

Abstract
We report a concise, enantioselective synthesis of the yohimbine alkaloids (−)-rauwolscine and (−)-alloyohimbane. The key transformation involves a highly enantio- and diastereoselective NHC-catalyzed dimerization and an amidation/N-acyliminium ion cyclization sequence to furnish four of the five requisite rings and three of the five stereocenters in two operations. This route also provides efficient access to all four diastereomeric arrangements of the core stereotriad of the yohimbine alkaloids from a common intermediate. This platform approach in combination with the ability to access both enantiomers from the carbene-catalyzed reaction is a powerful strategy that can produce a wide range of complex alkaloids and related structures for future biomedical investigations.
Graphical Abstract
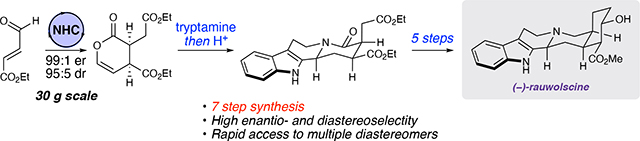
Alkaloids are secondary metabolites containing a basic nitrogen atom that are ubiquitous in nature and medicine.1 The prevalence of these molecules in pharmaceuticals and the increased desire to investigate sp3-carbon-rich molecules in drug discovery campaigns necessitates the efficient construction of complex nitrogen-containing heterocycles with high levels of enantiocontrol.2–3 Moreover, the design of adaptable syntheses that enable rapid access to a diverse array of structurally related molecules is of comparable importance. Over the past fifteen years, the field of N-heterocyclic carbene (NHC) catalysis has distinctively enabled the construction of enantioenriched heterocycles,4–6 and the power of cascade and one-pot processes has led to an ever-increasing level of synthetic efficiency in the assembly of complex architectures.7–9Herein we report the union of these strategies for the synthesis of yohimbine alkaloids.
The yohimbine natural products are a family of pentacyclic indole alkaloids derived from the amino acid tryptophan and the secoiridoid monoterpene secologanin (Scheme 1).10 Following the isolation of yohimbine (1) by Spiegel in 1900 and its structural determination by Witkop in 1943,11 a wide variety of yohimbine stereoisomers have been identified. These alkaloids can be divided into four different subfamilies, normal, allo, pseudo, or epiallo, which differ in their stereochemical arrangement around the D-ring; yohimbine (1), rauwolscine (2), pseudoyohimbine (3), and reserpine (4), respectively, are representative members, respectively (Scheme 1). These alkaloids show a wide range of activity within the central nervous system (CNS), and although the structural variations between diastereomeric subfamilies are modest, they lead to an impressive degree of divergence in their pharmacology.12–15 It is therefore of great interest to be able to access these alkaloids in a concise, modular, and stereoselective fashion to enable thorough investigation of structural analogs.
Scheme 1.

Representative yohimbine alkaloids and retrosynthetic approach.
The traditional approach to the yohimbines follows precedent set by Woodward and co-workers in their landmark synthesis of 4 in 1958, which involves construction of a fully-functionalized E-ring precursor followed by tethering of the tryptamine subunit and ring closure.16–17 This general strategy has been utilized in a myriad of total and formal syntheses, and continues to be the predominant approach to these natural products today.18–21 While this conventional approach has been successful in accessing specific natural products, the key β-carboline pharmacophore is not introduced until the latter stages of the synthesis.22 Consequently, efficient access to related analogs with biomedical potential is thereby limited and would require reworking of the early stages of the synthesis in order to introduce structural variation in the terpene portion of the molecule. Syntheses that introduce the E-ring last are comparatively less explored,18, 23–25 but have increased potential for access to a large array of related alkaloids.26–27 In the context of this broad synthetic landscape, we sought to develop a general approach for the synthesis of yohimbine alkaloids that would a) be fewer than 10 steps for maximum efficiency to drive future biomedical exploration, and b) be derived from a key intermediate that could be easily converted into the different subtype structures (see Scheme 1).
Our retrosynthetic plan is depicted in Scheme 1. We envisioned accessing (−)-rauwolscine (2) from the known β-ketoester 526 via diastereoselective reduction. β-Ketoester 5 would be obtained by reduction and homologation of diester 6 followed by ring closure. This key intermediate would arise from an amidation/N-acyliminium ion cyclization sequence with enol lactone 7,28–31 which could be accessed via NHC-catalyzed dimerization of commercially-available aldehyde 8.32 Additionally, we envisioned allo-configured tetracycle6 as a potential precursor to access the diastereomeric normal, pseudo, and epiallo yohimbine cores by selective stereochemical inversions at the C/D and D/E ring junctions.
