associated with many lung viral infections. The regulation or inhibition of T cells might, therefore, be of therapeutic potential during respiratory infection.
T-cell responses can be separated into several discrete phases: activation, expansion, effector and memory [13]. The point at which T cells perform their effector actions might occur during the expansion and effector phases of the T-cell response, whereas the induction of memory occurs during contraction in the late effector phase. T-cell receptors (TCRs) recognize small peptide fragments of antigen that are presented by major histocompatibility molecules (MHCs); MHC class I for CTLs and MHC class II for Th cells. MHC molecules are constitutively expressed at low levels on ‘professional’ antigen-presenting cells (APCs), such as B cells, dendritic cells and macrophages, but are also upregulated or induced on a variety of other cell types during inflammation. The progression of T cells to full effector function, and especially entry into the memory pool, requires a variety of additional signals. The precise requirement, sequence and cooperation among such signals to maintain T-cell responses and to prevent cell death are beginning to be determined. Cytokines, such as IL-2, assist initial T-cell proliferation, but a variety of additional co-stimulatory signals between the T cell and APCs are required for expansion, survival and entry into the memory pool (Figure 1 ).
Figure 1.
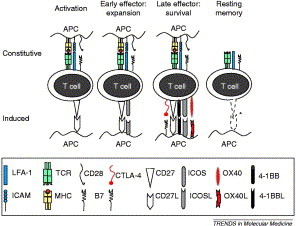
The coordinated sequence of T-cell activation. T-cell activation requires an ordered sequence of events. T cells scan the surface of antigen-presenting cells (APCs) for specific antigenic peptides that are expressed by major histocompatibility complex (MHC) class I or class II molecules. This is facilitated by the interaction of leukocyte function-associated antigen (LFA)-1 with T-cell binding to intracellular adhesion molecule (ICAM)-1 on the APC. Recognition of peptide–MHC by the T-cell receptor (TCR) is not sufficient to fully activate the T cell. A second co-stimulatory interaction between CD28 on the T cell and CD80 and/or CD86 (B7) on the APC is required. Expansion of activated T cells is then facilitated by additional co-stimulatory signals. CD27 is upregulated on activated T cells, as is inducible co-stimulatory molecule (ICOS). During the later stages of the effector response, T-cell activation is reduced by the expression of CTLA-4, which competes with CD28 for binding to CD80 and/or CD86 (B7). To maintain T-cell activation and form the memory pool, additional signals are required. OX40, 4-1BB and CD30 are induced on T cells and bind to their respective ligands on APCs, providing a survival advantage. The interaction of OX40 with OX40 ligand also reduces the expression of CTLA-4. It is not yet known how long ICOS persists on T cells or whether co-stimulatory molecules are present on memory cells (studies suggest that although CD27 is constitutive, it is downregulated on memory T cells). It is also unclear whether multiple co-stimulatory molecules exist on the same T cell.
2. Co-stimulatory molecules and their role in T-cell responses
There are three broad groups of molecules that are pivotal for T-cell responses: the cytokine receptor family that controls various aspects of T-cell function, the immunoglobulin (Ig) superfamily members (CD28, ICOS, PD-1 and CD2) and the TNF receptor superfamily (for example, OX40, 4-1BB, CD27 and CD30).
CD28 is constitutively expressed on resting T cells, binds to B7.1 or B7.2 on APCs and transmits a signal that (i) synergises with TCR signalling to promote a sustained T-cell response, (ii) decreases the number of TCR engagements that are required for effective T-cell activation and (iii) induces the expression of several cytokines 14, 15. Although CD28 co-stimulation is essential for the activation of naïve T cells that have entered the cell cycle, its requirement for memory T-cell responses depends on the model system 16, 17. CD27 (which is expressed on resting T cells and binds to CD70 expressed on APCs and activated T cells) might also function during early T-cell activation. Recent evidence in CD28-knockout mice suggests that CD27 does not affect the cell cycle but sustains T-cell survival through several divisions, rather than influencing the number of dividing cells 17, 18. Inhibitory molecules, such as CTLA-4 and PD1, are upregulated to limit clonal expansion (reviewed in 19, 20, 21). The majority of cells at this stage die by apoptosis, and only if they receive further survival signals do they progress into the memory pool (from which they can be rapidly reactivated to counter future antigenic insults).
