

Abstract
E2F8 is a transcriptional repressor that antagonizes E2F1 at the crossroads of the cell cycle, apoptosis, and cancer. Previously, we discovered that E2F8 is a direct target of the APC/C ubiquitin ligase. Nevertheless, it remains unknown how E2F8 is dynamically controlled throughout the entirety of the cell cycle. Here, using newly developed human cell-free systems that recapitulate distinct inter-mitotic and G1 phases and a continuous transition from prometaphase to G1, we reveal an interlocking dephosphorylation switch coordinating E2F8 degradation with mitotic exit and the activation of APC/CCdh1. Further, we uncover differential proteolysis rates for E2F8 at different points within G1 phase, accounting for its accumulation in late G1 while APC/CCdh1 is still active. Finally, we demonstrate that the F-box protein Cyclin F regulates E2F8 in G2-phase. Altogether, our data define E2F8 regulation throughout the cell cycle, illuminating an extensive coordination between phosphorylation, ubiquitination and transcription in mammalian cell cycle.
INTRODUCTION
The E2F family of transcription factors plays a pivotal role in regulating pro- and anti-proliferative processes, with implications in tissue homeostasis and human disease, most notably cancer (Chen et al., 2009).
E2F1, the canonical member of the E2F family, is at the cross-roads of the cell cycle and cell death, triggering the gene program dictating S-phase and mitotic entry on the one hand and, on the other hand, apoptosis (Polager and Ginsberg, 2008; Hallstrom and Nevins, 2009; Thurlings and de Bruin, 2016).
The activity of E2F1 is balanced by two of its direct targets E2F7 and E2F8, also known as atypical E2Fs (de Bruin et al., 2003; Di Stefano et al., 2003; Christensen et al., 2005; Maiti et al., 2005). E2F7 and E2F8 are structurally and functionally related (Lammens et al., 2009); they cooperate in repressing the transcription of E2F1 and its downstream target genes by binding consensus E2F motifs along their promoters. The result is a negative feedback circuit whose dynamics control cell fate, tissue homeostasis, and development by mechanisms that are still not entirely clear (Li et al., 2008; Ouseph et al., 2012; Liu et al., 2013). Despite being part of the “repressive” branch of E2F proteins, E2F7 and E2F8 belong to the pro-proliferative gene network underlying cell proliferation (Cohen et al., 2013).
E2F1, as well as E2F7 and E2F8, are regulated posttranslationally via temporal proteolysis. The anaphase-promoting complex/cyclosome (APC/C) is a multisubunit cell cycle ubiquitin (Ub) ligase and core component of the cell cycle machinery (King et al., 1995; Sudakin et al., 1995). The APC/C uses two related coactivators termed Cdc20 and Cdh1, which bind substrates and recruit them to the APC/C for ubiquitination and subsequent degradation (Kernan et al., 2018). We previously identified both E2F7 and E2F8 as targets of the Cdh1-bound form of APC/C (APC/CCdh1) (Cohen et al., 2013). These findings shifted the model by which the E2F1-E2F7-E2F8 circuitry communicates with the cell cycle clock to regulate the transition from the G1-phase of the cell cycle into S-phase. Nevertheless, the exact interdynamics of E2F1-E2F7-E2F8 circuitry throughout G1 and the mechanism by which they are achieved are not entirely resolved. No less obscure is the interplay between E2F1 and atypical E2Fs during G2-phase and mitosis. Dissecting these complex signaling circuits is important for understanding the decision-making mechanisms at two critical points in the life of a proliferating cell—commitment to DNA replication and division.
Cell-free systems are known for their capacity to reproduce complex cellular processes in vitro while maintaining a physiologically relevant context, thereby bridging the gap between in vivo and in vitro. These systems are optimal for direct and quantitative analysis of time-specific molecular events, including phosphorylation, ubiquitination, and proteolysis, circumventing caveats associated with long-term in vivo manipulations (Murray and Kirschner, 1989; Murray et al., 1996; Funabiki and Murray, 2000; Rape and Kirschner, 2004; Ayad et al., 2005; Telley et al., 2012; Nguyen et al., 2014). Here, we developed and utilized a panel of human cell-free systems with which we disentangle the mechanism underlying temporal dynamics of E2F8 protein from prometaphase to late S-phase. In vivo studies addressing the regulation of E2F8 during G2-phase complete the missing piece of the puzzle.
RESULTS
Temporal dynamics of E2F8 versus E2F1 across the cell cycle
E2F8 and E2F1 protein-levels were analyzed during cell cycle progression at 30- to 60-min time intervals in synchronous HeLa S3 cell populations released from prometaphase or early S-phase block. This analysis highlighted three important points. First, E2F1 begins to accumulate during early-mid G1 phase, coinciding with the peak of APC/CCdh1 activity (Figure 1A). The established APC/CCdh1 target Kifc1 is shown as a control (Figure 1A and Supplemental Figure S1). Second, E2F8 accumulation commences approximately 2.5–3 h after E2F1 initially appears. Third, E2F8 accumulates prior to the G1-S transition, well before the rise of canonical APC/C targets and the start of S-phase. At 7 h after release from mitotic block, increasing E2F8 levels are evident, yet cells have not yet entered S-phase based on DNA content measurements and before Kifc1 accumulation (Figure 1A).
FIGURE 1:
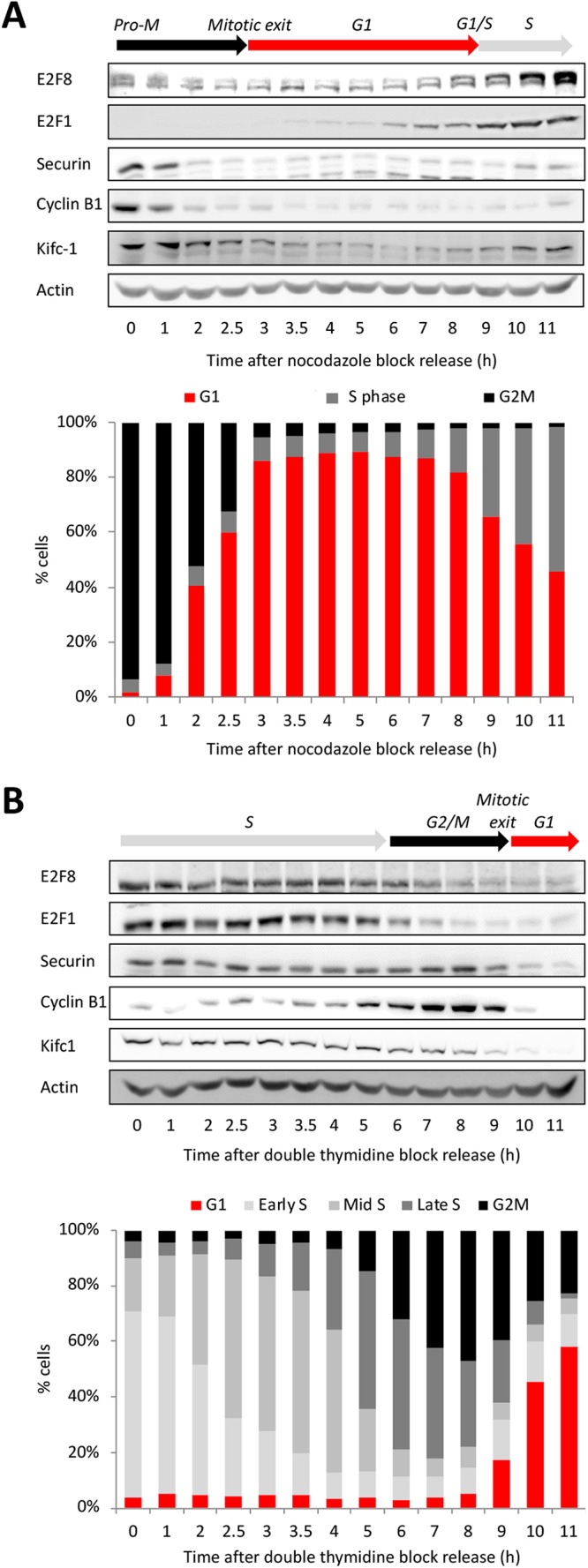
Temporal dynamics of E2F8 across the cell cycle. Western blot analyses of E2F8, E2F1, and canonical APC/C targets in synchronous S3 cells (DNA distributions are shown). Synchronization methods: release from thymidine-nocodazole block (A) and release from double thymidine block (B).