Our synthesis commenced with the construction of enantioenriched enol lactone 7 from aldehyde 8 (Scheme 2). After evaluating various conditions (see the Supporting Information for details), it was found that the use of 1 mol % of NHC pre-catalyst 9 delivered 7 in 83% yield with excellent control of the enantio- and diastereselectivity; notably, this transformation could be conducted on 30-gram scale without loss of efficiency. With ample quantities of 7, we began to explore the key amidation/N-acyliminium ion cyclization sequence with tryptamine to access 6. Initial experiments revealed that the acylation of tryptamine by 7 was rapid, but intramolecular N-acyliminium ion formation was inhibited by intermolecular imine formation with an additional equivalent of tryptamine. This side reaction proved to be minimally reversible even in the presence of water, as only 55% conversion of 7 was seen after 3 days when using equimolar quantities of 7 and tryptamine (see Figure S1). However, we were pleased to find that acidification of the reaction mixture triggered facile imine hydrolysis, and N-acyliminium ion formation and cyclization occurred readily upon addition of TFA. Additional optimization revealed that treatment of 7 with two equivalents of 10 in a biphasic solvent system of CH2Cl2 and aqueous Na2CO3 followed by acidification and TFA-promoted cyclization delivered 6 as a separable 80:20 mixture of diastereomers in 74% yield on decagram scale.30 Selective reduction of the lactam with BH3·DMS then provided tertiary amine 11 in 94% yield, whose structure was confirmed by X-ray crystallography of its TFA salt.
Scheme 2.

Synthesis of (−)-rauwolscine 2 and (−)-alloyohimbane 17.
With the ABCD ring system assembled, we turned our attention to construction of the E-ring, which required the homologation of both ester sidechains by one carbon, followed then by ring closure. Unfortunately, attempts to directly homologate 11 using Arndt-Eistert33 or Kowalksi34 conditions returned starting material or decomposition products, respectively, and LAH reduction to the diol followed by various logical conditions for SN2 with cyanide anion resulted only in quaternization of the tertiary amine.35 We then turned our attention to Wittig-Horner-type homologation conditions. Since an ester oxidation state was desired for the ring closure step, known phosphonate 12 reported by Mikołajczykand co-workerswas selected because the resulting ketene dithioacetal can be easily converted to an ester.36 We also elected to employ conditions disclosed by Takacs in our experimental design, wherein the partial reduction is performed in the presence of the metallated phosphonate to mitigate overreduction and ensure high efficiency in this transformation.37 A temperature and reducing reagent screen revealed that using lithium diisobutyl-tert-butoxyaluminum hydride (LDBBA)38 at 0 °C in the presence of excess 12 afforded 13 in 70% yield on multigram scale. Although 13 could be converted to the bis(methyl ester) under forcing acidic conditions [p-TsOH (10 equiv), MeOH, 80 °C], its cyclization under the Dieckmann conditions reported previously led to a mixture of 14a, 14b.26 Even more confounding was that these compounds and their enol isomers proved inseparable under multiple conditions and derivatizations. These roadblocks necessitated an alternative tactic for cyclization. We soon discovered that using a milder set of conditions [p-TsOH (2 equiv), DCM/MeOH, 0–45 °C] delivered an easily separable 62:38 mixture of α-oxo ketene dithioacetal 15 (major) and its regioisomer (not shown) in 80% combined yield. This transformation not only provided the ring closed product, but also showed the desired regioselectivity along with much needed separability. The unmasking of the resultant α-oxo-ketenedithioacetal of 15 with HgCl2 and BF3·OEt2 in methanol then provided the desired methyl ester 5 in 64% yield on >100 mg scale. Temperature proved to be critical to the success of this transformation; reactions conducted below 40 °C were exceedingly sluggish, while those in excess of 50 °C produced significant amounts the ring-opened bis(methyl ester).