T-cell-expressed OX40 and 4-1BB are known to provide the necessary signal for continued T-cell survival. Unlike CD28 and CD27, OX40 and 4-1BB are not present on naïve T cells but are expressed within 24 h of T-cell activation. Expression requires TCR–MHC–peptide interaction and, although CD28 signalling enhances expression, both OX40 and 4-1BB can be expressed in its absence 22, 23, 24, 25, 26. The re-expression of OX40 (data are not yet available for 4-1BB) is much faster (within 4 h) on resting, previously activated T cells [27]. OX40 and 4-1BB can be expressed on both CD4+ and CD8+ T cells, depending on the experimental system used 28, 29, 30, 31. It should be noted, however, that other cell types, such as macrophages, dendritic cells, NK cells and eosinophils, might also express 4-1BB (Table 1 ) (reviewed in [13]).
Table 1.
Characteristics and distribution of T cell co-stimulatory moleculesa
| Name | Cell expression | Naïve T-cell expression? | Inducible expression? | Ligand |
|---|---|---|---|---|
| CD28 | T cells | Yes | Yes | B7.1 or B7.2 |
| ICOS | T cells, NK cells | Yes | ICOSL | |
| OX40 | T, B cells, DCs, eosinophils | Yes | OX40L | |
| 4-1BB | T cells, DCs, macrophages, NK cells, eosinophils | Yes | 4-1BBL | |
| CD27 | T and B cells | Yes | Yes | CD70 |
| CD30 | T and B cells | Yes | CD30L | |
| CD40L | T and B cells, NK cells, DCs basophils, mast cells, eosinophils | Yes | CD40 | |
| HVEM | T and B cells, monocytes, DCs | Yes | Nob | LIGHT |
| CTLA-4 | T cells | Yes | B7.1 or B7.2 | |
| PD-1 | T and B cells, myeloid cells | Yes | PD-L1 or PD-L2 |
Abbreviations: CTLA-4, cytotoxic T lymphocyte antigen-4; DCs, dendritic cells; HVEM, herpesvirus entry mediator; ICOS, inducible co-stimulatory molecule; L, ligand; NK, natural killer; PD-1, program death-1.
Downregulated.
The ligands (L) for these late co-stimulatory molecules are also inducible on professional APCs by lipopolysaccharide, CD40 and Ig cross-linking (on B cells) 32, 33, 34, 35, 36, 37. Some cell-surface expression of OX40L and 4-1BBL is reported on T cells 38, 39, 40, 41. More importantly, OX40L is also present on inflamed endothelial cells 42, 43 and might, therefore, mediate the migration of OX40-expressing cells into inflammatory sites. Another inducible co-stimulator, ICOS (inducible co-stimulatory molecule), is also induced within 12–24 h after T-cell priming and binds to B7 h or B7RP-1 on APCs. However, the expression of ICOS appears slightly earlier and requires less stringent signals than do OX40 and 4-1BB [19].
OX40, 4-1BB and CD27 bind to TNF-receptor-associated factors (TRAFs), which then initiate several signalling pathways. 4-1BB can also bind to TRAF1 but, unlike other TNF receptor family members, not to TRAF5. For OX40 and 4-1BB, binding to TRAF2 activates phosphatidylinositol-3-kinase and AKT [44], which upregulate the anti-apoptotic genes Bcl-2, Bcl-xL and Bfl-1 [45]. All co-stimulatory TNF-receptor molecules induce cell survival through NF-κB, Jun N-terminal kinase and AP1 (reviewed in 13, 37, 46). The precise signalling pathways involved are still being elucidated. At present, co-stimulatory TNF-receptor molecules seem to share common signalling events, although further analysis might show some specificity through the precise combination of signals and the time at which they occur, relative to T-cell activation.
3. Where does this happen in the context of the lung microenvironment?
Naïve T cells do not initially enter the inflamed sites directly, but circulate through secondary lymphoid organs and the blood. Once activated by antigenic peptides presented by MHC molecules in the lymph nodes that are associated with the inflammatory site, T cells acquire chemokine receptors and homing molecules that enable entry into non-lymphoid inflamed tissues. The respiratory tract contains lymphoid tissue in the nose itself (nasal-associated lymphoid tissue, NALT), small foci of follicles (predominantly B cells) that are associated with the bronchi (although this reduces with age) and scattered lymphocytes throughout the lung parenchyma (although these are virtually absent in a healthy individual). The lower airways are not associated with constitutive secondary lymph nodes, nor do they possess classical afferent and efferent lymphatics. Mediastinal lymph nodes are induced following infection, display typical characteristics of secondary lymphoid tissue and retract once inflammation has subsided. The lung encounters a vast array of potentially antigenic material. A significant role for lung APCs is, therefore, the induction of tolerance, which is mediated by lung dendritic cells producing IL-10 (which induces the development of regulatory T cells) and is dependent on co-stimulation by CD86 (B7–2) and ICOS–ICOSL interactions [47]. The propensity for IL-10 production, however, might inadvertently induce allergic Th2 cells [48]. Despite the expression of B7 molecules, alveolar macrophages are poor at co-stimulating T cells [49] and also produce significant quantities of IL-10 [50]. Vigorous T-cell responses can occur if the threat exceeds the immune-suppressive environment. Nasal and airway epithelial cells express B-7 molecules, implying that they participate in naïve T-cell priming [51]. However, the majority of T-cell activation is thought to occur in lung-associated lymph nodes. Beyer et al. [52] showed that, during viral infection, the number and activation of pulmonary APCs increases [53].