We previously showed that APC/CCdh1 drives E2F8 degradation in early G1 (Cohen et al., 2013). However, it is unclear how E2F8 levels increase in late G1 while APC/CCdh1 is still active. Finally, E2F8 levels decrease in G2-phase, prior to mitosis, through an entirely unknown mechanism (Figure 1, A and B).
Temporal proteolysis of E2F8 in “dynamic” mitotic extracts progressing from prometaphase to G1-phase
APC/CCdc20 is activated and triggers degradation of Securin and Cyclin B after the spindle checkpoint is satisfied in mitotic metaphase. Activation of APC/CCdh1 begins as cells enter the subsequent G1, initiating degradation of Cdc20, Tome-1, and other Cdh1-specific substrates (illustrated in Figure 2A). These are major milestones along mitotic progression and exit that can be monitored by the sequential onset of Securin and Tome-1 degradation at the metaphase-to-anaphase transition and G1 entry, respectively (Zou et al., 1999; Zur and Brandeis, 2001; Hagting et al., 2002; Ayad et al., 2003). Mitotic exit is also characterized by a dephosphorylation wave orchestrated by the inactivation of Cdk1 and activation of phosphatases during mitotic exit (Wurzenberger and Gerlich, 2011; Grallert et al., 2015; Powers and Hall, 2017).
FIGURE 2:

Temporal proteolysis of E2F8 in dynamic mitotic extracts progressing from prometaphase to G1. (A) Schematics of molecular milestones along mitosis. At prometaphase (Pro-Met) and metaphase (Met), the mitotic checkpoint complex (MCC) prevents from APC/CCdc20 to ubiquitinate Securin. MCC removal induces Securin and Cyclin B1 degradation and the metaphase-to-anaphase (Met-to-Ana) transition. The drop in Cyclin B1 levels induces the switch from APC/CCdc20 to APC/CCdh1 activity, the degradation of Tome-1 and other Cdh1-sepcific targets, and mitotic exit into G1. (B–E) Cell extracts were made from thymidine/nocodazole-arrested S3 cells. MCC removal and mitotic exit occur spontaneously in these extracts in a temperature-controlled manner or by adding recombinant UbcH10. The addition of dominant negative UbcH10 (UbcH10DN) blocks mitotic progression and exit (B). Temporal proteolysis and (de)phosphorylation-induced electrophoretic mobility shifts of Securin and Tome-1 are shown (B and C). At optimal reaction condition (UbcH10; 28°C), extracts eventually reach a G1-like state in which APC/CCdh1 is active and Tome-1 is degraded (C). Time-dependent degradations of Securin (B), Tome-1 (C), and E2F8 (D) (35S-labeled IVT products) were assayed by SDS–PAGE and autoradiography. Source data (B–D) and quantification (E) are shown (* marks a deformed band). Temporal proteolysis and electrophoretic mobility shifts of E2F8 and Tome-1 are highly similar.
Prometaphase extracts can be made from nocodazole-arrested HeLa S3 (S3) cells (Rape and Kirschner, 2004; Ayad et al., 2005). We found that this system could be dynamically controlled to recreate the Ub and phosphorylation signaling milestones that occur from prometaphase to G1-phase. The spindle checkpoint could be inactivated in prometaphase extracts through an increase in temperature, or more efficiently, by adding the APC/C E2 enzyme UbcH10. Conversely, dominant negative UbcH10 (UbcH10DN) blocks extracts in a prometaphase-like state. These features are manifested by the temporal proteolysis and Cdk1-induced electrophoretic mobility shift of the APC/CCdc20 substrate, Securin (Holt et al., 2008) (Figure 2B). Specifically, shifting temperature from 25°C to 28°C shortened the half-life of 35S-labeled Securin in extracts from ∼80 to ∼60 min. The addition of UbcH10 further decreased Securin half-life under both conditions (Figure 2B).
The APC/CCdh1 substrate Tome-1 also exhibited an electrophoretic mobility shift, consistent with its mitotic phosphorylation (Ayad et al., 2003; Pe’er et al., 2013). In contrast to Securin, Tome-1 was not immediately degraded under these conditions. Tome-1 remains stable and at a high electrophoretic mobility throughout the experiment (Figure 2C). Importantly, the addition of UbcH10 triggered orderly Tome-1 dephosphorylation followed by its degradation in a temperature-controlled manner. These dynamics were evident by the gradual shift of Tome-1 back to its basal electrophoretic mobility form and the subsequent reduction of the 35S signal, recapitulating the activation of APC/CCdh1 during mitotic exit and G1 entry. Thus, at optimal reaction conditions, human cell extracts can recapitulate the temporal cell cycle milestones of mitotic phosphorylation and dephosphorylation, APC/CCdc20 activation, and the subsequent shift to APC/CCdh1, representing a compete transition from prometaphase to a G1-like state in vitro.
Leveraging this system, we investigated the behavior of E2F8. Degradation of E2F8 was evident only when mitotic exit was accelerated by UbcH10. Importantly, E2F8 destruction commenced considerably after Securin disappearance, and was nearly identical to that of Tome-1. These results suggest that E2F8 degradation is coordinated with mitotic exit and activation of APC/CCdh1, but not APC/CCdc20 (Figure 2, C–E). Since the electrophoretic mobility display of E2F8 and Tome-1 shared a remarkable resemblance, this also suggested an unappreciated role for orderly E2F8 (de)phosphorylation during mitotic progression and exit.
