

Abstract
In the postgenomic era, computer-aided drug design (CADD) has considerably extended its range of applications, spanning almost all stages in the drug discovery pipeline, from target identification to lead discovery, from lead optimization to preclinical or clinical trials. Two new technologies of CADD associated with target identification and new chemical entity discovery will be the focus of this review.
Li-he Zhang – School of Pharmaceutical Science, Peking University, Beijing, China
Kaixian Chen – Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
Introduction
Drug research and development (R & D) is comprehensive, expensive, time-consuming and full of risk. It is estimated that a drug from concept to market would take ∼12 years and cost more than US$800 million on an average [1]. Several new technologies have hence been developed and applied in drug R & D to shorten the research cycle and to reduce the expenses. Computer-aided drug design (CADD) is one of such evolutionary technologies [2].
Having emerged as a quantitative structure–activity relationship (QSAR) analysis in the early 1960s, the concept of CADD has evolved very quickly, especially in the recent decade as an unprecedented development of structural biology and computer capabilities. CADD technologies including molecular modeling and simulation have become promising in drug discovery. Recently, CADD has even been used in designing highly selective ligands for a certain target that shares very similar structures with many proteins, which is difficult to be done by other methods. One such example is the rational design of selective inhibitors of p90 ribosomal protein S6 kinase [3]. In the postgenomic era, owing to the dramatic increase of small molecule and biomacromolecule information, CADD tools have been applied in almost every stage of drug R & D, greatly changing the strategy and pipeline for drug discovery [2]. As indicated in Fig. 1 , CADD, from its traditional application of lead discovery and optimization, has extended toward two directions: upstream for target identification and validation, and downstream for preclinical study (ADMET prediction). In this review, we highlight some recent advances of CADD technologies; emphases are put on computational tools for target identification and new chemical entity discovery.
Figure 1.
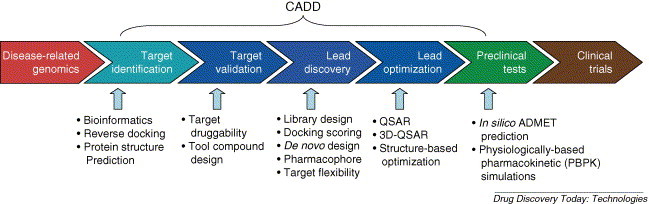
Drug discovery pipeline vs. computer-aided drug design (CADD) tools. CADD tools have been initially developed for lead optimization (such as QSAR) and then expanded for lead discovery (such as virtual screening). Now CADD tools have quickly extended toward both upstream and downstream directions along the drug discovery pipeline. In upstream direction, bioinformatics and reverse docking methods are usually used for target identification; once a target is identified, in silico methods are also developed to predict its 3D structures before experimental determination; computational methods can be used to predict target druggability and to design compounds before further experiments. In downstream direction, in silico ADMET prediction and physiologically based pharmacokinetic simulations can be conducted to model the preclinical test, which is usually integrated into in silico lead discovery stage to reduce the costs.
Target identification
Target identification and validation is the first key stage in the drug discovery pipeline (Fig. 1). However, identification and validation of druggable targets from among thousands of candidate macromolecules is still a challenging task [4]. Numerous technologies for addressing the targets have been developed recently. Genomic and proteomic approaches are the major tools for target identification. For example, a proteomic approach for identification of binding proteins for a given small molecule involves comparison of the protein expression profiles for a given cell or tissue in the presence or absence of the given molecule. This method has not been proved very successful in target discovery because it is laborious and time-consuming [5]. Therefore, complementary to the experimental methods, a series of computational (in silico) tools have also been developed for target identification. They can be cataloged into sequence-based approach and structure-based approaches.
Sequence-based approach contributes to the processes of target identification by providing functional information about target candidates and positioning information to biological networks. For those diseases caused by external pathogens such as bacteria and viruses, unique targets might be found in the pathogens by comparing functional genomics from humans with the corresponding genomics from pathogens [6]. For example, Dutta et al. used a subtractive genomic method to analyze the completed genome of Helicobacter pylori (H. pylori) and identified a set of genes that are likely to be essential to the pathogen but are absent in humans [7]. In theory, it is possible to recognize all the targets in the pathogen in this way; whereas for endogenous diseases, targets could be discovered by analyzing the differences of genomics between normal and abnormal tissues. A good example of this issue is that several novel steroid targets were identified by combinative use of bioinformatics and functional analysis of hormone response elements [8].