Despite the heterogeneity of various surface molecules on lung dendritic cells [54], virtually nothing is known about the expression of the more recently discovered co-stimulatory molecules. The proposed events regarding co-stimulatory-molecule kinetics during lung viral infection are shown in Figure 2 . Despite the near absence of knowledge on the temporal and spatial expression of co-stimulatory molecules and their ligands, several researchers have investigated the effects of their manipulation.
Figure 2.
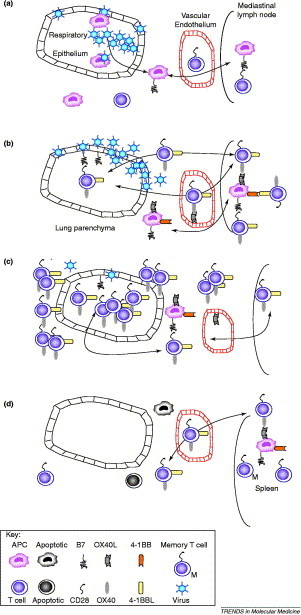
The coordinated activation of T lymphocytes during acute pulmonary viral infection. (a) Infection of the respiratory epithelium, alveolar macrophages and dendritic cells (e.g. with influenza virus or respiratory syncytial virus) causes the release of inflammatory cytokines and chemokines that recruit immune cells to the lung. Some epithelial cells that are killed during replication of the virus are ingested by antigen presenting cells (APCs). These, together with directly infected APCs, migrate to the expanding lung-associated lymph nodes (such as the mediastinal lymph node). Naïve T cells enter the lymph node from the blood and are activated by the viral antigens that are presented on APCs. The activation of T cells depends on the co-stimulatory interaction between CD28 and B7, and results in the expression of alternative chemokine receptors and adhesion molecules that facilitate the entry of activated T cells into the inflamed lung. (b) From 2–4 days after lung infection, T cells are further expanded and upregulate late co-stimulatory molecules (OX40 and possibly 4-1BB). These cells migrate to the site of viral replication via the blood. Note that inflamed endothelial cells can also express OX40L, which might facilitate the movement of OX40-expressing T cells into the lung. Once in the lung, further activation occurs through the interaction with activated APCs or epithelial cells (which can upregulate co-stimulatory molecules). (c) By days 4–8, the level of T-cell recruitment and activation is maximal. OX40 (and probably 4-1BB)-expressing T cells accumulate in and around the airways and blood vessels, disrupting their function. This, together with excessive inflammatory cytokine production, causes the clinical symptoms of infection (sweating, weight loss, cachexia and appetite suppression). (d) If the virus is eliminated and the patient survives, by day 8–18, the inflammatory infiltrate subsides (by activation-induced cell death) and the mediastinal lymph node reduces in size. Some antigen-specific cells survive this contraction process and migrate to the spleen as memory T cells. For clarity, we have omitted inducible co-stimulatory molecule (ICOS), which is induced much faster than OX40. The precise timing of co-stimulatory molecule expression is affected by the precise pathogen and dose.
4. Late co-stimulatory molecule manipulation and immunopathology
The restricted expression of late co-stimulatory molecules on antigen-activated T cells makes them ideal therapeutic targets for immune intervention. Manipulation will only affect those T cells recently activated by antigen, leaving the naïve repertoire intact (reviewed in [13]). Here, we only discuss the manipulation of co-stimulatory molecules during infection of the respiratory tract (for other diseases or lung conditions, see 37, 55).