At this stage, we were poised to investigate the final disconnection from our retrosynthetic analysis, the diastereoselective reduction of β-ketoester 5. Using NaBH4, 17-epi-rauwolscine 16 was furnished in good yield, with no detectable amount of 2 produced.26To circumvent this, we sought out reagents that would provide some semblance of the presumed thermodynamic control. A survey of the literature suggested that SmI2 would be well suited for this application.39 Initial experiments using alcoholic additives such as MeOH, iPrOH, or tBuOH returned the starting material unchanged. However, when H2O was used as the protic additive, SmI2 rapidly delivered (−)-rauwolscine 2 as a single diastereomer in 65% yield. Additionally, β-ketoester 5 could be decarboxylated to ketone 16 using LiOH followed by a modified Wolff-Kishner deoxygenation to provide the unsubstituted E-ring product (−)-alloyohimbane 17 in 45% yield over 2 steps. Notably, the step economy of our syntheses of 2, 16,and 17 compare favorably to previously reported asymmetric syntheses.40–43
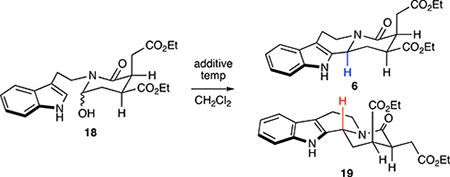
With an efficient and general route to allo configured yohimbines secured, we began investigating methods to access other diastereomericyohimbine frameworks. We began by exploring conditions to control the diastereoselectivity of the key N-acyliminium ion cyclization step. To fully examine activating agents for this transformation, we elected to conduct the N-acyliminium ion cyclization under anhydrous conditions. Hence, the intermediate hydroxylactam 18 (~85:15 d.r.) was isolated by extraction following the imine hydrolysis step in the initially optimized conditions (Table 1). In this new two-step process, employing acetyl chloride at cryogenic temperatures further improved selectivity for 6 over the previous one-pot conditions (entry 1). At higher temperatures (entry 2) or with the use of alternative acetate activating agents (entry 3), the reaction efficiency and selectivity decreased markedly. Lewis and Brønsted acids could also be employed successfully (entries 4–6), with high levels of selectivity and efficiency for 6 being observed with TMSCl at cryogenic temperatures (95:5 d.r. and 78% yield). However, performing the reaction at 40 °C rather than −78 °C, lead to reversal of diastereoselectivity, providing 19, a potential precursor for epiallo yohimbines, in 69% yield and 73:27 d.r. (entry 7).
Table 1.
Effect of Additives and Temperature on Cyclization Diastereoselectivity
 | ||||
|---|---|---|---|---|
| entry | additive | temp (°C) | d.r. (6:19) | yield (%)a |
| 1 | AcCl | −78 | 87:13 | 68% |
| 2 | AcCl | 20 | 60:40 | n.d. |
| 3 | Ac2O | −78 | –b | –b |
| 4 | BF3·Et2O | −78 | 84:16 | 72 |
| 5 | TMSCl | −78 | 95:5 | 78 |
| 6 | HCl | −78 | 95:5 | 70 |
| 7c | TMSCl | 40 | 27:73 | 69 |
| 8d,e | TMSCl | 40 | 27:73 | n.d. |
| 9 | HCl | 40 | 66:34 | n.d. |
Isolated yield over 2 steps starting from 7
no reaction
reaction time of 2 days
starting material was pure 6
reaction time 4 days.
n.d. = not determined.
To gain further understanding of the diastereoselectivity, pure 6 was subjected to the reaction conditions employed in entry 7, and the same diastereomeric ratio (6:19 = 27:73) was obtained after 4 days at 40 °C (entry 8); extended reaction times or the use of chiral additivies (thioureas or phosphric acids)31, 44 did not alter this ratio. Additionally, treatment of 18 with anhydrous HCl instead of TMSCl at 40 °C did not provide the same product distribution (entry 9), indicating that acid catalysis is not responsible for equilibration. Our current working model based on this data is that the high diastereoselectivity observed at cryogenic temperatures is the result of a kinetically-controlled ring closure,27 whereas at elevated temperatures, an equilibrating mechanism is operative,45 delivering the more thermodynamically stable tetracycle 19 as the major diastereomer.
Using the new conditions outlined above (Table 1), the allo configured product 6 could be readily secured with high levels of diastereoselectivity (Scheme 3). The subjection of 6 to NaOEt in THF-EtOH (1:1) at −20 °C led to selective epimerization of the C-15 (yohimbine numbering) stereocenter, producing the pseudo configured product 20 in 52% yield. Alternatively, the oxidation of 20 to the enamide using copper(II) acetate in an atmosphere of molecular oxygen followed by reduction with NaBH4 in AcOH provided the normal configured tetracycle 21 in 72% yield (2 steps) as a single diastereomer.46 Finally, as shown in Table 1 above, simple modulation of the conditions for cyclization delivered the epiallo configured product 19 in 69% yield with modest control of the diastereoselectivity (73:27 d.r.). Combined, these connected approaches provide concise entry into multiple yohimbine stereoisomer subfamilies.