E2F8 dynamics in APC/C Cdc20-active extracts
Cdc20 versus Cdh1 specificity of APC/C targets is a key element in the overall mechanism underlying orderly cell division in all eukaryotes (Kernan et al., 2018). The kinetics of E2F8 degradation observed above (Figure 2D) are consistent with regulation by APC/CCdh1, despite prior in vivo findings suggesting destruction by APC/CCdc20 (Boekhout et al., 2016). To address this discrepancy, we developed a mitotic cell-free system with stable APC/CCdc20 activity. Cdk1 activity associated with early mitosis prevents Cdh1 from interacting with APC/C (Zachariae et al., 1998; Jaspersen et al., 1999; Kramer et al., 2000; Listovsky et al., 2000). We derived extracts from mitotic 293-T-REx cells where Cdk1 is constitutively active, due to tetracycline (tet)-induced expression of a nondegradable allele of Cyclin B1 (hereafter referred to as NDB cells/system) (Figure 3A). Nondegradable Cyclin B1 expression blocks cells in an anaphase-like state, that is, post–spindle assembly checkpoint inactivation (Wheatley et al., 1997; Zur and Brandeis, 2001, 2002; Pe’er et al., 2013). In the absence of tet, the selected NDB colony tolerates the basal expression of nondegradable Cyclin B1, as evidenced by the normal cell cycle profile and cell size range (Figure 3, B and C). Following treatment with tet, virtually all cells arrest in mitosis, displaying a typical round shape and separated sister chromatids (Figure 3, D–G).
FIGURE 3:

E2F8 dynamics in APC/CCdc20-active extracts. (A) Human Cyclin B1 in which Arg-42 and Leu-45 are substituted with Gly and Val is nondegradable (ND). A colony of 293-T-REx cells stably expressing ND-Cyclin B1 (NDB) under tet-regulated CMV promoter was generated (i.e., NDB cells). (B) DNA distribution of asynchronous NDB cell population. (C) Median cell size (fL: femtoliter) of NDB cells vs. parental 293-T-REx cells. (D) DNA distribution of NDB cells following 22 h treatment with tet. Cell cycle phase distributions of NDB cells pre- and postinduction with tet are shown. (E) A panel of light phase images of NDB cells pre- and postincubation with nocodazole (Noc) or tet. (F) Noc- or tet-treated NDB cells were harvested for chromosome spreads. Representative fluorescent images of DAPI stained chromosomes are shown. (G) An image series of live NDB cells stably expressing histone-H2B following tet treatment. A higher resolution image of a live, tet-treated NDB cell with typical disorganized chromatids is shown on the right. (H) Noc- or tet-treated NDB cells were harvested for Western blotting. (I) NDB cells showing the lowest 10% forward scatter width (FSC-W) signal were sorted. These G1 cells (Vecsler et al., 2013), as well as tet-treated NDB cells, were harvested for Western blotting with the depicted antibodies. (J) Mitotic cell extracts were generated from tet-induced NDB cells. Degradation and electrophoretic mobility-shift of 35S-labeled Securin, Tome-1, and E2F8 (IVT products) were assayed in mitotic NDB extracts supplemented with mock, UbcH10DN, or APC/C inhibitor Emi1 (C-terminal fragment; recombinant). (M–P) Tet-induced NDB cells/extracts can exit mitosis into a G1-like state by blocking Cdk1 activity. (K, L) DNA distributions, light phase images (M), and immunoblot (IB) (N) of tet-induced NDB cells pre- and postincubation with RO-3306 (135 min). The mitotic vs. G1 electrophoretic mobility-shift of Cdc27 is shown. (M) Mitotic extracts were generated from tet-induced NDB cells. Coimmunoprecipitation (IP) of Cdc27 was performed pre- and posttreatment with RO-3306 (30 min). (N) Time-dependent dynamics of endogenous Cdc20 in mitotic NDB extracts following incubation with DMSO, RO-3306 and/or UbcH10. Cdc20 levels were detected by Western blotting. Loading control (Tubulin) is also shown. (O) Time-dependent degradation of Tome-1 and E2F8 (35S-labeled IVT products) in NDB mitotic extracts preactivated with RO-3306 (15 min). Reactions were mock treated or supplemented with UbcH10, UbcH10DN, or MG132.
Importantly, APC/CCdc20, but not APC/CCdh1, is active in tet-treated NBD cells. Consequently, Geminin, an APC/CCdc20 substrate (McGarry and Kirschner, 1998), but not Cdc20, an APC/CCdh1 substrate (Pfleger and Kirschner, 2000), is reduced in tet-induced NDB cells relative to prometaphase arrested mitotic cells (Figure 3H). A reduction in Cdc20 could be observed only in G1 cells (Figure 3I). Consistent with the expression of nondegradable Cyclin B1, mitotic extracts derived from tet-induced NDB cells exhibit high Cdk1 activity, highlighted by electrophoretic mobility shifts of Securin and Tome-1 (Figure 3J). Moreover, APC/CCdc20 is highly active, evidenced by the degradation of Securin but not Tome-1 (Figure 3J). As opposed to “dynamic” prometaphase extracts made from nocodazole-arrested cells (Figure 2), NDB mitotic extracts are “static,” trapped in an anaphase-like state due to high Cdk1 activity. E2F8 was shifted to a high electrophoretic mobility form following incubation in NDB mitotic extract, further supporting the idea that E2F8 is phosphorylated by mitotic kinases. Importantly, unlike Securin, E2F8 levels remained nearly unchanged in NDB extracts (Figure 3J). To rule out the possibility that the stability of E2F8 and Tome-1 in extracts derived from HEK293 is cell line specific, we inactivated Cdk1 using the small-molecule inhibitor RO-3306. This treatment successfully overrides the mitotic arrest induced by nondegradable Cyclin B1, resulting in mitotic exit, evidenced by cell morphology, DNA profiling, and the canonical dephosphorylated state of the APC/C subunit Cdc27 (Kramer et al., 2000) (Figure 3, K and L). Furthermore, Cdk1 inhibition in NDB mitotic extracts causes APC/C to dissociate from Cdc20, bind Cdh1, and degrade endogenous Cdc20 (an APC/CCdh1 substrate), recapitulating the canonical APC/CCdc20-to-APC/CCdh1 switch associated with mitotic exit (Figure 3, M and N). Neither Tome-1 nor E2F8 was mobility shifted in Cdk1-inhibited extracts (Figure 3O). Most importantly, both proteins were degraded in RO–3306-treated NDB cell extracts where APC/CCdh1 has been activated. This degradation was blocked by dominant negative UbcH10 and is thus APC/C dependent (Figure 3O). Altogether, results in Figures 2 and 3 demonstrate that E2F8 proteolysis is coordinated with mitotic exit and APC/CCdh1 activation.
E2F8 ubiquitination in G1 is primarily Lys11 linked
Cell extracts generated from synchronous S3 cells 3–3.5 h after release from a nocodazole block exhibit homogenous and optimal APC/CCdh1 activity. These cell extracts, defined here as “G1 extracts,” have been used for the discovery of E2F8, E2F7, and other cell cycle proteins as APC/CCdh1 targets (Cohen et al., 2013; Pe’er et al., 2013; Singh et al., 2014). While Lys(K)48-linked ubiquitination is considered the main signal for proteasomal protein degradation (Kravtsova-Ivantsiv et al., 2013), APC/C preference for K11-linked Ub chains has been demonstrated for an increasing number of substrates, including Securin and Kifc1 (Jin et al., 2008; Wu et al., 2010; Noach-Hirsh et al., 2015). However, it remains unclear if APC/C uses K11-linked chains to mediate the degradation of all substrates. To test whether this feature applies to E2F8, we utilized a designated microfluidic platform with which ubiquitination of freshly expressed proteins can be tested in G1 extract (Noach-Hirsh et al., 2015). The assay is based on in situ detection of EGFP-tagged substrates (Supplemental Figure S2) and rhodamine-labeled Ub (Rd-Ub) (illustrated in Figure 4, A and B). Once ubiquitination of E2F8-EGFP was validated on-chip to be APC/C specific (Figure 4C), we tested the displacement of Rd-Ub from E2F8 by 10-fold excess of unlabeled K-to-Arg(R) mutant Ub variants. As expected, excess of unlabeled WT Ub in the reaction mix outcompeted Rd-Ub, evidenced by the sharp drop of net Rd signal. Similar results were obtained when K48R- or K63R-Ub mutants (UbK48R, UbK63R) were added. In contrast, the impact of K11R-Ub mutant (UbK11R) on Rd-Ub signal was minimal (Figure 4D), reflecting the low capacity of this particular mutant to form Ub chains on E2F8. Consistent with APC/C forming K11-linked Ub chains on E2F8, E2F8 proteolysis in G1 extracts is inefficient when UbK11R is supplemented to the reaction (Figure 4E). Supplementing extracts with UbK48R or UbK63R had no effect on E2F8 degradation. Thus, E2F8 ubiquitination and degradation at G1 are primarily mediated by K11-linked Ub chains.