The strategies used to combat respiratory infection are mostly dictated by the pathogen. Pathogens that cause direct pathology might be more suited to strategies that enhance immunity. A reduction in immunity, however, could benefit individuals who are infected with pathogens that induce an excessive inflammatory infiltrate. The promotion of T-cell-mediated immunity can be achieved using agonist antibodies against co-stimulatory molecules and soluble or vector-expressed forms of the ligand. For example, adenoviral-expressed 4-1BBL (which binds to 4-1BB on activated T cells) or B7.1 (which binds to CD28 on all T cells) enhances influenza-specific human CD8+ T cells [56]. Although agonist antibodies against 4-1BB promote influenza specific CD8+ T cells, the effect on illness or lung inflammation [57] is not known. Similarly, an OX40L–Ig fusion protein, which binds to OX40 on activated T cells, promotes immunity against lung Cryptococcus neoformans infection, enhances pathogen clearance and reduces pulmonary eosinophilia; these effects are dependent on the induction of IL-12 and/or IFN-γ [58]. Therefore, the administration of co-stimulatory molecule agonists provides a novel adjuvant role, which is specific to those cells that respond to vaccination or infection. It might be more appropriate to promote co-stimulatory molecules that are upregulated on antigen-activated T cells (especially OX40 and 4-1BB) because targeting constitutively expressed molecules (CD28 and, possibly, CD27 and ICOS) could promote the survival of a larger proportion of cells, which might cause immunopathology or auto-immunity.
Data regarding the inhibition of co-stimulatory molecules during infection in the lung are more frequent, as a result of the presence of several reagents and knockout mice. In order of importance following T-cell activation:
4.1. CTLA-4
In murine models, CTLA-4–Ig fusion proteins (which bind to B7 molecules on the APC and compete for binding with CD28 and CTLA4) prevent the pneumonitis that is caused by Saccharopolyspora rectivirgula [59] and reduce influenza-virus-specific IFN-γ production, antibody titres and the number of virus-specific CD8+ T cells. Although influenza virus is still eliminated, the kinetics of clearance is delayed [60]. The complication with using soluble CTLA-4 is that there are two forms of the receptor [B7.1 (CD80) and B7.2 (CD86)], which seem to have subtly different roles. However, this might provide more subtle intervention opportunities, because mutated CTLA-4–Ig inhibits influenza-induced inflammation without preventing viral clearance [60]. The possibility of interfering with non-pathogen-specific T cells is another potential drawback.
4.2. CD27
Intranasal influenza virus infection of CD27-knockout mice results in reduced anti-viral CD4+ and CD8+ T cells in the acute phase and the memory pool [61]. The effect on both primary and memory responses lends further support to the hypothesis that CD27 acts concurrently with, or at least very close to, CD28 signals. Recent data from Hendriks et al. [17] show that CD27 and CD28 cooperate in the CD8+ T-cell response to influenza virus infection. CD27 can compensate for the lack of CD28 during influenza-specific T-cell responses in the lung and for T-cell priming in the mediastinal lymph nodes, whereas in mice lacking both molecules, T-cell responses are virtually absent. The influence of CD27 on virus-specific T-cell responses appears to be restricted to the site of viral replication because splenic CD8+ T cells are unaffected. In addition, mice lacking CD27 or CD28, or both, show reduced recall CD8+ T-cell responses to an influenza virus challenge. The mode of action of each molecule might differ subtly depending on the site studied. CD28, but not CD27, affects the cell cycle, whereas CD27, particularly in the lung, assists with the survival of antigen-activated CD8+ T cells.
4.3. ICOS
The majority of studies on the role of ICOS inhibition in the lung have been conducted in models of asthma and allergy 19, 21. However, one study by Bertram et al. [62] reported anti-influenza responses after the intraperitoneal infection of ICOS-knockout mice. An absence of ICOS reduces antibody responses during primary infection and during re-challenge. By day 38 of primary infection, influenza-specific CD8+ T cell levels are half those observed in wild-type mice, which might be associated with a lack of IL-2 production by CD4+ T cells in the knockout mice [62]. Because of the route of infection, no data are available on viral clearance or immune pathology.
4.4. 4-1BB and OX40
4-1BB appears to be crucial for late or secondary CD8+ T-cell responses to influenza virus infection. 4-1BB-knockout mice display normal CD8+ T-cell responses during a primary infection, which are impaired during secondary infections. Additionally, soluble 4-1BB inhibits T-cell responses to influenza peptide in CD28-deficient mice. The effect on T-cell populations is more dramatic when both CD28 and 4-1BB are absent 63, 64. Agonistic antibodies against 4-1BB restore the defect in secondary CD8+ T cells in influenza-infected 4-1BB-knockout mice, but only when administered during secondary, and not primary, responses. Interestingly, agonistic antibodies against 4-1BB can restore the defective primary CD8+ T-cell response in CD28-knockout mice when administered early during infection [65]. 4-1BB is, therefore, important for the generation of influenza-specific CD8+ T cells. Data so far suggest that anti-viral CD4+ T cells and antibodies are not affected by the manipulation of 4-1BB [64]. However, these manipulations have only been tested in an intraperitoneal infection model, and there are no data available on virus clearance or immunopathology.
