

Abstract
Life-threatening cytokine release syndromes include primary (p) and secondary (s) forms of hemophagocytic lymphohistiocytosis (HLH). Below detection in healthy individuals, interferon γ (IFNγ) levels are elevated to measurable concentrations in these afflictions suggesting a central role for this cytokine in the development and maintenance of HLH. Mimicking an infection-driven model of sHLH in mice, we observed that the tissue-derived levels of IFNγ are actually 500- to 2000-fold higher than those measured in the blood. To identify a blood biomarker, we postulated that the IFNγ gene products, CXCL9 and CXCL10 would correlate with disease parameters in the mouse model. To translate this into a disease relevant biomarker, we investigated whether CXCL9 and CXCL10 levels correlated with disease activity in pediatric sHLH patients. Our data demonstrate that disease control in mice correlates with neutralization of IFNγ activity in tissues and that the 2 chemokines serve as serum biomarkers to reflect disease status. Importantly, CXCL9 and CXCL10 levels in pediatric sHLH were shown to correlate with key disease parameters and severity in these patients. Thus, the translatability of the IFNγ-biomarker correlates from mouse to human, advocating the use of serum CXCL9 or CXCL10 as a means to monitor total IFNγ activity in patients with sHLH.
Abbreviations: HLH, hemophagocytic lymphohistiocytosis; pHLH, primary hemophagocytic lymphohistiocytosis; sHLH, secondary hemophagocytic lymphohistiocytosis; IFNγ, interferon γ; TLRs, Toll-like receptors; TNFα, tumor necrosis factor α; mIFNγ, mouse IFNγ; ALT, alanine transaminase; LDH, lactate dehydrogenase; mRNA, messenger RNA; qPCR, quantitative PCR; IL-6, interleukin 6
At a Glance Commentary.
Buatois V, et al.
Background
Mouse models show that interferon γ (IFNγ) is central to the immune pathogenesis in hemophagocytic lymphohistiocytosis (HLH). The ubiquitous expression of the IFNγ receptor, however, results in the cytokine acting locally in tissues making its spillover into the blood for detection of a challenge. Identifying IFNγ signature biomarkers in blood reflecting disease in organs, therefore, is pivotal.
Translational Significance
Using a mouse model mimicking infection-driven HLH, we identified IFNγ-driven chemokines, CXCL9 and CXCL10, as blood biomarkers of IFNγ activity in tissues. Levels of these chemokines correlated with disease parameters in mice and, importantly, to disease parameters and severity in pediatric patients with secondary HLH.
Introduction
Hypercytokinemia, the hallmark of cytokine release syndromes, is an excessive, uncontrolled release of proinflammatory mediators resulting in overwhelming systemic inflammation and multiple organ failure that, if left uncontrolled, can lead to death.1 Originally described in graft-versus-host disease,2 hypercytokinemia has more recently been identified as a fundamental characteristic in immunopathologies of a variety of infectious diseases including primary influenza,3 Ebola virus disease,4 malaria,5 and virus-associated hemophagocytic lymphohistiocytosis (HLH).6 Hypercytokinemia can also be observed during sepsis,7 and in primary (p) and secondary (s) HLH independently of the disease trigger.8, 9, 10, 11, 12 These diseases share clinical and laboratory manifestations including fever, malaise, organomegaly, leukopenia, thrombocytopenia, and hyperferritinemia.1
Hypercytokinemia, commonly referred to as a cytokine storm, is often consequent to an infectious trigger, presenting as an exaggerated activation of the innate immune system.1, 13 Toll-like receptors (TLRs), a class of proteins that serve to trigger innate immune responses against numerous pathogens, are considered to be key instigators leading to hypercytokinemia. TLR4 signaling, for example, is thought to be important for the hypercytokinemia seen during gram-negative bacteria-induced sepsis.14 In mice, repeated stimulation of TLR9, a receptor for unmethylated 5′-C-phosphate-G-3′ (CpG) DNA of numerous bacteria and viruses,15 leads to a cytokine storm that triggers clinical and laboratory features resembling HLH secondary to infections.16, 17
Cytokines such as interferon γ (IFNγ), tumor necrosis factor α (TNFα), interleukin-1 β, and interleukin-18 have been extensively studied in the context of clinical hypercytokinemia.18, 19, 20 Of these, IFNγ is considered to be the dominant cytokine affording biological effects on potentially all cells by virtue of the ubiquitous expression of the cognate receptor.21 Produced by cells of the innate and adaptive immune systems, including natural killer (NK) cells, natural killer T cells, T cells, neutrophils and dendritic cells,16, 22, 23 IFNγ is well place to serve as the instigating mediator of cytokine release syndromes. Indeed, substantially elevated levels of IFNγ are measured in patients with Severe Acute Respiratory Syndrome24, 25 and HLH,26, 27, 28 both cytokine driven diseases. Furthermore, overexpression of IFNγ in transgenic mice is sufficient to drive systemic and organ inflammation,29 and in animal models of pHLH30, 31 and sHLH,16, 17 cytokine neutralization studies revealed the pivotal role of IFNγ. In the model of TLR9-induced cytokine release syndrome, IFNγ was shown to be critical for the development of some features of sHLH including cytopenias and hepatic dysfunction.16, 17 Although these findings are intriguing, their relevance to the pathophysiology of human disease remains to be proven.
In this study, our objective was to dissect the role of IFNγ using a mouse model of sHLH and translate the findings to correlate an IFNγ signature to disease parameters in patients. Using a model that mimics infection-driven sHLH (by giving mice repeated injections of CpG-ODN), for the first time, we quantify the magnitude of IFNγ produced as compared to steady-state conditions. Neutralization at various points of disease demonstrated that the complete neutralization of IFNγ activity in organs was necessary to reverse disease parameters. This abrogation of IFNγ correlated with the decrease of 2 IFNγ signature chemokines, CXCL9 and CXCL10, in blood, which served as robust serum biomarkers to reflect disease status. These CXCL9 and CXCL10 levels positively correlated with sHLH parameters, including thrombocytopenia, hyperferritinemia, and lymphopenia. These results were in fact also clinically meaningful as they could be translated to human using data obtained from pediatric sHLH patients. In these children, circulating CXCL9 and CXCL10 concentrations were found to be proportional to IFNγ levels and correlated with active sHLH parameters (ie, neutropenia, thrombocytopenia, hyperferritinemia, and hepatic dysfunction). Taken together, the results of this translational study support the expansion of clinical trials beyond blockade of IFNγ in pHLH to other cytokine release syndromes. Furthermore, these observations support the use of CXCL9 and CXCL10 as markers of Type II interferonopathies as well as indicators of response to therapeutic intervention.