Structure-based approach that has shown promise in recent years is to use computational methods to find putative binding proteins for a given compound from either genomic or protein databases, and to subsequently use experimental procedures to validate the computational result [9]. One such computational approach, which is the reverse of docking a set of ligands into a given target, is to dock a compound with a known biological activity into the binding sites of all the three-dimensional (3D) structures in a given protein database. Protein ‘hits’ identified in this manner can then serve as potential candidates for experimental validation. Accordingly, this approach is referred to as reverse docking (or inverse docking) [10, 11]. The general procedure of target identification by using reverse docking integrating with biological technologies is shown in Fig. 2 . It includes four steps: reverse docking of a small molecule to select hit proteins; hit proteins postprocessing of through bioinformatic analysis to select candidates; experimentally validating by using biochemical and/or cellular assays; and finally, if it is possible and necessary, determining the X-ray crystal (or NMR) structures of the small molecule-protein complexes to verify the target at the atomic level. This approach requires a sufficient number of known protein structures covering a diverse range of drug targets (Fig. 2). The protein structures are usually selected from the protein data bank (PDB) [12] or constructed with protein structure prediction method. Using a subset of PDB as an example, Paul et al. successfully recovered the corresponding targets of four unrelated ligands with the help of reverse docking method [13].
Figure 2.
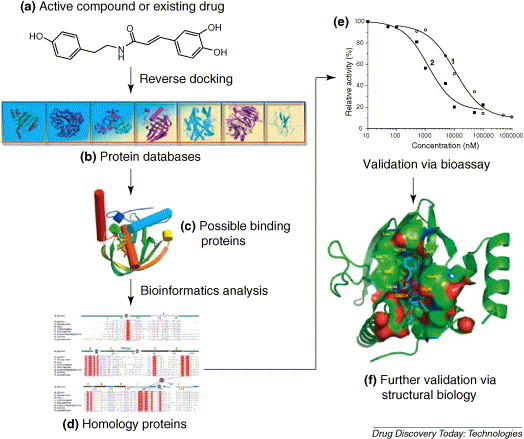
A schematic diagram for target identification by using reverse docking approach in conjunction with bioassay and structural biology. (a) Chemical structure of N-trans-caffeoyltyramine, a natural product isolated from the folk medicine Ceratostigma willmottianum. (b) A schematic representation of the in-house potential drug target database (PDTD). Other protein structure databases, like the PDB, can also be used for reverse docking. (c) Binding mode of the natural product with the protein target hits produced by reverse docking and scoring. (d) Because of the limitation of the protein entries in the databases, the target hits may not be the proteins encoded in the pathogenic genome (e.g. bacteria or virus). On the contrary, for a special species (e.g. SARS coronavirus), there are not enough 3D structures of proteins. Therefore, bioinformatic tools were used to search homology proteins of the target hits from the pathogenic genome. The homology proteins will be validated experimentally. (e) The target hits (HpDC and HpPDF) were expressed, and the binding affinities and/or inhibition or activation activities of the probes to the target hits were determined. In this way, the target candidates can be selected for further functional validation. (f) The crystal structure of natural product-target candidate (HpPDF) complex. This means that the binding between the natural product and target candidate was verified at the atomic level. The images of the proteins were generated using the PyMol program (http://pymol.sourceforge.net/).