The inhibition of OX40 produces a similar effect to 4-1BB, but with reduced CD4+ and CD8+ anti-viral T cells. Using an OX40–Ig fusion protein to block the interaction of OX40 with its ligand on APCs, influenza-induced weight loss and cachexia are eliminated and the virus is still removed from the lung. More importantly, such treatment is beneficial three days after the onset of clinical symptoms [29]. The reduction of virus-associated illness is accompanied by a reduced cellular infiltrate into the airways and lung parenchyma. A reduction in the proliferation and an enhancement in the apoptosis of lung cells accounts for this reduced cellularity. These data are partially supported by studies in OX40-knockout mice, in which CD4+ T-cell infiltrates were reduced. However, no effect was observed on CD8+ T-cell responses [66]. Based on these findings with OX40, it is tempting to speculate that the inhibition of 4-1BB or its ligand would produce similar effects.
It is important to appreciate that a reduction in the immunopathology resulting from infection must be balanced by the ability of the immune system to clear the virus and prevent illness. For OX40 inhibition, although MHC tetramer binding to CD8+ T cells is reduced, it is not entirely eliminated. Influenza virus is inflammatory and, therefore, a moderate reduction in the vigour of the inflammatory response might be beneficial without affecting virus clearance. The ability to clear viral infection despite significantly reduced lung inflammation might also reflect redundancy among late co-stimulatory signals and the presence of intact innate and antibody-mediated immune mechanisms. For example, the inhibition of OX40 leaves the 4-1BB co-stimulatory pathway intact. This hypothesis is consistent with the efficient control of other infections in OX40-knockout mice 66, 67. CD27-knockout mice similarly recover from influenza virus infection without displaying any signs of illness [61]. The interruption of CD30, which is also expressed on effector and memory T cells, has yet to be tested in models of lung viral infection. To understand the synergy and redundancy it will be important to examine anti-viral immunity in an environment that is deficient for multiple late co-stimulatory molecules (e.g. 4-1BB and OX40).
5. Concluding remarks
A clear conclusion from in vivo lung infection models is that a reduction, but not an elimination, of inflammatory infiltrate reduces the clinical manifestations without compromising pathogen clearance. The manner in which this reduction is achieved is important. The manipulation of late co-stimulatory molecules is tempting because of their predominant expression on effector, but not naïve, T cells. It is likely that the inhibition of OX40, 4-1BB or CD30 would serve this purpose, although studies on the protection from re-infection and clinical disease are required following the manipulation of 4-1BB and CD30. Other co-stimulatory molecules, such as herpesvirus entry mediator (HVEM, which is expressed on naïve, and some memory T cells) and CD30 (which is expressed on effector cells and some memory cells), have yet to be tested. Similarly, the engagement of PD-1 on activated T cells by either of the two ligands (PDL-1 and PDL-2) inhibits T-cell proliferation and cytokine secretion 20, 21. Soluble forms of these ligands or agonistic antibodies against PD-1 might, therefore, limit excessive T-cell expansion, although this has yet to be tested in models of inflammatory lung disease. It might also be possible to manipulate late co-stimulatory molecules by interfering with the signalling pathway. TNF receptor family members have been shown to bind to TRAFs [37]. At present, it appears that the inhibition of signalling might concurrently affect too many co-stimulatory molecules. By manipulating molecules on the cell surface, the redundancy that exists could be advantageous. Although the outcome of inhibiting co-stimulatory molecules is clear, the mechanism is not. Therefore, further studies will need to address whether manipulation alters T-cell survival, cytokine production and/or chemokine receptor expression patterns [22].
The manipulation of late co-stimulatory signals has clear therapeutic potential: on one hand we can reduce immune-based disorders such as asthma, bronchiolitis and pneumonia, and on the other we can promote immunity against the lung infections in which the pathogen is the problem. Treatment to inhibit late co-stimulatory signals can also be extended to autoimmune conditions, whereas the promotion of these signals enhances anti-tumour immunity [68] and might improve the efficacy of vaccines 69, 70. The nature of the compound that is used to manipulate co-stimulatory signals will require significant thought, because the production of fusion proteins and antibodies is expensive. Future analysis of the exact binding regions between co-stimulatory molecules and their ligands might highlight specific epitopes that could be used as inhibitors (Box 1).