Scheme 3.

Access to All Yohimbine Stereoisomeric Cores
In conclusion, we have developed a concise synthesis of (−)-rauwolscine2 and (−)-alloyohimbane 17 from commercially available ethyl 4-oxobutenoate 8. The key NHC catalyzed annulation product 7 is readily accessible in enantiopure form and can be converted to the complex tetracyclic lactam 6 in a single operation on multi-gram scale, which allows facile access to allo-configured yohimbines. Selective manipulation of this intermediate also facilitates entry into the normal, pseudo, and epiallo classes of this important natural product family. Additionally, by employing either enantiomeric NHC catalyst (+ or −), it is feasible that all eight possible stereoisomeric cores of these alkaloids are accessible, enabling production of a wide range of molecules to drive the discovery of new biologically active molecules.
Supplementary Material
Acknowledgements
Financial support from the National Institutes of Health and Northwestern University is acknowledged (NIGMS R01 GM073072 and GM131431). The authors thank Keegan Fitzpatrick (Northwestern) for X-ray crystallographic analysis of 11 and 2 and Allison Devitt (Northwestern) for assistance with NMR characterization of 20.
Footnotes
The authors declare no competing financial interests.
Supporting Information
Supporting information available free of charge on the ACS publication website, including experimental procedures and spectroscopic data for compounds (PDF) and X-ray data (CIF)
References
- 1.Roberts MF; Wink M, Alkaloids: Biochemistry, Ecology, and Medicinal Applications. Plenum: 1998. [Google Scholar]
- 2.Lovering F; Bikker J; Humblet C, Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 3.Vitaku E; Smith DT; Njardarson JT, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 4.Hopkinson MN; Richter C; Schedler M; Glorius F, An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [DOI] [PubMed] [Google Scholar]
- 5.Flanigan DM; Romanov-Michailidis F; White NA; Rovis T, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev 2015, 115 (17), 9307–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izquierdo J; Hutson GE; Cohen DT; Scheidt KA, A Continuum of Progress: Applications of N-Hetereocyclic Carbene Catalysis in Total Synthesis. Angew. Chem. Int. Ed 2012, 51, 11686–11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolaou KC; Chen JS, The art of total synthesis through cascade reactions. Chem. Soc. Rev 2009, 38, 2993–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grondal C; Jeanty M; Enders D, Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem 2010, 2, 167. [DOI] [PubMed] [Google Scholar]
- 9.Coulthard G; Erb W; Aggarwal VK, Stereocontrolled organocatalytic synthesis of prostaglandin PGF2α in seven steps. Nature 2012, 489, 278. [DOI] [PubMed] [Google Scholar]
- 10.O’Connor SE; Maresh JJ, Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep 2006, 23, 532–547. [DOI] [PubMed] [Google Scholar]
- 11.Witkop B, Zurkonstitution des yohimbins und seiner abbauprodukte. Liebigs Ann. 1943, 554, 83–126. [Google Scholar]
- 12.Saxton JE, Chapter 10 The Indole Alkaloids**This material is supplementary to Volume II, Chapter 13 In The Alkaloids: Chemistry and Physiology, Manske RHF, Ed. Academic Press: 1960; Vol. 7, pp 1–199. [Google Scholar]
- 13.Szántay C; Blaskó G; Honty K; Dörnyei G, Chapter 2 Corynantheine, Yohimbine, and Related Alkaloids In The Alkaloids: Chemistry and Pharmacology, Brossi A, Ed. Academic Press: 1986; Vol. 27, pp 131–268. [Google Scholar]