FIGURE 4:

Ubiquitination of E2F8 by APC/CCdh1 is primarily via K11-linked Ub chains. (A) Image of an integrated microfluidic platform comprising microcompartments isolated by pneumatic valves. (B) Each microcompartment has two chambers. Fresh E2F8-EGFP IVT product was applied to the chip and immobilized to the “Protein chamber” via anti-GFP antibodies (Abs) and a designated surface chemistry (i). Next, G1 extracts supplemented with Rd-Ub were applied to the second chamber (ii). The opening of the valve allows reaction mix to diffuse into protein chambers, enabling ubiquitination of the immobilized substrate (iii). After 10 min incubation, protein chambers are washed (iv). Rd-Ub moieties attached to E2F8-EGFP at the protein chamber are quantified by a fluorescence imaging. Rd-Ub signal in each protein chamber is normalized to E2F8-EGFP levels, i.e., “Protein signal” (v). (C) APC/CCdh1-mediated ubiquitination of E2F8 on-chip. E2F8-EGFP was expressed in reticulocyte lysate, deposited on the chip surface, and incubated with G1 extracts supplemented with mock, UbcH10DN, or Emi1. Normalized Rd-Ub signals were calculated from 20 microcompartments (mean [X], median [–], and four quantiles (box and whiskers) are indicated; *p < 0.001). Array sections showing “raw” Rd-Ub signals of six microreactions for each of the three conditions are shown (red dots). A representative image of immobilized E2F8-EGFP is also shown (green dots). (D) Ubiquitination of E2F8-EGFP was assayed in the presence of G1 extracts, Rd-Ub, and excess of unlabeled WT or mutant Ub in which Lys 11 (UbK11R), Lys 48 (UbK48R), or Lys 63 (UbK63R) was substituted with Arg. Plots average 18 microreactions. Array sections showing raw Rd-Ub signals are depicted. (E) Degradation of 35S-labeled E2F8 (IVT product) was assayed in G1 extracts supplemented with WT or mutant Ub. Time-dependent degradation was assayed by SDS–PAGE and autoradiography. Mean and SE values are plotted (n = 3). 35S-E2F8 signals are normalized to t = 0. A set of source data is shown.
Multiple degron motifs coordinate E2F8 proteolysis in G1
Direct assays in G1 extracts have been proven informative in mapping and characterizing destruction motifs of APC/C substrates (Jin et al., 2008; Wu et al., 2010; Pe’er et al., 2013; Singh et al., 2014). E2F8 carries multiple motifs potentially associated with APC/C-mediated degradation—three KEN sequences and two D-box (R-X-X-L) sequences (Figure 5A). We generated point mutations in each of these motifs (Figure 5B) and quantified the kinetics of E2F8 proteolysis in G1 extracts. Mutation of the second D-box motif, RXXL-to-GXXV mutation at position 183 (DM2) inhibited E2F8 degradation (Figure 5C). Mutations in each of the previously reported KEN boxes at positions 5 and 375 (KM1 and KM2) (Boekhout et al., 2016) had a minimal impact on E2F8 proteolysis. Remarkably, the two KEN box mutations (KM1 and KM2) had a significant impact on E2F8 degradation when combined with DM1 or with each other (Figure 5D). Mutations in RXXL 87 (DM1) and KEN 637 (KM3) had no inhibitory effect (Figure 5, C and D). The effect of KM1 and KM3 was further assayed in the context of short N- and C-terminal fragments (E2F8-N80 and E2F8-C; Figure 5E), reasoning that this approach could highlight the potential potency of single elements in driving E2F8 degradation. The E2F8 C-terminus was stable, consistent with this region not regulating proteolysis. Instead, degradation of E2F8-N80 was efficient and KEN specific (Figure 5F). Overall, we conclude that DM2, KM1, and KM2 but not DM1 and KM3 mediate E2F8 degradation by APC/CCdh1. This result also highlights the ability of multiple degrons to contribute cooperatively to substrate proteolysis, perhaps through multivalent APC/C binding (Watson et al., 2019).
FIGURE 5:

Multiple functional motifs coordinate E2F8 proteolysis in G1. (A) Schematics of human E2F8. KEN, and RXXL motifs are shown alongside the conserved DNA-binding (RRXYD) and dimerization (DD1, DD2) domains. (B) List of E2F8 mutant variants generated by site-directed mutagenesis. Amino acid substitutions are indicated for each of the five KEN/RXXL motifs. KM1/2/3: KEN-box mutant 1/2/3; DM1/2: Destruction-box mutant 1/2. (C) Degradation of 35S-labeled E2F8 variants (IVT products) was tested in G1 extracts supplemented with UbcH10 or UbcH10DN. Time-dependent degradation was assayed by SDS–PAGE and autoradiography. Representative raw data and quantifications are shown. Mean E2F8 levels (35S signals) normalized to max signal at t = 0 are shown (n = 3–4). Bars represent SE. (D) E2F8 double mutants were analyzed as described in C. (E) Schematics of N- and C-terminal fragments of E2F8 (E2F8-N80/C) carrying a single KEN motif. (F) Time-dependent degradation of E2F8 fragments (see details in C).