Materials and Methods
Mice
Female C57BL/6 mice (7–8 weeks) were purchased from Charles River Laboratories (L'Arbresle, France). All experiments were performed in accordance with the Swiss Veterinary Office for animal experimentation.
Patients
Serum samples were collected from 14 patients with sHLH (age at onset, 8.6 years; interquartile range [IQR], 4.1–12.9 years; and female 36%), seen at the Ospedale Pediatrico Bambino Gesù, Italy. All patients met the 2004-HLH diagnostic guidelines: 7 patients met 5 and 7 patients met 4 criteria. It should be noted that levels of sCD25 in U/mL were not available as the test is not performed routinely in the institution where these patients were recruited. Primary HLH was excluded based on the absence of family history, absence of mutations in the genes known to cause HLH, and presence of normal functional studies (including NK activity, perforin expression, and CD107 degranulation assay). An underlying infection was demonstrated in 7 patients (Ebstein-Barr virus in 2, cytomegalovirus in 1, rotavirus in 1, human herpes virus 6 in 1, propionibacteria sepsis in 1, and morganella morganii osteomyelitis in 1). Serum samples were obtained during active disease at presentation (n = 11) and after achieving remission, according to the definition of nonactive disease used in the HLH-2004 protocol (11 patients). Multiple samples were available from most patients and were used for the analysis of correlations. Laboratory features, including neutrophil and platelet blood count, levels of ferritin, alanine transaminase, and lactate dehydrogenase at time of sampling were collected in a database. The study was approved by the institutional ethics committee, and informed consent was obtained from parents or patients as appropriate.
TLR9–mediated sHLH in mice
sHLH was induced as previously described.16 In brief, C57BL/6 female mice were injected intraperitoneally on days 0, 2, 4, 7, and 9 with 50 μg of CpG-ODN 1826 (Invivogen). At each indicated time point, mice were euthanized by CO2 inhalation. Body and spleen weights were recorded. Peripheral blood was collected in Microtainer BD tubes containing EDTA anticoagulant (Becton Dickinson). A complete blood count was performed on a Hemavet analyzer (Hematology Analyzer [950FS], Drew Scientific).
Antibodies for in vivo use
Anti-mouse IFNγ (mIFNγ) neutralizing mAb (XMG1.2) and rat IgG1 isotype-matched control mAb (mAb35) were produced and purified at NovImmune from hybridoma culture supernatants or were purchased from BioXCell. Antibodies were injected intravenously at a dose of 100 mg/kg.
Measurement of serum cytokines, chemokines, and ferritin levels
Mouse and human cytokine and chemokine levels were determined using MILLIPLEX MAP multiplex immunodetection kits (Millipore) and analyzed on the Luminex instrument. Ferritin concentrations were determined using enzyme-linked immunosorbent assay (ELISA) kits (ALPCO Diagnostics) following the manufacturer's instructions.
Measurement of total mIFNγ
An ELISA-based assay was developed and adapted from the method described by Finkelman and Morris,32 to allow for the quantification of total IFNγ (free and bound to XMG1.2). Differing XMG1.2:IFNγ ratios present in the serum samples (found at various time points) significantly affected assay performance. Thus, a procedural modification was required to compensate for this variability, which included the addition of an excess of XMG1.2 (100 μg/mL) to the samples ex vivo. This step ensured the stability of the mAb-cytokine complex allowing for a more precise quantification of the total IFNγ. Briefly, MaxiSorp 96-well plates (Nunc) were coated with an anti-mIFNγ mAb (1 μg/mL, AN-18, BD Biosciences). Serum samples were added to each well. mIFNγ was then detected using biotin-conjugated anti-mIFNγ mAb (8 ng/mL, R4-6A2, BD Biosciences), Peroxidase-Streptavidin (JacksonImmunoResearch), and Tetramethylbenzidine substrate (Sigma–Aldrich). Optical density was read at 450 nm using Epoch microplate reader (BioTek), and data were analyzed with Gen5 software.
Gene expression
Total RNA from spleen and liver was extracted with the RNeasy mini Kit (Qiagen) according to the manufacturer's instructions. Reverse transcription was performed by using High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative PCR (qPCR) was performed in triplicates using SYBR Green Master Mix (Applied Biosystems). The primer sequences were generated using Primer Express software version 3.0 (Applied Biosystems). Sequences primers are: β-actin, forward: AGCCTTCCTTCTTGGGTATGG and reverse: CAACGTCACACTTCATGATG GAAT; IFNγ, forward: CAACAGCAAGGCGAA AAA GG and reverse: CCTGTGGGTTGTTGA CCTCAA; CXCL9, forward: TGCACGATGCTCCTGCA and reverse: AGGTCTTTGAGGGATTT GTAGTGG; CXCL10, forward: GACGGTCCGCTGCAACTG and reverse: GCTTCCCTATGG CCCTCATT; MHCII transactivator (CIITA) form pIV, forward: CATGCAGGCAGCACTCAGAA and reverse: ATCCAT GGTGGCACACAGACT; IL-6: forward, TCGGAGGCTTAATTACAC ATGTTC and reverse: TGCCATTGCACAACTCTTTTCT; and TNFα, forward: GCCACCACG CTCTTCTGTCT and reverse: GGTCTGGGCCATAGAACTGATG. The relative expression levels were calculated as 2ˆ(Ct gene−Ct β-actin) with β-actin RNA as the endogenous control.
Histology
Livers were fixed overnight in 10% neutral buffered formalin (Sigma–Aldrich), rinsed in phosphate-buffered saline, dehydrated, and embedded in paraffin (Leica Microsystems). Sections were stained with hemalun and eosin (VWR) and analyzed for cell infiltration by calculating the area that contained foci of leucocytes infiltration using ImageJ software (NIH).
Statistical analysis
GraphPad Prism 6 was used for all statistical analysis. The 1-tailed nonparametric Mann–Whitney U t test was used for statistical comparisons between 2 groups. Data are expressed as mean ± standard error of the mean. All statistical tests have been indicated in the figure legends. P values of ≤0.05 were considered significant. When indicated, statistical analysis was performed using the Spearman test.