- 14.Shore PA; Giachetti A, Reserpine: Basic and Clinical Pharmacology In Handbook of Psychopharmacology: Volume 10: Neuroleptics and Schizophrenia, Iversen LL; Iversen SD; Snyder SH, Eds. Springer US: Boston, MA, 1978; pp 197–219. [Google Scholar]
- 15.Hatch RC; Kitzman JV; Zahner JM; Clark JD, Antagonism of xylazine sedation with yohimbine, 4-aminopyridine, and doxapram in dogs. Am. J. Vet. Res 1985, 46, 371–375. [PubMed] [Google Scholar]
- 16.Woodward RB; Bader FE; Bickel H; Frey AJ; Kierstead RW, The total synthesis of reserpine. Tetrahedron 1958, 2, 1–57. [Google Scholar]
- 17.Woodward RB; Bader FE; Bickel H; Frey AJ; Kierstead RW, The Total Synthesis of Reserpine. J. Am. Chem. Soc 1956, 78, 2023–2025. [Google Scholar]
- 18.Baxter EW; Mariano PS, Recent Advances in the Synthesis of Yohimbine Alkaloids In Alkaloids: Chemical and Biological Perspectives, Pelletier SW, Ed. Springer New York: New York, NY, 1992; pp 197–319. [Google Scholar]
- 19.Stork G; Tang PC; Casey M; Goodman B; Toyota M, Regiospecific and stereoselective syntheses of (±)-reserpine and (−)-reserpine. J. Am. Chem. Soc 2005, 127, 16255–16262. [DOI] [PubMed] [Google Scholar]
- 20.Chen F-E; Huang J, Reserpine: a challenge for total synthesis of natural products. Chem. Rev 2005, 105, 4671–4706. [DOI] [PubMed] [Google Scholar]
- 21.Lebold TP; Wood JL; Deitch J; Lodewyk MW; Tantillo DJ; Sarpong R, A divergent approach to the synthesis of the yohimbinoid alkaloids venenatine and alstovenine. Nat. Chem 2013, 5, 126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maity P; Adhikari D; Jana AK, An overview on synthetic entries to tetrahydro-β-carbolines. Tetrahedron 2019, 75, 965–1028. [Google Scholar]
- 23.Herlé B; Wanner MJ; van Maarseveen JH; Hiemstra H, Total Synthesis of (+)-Yohimbine via an Enantioselective Organocatalytic Pictet–Spengler Reaction. J. Org. Chem 2011, 76, 8907–8912. [DOI] [PubMed] [Google Scholar]
- 24.Barcan GA; Patel A; Houk KN; Kwon O, A torquoselective 6π electrocyclization approach to reserpine alkaloids. Org. Lett 2012, 14, 5388–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajapaksa NS; McGowan MA; Rienzo M; Jacobsen EN, Enantioselective total synthesis of (+)-reserpine. Org. Lett 2013, 15, 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toke L; Gombos Z; Blasko G; Hont K; Szabo L; Tamas J; Szantay C, Synthesis of yohimbines. II. Alternative route to alloyohimbine alkaloids. J. Org. Chem 1973, 38, 2501–2509. [Google Scholar]
- 27.Zhang W; Bah J; Wohlfarth A; Franzén J, A stereodivergent strategy for the preparation of corynantheine and ipecac alkaloids, their epimers, and analogues: efficient total synthesis of (−)-dihydrocorynantheol, (−)-corynantheol, (−)-protoemetinol, (−)-corynantheal, (−)-protoemetine, and related natural and nonnatural compounds. Chem. Eur. J 2011, 17, 13814–13824. [DOI] [PubMed] [Google Scholar]
- 28.Maryanoff BE; Zhang H-C; Cohen JH; Turchi IJ; Maryanoff CA, Cyclizations of N-Acyliminium Ions. Chem. Rev 2004, 104, 1431–1628. [DOI] [PubMed] [Google Scholar]
- 29.Wu P; Nielsen TE, Scaffold Diversity from N-Acyliminium Ions. Chem. Rev 2017, 117, 7811–7856. [DOI] [PubMed] [Google Scholar]
- 30.Franzén J; Fisher A, Asymmetric alkaloid synthesis: a one-pot organocatalytic reaction to quinolizidine derivatives. Angew. Chem. Int. Ed 2009, 48, 787–791. [DOI] [PubMed] [Google Scholar]