E2F8 proteolysis in G1 is mediated by N-terminal Cdk1 sites
The temporal electrophoretic mobility of E2F8 in mitotic extracts (Figures 2 and 3) can be explained by orderly phosphorylation and dephosphorylation during mitotic progression and exit. There are four T/SP sites in E2F8-N80 fragment, two of which are TPxK, that is, the canonical Cdk1 consensus sites (Figure 6A). Both full-length E2F8 and N80-E2F8 fragments were stable and mobility shifted in mitotic NDB extracts (Figure 6B). These mobility shifts were blocked by Cdk1 inhibitor. Individual Thr(T)-to-Ala(A) mutations in position 20 or 44 reduced the mobility shift of E2F8-N80 and to a greater extent when combined (Figure 6C). We concluded that Cdk1/Cyclin B1 phosphorylates E2F8 in mitosis at positions T20 and T44. Phosphorylation in proximity to destruction motifs can regulate APC/C-mediated ubiquitination (Holt et al., 2008, Singh et al., 2014). This mechanism helps to coordinate degradation with the cell cycle clock (Holt et al., 2008). We, therefore, tested the potential link between E2F8 phosphorylation and degradation. The E2F8-N80 fragment (Figure 6C) allowed us to focus on the potential relationship between T20/T44 phosphorylation and KEN box at position 5. Alanine mutations at T20 and/or T44 had no impact on the degradation of E2F8-N80 in G1 extracts (Figure 6D). In contrast, phosphomimetic mutation (T-to Asp(D)) at T44 partially impeded E2F8-N80 degradation. This inhibitory effect was vastly increased when both T20 and T44 were substituted with D (Figure 6D). Most importantly, T20D/T44D mutation strongly impaired the proteolysis of full-length E2F8 in G1 extracts (Figure 6C). In fact, the impact of T20D/T44D mutation on E2F8 half-life in G1 extracts was greater than any of the single KEN or RXXL mutations (Figure 5C). Altogether, our findings couple E2F8 ubiquitination by APC/CCdh1 with an unphosphorylated state of T20 and T44. This molecular switch can restrict E2F8 degradation by APC/C to times of low Cdk1 activity.
FIGURE 6:
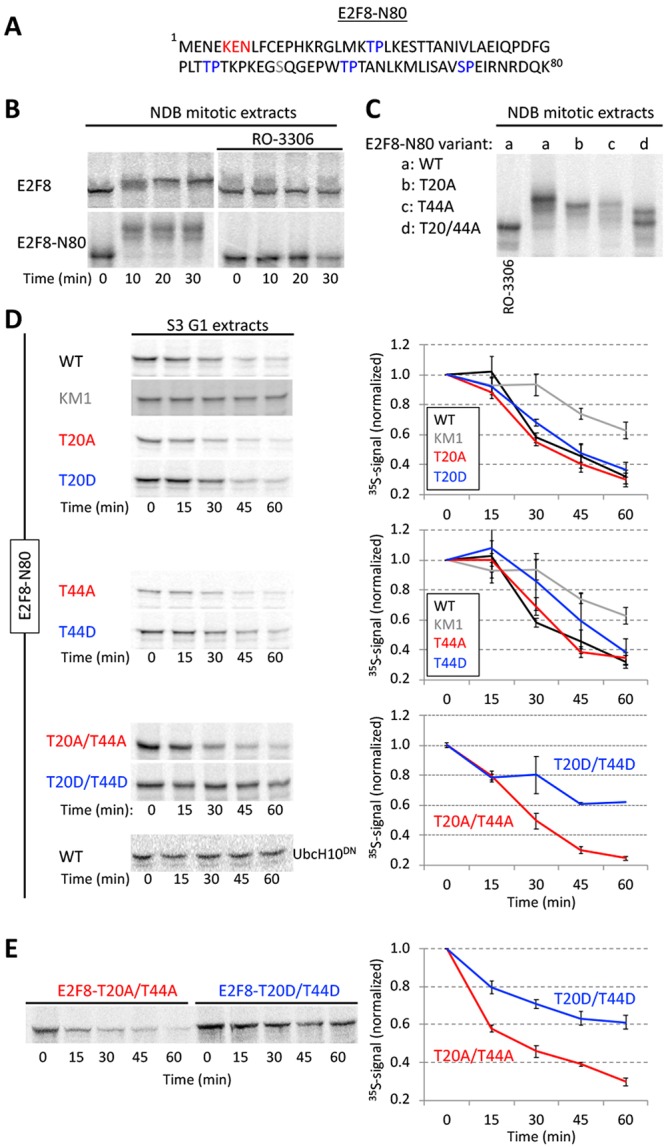
Phosphomimetic Cdk1 sites stabilized E2F8 in G1 extracts. (A) E2F8 N-terminal fragment of 80 amino acids (E2F8-N80). KEN box and four canonical Cdk1 consensus phosphorylation sites are colored. (B) Time-dependent electrophoretic mobility shift of full length- and E2F8-N80 (35S-labeled IVT products) in NDB mitotic extracts supplemented with mock or the Cdk1 inhibitor RO-3306. (C) Thr (T)20 and/or T44 of E2F8-N80 were substituted with Ala (A). Mobility shifts of WT vs. mutant E2F8-N80 variants are shown. (D) Time-dependent degradation in G1 extracts of E2F8-N80 and the following variants: KEN box mutant (KM1), single/double phosphomimetic mutants (T-to-Asp [D]), and single/double phospho-dead mutants (T-to-A). (E) Time-dependent degradation of full-length E2F8 carrying double phosphomimetic or double phospho-dead mutations. (B–E) Protein degradations and electrophoretic mobility-shifts were assayed by SDS–PAGE and autoradiography. A set of source data is shown. Mean and SE calculated from three degradation assays are plotted.
Temporal proteolysis of E2F8 across the cell cycle
The contribution of temporal proteolysis to the overall dynamics of E2F8 across the cell cycle (Figure 1) has yet to be addressed systematically. To this end, seven cell-free systems were generated, recapitulating cell cycle milestones from prometaphase to late S-phase. Degradation of E2F8 was assayed alongside Securin and p27/Kip1. The latter was used as a positive control for SCFSkp2 activity (Carrano et al., 1999). The stability of E2F8 and delayed proteolysis of Securin in prometaphase extracts (Pro-M) were as expected (Figures 7A and 2, B and D). The stability of E2F8 in G1-S extracts and in all the three S-phase extracts was in agreement with the temporal dynamics of the protein at these stages (Figures 7A and 1). The p27 proteolysis validated the specific activity of mid- and late S-phase extracts. Proteolysis of both E2F8 and Securin in early-mid G1 extracts was optimal, reflecting the high APC/CCdh1 activity and lowest abundance of E2F8 associated with this stage (Figure 7A). The degradation of Securin was slightly more efficient than E2F8 (Figure 7, A and B), potentially reflecting the high ubiquitination efficiency of the former protein (Rape et al., 2006). More importantly, APC/C activity has been shown to weaken during late G1 (Huang, Park et al., 2001; Rape and Kirschner, 2004). Accordingly, the half-life of Securin was indeed slightly longer in late G1 extracts, prepared 6 h after the nocodazole release. However, while the overall degradation of Securin remained potent in late G1 extracts, E2F8 proteolysis nearly ceased (Figure 7, A and B). The differential stability of E2F8 versus Securin in late G1 extracts effectively demonstrates an intrinsic mechanism by which E2F8 can accumulate while APC/C is still active, providing a mechanism for E2F8 buildup during late G1 (Figure 1A).
FIGURE 7:

Temporal proteolysis of E2F8 across the cell cycle. (A) Degradations of 35S-labeled E2F8, Securin, and p27 (IVT products) were tested in seven cell extracts generated from synchronous S3 cells at seven points across the cell cycle (R.F.Noc: release from a thymidine-nocodazole block; R.F.DTB: release from a double thymidine block). DNA distributions are shown. Extracts were supplemented only with Ub and energy-regeneration mix. Time-dependent degradation was assayed by SDS–PAGE and autoradiography. A set of source data and quantifications is shown. (B) Average, SE, and polynomial fit (dotted line) calculated from three degradation assays of E2F8 and Securin in early-mid- and late-G1 extracts. 35S signals were normalized to t0.