A reverse docking web server, Target Fishing Dock (TarFisDock), was also developed for identifying new drug targets [11]. For this server, a potential drug target database (PDTD) was constructed. The target proteins collected in PDTD were selected from the literatures and from several online databases, such as DrugBank. The structures of proteins were from PDB. Recently, TarFisDock has been proved as a tool of great potential value for identifying the target of anti-H. pylori natural product [14]. Colonization of the human stomach by the bacterium H. pylori is a major causative factor for gastrointestinal illnesses and gastric cancers. However, discovery of anti-H. pylori agents is a difficult task because of lack of mature protein targets. Therefore, identifying new molecular targets for developing new drugs against H. pylori is obviously necessary. The in-house potential drug target database was searched by using the reverse docking tool TarFisDock, taking the active natural product (Fig. 2a) discovered by anti-H. pylori screening as a probe. Homology search revealed that among the 15 candidates discovered by reverse docking, only diaminopimelate decarboxylase and peptide deformylase (PDF) have homologous proteins in the genome of H. pylori. Enzymatic assay demonstrated the natural product and one of its derivatives are the potent inhibitors against the H. pylori PDF (HpPDF) with IC50 values of 10.8 and 1.25 μM, respectively (Fig. 2e). X-ray crystal structures of HpPDF and the complexes of HpPDF with the natural product and its analog were also determined (Fig. 2f), demonstrating at the atomic level that HpPDF is a potential target for screening new anti-H. pylori agents.
The advantage of reverse docking is obvious: in addition to identifying target candidates for active compounds, it is also possible to identify potential targets responsible for toxicity and/or side effects of a drug supposing that the target database contains all the possible targets [15]. However, reverse docking still has certain limitations. The major one is that the protein entries in the protein structure databases, like the PDB, are not enough for covering all the protein information of disease-related genomes. The second one is that this approach has not considered the flexibility of proteins during docking simulation. These two aspects will produce negative false. Another limitation is that the scoring function for reverse docking is not accurate enough, which will produce positive false [11]. One tendency to overcome these shortages is to develop new docking programs including protein flexibility and accurate scoring function. Another tendency is to integrate sequence-based and structure-based approaches [4].
New chemical entity discovery
Drug discovery and development in the past 100 years has been performed only against approximately 500 targets; and in the same period, about 20,000,000 organic compounds including natural products have been synthesized or isolated. However, the use of organic chemicals in drug discovery seems to be out of favor because the existing targets have not been screened by all the available compounds. In addition to this, the completion of the human genome suggests that there are 600–1500 druggable targets for drug intervention to control human diseases [16]. Therefore, it is believable that a large number of new drugs, at least many leads or hits, are hiding in the existing chemical mine. However, digging out this source is a hard task. Collecting all the existing compounds and screening them randomly are extremely unpractical, because it is intolerably expensive and time-consuming although virtual screening shows a dawning to satisfy this requirement [17]. Indeed, recent promising advancement in virtual screening has demonstrated the efficiency of this approach in discovering lead (active) compounds. Virtual screening enriched the hit rate (defined as the number of compounds that bind at a particular concentration divided by the number of compounds experimentally tested) by about 100–1000-fold over random screening. Accordingly, virtual screening has been involved in the pipeline of drug discovery as a practical tool [18, 19].
Nevertheless, as mentioned above, all hits produced from virtual screening are existing compounds or old drugs, that is, virtual screening can only find the new medical usages for the existing compounds or drugs. What big pharmas and medicinal chemists are seeking is new chemical entities (NCEs), which can be strictly protected by the compound patents. There are at least two kinds of CADD methods for NCE discovery, de novo drug design [20, 21, 22] and combinatorial library design [23].
The de novo drug design does not start from a database of complete molecules but aims at building a complete molecule from molecular bricks (‘building blocks’) to chemically fill the binding sites of target molecule [24]. The complete chemical entries could be constructed through linking the ‘building blocks’ together, or by growing from an ‘embryo’ molecule with the guidance of evaluation of binding affinity. The ‘building blocks’ could be either atoms or fragments (functional groups or small molecules). But using atoms as ‘building blocks’ is thought to be inefficient, therefore, it is seldom used nowadays. In the fragment linking approach, the binding site is mapped to identify the possible anchor points for functional groups. These groups are then linked together, and they form a complete molecule. In the sequential-growing approach, the molecule grows in the binding site controlled by an appropriate search algorithm, which evaluates each growing possibility with a scoring function. Different from docking-based virtual screening, fragment-based de novo design can perform sampling in the whole compound space, obtaining novel structures that are not limited in available databases. But the quality of a growing step strongly depends on the previous steps. Any step chemically going wrong would lead to an unacceptable result. For the fragment linking approach, choosing linkers to connect fragments together as complete structures is a problem. The most remarkable drawback of this approach might be the synthetic accessibility of the designed structures.