- 31.Muratore ME; Holloway CA; Pilling AW; Storer RI; Trevitt G; Dixon DJ, Enantioselective Brønsted Acid-Catalyzed N-Acyliminium Cyclization Cascades. J. Am. Chem. Soc 2009, 131, 10796–10797. [DOI] [PubMed] [Google Scholar]
- 32.Latendorf K; Mechler M; Schamne I; Mack D; Frey W; Peters R, Titanium Salen Complexes with Appended Silver NHC Groups as Nucleophilic Carbene Reservoir for Cooperative Asymmetric Lewis Acid/NHC Catalysis. Eur. J. Org. Chem 2017, 2017, 4140–4167. [Google Scholar]
- 33.Arndt F; Eistert B, Ein Verfahren zur Überführung von Carbonsäuren in ihre höheren Homologen bzw. deren Derivate. Chem. Ber 1935, 68, 200–208. [Google Scholar]
- 34.Kowalski CJ; Haque MS; Fields KW, Ester homologation via α-bromo α-keto dianion rearrangement. J. Am. Chem. Soc 1985, 107, 1429–1430. [Google Scholar]
- 35.Kalaus G; Malkieh N; Katona I; Kajtar-Peredy M; Koritsanszky T; Kalman A; Szabo L; Szantay C, Synthesis of Vinca alkaloids and related compounds. 21. Preparation of (±)-eburnamonine, (±)-3-epieburnamonine, and (±)-C-norquebrachamine from a common intermediate. J. Org. Chem 1985, 50, 3760–3767. [Google Scholar]
- 36.Mikołajczyk M; Grzejszczak S; Zatorski A; Mlotkowska B; Gross H; Costisella B, Organosulphur compounds—XVIII: a new and general synthesis of ketene S,S- and O,S-thioacetals based on the Horner-Wittig reaction. Tetrahedron 1978, 34, 3081–3088. [Google Scholar]
- 37.Takacs JM; Helle MA; Seely FL, An improved procedure for the two carbon homologation of esters to α,β-unsaturated esters. Tetrahedron Lett. 1986, 27, 1257–1260. [Google Scholar]
- 38.Kim MS; Choi YM; An DK, Lithium diisobutyl-t-butoxyaluminum hydride, a new and efficient reducing agent for the conversion of esters to aldehydes. Tetrahedron Lett. 2007, 48, 5061–5064. [Google Scholar]
- 39.Evans DA; Kaldor SW; Jones TK; Clardy J; Stout TJ, Total synthesis of the macrolide antibiotic cytovaricin. J. Am. Chem. Soc 1990, 112, 7001–7031. [Google Scholar]
- 40.Feng W; Jiang D; Kee C-W; Liu H; Tan C-H, Bicyclic guanidine catalyzed asymmetric tandem isomerization intramolecular-Diels–Alder reaction: the first catalytic enantioselective total synthesis of (+)-alpha-yohimbine. Chem. Asian. J 2016, 11, 390–394. [DOI] [PubMed] [Google Scholar]
- 41.Aube J; Ghosh S; Tanol M, Symmetry-driven synthesis of indole alkaloids: asymmetric total syntheses of (+)-yohimbine, (−)-yohimbone, (−)-yohimbane, and (+)-alloyohimbane. J. Am. Chem. Soc 1994, 116, 9009–9018. [Google Scholar]
- 42.Wang X; Xia D; Qin W; Zhou R; Zhou X; Zhou Q; Liu W; Dai X; Wang H; Wang S; Tan L; Zhang D; Song H; Liu X-Y; Qin Y, A Radical Cascade Enabling Collective Syntheses of Natural Products. Chem 2017, 2, 803–816. [Google Scholar]
- 43.Miyafuji A; Ito K; Katsuki T, Application of Oxidative Desymmetrization of meso-Tetrahydrofurans: Syntheses of Functionalized Chiral Building Blocks and of (−)-Alloyohimbane. Heterocycles 2000, 52, 261–272. [Google Scholar]
- 44.Taylor MS; Jacobsen EN, Highly Enantioselective Catalytic Acyl-Pictet−Spengler Reactions. J. Am. Chem. Soc 2004, 126, 10558–10559. [DOI] [PubMed] [Google Scholar]
- 45.Lounasmaa M; Berner M; Brunner M; Suomalainen H; Tolvanen A, Acid-catalysed epimerization of indolo[2,3-a]quinolizidine derivatives: role of the nitrogen lone pairs in the mechanism. Tetrahedron 1998, 54, 10205–10216. [Google Scholar]
- 46.Bohlmann C; Bohlmann R; Rivera EG; Vogel C; Manandhar MD; Winterfeldt E, Reaktionen an Indolderivaten LIII. Enantioselektive Totalsynthese von (+)-Geissoschizin und (−)-Geissoschizol. Liebigs Ann. 1985, 1752–1763. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.