E2F8 is regulated by the F-box protein Cyclin F
The reduction in E2F8 levels seen premitosis (Figure 1) is likely to be regulated at both transcriptional (E2F1) and posttranslational levels. The SCF-family of Ub ligases represent attractive candidates for mediating E2F8 destruction owing to their key roles in the cell cycle (Silverman et al., 2012). SCF ligases utilize substrate adaptor F-box proteins that bind specific substrates and mediate their ubiquitination and degradation. SCFSkp2, SCFβ-TrCP, and SCFCyclin F have all been implicated in cell cycle and are candidates for driving E2F8 down-regulation premitosis. Among those three, several pieces of evidence nominate Cyclin F as the most likely candidate. First, SCFSkp2 is active when E2F8 levels peaks, and E2F8 is stable in S-phase extracts that recapitulate p27 degradation, a well-established SCFSkp2 substrate (Figure 7A). Second, SCFβ-TrCP binds to a well-defined recognition sequence (DSGXX(XX)S) that is absent in E2F8 (Yaron et al., 1998; Orian et al., 2000). Third, Cyclin F peaks at G2 phase (D’Angiolella et al., 2013; Choudhury et al., 2016; Galper et al., 2017; Mavrommati et al., 2018), concomitantly with E2F8 down-regulation (Figure 1). And fourth, E2F8 carries putative RxL motifs (Cy box) (Figure 8A) that are known to mediate ubiquitination by SCFCyclin F (D’Angiolella et al., 2010). There is no in vitro extract assay for Cyclin F-mediated ubiquitination. Therefore, we examined the functional relationship between E2F8 and Cyclin F in vivo.
FIGURE 8:

Cyclin F mediates the degradation of E2F8 in G2-phase. (A) Schematic depiction of the RxL motifs in E2F8. (B) The abundance of E2F8 in control Cyclin F KO HeLa cells was analyzed by Western blot. (C) HEK293T cells were transiently transfected with HIS-E2F8-HA with and without FLAG-Cyclin F. Cells were analyzed by Western blot 48 h posttransfection. SE, short exposure; LE, long exposure. (D) MCF7 and T47D were engineered to express doxycycline (Dox) inducible Cyclin F. Cells were treated with doxycycline at increasing concentrations for 48 h and then endogenous E2F8 was analyzed by Western blot. (E) HEK293T cells were transiently transfected with FLAG-Cyclin F or its ΔF-box variant and analyzed by Western blot 48 h posttransfection. (F) MCF7 cells were treated with doxycycline to induce expression of Cyclin F. Eight hours prior to harvesting for Western blot, cells were treated with either of two proteasome inhibitors, MG132 and bortezomib. Endogenous E2F8 was analyzed by Western blot. (G) Control and Cyclin F KO HeLa cells were synchronized at the G1-S boundary by a double thymidine block. Following release from the second thymidine block, samples were collected for Western blot analysis at the indicated time points. (H) HEK293T cells were transfect with wild-type or mutant versions of HIS-E2F8-HA harboring alanine substitutions at the indicated RxL motifs shown in A. Their response to ectopic coexpression was analyzed by Western blot 48 h after transfection. (I, J) HIS-E2F8-HA and FLAG-Cyclin F were cotransfected into HEK293T cells. Cell lysates were subjected to co-IP with either anti-HA or anti-FLAG antibodies.
Endogenous E2F8 levels increased in cells treated with MLN4924, an indirect inhibitor of SCF. As expected, E2F8 levels also rose upon inhibition of ubiquitination (MLN7243) and proteasomal (Bortezomib) but not lysosomal (Chloroquine) degradation (Supplemental Figure S3A). These results, however, may simply reflect drug-induced enrichment of cells at phases where E2F8 levels are relatively high (Supplemental Figure S3B).
E2F8 levels were elevated in cells where Cyclin F has been knocked out (KO) using CRISPR-Cas9 (Figure 8B). In addition, Cyclin F overexpression reduced the levels of ectopically expressed E2F8, suggesting that Cyclin F promotes E2F8 degradation (Figure 8C). To analyze endogenous E2F8, we constructed cells with doxycycline-inducible Cyclin F expression in MCF7 and T47D cell lines. In both, Cyclin F expression caused a dose-dependent decrease in E2F8 abundance (Figure 8D). Furthermore, down-regulation of E2F8 was less profound in HEK293 cells overexpressing Cyclin F variant lacking the F-box domain (Figure 8E) that is critical for Cyclin F assembly into productive SCF complexes (Choudhury et al., 2016). Together, these results suggests that Cyclin F regulates E2F8 degradation via Ub-mediated proteolysis. Accordingly, treatment with proteasome inhibitors MG132 and bortezomib prevented the degradation of endogenous E2F8 in MCF7 cells expressing doxycycline-inducible Cyclin F (Figure 8F). We conclude that Cyclin F promotes degradation of E2F8 through the proteasome.
Next, we examined E2F8 abundance in control and Cyclin F KO cells traversing G2. Cells were released from synchronization at G1/S by double thymidine block and analyzed by immunoblotting. A reduction in E2F8 was evident in late S and G2-phase, prior to mitotic entry, in control cells (Figure 8G). However, E2F8 levels persisted into mitosis in Cyclin F KO cells, strongly suggesting that the degradation of E2F8 observed in G2 is Cyclin F dependent. Owing to the role that RxL motifs often play in SCFCyclin F-mediated ubiquitination, we analyzed E2F8 variants in which the Arg of each of the four RxL motifs was substituted with Ala (RxL-to-AxL). Similar to the result above, Cyclin F overexpression strongly down-regulated the abundance of wild type, exogenously expressed E2F8. Mutation of R313 resulted in limited sensitivity to Cyclin F overexpression, whereas the three other E2F8 alleles (R15A, R81A, and R587A) behaved similar to the wild-type protein (Figure 8H). It is noteworthy, however, that the expression of R313 mutant was consistently lower compared with all other mutated variants (Figure 8H). RRL 313 is within the E2F8 DNA-binding domain (Figure 5A). Thus, the impact of the R313A mutation on E2F8 dynamics may simply reflect a global structural failure. In spite of this potential caveat, E2F8 carrying mutation in both DNA-binding domains was degraded by APC/CCdh1 (Supplemental Figure S4).
We next expressed FLAG-Cyclin F and 6HIS-E2F8-HA in HEK293T cells and analyzed their interaction by co-IP. We detected an interaction between the two proteins, irrespective of which protein was precipitated (Figure 8, I and J), suggesting that E2F8 binds Cyclin F. Taken together, these data suggest that E2F8 regulation by Cyclin F is direct and that SCFCyclin F activity down-regulates E2F8 prior to M-phase entry. These data (see also Wasserman et al., 2019) complement three parallel studies connecting SCFCyclin F-mediated proteolysis to temporal dynamics of E2F members (Burdova et al., 2019; Clijsters et al., 2019; Yuan et al., 2019).
DISCUSSION
In this study, we developed and utilized human cell-free systems that recapitulate the phosphorylation and Ub signaling events that underlie the core of the cell cycle oscillator. We discovered multiple mechanisms controlling temporal proteolysis of E2F8 in proliferating cells, with relevance to the overall coregulation among E2F8, E2F1, and the cell cycle clock.