The advent of combinatorial chemistry is one of the most exciting developments in medicinal chemistry in the last decade. Coupled with automation technologies and high-throughput screening (HTS), it offers great potential for discovering new drug leads. This technology allows thousands or even millions of compounds to be synthesized at the same time. However, many products in the huge library are redundant. It also does not make sense to validate and assay millions of compounds. In addition to this, it was found that the large number of compounds synthesized did not result in the remarkable increase in drug candidates though the number of compounds synthesized and screened has increased by several orders of magnitude [25]. Initially, the focus of combinatorial library design was on selecting diverse sets of compounds on the assumption that maximizing diversity would result in a broad coverage of bioactivity space and hence would maximize the chances of finding drug leads. The creation of diversity through compound libraries has been a central claim and task of combinatorial chemistry since its inception. Suggestions and assumptions on how to assess diversity have been studied during the last decade. To synthesize a chemical library with reasonable size and considerable hit rate, 3D structural information and properties of a studied target should be taken into consideration to filter out redundant compounds [23]. Thus, the critical challenges are firstly to select sets of fragments that have the best potential to be parts of new drug leads for a given target; and secondly to set up proper criteria for product judgment (screening). To overcome the first challenge, three types of virtual libraries have been suggested. They are focused libraries, targeted libraries and primary screening libraries. A focused library is built on the basis of a lead molecule or pharmacophore and is geared toward one particular molecular target. A targeted library is designed for finding drug leads against specific targets. A primary screening library is a large combinatorial library used to randomly find new hits or to design novel scaffolds. To solve the second problem, druglikeness (ADMET) and structural diversity have been introduced into library design to reduce its size and increase its efficiency.
Adopting the advantages of focused library and targeted library, as well as integrating technologies of docking-based virtual screening and druglike (ADMET) analysis, a target-focused library design method was developed, based on which a software package, called LD1.0, was also developed [23]. The flowchart of LD1.0 is shown in Fig. 3 . Starting with the structures of hits and therapeutic target, the overall skeleton of potential ligands is schematically split into several fragments according to the interaction mechanism and the physicochemical properties of the binding site. Individual fragment library is constructed for each fragment, taking into account the binding features of the fragments to the binding site. Finally, target-focused libraries on the studied target are constructed with the judgments from structural diversity, druglikeness (ADMET) profiles and binding affinities [23]. During the target-focused library design, library-based genetic algorithm was applied to optimize the focused library, and the newly developed druglikeness filter was used to predict the druglike profile of the library [26]. Molecular docking approach was employed to predict the binding affinities of the library molecules with the target.
Figure 3.
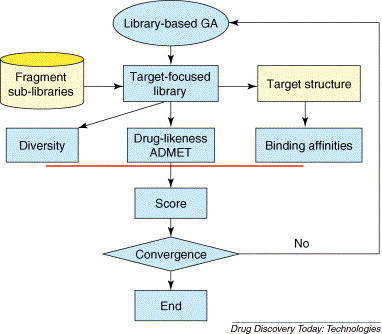
Flowchart of the software LD1.0 for target-focused library design. A target-focused library is built based on target protein (yellow colored) and corresponding fragment sub-libraries (yellow colored). Firstly, the scaffold of ligands is divided into several fragments with the guidance of the ligand structures, the feature of binding site and interaction mechanism between the ligands and the site. Secondly, corresponding fragment sub-libraries are constructed for building initial focused libraries. Thirdly, the initial libraries are scored with the criteria for molecular structural diversity, druglikeness, ADMET profiles and binding affinity of compounds in the libraries. Lastly, the best target-focused library is obtained through library-based GA optimization approach.