Results
IFNγ, CXCL9, and CXCL10 levels track together and accompany the deteriorating presentation of laboratory and clinical features of sHLH in mice
An increase of circulating cytokines has been described in the TLR9-induced sHLH model.16 To strengthen these data, we conducted experiments which followed the kinetics of IFNγ and IFNγ-dependant chemokines (CXCL9 and CXCL10) in blood and in tissues. The stimulation of TLR9, provoked by DNA rich in CpG-ODN motifs repeatedly injected led to hypercytokinemia (Fig 1 A). Within 6 hours after the first TLR9 stimulation, IFNγ, IL-6, TNFα, CXCL9, and CXCL10 were elevated in the blood. This was accompanied by de novo messenger RNA (mRNA) synthesis of proinflammatory mediators in the liver (Fig 1 B) and spleen (Supplementary Fig 1). After subsequent TLR9 stimulations, IFNγ, CXCL9, and CXCL10 levels oscillated similarly suggesting that these chemokines may be biomarkers of fluctuations in IFNγ activity. Conversely, IL-6 and TNFα levels were measured at a more stable rate (Fig 1).
Fig 1.

CpG-ODN–injected mice display a striking elevation of inflammatory mediators in serum and tissue. Mice (n = 4–5 per time point) were injected with 50 μg of CpG-ODN at days 0, 2, 4, 7, and 9 (gray filled triangles). (A) Serum levels of IFNγ, IL-6, TNFα, CXCL9, and CXCL10 were measured in a multiplex assay using the Luminex technology. (B) Quantification of liver derived mRNA was obtained by qPCR. On days when CpG-ODN was administered, samples were collected 6 hours after injection. Values are the mean ± standard error of the mean. Data are representative of 2 experiments. AU, arbitrary unit; IFNγ, interferon γ; IL-6, interleukin 6; TNFα, tumor necrosis factor α; mRNA, messenger RNA; qPCR, quantitative PCR.
Development of hypercytokinemia leads to weight loss and alterations in hematological parameters. Within 24 hours after the first CpG-ODN injection, mice lost a significant amount (7%) of their starting weight (Fig 2 ). Lymphocyte counts in the blood also decreased rapidly and remained low throughout the study (Fig 2). With repeated TLR9 stimulation, platelet and erythrocyte counts steadily decreased, whereas splenomegaly was increasingly evident (Fig 2). Interestingly, serum ferritin, a marker of an inflammatory response, spiked at day 7, after the 4th CpG-ODN injection (Fig 2), which correlated with the observed maximal levels achieved in blood for the inflammatory mediators studied (Fig 1 A). Taken together, we observed that repeated TLR9 stimulation with CpG-ODN in mice recreates a hypercytokinemia albeit with peaks and troughs that manifest as a steady deterioration in laboratory and clinical parameters.
Fig 2.

CpG-ODN–injected mice present clinical and laboratory features of sHLH. Mice (n = 4–5 per time point) were injected with 50 μg of CpG-ODN at days 0, 2, 4, 7, and 9 (gray filled triangles). Body and spleen weights were monitored, and blood parameters including lymphocyte, platelet, and red blood cell counts were measured using a hemavet analyzer. Ferritin serum concentrations were measured by ELISA. On days when CpG-ODN was administered, samples were collected 6 hours after injection. Values are the mean ± standard error of the mean. Data are representative of 2 experiments. Statistics were performed between the indicated time point and the baseline (ie, at day −1) values. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 were obtained using the 1-tailed nonparametric Mann–Whitney U t test. sHLH, secondary hemophagocytic lymphohistiocytosis.
IFNγ production in tissue compartment correlates with TLR9-induced hypercytokinemia
Owing to our detailed study of blood cytokines, we revealed a dynamic fluctuation of IFNγ in the blood while disease progression was sustained. We hypothesized that most IFNγ may be produced and consumed in tissues, whereas the fluctuating serum levels represent a spillover of the cytokine into the blood. To investigate this possibility, we set out to quantify the amount of IFNγ produced in tissues, by injecting the anti-mIFNγ mAb, XMG1.2, to mice while receiving the disease inducing CpG-ODN. We took advantage of a previously described phenomena,32 in which IFNγ in tissues is captured by XMG1.2, that then drains via the lymphatics into the blood leading to accumulation of the cytokine as a complex to the mAb. Thus, this cytokine-mAb complex measured ex vivo in blood samplings is thus directly proportional to the total amount of IFNγ produced by the organism.
Administration of XMG1.2 before the first CpG-ODN injection resulted in IFNγ measured at 1.7 ng/mL in serum, reflecting the basal level of production in naive mice (Fig 3 A). After CpG-ODN injection, a marked increase in total IFNγ reaching >100 ng/mL (Fig 3 A) was measured in the XMG1.2–injected mice as compared to the levels measured without cytokine-mAb complex stabilization (∼0.6 ng/mL; Fig 1 A). Peak levels of total IFNγ reached 280 ng/mL (Fig 3 A), a 169-fold increase over the basal total IFNγ levels in naive mice and a 470-fold increase over levels detected in serum of CpG-ODN treated mice without XMG1.2 (Fig 1 A), demonstrating the considerable production of IFNγ in tissues from mice undergoing TLR9-induced hypercytokinemia. Owing to these levels of total IFNγ produced in this model and our experience with other IFNγ-induced disease models, for example, pHLH,31 we next evaluated if the dose of 100 mg/kg of XMG1.2 was sufficient to neutralize IFNγ tissue activity. To this end, mice were injected with XMG1.2 24 hours post CpG-ODN administration, and the mRNA expression in liver of the IFNγ-dependent gene, CIITA pIV, was evaluated over time. As a robust induction of CIITA pIV, mRNA was observed after the first CpG-ODN injection, the elimination of CIITA pIV expression after XMG1.2 administration demonstrated that complete neutralization of IFNγ in tissues was achieved (Fig 3 B).
Fig 3.