The dynamic mitotic extracts developed and described here revealed orderly phosphorylation, dephosphorylation, and proteolysis of E2F8 while traversing spindle checkpoint inactivation, APC/CCdc20 activation, the APC/CCdc20-to-APC/CCdh1 switch, and G1 entry (Figure 2). To the best of our knowledge, a complete transition of cell extracts from prometaphase to G1 has not been demonstrated in somatic cell systems. A complementary system generated from anaphase-like cells (NDB) allowed us to investigate E2F8 under conditions of high Cdk1 and APC/CCdc20 activity (Figure 3). Using these two cell-free systems, we identified T20 and T44 in E2F8 as Cdk1 phosphorylation sites (Figure 6) and coupled temporal proteolysis of E2F8 with mitotic exit dephosphorylation and APC/CCdh1 activation (Figure 2). In contrast to our initial expectation, early mitotic E2F8, although low leveled (Figure 1), did not exhibit a short half-life (Figure 3). This pattern of temporal proteolysis is difficult to detect in vivo for a protein that is degraded during both G2 and G1 (Figures 1, 7, and 8). While Boekhout et al. (2016) provided in vivo evidence in support of our discovery that E2F8 is an APC/CCdh1 target (Cohen et al., 2013), they concluded that E2F8 proteolysis is mediated by APC/CCdc20. Their conclusion relied on in vivo perturbation of Cdc20, which severely perturbs cell cycle progression by forcing mitotic arrest, complicating their interpretation, particularly in light of these new data.
Using G1 extracts, we found E2F8 ubiquitination to be primarily K11 linked (Figure 4), mapped three destruction motifs in E2F8, and ranked their individual and cooperative contribution to APC/CCdh1-mediated proteolysis (Figure 5). Importantly, we discovered that phosphomimetic E2F8 is stable in G1 (Figure 6). This finding, together with the dephosphorylation dynamics revealed prior to E2F8 proteolysis during mitotic exit (Figure 2), suggests that dephosphorylation of E2F8 facilitates recognition and degradation by APC/CCdh1. That S/T20 and T44 Cdk1 phosphorylation sites are conserved in atypical E2Fs across vertebrates further suggests that this molecular switch likely applies to E2F7. Experiments in G1 extracts also illuminated an unexpected role for dimerization domains in E2F8 proteolysis (Supplemental Figure S4). Whether these results genuinely couple E2F8 dimerization with degradation or, alternatively, mirror a global structural change with implications on folding, function, and regulation of E2F8, awaits further investigation (see the Supplemental Information for more details).
The half-life of E2F8 in G1 was analyzed in two distinct points (Figure 7). In early-mid G1 extracts, E2F8 proteolysis was optimal. In late G1, however, E2F8 proteolysis nearly ceased while the overall degradation of Securin stayed potent (Figure 7B). These dynamics demonstrate that E2F8, unlike Securin and probably other APC/C substrates, is differentially stabilized during late G1, concomitantly with the declining activity of APC/C (Rape and Kirschner, 2004; Meyer and Rape, 2011). Because E2F1 is already active at this stage, this feature, by itself, allows E2F8 to accumulate and potentially coregulate S-phase entry while APC/CCdh1 is still active (Figure 1). Enhanced stability under suboptimal APC/C activity is a feature of “distributive” APC/CCdh1 targets, that is, substrates that must associate with the APC/C multiple times to obtain a proteolytic Ub chain, for example, Cyclin A (Rape et al., 2006). Consequently, distributive substrates are the first to become stable under limited APC/C activity because of 1) the higher chance of deubiquitinating isopeptidases to strip the emerging Ub chains and 2) the competition with “processive” substrates like Securin, which undergoes multiubiquitination in a single binding event (Meyer and Rape, 2011). Interestingly, both E2F8 and Cyclin A are E2F1 targets involved in the G1-S transition. The early rise of E2F1 in G1 (Figure 1) is hard to reconcile with previous in vivo studies, which suggested that E2F1 is an APC/CCdh1 target (Budhavarapu et al., 2012). We further note that E2F1, unlike E2F8 and E2F7, is stable in G1 extracts (Supplemental Figure S1).
E2F1 and E2F8 levels diminish before prometaphase (Figure 1). This temporal dynamic is rare among APC/C targets (Meyer and Rape, 2011). While SCFCyclin F-mediated proteolysis accounts for E2F1 reduction during late S and G2 phases (Clijsters et al., 2019), how atypical E2Fs are regulated in G2 was unknown. Here, we show that Cyclin F also down-regulates E2F8 in a proteasome-dependent manner (Figure 8), implying a direct link between E2F8 and SCFCyclin F-mediated ubiquitination. The elevated levels of E2F8 observed in Cyclin F KO cells during G2, where Cyclin F protein peaks, emphasize the role of SCFCyclin F in down-regulating E2F8 premitosis. Whereas in vivo ubiquitination assays of Cyclin F substrates are possible (Choudhury et al., 2016) and proven informative for E2F1 (Clijsters et al., 2019), equivalent assays with E2F8 were ambiguous and uninformative (unpublished data; Yuan et al., 2019). This could reflect an unappreciated difficulty to assay proteins with dual modes of degradation (Choudhury et al., 2016). This conception highlights another layer of complexity; E2F7 and E2F8 are both direct targets of E2F1. E2F1, as well as E2F2 and E2F3, is an SCFCyclin F substrate. This strongly argues against straightforward interpretations of E2F7/8-related phenotypes in Cyclin F KO/knockdown cells (Yuan et al., 2019).
Altogether, our findings support a model, depicted in Figure 9, wherein SCFCyclin F down-regulates E2F8 in G2, following E2F1 reduction. Since Cyclin F is degraded in mitosis, low-leveled E2F8 remains stable during early mitosis when APC/CCdc20 is active. Conceptually, this mechanism can allow E2F8 to block residual, unwanted E2F1 activity during mitosis, but before cell division is successfully completed, and is consistent with overactivation of E2F1 having negative consequences on chromosome segregation fidelity (Manning et al., 2010; Pfister et al., 2018). Cdk1 phosphorylates E2F8 on Thr-20 and Thr-44 during mitotic entry. This module has a stabilizing effect on E2F8. Cdk1 activity dropped during mitotic exit. Subsequent dephosphorylation of both E2F8 and Cdh1 triggers E2F8 degradation by APC/CCdh1. E2F1 accumulates in early-mid-G1 under high APC/CCdh1 activity, inducing its own expression alongside expression of E2F8, E2F7 and other target genes that promote S-phase entry (e.g., Cdc6, Cyclin E). As long as E2F7 and E2F8 are destabilized by APC/CCdh1, E2F1 activity is unrestrained. Maximizing transcription capacity at this stage might be critical for low-leveled E2F1 to ignite the positive feedback required for its autocatalytic increase that drives cells into S-phase (Johnson et al., 1994). E2F8 is differentially stabilized under limited APC/CCdh1 activity associated with late G1. This feature allows E2F8 to accumulate during late G1, while APC/CCdh1 is still active and mediates proteolysis of Securin and probably other substrates. Because E2F1 levels are already high, the E2F1-E2F8 negative feedback can be formed already in G1 to balance the transcriptional activity of E2F1. Overall, these interdynamics ensure a safe transition into S-phase (Figure 9). In this study, we focused on E2F8. However, owing to the overall similarity of atypical E2Fs, it is easy to speculate that the model herein applied to E2F7 as well.