The quality of fragment libraries is critical to the final focused library. There are at least three ways to construct the fragment libraries [23]. Extracting fragments from known drugs or ligands (inhibitors or activators) of the studied target is an effective approach for collecting building blocks. Homology proteins usually share similar structural features and characteristics, especially at the binding site or active site. Therefore, the ligands for different targets belonging to the same family should share some common fragments. Thus, the fragments for constructing target-focused library could be designed by referring to the structures of the ligands of the homology proteins of the target. Also, fragments could be isolated from the active hits produced through primary screening (HTS and virtual screening).
The efficiency of the strategy for target-focused library design and screening has been demonstrated by our recent example of discovering human cyclophilin A (CypA) inhibitors [27, 28]. By employing docking-based virtual screening in conjunction with chemical synthesis and bioassay, 14 binders of CypA were discovered, and four of them showed high CypA PPIase inhibition activities with IC50 values of 2.5–6.2 μM [27]. To discover new chemical entities of CypA inhibitors with more potent activities, a target-focused library was designed based on the structures of the 14 hits and their binding modes to CypA by using the program LD1.0. The binding modes indicated that the small molecular CypA binders can be divided into three parts: part A interacts with the small pocket of CypA (pocket A), part B is located in the large pocket (pocket B) and part L is a linker between A and B, interacting with the ‘saddle’ pocket between sites A and B (Fig. 4 ). LD1.0 selected 5 fragments for part A, 17 fragments for part B and three linkers for part L, thus a 5 × 3 × 17 target-focused library could be generated. This library can be synthesized using either traditional organic synthesis or combinatorial chemistry. But it was not done in this manner. Sixteen compounds were selected out from the designed library via virtual screening before synthesis. Bioassay revealed that all these 16 molecules were CypA binders with binding affinities (K D values) ranging from 0.076 to 41.0 μM, and five of them were potent CypA inhibitors with PPIase inhibitory activities (IC50 values) of 0.25–6.43 μM. The hit rates for binders and inhibitors were as high as 100% and 31.25%, respectively. Remarkably, both the binding affinity and the inhibitory activity of the most potent compound increased ∼10 times than that of the most active compound discovered in the first cycle of discovery [28].
Figure 4.
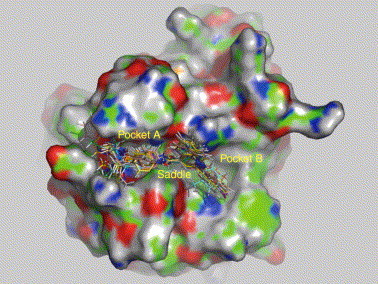
Three-dimensional structural modes of CypA inhibitors with CypA derived from the docking simulations. Fragments for target-focused library design can be selected according to the interaction modes between primarily screened hits (binders) and target. Accordingly, these fragments are leadlike building blocks for constructing library. When the fragments are connected into whole molecules, they are optimized again for the interaction poses and binding affinities by using docking. This image was generated using the PyMol program.
Conclusions and outlook
The technological progress of CADD brought a paradigm change to both pharmas and research institutions: it was now possible to obtain appropriate hits within several weeks because of the contribution of CADD [29]. Traditionally, structure-based and pharmacophore techniques and QSAR are major tools for CADD. Parallel to the development of combinatorial chemistry and HTS since more than a decade ago, several new technologies, such as library design, virtual screening, druglike analysis and ADMET prediction, have become important tools in the computer-aided discovery of new drugs. In the coming future, in addition to improving individually existing CADD techniques, such as increasing the accuracy and effectiveness of virtual screening, one major tendency of CADD technology development will be to integrate computational chemistry and biology together with chemoinformatics and bioinformatics. This will leading to a new topic known as pharmacoinformatics, which will impact the pharmaceutical development process and increase the success rate of development candidates [30].
Another tendency is that CADD technologies have been entering into the functional genomic studies and target identification in particular. After the completion of the human genome and numerous pathogen genomes, efforts are underway to understand the role of gene products in biological pathways and human diseases and to exploit their functions for the sake of discovering new drug targets [31]. Small and cell-permeable chemical ligands are used increasingly in genomic approaches to understand the global functions of genomes and proteomes. This approach is referred to as chemical biology (or chemogenomics) [32]. As such, reverse docking can be referred to as computational chemical biology, which has been proven to be an effective way in finding clues of new targets [11, 12, 13, 14, 15]. On the contrary, the CADD techniques like virtual screening and library design can also be used to design small molecule probes for illuminating the molecular mechanisms underlying biological processes through altering or perturbing the functions of target proteins by inhibiting or activating their normal functions [17, 32].