Neutralization of total IFNγ production in CpG-ODN–injected mice controls the cytokine storm. (A) Mice (n = 8 per time point) were injected with 100 mg/kg of the anti-mIFNγ mAb, XMG1.2, at days −1, 1, 3, and 6 (black filled triangles) and 50 μg of CpG-ODN at days 0, 2, 4, 7, and 9 (gray filled triangles). Serum was obtained predose, at day 10 and 1 hour before each CpG-ODN injection. Total IFNγ was measured using an ELISA assay. Values are the mean ± standard error of the mean (SEM). Data are representative of 2 experiments. (B–E) Mice (n = 8 per time point) were injected with 50 μg of CpG-ODN at days 0, 2, 4, and 7 (gray filled triangles) and administered 100 mg/kg of the anti-mIFNγ mAb, XMG1.2 (dotted line), or an isotype control mAb (solid line), on days 1, 3, and 6 (black filled triangles). Livers were obtained to evaluate CIITA pIV mRNA, an IFNγ-inducible gene, by qPCR (B) IL-6 and TNFα serum concentrations were quantified in a multiplex assay using the Luminex technology (C). Quantification of tissue-derived cytokine mRNA was evaluated by qPCR from the liver (D) and the spleen (E). The samples from day 0 were collected before the CpG-ODN injection, whereas samples from day 4 and day 7 were collected 6 hours after CpG-ODN injection. Samples from the day of a mAb administration were collected before the injection. Values are the mean ± SEM. Data are representative of 2 experiments. Statistics were performed to compare the values at each time point between isotype control and XMG1.2 treated groups. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 were obtained using the 1-tailed nonparametric Mann–Whitney U t test. IFNγ, interferon γ; IL-6, interleukin 6; TNFα, tumor necrosis factor α; mRNA, messenger RNA; qPCR, quantitative PCR.
Tissue-derived IFNγ is responsible for clinical and laboratory features in TLR9-mediated cytokine release syndrome
Having made the novel observation that high levels of IFNγ originate from the tissues after TLR9 stimulation, we performed a kinetic study to determine the degree to which the cytokine was associated with the features of sHLH throughout the duration of the model. XMG1.2 was administered on day (d) 1 (post the peak of total IFNγ), d3, and d6 to mice challenged with CpG-ODN on d0, 2, 4, and 7. Serum levels for both IL-6 and TNFα were significantly lower and maintained close to baseline (Fig 3 C). Interestingly, neutralization of IFNγ in tissues resulted in the decrease in mRNA levels for IL-6 in the liver (Fig 3 D) and spleen (Fig 3 E), and for TNFα in the liver (Fig 3 D). IFNγ neutralization significantly reduced body weight loss, splenomegaly, lymphopenia, thrombocytopenia, anemia, and hyperferritinemia (Fig 4 A). Liver inflammation, illustrated by cellular infiltration creating inflammatory cell foci, was also reduced after administration of XMG1.2 (Fig 4 B). The role for IFNγ at a later stage of disease was also investigated, that is, when clinical features and laboratory abnormalities were established (Fig 2), and total IFNγ production was at a plateau (Fig 3 A). Neutralization of IFNγ from d4 resulted in the abrogation of the hyper-release of IL-6 and TNFα to the blood (Fig 5 A). Moreover, this delay IFNγ neutralization resulted in a reduction of body weight loss and an improvement in splenomegaly (Fig 5 A). Thrombocytopenia, anemia, and hyperferritinemia were ameliorated although lymphopenia remained unchanged (Fig 5 A). The delayed inhibition also controlled liver inflammation as a significant drop in cellular infiltration as well-decreased IL-6 and TNFα mRNA expression were observed (Fig 5 B). Taken together, the efficient neutralization of the high rate of tissue production of IFNγ was required to induce a significant amelioration of the cytokine release syndrome parameters.
Fig 4.

IFNγ is required for hematological and tissue parameters in the model of sHLH. Mice (n = 8 per time point) were injected with 50 μg of CpG-ODN at days 0, 2, 4, and 7 (gray filled triangles) and administered 100 mg/kg of the anti-IFNγ mAb, XMG1.2 (dotted line), or with an isotype control mAb (solid line), on days 1, 3, and 6 (black filled triangles). (A) Body and spleen weights were monitored, and blood parameters, including lymphocyte, platelet, and red blood cell counts, were measured using a hemavet analyzer. Serum ferritin was measured by ELISA. The samples from day 0 were collected before the CpG-ODN injection, whereas samples from day 2, 4, and 7 were collected 6 hours after CpG-ODN injection. Samples from the day of a mAb administration were collected before the injection. Values are the mean ± standard error of the mean. Data are representative of 2 experiments. Statistics were performed at each time point between isotype control and XMG1.2-treated group values. (B) Liver inflammation was evaluated on day 8 by calculating the area that contained foci of leukocyte infiltration as illustrated by the outlined areas in the representative photomicrographs of H and E-stained sections (Zeiss Axiovert 40 CFL; Zeiss AxioCam MRc Rev.3; original magnification ×100) from an isotype control or XMG1.2-treated mouse. The graph represents the quantitative analysis of at least 9 fields per liver capturing the fold increase of area as compared to untreated mice (ie, no CpG injection); each symbol represents an individual mouse; horizontal lines represent the mean values. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 were obtained using the 1-tailed nonparametric Mann–Whitney U t test. IFNγ, interferon γ; sHLH, secondary hemophagocytic lymphohistiocytosis.
Fig 5.

Late intervention in the sHLH continues to afford clinical benefit. Mice (n = 8 per time point) were injected with 50 μg of CpG-ODN at days 0, 2, 4, and 7 (gray filled triangles) and administered 100 mg/kg of the anti-IFNγ mAb, XMG1.2 (dotted line), or an isotype control mAb (solid line), on days 4 and 6 (black filled triangles). (A) IL-6 and TNFα serum concentrations were quantified in a multiplex assay using the Luminex technology; body and spleen weights were monitored and blood parameters including lymphocyte, platelet and red blood cell counts were measured using a hemavet analyzer and serum ferritin was measured by ELISA. (B) Liver inflammation was evaluated on day 8 by calculating the area that contained foci of leukocyte infiltration as illustrated in (B). The graph represents the quantitative analysis of at least 9 fields per liver capturing the fold increase of area as compared to untreated mice (ie, no CpG injection); each symbol represents an individual mouse; horizontal lines represent the mean values. IL-6 and TNFα mRNA levels were evaluated by qPCR in the liver. The samples from day 0 were collected before the CpG-ODN injection, whereas samples from day 4 and day 7 were collected 6 hours after CpG-ODN injection. Samples from the day of a mAb administration were collected before the injection. Values are the mean ± standard error of the mean. Data are representative of 1 experiment. Statistics were performed at each time point between isotype control and XMG1.2-treated groups. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 were obtained using the 1-tailed nonparametric Mann–Whitney U t test. AU, arbitrary unit; IL-6, interleukin 6; TNFα, tumor necrosis factor α; mRNA, messenger RNA; qPCR, quantitative PCR; sHLH, secondary hemophagocytic lymphohistiocytosis.