Box 1. Future research.
• Determine which molecules are involved in tumour necrosis factor receptor superfamily signaling, and their potential for intervention.
• Determine the effect of 4-1BB and CD30 inhibition on immunity to infection in the lung.
• Analyse the coordinated expression of co-stimulatory molecule expression in vivo.
• Investigate the potential of inhibition through PD-1 to inhibit inflammatory disease.
• Elucidate the role of co-stimulatory molecules in cell trafficking, positioning and development.
• Test co-stimulatory agonists and antagonists in human inflammatory disorders.
Acknowledgements
The authors would like to thank the Medical Research Council, The National Asthma Campaign and the BBSRC for their continued support.
References
- 1.Murray C.J., Lopez A.D. Mortality by cause for eight regions of the world: global burden of disease study. Lancet. 1997;349:1269–1276. doi: 10.1016/S0140-6736(96)07493-4. [DOI] [PubMed] [Google Scholar]
- 2.Lee N. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 3.Poutanen S.M. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 4.Ksiazek T.G. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 5.Peiris J.S. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McIntosh K., Fishaut J.M. Immunopathologic mechanisms in lower respiratory tract disease of infants due to respiratory syncytial virus. Prog. Med. Virol. 1980;26:94–118. [PubMed] [Google Scholar]
- 7.Openshaw P.J.M. Pulmonary eosinophilic response to respiratory syncytial virus infection in mice sensitized to the major surface glycoprotein G. Int. Immunol. 1992;4:493–500. doi: 10.1093/intimm/4.4.493. [DOI] [PubMed] [Google Scholar]
- 8.Bembridge G.P. Recombinant vaccinia virus co-expressing the F protein of respiratory syncytial virus (RSV) and interleukin-4 (IL-4) does not inhibit the development of RSV-specific memory cytotoxic T lymphocytes, whereas priming is diminished in the presence of high levels of IL-2 or γ interferon. J. Virol. 1998;72:4080–4087. doi: 10.1128/jvi.72.5.4080-4087.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussell T. IL-12 treatment attenuates Th2 and B cell responses but does not improve vaccine-enhanced lung illness. J. Immunol. 1997;159:328–334. [PubMed] [Google Scholar]
- 10.Hussell T. Inhibition of tumour necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001;31:2566–2573. doi: 10.1002/1521-4141(200109)31:9<2566::aid-immu2566>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 11.Neurath M.F. The role of Th1/Th2 polarization in mucosal immunity. Nat. Med. 2002;8:567–573. doi: 10.1038/nm0602-567. [DOI] [PubMed] [Google Scholar]
- 12.Doherty P.C. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 1997;159:105–117. doi: 10.1111/j.1600-065x.1997.tb01010.x. [DOI] [PubMed] [Google Scholar]
- 13.Croft M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003;14:265–273. doi: 10.1016/s1359-6101(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 14.Linsley P.S., Ledbetter J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 15.Lanzavecchia A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96:1–4. doi: 10.1016/s0092-8674(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 16.Croft M. Naïve versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen-presenting cell types including resting B cells. J. Immunol. 1994;152:2675–2685. [PubMed] [Google Scholar]
- 17.Hendriks J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J. Exp. Med. 2003;198:1369–1380. doi: 10.1084/jem.20030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hintzen R.Q. CD27: marker and mediator of T-cell activation? Immunol. Today. 1994;15:307–311. doi: 10.1016/0167-5699(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 19.Coyle A.J., Gutierrez-Ramos J.C. The role of ICOS and other costimulatory molecules in allergy and asthma. Springer Semin. Immunopathol. 2004;25:349–359. doi: 10.1007/s00281-003-0154-y. [DOI] [PubMed] [Google Scholar]
- 20.Sharpe A.H., Freeman G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 21.Carreno B.M., Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- 22.Walker L.S. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J. Exp. Med. 1999;190:1115–1122. doi: 10.1084/jem.190.8.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers P.R. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 24.Gramaglia I. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000;165:3043–3050. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- 25.Diehl L. In vivo triggering through 4-1BB enables Th-independent priming of CTL in the presence of an intact CD28 costimulatory pathway. J. Immunol. 2002;168:3755–3762. doi: 10.4049/jimmunol.168.8.3755. [DOI] [PubMed] [Google Scholar]