Links
-
•
Universal Protein Resource: http://www.uniprot.org/
-
•
Swiss_Model: http://swissmodel.expasy.org//SWISS-MODEL.html
-
•
National Center for Biotechnology Information: http://www.ncbi.nlm.nih.gov/
-
•
Protein Data Bank: http://www.rcsb.org/pdb/
-
•
TarFisDock web server: http://www.dddc.ac.cn/tarfisdock/
-
•
Virtual Computational Chemistry Laboratory: http://www.virtuallaboratory.org/
- •
-
•
PDBbind Database: http://www.pdbbind.org/
-
•
The Binding Database: http://www.bindingdb.org/bind/index.jsp
-
•
ZINC is not commercial: http://blaster.docking.org/zinc/
Outstanding issues
-
•
In postgenomic era, the concept of computer-aided drug design (CADD) has extended from lead discovery to target identification, from lead optimization to preclinical or clinical trials.
-
•
Two approaches for in silico target identification: sequence-based and structure-based. In sequence-based approach, bioinformatic methods are applied to analyze and compare multiple sequences and identify potential targets from scratch; whereas in structure-based approach, reverse-docking methods might be helpful to identify target candidates for active compounds.
-
•
At least two in silico strategies for the discovery of NCE: de novo drug design and combinatorial library design, especially target-focused library design, which has been demonstrated by our recent example of discovering human cyclophilin A inhibitors. Druglikeness or ADMET properties can be considered in library design.
Related articles
Jorgensen, W.L. (2004) The many roles of computation in drug discovery. Science 303, 1813–1818
Shoichet, B.K. (2004) Virtual screening of chemical libraries. Nature 432, 862–865
Schneider, G. and Fechner, U. (2005) Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 4, 649–663
Hajduk, P.J. et al. (2005) Predicting protein druggability. Drug Discov. Today 10, 1675–1682
Van de Waterbeemd, H. and Gifford, E. (2003) ADMET in silico modeling: towards prediction paradise? Nat. Rev. Drug Discov. 2, 192–204
Acknowledgements
We are grateful for the financial supports from the National Natural Science Foundation of China No. 20572023 (Y.T.), Shanghai Pujiang Program No. 05PJ14034 (Y.T.), Shanghai Key Basic Research Project No. 05JC14092 (W.Z.) and the State Key Program of Basic Research of China No. 002CB512802 (H.J.).
References
- 1.DiMasi J.A. The price of innovation: new estimates of drug development costs. J. Health Economics. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 2.Jorgensen W.L. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 3.Cohen M.S. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajduk P.J. Predicting protein druggability. Drug Discov. Today. 2005;10:1675–1682. doi: 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]
- 5.Huang C.M. Proteomics reveals that proteins expressed during the early stage of Bacillus anthracis infection are potential targets for the development of vaccines and drugs. Genomics Proteomics Bioinformatics. 2004;2:143–151. doi: 10.1016/S1672-0229(04)02020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Lara J. Staphylococcus aureus: the search for novel targets. Drug Discov. Today. 2005;10:643–651. doi: 10.1016/S1359-6446(05)03432-X. [DOI] [PubMed] [Google Scholar]
- 7.Dutta A. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In Silico Biol. 2006;6:0005. [PubMed] [Google Scholar]
- 8.Horie-Inoue K. Identification of novel steroid target genes through the combination of bioinformatics and functional analysis of hormone response elements. Biochem. Biophys. Res. Commun. 2006;339:99–106. doi: 10.1016/j.bbrc.2005.10.188. [DOI] [PubMed] [Google Scholar]