Blood and tissue CXCL9 and CXCL10 levels are associated with IFNγ activity in TLR9-induced cytokine release syndrome
To further support the hypothesis that these 2 chemokines mirror IFNγ activity, we demonstrated that IFNγ neutralization (starting at either at d1 or d4 of disease onset), resulted in a significant decrease in liver mRNA levels of CXCL9 and CXLC10 levels (Fig 6 A). The significant decrease in CXCL9 and CXCL10 in blood (Fig 6 B) is further testament to the shutdown of chemokine production in tissues. These results suggest that serum levels of CXCL9 and CXCL10 reflect IFNγ activity in TLR9-induced sHLH. Confirming this association, a significant correlation of serum IFNγ levels with serum concentration of CXCL9 and CXCL10 was observed in CpG-treated mice in absence of XMG1.2 (Fig 6 C). Finally, correlative analysis was performed and demonstrated that levels for the IFNγ-dependant chemokines in blood were associated to syndrome parameters including thrombocytopenia, hyperferritinemia, and lymphopenia (Fig 7 ). However, no significant correlation was seen between serum IFNγ and the previously parameters, excepting for lymphocyte counts (P = 0.0022, Fig 7). These data reinforce that circulating CXCL9 and CXCL10 levels mirror tissue IFNγ activity and, importantly, disease parameters in blood.
Fig 6.

Circulating CXCL9 and CXCL10 production correlate with IFNγ tissue activity in the model of TLR9-induced sHLH. (A, B) Mice (n = 7 mice per time-point) were injected with 50 μg of CpG-ODN on days 0, 2, 4, and 7 (gray filled triangles) and administered 100 mg/kg of the anti-IFNγ mAb, XMG1.2 (dotted line) or an isotype control mAb (solid line) at days 1, 3, and 6 (A and B, upper panels; black filled triangles) or at days 4 and 6 (A and B, lower panels; black filled triangles). mRNA quantification of cytokines from livers was obtained by qPCR, and serum concentrations of CXCL9 and CXCL10 were quantified in a multiplex assay using the Luminex technology. The samples from day 0 were collected before the CpG-ODN injection, whereas samples from day 4 and day 7 were collected 6 hours after CpG-ODN injection. Samples from the day of a mAb injection were collected before the injection. Values are the mean ± standard error of the mean. Data are representative of 2 experiments. Statistics were performed to compare the values at each time point between isotype control and XMG1.2-treated groups. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ****P < 0.0001 were obtained using the 1-tailed nonparametric Mann–Whitney U t test. (C) Mice were injected with 50 μg of CpG-ODN at days 0, 2, 4, 7, and 9. Serum levels of IFNγ, CXCL9, and CXCL10 were quantified in a multiplex assay using the Luminex technology. Correlations were performed between CXCL9 and IFNγ or CXCL10 and IFNγ levels at different time point as HLH develops (depicted in Fig 1). Statistics were performed and P values were obtained using the Spearman test. The line of best fit was generated using the nonlinear regression analysis using GrapPad Prism software. IFNγ, interferon γ; mRNA, messenger RNA; qPCR, quantitative PCR; sHLH, secondary hemophagocytic lymphohistiocytosis; TLR9, Toll-like receptor 9.
Fig 7.

Circulating CXCL9 and CXCL10 levels correlate with disease parameters in the model of TLR9-induced sHLH. Mice were injected with 50 μg of CpG-ODN at days 0, 2, 4, 7, and 9. Serum levels of CXCL9, CXCL10, and IFNγ were quantified in a multiplex assay using the Luminex technology. Blood parameters including platelet and lymphocyte counts were measured using a hemavet analyzer, and serum ferritin was measured by ELISA. Correlations were performed between CXCL9 and disease parameters, CXCL10 and disease parameters, and IFNγ and disease parameters at different time point as HLH develops (depicted in Fig 1). Statistics were performed and P values were obtained using the Spearman test. *P < 0.05, **P < 0.01, and ***P < 0.001. The line of best fit was generated using the nonlinear regression analysis using GrapPad Prism software. TLR9, Toll-like receptor 9; sHLH, secondary hemophagocytic lymphohistiocytosis; IFNγ, interferon γ.
Blood levels of CXCL9 and CXCL10 are indicative of disease in pediatric sHLH
The pathogenic role of IFNγ and the correlation with CXCL9 and CXCL10 in the TLR9-induced model of sHLH led us to further explore this association in a patient setting. In pediatric sHLH patients with active full blown disease (Supplementary Table I), blood levels of IFNγ (median 34.7 pg/mL, IQR 23.9–170.1), CXCL9, and CXCL10 (CXCL9: 33,598 pg/mL, IQR 3083–127,687; CXCL10; 4420 pg/mL, IQR 799–8226) were markedly higher than those of patients sampled during nonactive disease following effective treatment (IFNγ <3.5 pg/mL, IQR <3.5–6.5; CXCL9: 745 pg/mL, IQR 469–1098; and CXCL10; 132 pg/mL, IQR 74–157) (Fig 8 A). From 20 blood samples obtained at different times during active sHLH disease, IFNγ levels significantly correlated with levels of CXCL9 and, to a lesser extent, of CXCL10 (Fig 8 B). We also investigated the correlation of circulating levels for IFNγ, CXCL9, and CXCL10, with laboratory parameters of sHLH including neutrophil and platelet counts, ferritin, lactate dehydrogenase, and alanine transaminase levels. We found that blood CXCL9 levels were significantly correlated with all the 5 laboratory parameters studied (Fig 8 D). IFNγ and CXCL10 levels were also significantly correlated with the previously mentioned disease parameters, with the exception being between IFNγ and platelet counts (Fig 8 C), and CXCL10 and ferritin levels (Fig 8 E), not reaching significance. Taken together, our findings in patients further suggest the pathogenic role for IFNγ in sHLH and advocate the use of blood levels for CXCL9 and CXCL10 as potential biomarkers of IFNγ activity in sHLH.
Fig 8.