- 26.Akiba H. CD28-independent costimulation of T cells by OX40 ligand and CD70 on activated B cells. J. Immunol. 1999;162:7058–7066. [PubMed] [Google Scholar]
- 27.Gramaglia I. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 28.Weinberg A.D. OX-40: life beyond the effector T cell stage. Semin. Immunol. 1998;10:471–480. doi: 10.1006/smim.1998.0146. [DOI] [PubMed] [Google Scholar]
- 29.Humphreys I.R. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J. Exp. Med. 2003;198:1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dawicki W., Watts T.H. Expression and function of 4-1BB during CD4 versus CD8 T cell responses in vivo. Eur. J. Immunol. 2004;34:743–751. doi: 10.1002/eji.200324278. [DOI] [PubMed] [Google Scholar]
- 31.Bansal-Pakala P. Costimulation of CD8 T cell responses by OX40. J. Immunol. 2004;172:4821–4825. doi: 10.4049/jimmunol.172.8.4821. [DOI] [PubMed] [Google Scholar]
- 32.Brocker T. CD4 T cell traffic control: in vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B follicles. Eur. J. Immunol. 1999;29:1610–1616. doi: 10.1002/(SICI)1521-4141(199905)29:05<1610::AID-IMMU1610>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Weinberg A.D. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J. Immunol. 1999;162:1818–1826. [PubMed] [Google Scholar]
- 34.Stuber E. Cross-linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity. 1995;2:507–521. doi: 10.1016/1074-7613(95)90031-4. [DOI] [PubMed] [Google Scholar]
- 35.Futagawa T. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 2002;14:275–286. doi: 10.1093/intimm/14.3.275. [DOI] [PubMed] [Google Scholar]
- 36.Goodwin R.G. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. Eur. J. Immunol. 1993;23:2631–2641. doi: 10.1002/eji.1830231037. [DOI] [PubMed] [Google Scholar]
- 37.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat. Rev. Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 38.Baum P.R. Molecular characterization of murine and human OX40/OX40 ligand systems: identification of a human OX40 ligand as the HTLV-1-regulated protein gp34. EMBO J. 1994;13:3992–4001. doi: 10.1002/j.1460-2075.1994.tb06715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takasawa N. Expression of gp34 (OX40 ligand) and OX40 on human T cell clones. Jpn. J. Cancer Res. 2001;92:377–382. doi: 10.1111/j.1349-7006.2001.tb01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H.C., Klein J.R. Multiple levels of activation of murine CD8+ intraepithelial lymphocytes defined by OX40 (CD134) expression: effects on cell-mediated cytotoxicity, IFN-γ, and IL-10 regulation. J. Immunol. 2001;167:6717–6723. doi: 10.4049/jimmunol.167.12.6717. [DOI] [PubMed] [Google Scholar]
- 41.Palma C. CD137 and CD137 ligand constitutively coexpressed on human T and B leukemia cells signal proliferation and survival. Int. J. Cancer. 2004;108:390–398. doi: 10.1002/ijc.11574. [DOI] [PubMed] [Google Scholar]
- 42.Imura A. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J. Exp. Med. 1996;183:2185–2195. doi: 10.1084/jem.183.5.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Souza H.S. Expression of lymphocyte-endothelial receptor-ligand pairs, α4β7/MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut. 1999;45:856–863. doi: 10.1136/gut.45.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song J. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat. Immunol. 2004;5:150–158. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 45.Lee H.W. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J. Immunol. 2002;169:4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 46.Prell R.A. OX40-mediated memory T cell generation is TNF receptor-associated factor 2 dependent. J. Immunol. 2003;171:5997–6005. doi: 10.4049/jimmunol.171.11.5997. [DOI] [PubMed] [Google Scholar]
- 47.Macaubas C. Respiratory tolerance in the protection against asthma. Curr. Drug Targets Inflamm. Allergy. 2003;2:175–186. doi: 10.2174/1568010033484304. [DOI] [PubMed] [Google Scholar]
- 48.Stumbles P.A. Airway dendritic cells: co-ordinators of immunological homeostasis and immunity in the respiratory tract. APMIS. 2003;111:741–755. doi: 10.1034/j.1600-0463.2003.11107806.x. [DOI] [PubMed] [Google Scholar]