- 9.Rockey W.M., Elcock A.H. Rapid computational identification of the targets of protein kinase inhibitors. J. Med. Chem. 2005;48:4138–4152. doi: 10.1021/jm049461b. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y.Z., Zhi D.G. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins. 2001;43:217–226. doi: 10.1002/1097-0134(20010501)43:2<217::aid-prot1032>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 11.Li H. TarFisDock: a web server for identifying drug targets with docking approach. Nucl. Acids Res. 2006;34:W219–W224. doi: 10.1093/nar/gkl114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berman H.M. The protein data bank. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul N. Recovering the true targets of specific ligands by virtual screening of the protein data bank. Proteins. 2004;54:671–680. doi: 10.1002/prot.10625. [DOI] [PubMed] [Google Scholar]
- 14.Cai J. Peptide deformylase is a potential target for anti-Helicobacter pylori drugs: reverse docking, enzymatic assay and X-ray crystallography validation. Protein Sci. 2006;15:2071–2081. doi: 10.1110/ps.062238406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y.Z., Ung C.Y. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand-protein inverse docking approach. J. Mol. Graph. Model. 2001;20:199–218. doi: 10.1016/s1093-3263(01)00109-7. [DOI] [PubMed] [Google Scholar]
- 16.Caron P.R. Chemogenomic approaches to drug discovery. Curr. Opin. Chem. Biol. 2001;5:464–470. doi: 10.1016/s1367-5931(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 17.Shen J.H. Virtual screening on natural products for discovering active compounds and target clues. Curr. Med. Chem. 2003;10:2327–2342. doi: 10.2174/0929867033456729. [DOI] [PubMed] [Google Scholar]
- 18.Doman T.N. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 19.Shoichet B.K. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honma T. Recent advances in de novo design strategy for practical lead identification. Med. Res. Rev. 2003;23:606–632. doi: 10.1002/med.10046. [DOI] [PubMed] [Google Scholar]
- 21.Schneider G., Fechner U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005;4:649–663. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- 22.Heikkilä T. The first de novo designed inhibitors of Plasmodium falciparum dihydrooratate dehydrogenase. Bioorg. Med. Chem. Lett. 2006;16:88–92. doi: 10.1016/j.bmcl.2005.09.045. [DOI] [PubMed] [Google Scholar]
- 23.Chen G. Focused combinatorial library design based on structural diversity, druglikeness and binding affinity score. J. Comb. Chem. 2005;7:398–406. doi: 10.1021/cc049866h. [DOI] [PubMed] [Google Scholar]
- 24.Rotstein S.H., Murcko M.A. GroupBuild: a fragment-based method for de novo drug design. J. Med. Chem. 1993;36:1700–1710. doi: 10.1021/jm00064a003. [DOI] [PubMed] [Google Scholar]
- 25.Oprea T.I. Chemical space navigation in lead discovery. Curr. Opin. Chem. Biol. 2002;6:384–389. doi: 10.1016/s1367-5931(02)00329-0. [DOI] [PubMed] [Google Scholar]
- 26.Zheng S. A new rapid and effective chemistry space filter in recognizing a druglike database. J. Chem. Inf. Model. 2005;45:856–862. doi: 10.1021/ci050031j. [DOI] [PubMed] [Google Scholar]
- 27.Li J. Discovering novel chemical inhibitors of human cyclophilin A: virtual screening, synthesis and bioassay. Bioorg. Med. Chem. 2006;14:2209–2224. doi: 10.1016/j.bmc.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J. Strategy for discovering chemical inhibitors of human cyclophilin A: focused library design, virtual screening, chemical synthesis and bioassay. J. Comb. Chem. 2006;8:326–337. doi: 10.1021/cc0501561. [DOI] [PubMed] [Google Scholar]
- 29.Eringis D., Goldman B. Locus Discovery: from structure to hit in weeks. Drug Discov. Today. 2002;7:S16–S18. [Google Scholar]
- 30.Schuffenhauer A., Jacoby E. Annotating and mining the ligand-target chemogenomics knowledge space. Drug Discov. Today: BIOSILICO. 2004;2:190–200. [Google Scholar]
- 31.Kopec K.K. Target identification and validation in drug discovery: the role of proteomics. Biochem. Pharmacol. 2005;69:1133–1139. doi: 10.1016/j.bcp.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Stockwell B.R. Exploring biology with small organic molecules. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]