CXCL9 and CXCL10 production correlate with IFNγ activity in patients with sHLH. (A) Serum levels of IFNγ, CXCL9, and CXCL10 in patients with sHLH sampled during full-blown active or nonactive disease were quantified in a multiplex assay using the Luminex technology. Statistics were performed and P values were obtained using the Mann–Whitney U test. (B) Correlations were performed for IFNγ with CXCL9 or CXCL10 levels in sera of patients with active full-blown sHLH. Statistics were performed and P values were obtained using the Spearman test. (C–E). Correlations were performed between serum IFNγ (C), CXCL9 (D), or CXCL10 (E) levels with neutrophil count, platelet count, ferritin, LDH or ALT concentrations. Statistics were performed and P values obtained using the Spearman test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The line of best fit was generated using the nonlinear regression analysis using GrapPad Prism software. IFNγ, interferon γ; sHLH, secondary hemophagocytic lymphohistiocytosis; ALT, alanine transaminase; LDH, lactate dehydrogenase.
Discussion
HLH presents as a severe systemic inflammation accompanied by hypercytokinemia, a potentially fatal hyper-release of inflammatory mediators including cytokines and chemokines.1 Accurately measuring and relating the components of the hypercytokinemia in the blood to the clinical and laboratory manifestations during disease is complicated due to the changes of the cytokine signature over time and the short half-life of these proteins in vivo. Determining the accurate levels of cytokines and chemokines in patients with HLH is of particular importance for further investigation of their link to the pathophysiology; these data also contribute to our understanding of the potential relevance of their neutralization as a therapeutic target.32 Thus, using a mouse model mimicking an infection-driven sHLH with associated cytokine release syndrome,16 as well as assessing serum samples from patients, we have defined blood-borne inflammatory mediators of the hypercytokinemia causing disease in tissues.
The analyses afforded by the kinetic study we performed in the animal model of sHLH demonstrated a strong association of the levels of IFNγ, CXCL9, and CXCL10 in blood following each CpG-ODN injection. However, as the high-affinity receptor for IFNγ is ubiquitously expressed,33 facilitating a rapid elimination of the cytokine at sites of production, we hypothesized that the measured concentration of IFNγ in the blood is a poor indicator of the true quantity being produced. Indeed, our results support the notion that what is measured in blood can be considered as spillover from events occurring in tissues that have succumbed to hyperinflammation.1, 34, 35, 36 Adapting a method that exploits the ability of the anti-mouse IFNγ mAb, XMG1.2, to capture IFNγ present in organs and mobilize it as an immune complex into the blood compartment,32 we observed a rapid and dramatic increase in IFNγ above basal levels after the first CpG-ODN injection. Subsequent TLR9 stimulation resulted in incremental increases of total IFNγ levels, finally reaching steady-state concentrations 169-fold higher than those measured in mice under homeostatic conditions. Importantly, the ability of XMG1.2 to infiltrate organs was demonstrated by the abrogation in tissues of CIITA pIV expression (an IFNγ-inducible gene) and the diminished de novo transcription of CXCL9 and CXCL10 in the liver corresponding to the rapid disappearance of these IFNγ-inducible chemokines from the blood. These alterations in proteins regulated by IFNγ reinforce the notion that the source of the hypercytokinemia observed in sHLH occurs at the organ level. In addition, the results infer that cytokine levels detected in blood may fail to reflect the true magnitude of total cytokine production occurring in the body. This phenomenon is also exemplified in severe infections affecting the respiratory tract (eg, avian influenza or severe primary influenza virus infections), where the inflammatory cascade leading to hypercytokinemia and consequent immunopathology occurs in the deep tissues.1
Previously, Behrens et al16 had nicely demonstrated that neutralizing IFNγ ameliorates the HLH-like features induced by the repeated CpG injections given to BALB/c mice. However, the authors concluded that some disease parameters, leukopenia and hyperferritinemia, were IFNγ-independent. Here, we demonstrated that the neutralization of IFNγ in organs controlled the hypercytokinemia and ameliorated the laboratory and clinical sHLH parameters in this mouse model including body weight loss, anemia, lymphopenia, hyperferrinitinemia, splenomegaly, and hepatic inflammation. We and others have shown that a range of 30–100 mg/kg of XMG1.2 is required to achieve a therapeutic benefit in a mouse model of pHLH, where perforin-deficient mice are infected with LCMV.30, 31 Our data help to explain the observation that when a lower dose of XMG1.2 (ie, 8 mg/kg) was administered in an alternative HLH model, involving perforin-deficient mice infected with mouse cytomegalovirus,37 it was not possible to significantly alter the disease. Taken together, these results underline how the local hyper-production of IFNγ drives the organ pathology, triggering inflammation and tissue damage, and ultimately disseminating systemically.
Others have shown that serum peaks of IFNγ after CpG injections, although modest, suggested an important role for this cytokine in driving tissue pathology seen in CpG-treated mice.16 Nonetheless, using IFNγ as a blood biomarker is complicated by the fact that the cytokine is rapidly catabolized and, or used in tissues. This limitation was addressed in our study by demonstrating that blood levels of the chemokines, CXCL9, and CXCL10, which are directly induced by IFNγ, were altered and correlated with HLH parameters including thrombocytopenia, hyperferritinemia, and lymphopenia. These abundantly produced chemokines serve as biomarkers for IFNγ activity in mouse organs. The clinical importance of this finding was borne out by the translation of these observations to pediatric patients with sHLH where high levels of IFNγ, CXCL9, and CXCL10 were detected in blood from patients with active disease. Although infections were demonstrable in only half of the patients, it is well known that the infectious trigger may not always be apparent at time of HLH diagnosis.38 Moreover, since the patients studied did not have mutations in known genes causing HLH, had normal CD107 degranulation, as well as perforin expression and NK cytotoxicity, did not experience HLH reactivation, and had a negative family history, it is highly unlikely that any patient with pHLH was included in the analysis. Our findings that sHLH patients with active disease have high circulating levels of IFNγ extend the data reported by Xu et al.39 Although they did not report levels of IFNγ specifically in patients with sHLH, they found high levels of IFNγ in a population of HLH patients comprising a relevant portion (ie, 50%) with documented infections. Moreover, our data show that levels of CXCL9 and, to a lesser extent CXCL10, in the blood correlated to those measured for IFNγ. As in the mouse model of sHLH, increased levels of CXCL9 correlated with several laboratory parameters of disease severity.