- 49.Blumenthal R.L. Human alveolar macrophages induce functional inactivation in antigen-specific CD4 T cells. J. Allergy Clin. Immunol. 2001;107:258–264. doi: 10.1067/mai.2001.112845. [DOI] [PubMed] [Google Scholar]
- 50.Soltys J. Functional IL-10 deficiency in the lung of cystic fibrosis (cftr−/−) and IL-10 knockout mice causes increased expression and function of B7 costimulatory molecules on alveolar macrophages. J. Immunol. 2002;168:1903–1910. doi: 10.4049/jimmunol.168.4.1903. [DOI] [PubMed] [Google Scholar]
- 51.Salik E. Antigen trafficking and accessory cell function in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999;21:365–379. doi: 10.1165/ajrcmb.21.3.3529. [DOI] [PubMed] [Google Scholar]
- 52.Beyer M. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J. Allergy Clin. Immunol. 2004;113:127–133. doi: 10.1016/j.jaci.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 53.Legge K.L., Braciale T.J. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 54.Xia W.J. Accessory cells of the lung. II. Ia+ pulmonary dendritic cells display cell surface antigen heterogeneity. Am. J. Respir. Cell Mol. Biol. 1991;5:276–283. doi: 10.1165/ajrcmb/5.3.276. [DOI] [PubMed] [Google Scholar]
- 55.Bertram E.M. Role of T cell costimulation in anti-viral immunity. Semin. Immunol. 2004;16:185–196. doi: 10.1016/j.smim.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 56.Bukczynski J. Costimulatory ligand 4-1BBL (CD137L) as an efficient adjuvant for human antiviral cytotoxic T cell responses. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1291–1296. doi: 10.1073/pnas.0306567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halstead E.S. In vivo stimulation of CD137 broadens primary antiviral CD8+ T cell responses. Nat. Immunol. 2002;3:536–541. doi: 10.1038/ni798. [DOI] [PubMed] [Google Scholar]
- 58.Humphreys I.R. OX40 ligation on activated T cells enhances the control of Cryptococcus neoformans and reduces pulmonary eosinophilia. J. Immunol. 2003;170:6125–6132. doi: 10.4049/jimmunol.170.12.6125. [DOI] [PubMed] [Google Scholar]
- 59.Israel-Assayag E. Blockade of T cell costimulation by CTLA4–Ig inhibits lung inflammation in murine hypersensitivity pneumonitis. J. Immunol. 1999;163:6794–6799. [PubMed] [Google Scholar]
- 60.Lumsden J.M. Differential requirement for CD80 and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J. Immunol. 2000;164:79–85. doi: 10.4049/jimmunol.164.1.79. [DOI] [PubMed] [Google Scholar]
- 61.Hendriks J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat. Immunol. 2000;1:433–440. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 62.Bertram E.M. Role of ICOS versus CD28 in antiviral immunity. Eur. J. Immunol. 2002;32:3376–3385. doi: 10.1002/1521-4141(200212)32:12<3376::AID-IMMU3376>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 63.DeBenedette M.A. Analysis of 4-1BB ligand (4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals a role for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J. Immunol. 1999;163:4833–4841. [PubMed] [Google Scholar]
- 64.Bertram E.M. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J. Immunol. 2002;168:3777–3785. doi: 10.4049/jimmunol.168.8.3777. [DOI] [PubMed] [Google Scholar]
- 65.Bertram E.M. A switch in costimulation from CD28 to 4-1BB during primary versus secondary CD8 T cell response to influenza in vivo. J. Immunol. 2004;172:981–988. doi: 10.4049/jimmunol.172.2.981. [DOI] [PubMed] [Google Scholar]
- 66.Kopf M. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL Responses after virus infection. Immunity. 1999;11:699–708. doi: 10.1016/s1074-7613(00)80144-2. [DOI] [PubMed] [Google Scholar]
- 67.Pippig S.D. Robust B cell immunity but impaired T cell proliferation in the absence of CD134 (OX40) J. Immunol. 1999;163:6520–6529. [PubMed] [Google Scholar]
- 68.Weinberg A.D. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J. Immunol. 2000;164:2160–2169. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 69.Taylor L. In vitro and in vivo activities of OX40 (CD134)–IgG fusion protein isoforms with different levels of immune-effector functions. J. Leukoc. Biol. 2002;72:522–529. [PubMed] [Google Scholar]
- 70.Weinberg A.D. OX40: targeted immunotherapy – implications for tempering autoimmunity and enhancing vaccines. Trends Immunol. 2002;23:102–109. doi: 10.1016/s1471-4906(01)02127-5. [DOI] [PubMed] [Google Scholar]