Targeting IFNγ as a treatment option for cytokine release syndromes was first proposed over 20 years ago.40 A number of studies, including our own, have demonstrated the pivotal role of IFNγ in models of pHLH.30, 31 Here, we demonstrate that a very high rate of IFNγ production in the tissues is central to the TLR9-mediated syndrome in mice and that blood levels of CXCL9 and CXCL10 are correlated to disease parameters in that animal model as well as in sHLH patients. Although IFNγ levels have been monitored in patients with sHLH, our observations support this cytokine as key driver of hypercytokenimia in HLH and advocate the use of CXCL9 and CXCL10 levels as biomarkers of IFNγ activity. We therefore anticipate that ongoing efforts investigating the blockade of IFNγ in patients with pHLH will ultimately support this strategy as a therapeutic approach in secondary forms of the disease.
Acknowledgments
Conflicts of Interest: All authors have read the journal's authorship agreement. As for competing interests, all authors have read the journal's policy and declare the following: V. Buatois, L. Chatel, L. Cons, F. Richard, F. Guilhot, Z. Johnson, C. de Min, M. H. Kosco-Vilbois, and W. Ferlin are employees of Novimmune, and F. de Benedetti is a consultant for Novimmune.
This work was supported by the Seventh Framework Program (FP7) European Union's Research and Innovation Program.
Author contributions are as follow: V. Buatois and W. Ferlin designed and coordinated the project, analyzed the data, and wrote the manuscript; L. Chatel and L. Cons performed the murine experiments; F. Richard and F. Guilhot performed multiplex human sample testing; C. Bracaglia and F. de.Benedetti recorded and generated database for human data; S. Lory provided technical advice; V. Buatois and L. Chatel prepared the figures for the manuscript; V. Buatois, W. Ferlin, M. H. Kosco-Vilbois, Z. Johnson, C. de.Min, and F. de Benedetti edited the manuscript.
The authors would like to acknowledge Serge Wolfersperger for animal husbandry.
Contributor Information
Vanessa Buatois, Email: vbuatois@novimmune.com.
Walter G. Ferlin, Email: wferlin@novimmune.com.
Appendix
Supplementary Fig 1.
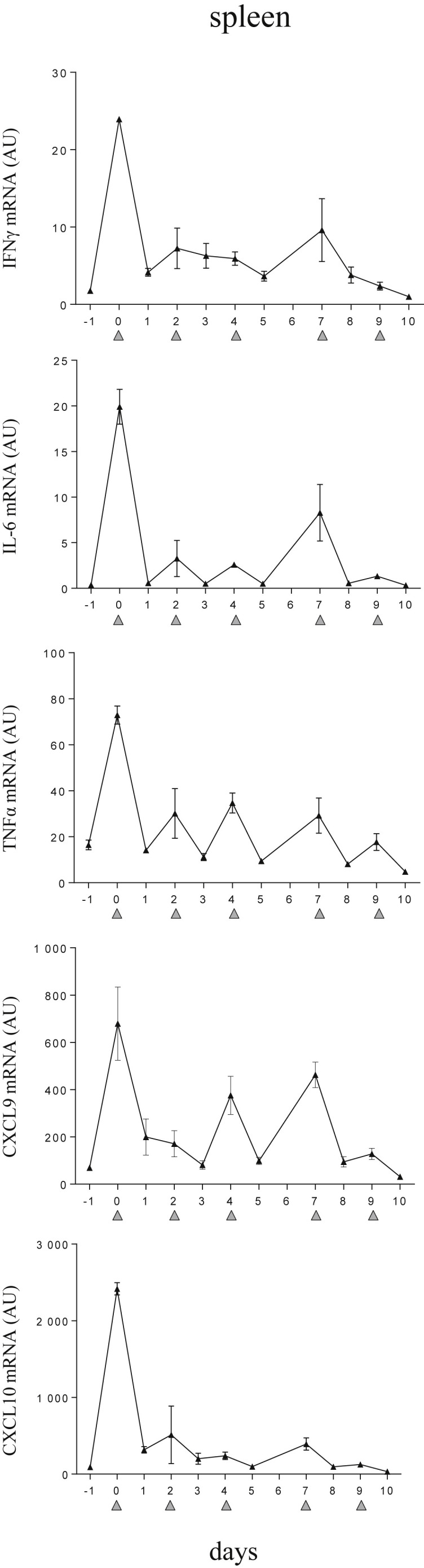
CpG-ODN–injected mice display a striking elevation of inflammatory mediators in spleen. Mice were injected with 50 μg of CpG-ODN at days 0, 2, 4, 7, and 9 (gray filled triangles). Quantification of splenic derived mRNA was obtained by qPCR. On days when CpG-ODN was administered, tissue samples were collected 6 hours after injection. Values are the mean ± standard error of the mean. Data are representative of 2 experiments. AU, arbitrary unit; mRNA, messenger RNA; qPCR, quantitative PCR; IFNγ, interferon γ; IL-6, interleukin 6; tumor necrosis factor α.
Supplementary Table I.
Clinical features at presentation of the patients with sHLH studied
| Patients, N | 14 |
|---|---|
| Fever, n/N (%) | 12/14 (85.7) |
| Splenomegaly, n/N (%) | 5/14 (35.7) |
| Hepatomegaly, n/N (%) | 7/14 (50) |
| CNS involvement, n/N (%) | 4/14 (28.6) |
| Haemorrhages, n/N (%) | 2/14 (14.3) |
| Anemia (<90 g/L), n/N (%) | 8/14 (57.1) |
| Trombocytopenia (<100 × 109/L), n/N (%) | 5/14 (35.7) |
| Leukopenia (<4 × 109/L), n/N (%) | 5/14 (35.7) |
| Neutropenia (<1 × 109/L), n/N (%) | 3/14 (21.4) |
| Hypertriglyceridemia (≥265 mg/dl), n/N (%) | 10/14 (71.4) |
| Hypofibrinogenemia (≤1.5 g/l), n/N (%) | 6/14 (42.9) |
| Hemophagocytosis, n/N (%) | 12/14 (85.7) |
| Ferritin (≥500 mcg/L), n/N (%) | 13/14 (92.8) |
Abbreviation: sHLH, secondary hemophagocytic lymphohistiocytosis.
14 patients (age at onset 8.6 years, interquartile range (IQR) 4.1–12.9 years; female 36%) met the 2004-HLH diagnostic guidelines: 7 patients met 5 and 7 patients met 4 criteria. The table shows the number and the proportion of patients who developed each of the diagnostic criteria.
References
- 1.Tisoncik J.R., Korth M.J., Simmons C.P. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76:16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrara J.L., Abhyankar S., Gilliland D.G. Cytokine storm of graft-versus-host disease: a critical effector role for interleukin-1. Transplant Proc. 1993;25:1216–1217. [PubMed] [Google Scholar]
- 3.Yokota S. Influenza-associated encephalopathy–pathophysiology and disease mechanismsNihon Rinsho. 2003;61:1953–1958. [PubMed] [Google Scholar]
- 4.Wauquier N., Becquart P., Padilla C., Baize S., Leroy E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl Trop Dis. 2010;4:e837. doi: 10.1371/journal.pntd.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark I.A., Cowden W.B. The pathophysiology of falciparum malaria. Pharmacol Ther. 2003;99:221–260. doi: 10.1016/s0163-7258(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 6.Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44:259–272. doi: 10.1016/s1040-8428(02)00117-8. [DOI] [PubMed] [Google Scholar]
- 7.Sriskandan S., Altmann D.M. The immunology of sepsis. J Pathol. 2008;214:211–223. doi: 10.1002/path.2274. [DOI] [PubMed] [Google Scholar]
- 8.Falini B., Pileri S., De Solas I. Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood. 1990;75:434–444. [PubMed] [Google Scholar]
- 9.Finkielman J.D., Grinberg A.R., Paz L.A. Case report: reactive hemophagocytic syndrome associated with disseminated strongyloidiasis. Am J Med Sci. 1996;312:37–39. doi: 10.1097/00000441-199607000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Gagnaire M.H., Galambrun C., Stephan J.L. Hemophagocytic syndrome: A misleading complication of visceral leishmaniasis in children–a series of 12 cases. Pediatrics. 2000;106:E58. doi: 10.1542/peds.106.4.e58. [DOI] [PubMed] [Google Scholar]
- 11.Janka G.E. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–246. doi: 10.1146/annurev-med-041610-134208. [DOI] [PubMed] [Google Scholar]
- 12.Ravelli A., Grom A.A., Behrens E.M., Cron R.Q. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13:289–298. doi: 10.1038/gene.2012.3. [DOI] [PubMed] [Google Scholar]
- 13.Harrison C. Sepsis: calming the cytokine storm. Nat Rev Drug Discov. 2010;9:360–361. doi: 10.1038/nrd3162. [DOI] [PubMed] [Google Scholar]
- 14.Poltorak A., He X., Smirnova I. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 15.Hemmi H., Takeuchi O., Kawai T. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 16.Behrens E.M., Canna S.W., Slade K. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 2011;121:2264–2277. doi: 10.1172/JCI43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canna S.W., Wrobel J., Chu N. Interferon-gamma mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. 2013;65:1764–1775. doi: 10.1002/art.37958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lv S., Han M., Yi R. Anti-TNF-alpha therapy for patients with sepsis: a systematic meta-analysis. Int J Clin Pract. 2014;68:520–528. doi: 10.1111/ijcp.12382. [DOI] [PubMed] [Google Scholar]
- 19.Pascual V., Allantaz F., Arce E., Punaro M., Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–1486. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu M., Nakagishi Y., Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61:345–348. doi: 10.1016/j.cyto.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 21.Billiau A., Matthys P. Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev. 2009;20:97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Ethuin F., Gerard B., Benna J.E. Human neutrophils produce interferon gamma upon stimulation by interleukin-12. Lab Invest. 2004;84:1363–1371. doi: 10.1038/labinvest.3700148. [DOI] [PubMed] [Google Scholar]
- 23.Schoenborn J.R., Wilson C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 24.Huang K.J., Su I.J., Theron M. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol. 2005;75:185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theron M., Huang K.J., Chen Y.W., Liu C.C., Lei H.Y. A probable role for IFN-gamma in the development of a lung immunopathology in SARS. Cytokine. 2005;32:30–38. doi: 10.1016/j.cyto.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henter J.I., Elinder G., Soder O. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78:2918–2922. [PubMed] [Google Scholar]
- 27.Osugi Y., Hara J., Tagawa S. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–4103. [PubMed] [Google Scholar]
- 28.Takada H., Takahata Y., Nomura A. Increased serum levels of interferon-gamma-inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol. 2003;133:448–453. doi: 10.1046/j.1365-2249.2003.02237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reinhardt R.L., Liang H.E., Bao K. A novel model for IFN-gamma-mediated autoinflammatory syndromes. J Immunol. 2015;194:2358–2368. doi: 10.4049/jimmunol.1401992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jordan M.B., Hildeman D., Kappler J., Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 31.Pachlopnik S.J., Ho C.H., Chretien F. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1:112–124. doi: 10.1002/emmm.200900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finkelman F.D., Morris S.C. Development of an assay to measure in vivo cytokine production in the mouse. Int Immunol. 1999;11:1811–1818. doi: 10.1093/intimm/11.11.1811. [DOI] [PubMed] [Google Scholar]
- 33.van Loon A.P., Ozmen L., Fountoulakis M. High-affinity receptor for interferon-gamma (IFN-gamma), a ubiquitous protein occurring in different molecular forms on human cells: blood monocytes and eleven different cell lines have the same IFN-gamma receptor protein. J Leukoc Biol. 1991;49:462–473. doi: 10.1002/jlb.49.5.462. [DOI] [PubMed] [Google Scholar]
- 34.Cilloniz C., Shinya K., Peng X. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009;5:e1000604. doi: 10.1371/journal.ppat.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S.M., Chan R.W., Gardy J.L. Systems-level comparison of host responses induced by pandemic and seasonal influenza A H1N1 viruses in primary human type I-like alveolar epithelial cells in vitro. Respir Res. 2010;11:147. doi: 10.1186/1465-9921-11-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinya K., Ebina M., Yamada S. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 37.van Dommelen S.L., Sumaria N., Schreiber R.D. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity. 2006;25:835–848. doi: 10.1016/j.immuni.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 38.Jordan M.B., Allen C.E., Weitzman S., Filipovich A.H., McClain K.L. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu X.J., Tang Y.M., Song H. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160:984–990. doi: 10.1016/j.jpeds.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 40.Matthys P., Dillen C., Proost P. Modification of the anti-CD3-induced cytokine release syndrome by anti-interferon-gamma or anti-interleukin-6 antibody treatment: protective effects and biphasic changes in blood cytokine levels. Eur J Immunol. 1993;23:2209–2216. doi: 10.1002/eji.1830230924. [DOI] [PubMed] [Google Scholar]