FIGURE 9:

Multiple mechanisms coordinate the dynamics of E2F8 in cycling mammalian cells. At the transcriptional level, E2F8 is primarily regulated by E2F1 via a negative feedback mechanism. Posttranslationally, E2F8 is controlled by temporal proteolysis orchestrated by multiple pathways. E2F8 peaks in S-phase. During G2-phase, E2F8 protein is down-regulated by SCFCyclin F activity. Although low-leveled, E2F8 proteolysis during early mitosis, while APC/CCdc20 is active, is inefficient. E2F8 is phosphorylated in mitosis by Cdk1. This phosphorylation has a stabilizing effect on the protein. During mitotic exit, Cdk1 is inactivated and both E2F8 and Cdh1 are dephosphorylated. This dual molecular switch initiates both the assembly of APC/CCdh1 and its ability to ubiquitinate E2F8. The levels of E2F8 remain minimal through G1 as long as APC/CCdh1 is fully active. During late G1, APC/CCdh1 activity weakens by an autonomous mechanism. The enhanced sensitivity of E2F8 to suboptimal APC/CCdh1 activity effectively stabilizes the protein while Securin and perhaps other APC/C targets are still degraded. Because E2F1 is already present, the negative feedback circuitry between E2F1 and E2F8 can be formed already in late G1 in ensuring a safe transition into S-phase.
MATERIALS AND METHODS
Plasmids
The following plasmids were used in previous studies: pCS2-FA-E2F8 and pCS2-FA-E2F7 (Cohen et al., 2013); pCS2-FA-Securin (Pe’er et al., 2013); pCS2-Tome-1 (Xenopus laevis) (Ayad et al., 2003); FLAG-tagged WT- and ΔF-Box-mutant Cyclin F (Choudhury et al., 2016). Histone H2B-YFP plasmid was a gift from Yaron Shav-Tal (Bar-Ilan University). The following plasmids were generated for this study: 1) pCS2-FA-E2F8-EGFP; E2F8 open reading frame (ORF) was amplified by PCR using pCS2-FA-E2F8 as a template and primers flanked with FseI (forward) and AgeI (reverse) restriction enzyme (RE) sites and cloned into pCS2-FA-EGFP vector; 2) pCS2-FA-P27; ORF of p27 was amplified from cDNA template (Open Biosystems) and cloned into pCS2-FA vector using FseI (5′) and AscI (3′) RE; 3) pCS2-FA-His-E2F8-HA; E2F8 ORF was amplified with a forward primer flanked with FseI RE site and a reverse primer flanked with HA tag followed by AscI RE site. The PCR product was cloned downstream to His tag; 4) pCS2-His-E2F8-N80-EGFP; the ORF encoding for amino acids 1–80 of E2F8 was amplified using pCS2-FA-E2F8 template and a primer set flanked with FseI (forward) and AgeI (reverse) RE sites. The PCR product was cloned into pCS2-FA-EGFP vector downstream to His tag and upstream to EGFP; 5) pCS2-E2F8-C; the ORF encoding for amino acids 607–867 of E2F8 was amplified using pCS2-FA-E2F8 template and a primer set flanked with FseI (forward) and AgeI (reverse) RE sites. The PCR product was cloned in frame with an upstream His tag and downstream EGFP tag in a pCS2-FA vector; 6) pcDNA-E2F1; the ORF of human E2F1 was amplified using E2F1 cDNA (a gift from Doron Ginsberg, Bar Ilan University) and a primer set flanked with KpnI (forward) and EcoRI (reverse) RE sites. The PCR product was cloned into pcDNA3.1(+) vector. The following E2F8 mutant variants were generated using pCS2-FA-E2F8 as a template: 1) KEN mutant (KM) E2F8-KM1 (5KEN7 to AAN); 2) KM E2F8-KM2 (375KEN377 to AAN); 3) KM E2F8-KM3 (657KEN659 to AAN); 4) RXXL mutant 1 (D-box mutant 1) E2F8-DM1 (87RXXL90 to GXXV); 5) RXXL mutant 2 (D-box mutant 2) E2F8-DM2 (183RXXL186 to GXXV); 6) E2F8-DM1-DM2; 7) E2F8-KM1-KM2; 8) E2F8-KM1-DM2; 9) E2F8-KM2-DM2; 10) E2F8-KM3-DM2; 11) DNA-binding domain (DBD) double mutant E2F8-DBD (155RRIYD159 and 313RRLYD317 to AAIYD and AALYD); 12) dimerization domain double mutant E2F8-DD (deletion of 162NVL164 and 320NVL322); 13) E2F8-DBD-DD (quadruple mutant); 14) E2F8-T20A (Thr-20 to Ala); 15) E2F8-T20D (Thr-20 to Asp); 16) E2F8-T44A (Thr-44 to Ala); 17) E2F8-T44D (Thr-44 to Asp); 18) E2F8-T20A-T44A (double mutant); 19) E2F8-T20D-T44D (double mutant); 20) Cy box mutant 1–E2F8-CB1 (15RGL17 to AGL); 21) Cy box mutant 2–E2F8-CB2 (81RGL83 to AGL); 22) Cy box mutant 3–E2F8-CB2 (313RRL315 to ARL); 23) Cy box mutant 4–E2F8-CB4 (857RKL859 to AKL). The following E2F8 variants were generated using pCS2-FA-His-E2F8-HA as a template: 1) E2F8-CB1; 2) E2F8-CB2; 3) E2F8-CB3; 4) E2F8-CB4. The following E2F8-N80 variants were generated using pCS2-His-E2F8-N80-EGFP as a template: 1) E2F8-N80-KM1 (5KEN7 to AAN); 2) E2F8-N80-T20A (Thr-20 to Ala); 3) E2F8-N80-T20D (Thr-20 to Asp); 4) E2F8-N80-T44A (Thr-44 to Ala); 5) E2F8-N80-T44D (Thr-44 to Asp); 6) E2F8-N80-T20A-T44A (double mutant); 7) E2F8-N80-T20D-T44D (double mutant). Overall, 40 new plasmids were generated. Aside from Tome-1, all ORFs were of human origin. All mutations were generated by site-directed mutagenesis (Agilent; #20521). Cloning and mutageneses were validated by Sanger sequencing. A full list of DNA oligos used for cloning is provided in Supplemental Table S1.
Cell culture
HeLa S3 (S3) and HEK293T cells were originally from the American Type Culture Collection. The 293-T-REx cells were a gift from Vamsi Mootha (Harvard Medical School). All cell lines were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 1% penicillin–streptomycin solution (Biological Industries; #03-020-1B, #03-031-1B). Cells were maintained at 37°C in a humidified 5% CO2-containing atmosphere. S3 cells were either cultured on plates or in 1-l glass spinner flasks in suspension (80 rpm). The 293-T-REx cells were cultured in the presence of 5 µg/ml blasticidin (Life Technologies; #A11139-03) to maintain the pcDNA6/TR plasmid carrying the ORF for tet repressor. The derivation of the Cyclin F KO cell lines using CRISPR/Cas9 gene editing technology was previously described (Choudhury et al., 2016).
Generation of NDB cell system
The 293-T-REx cells were stably transfected with pcDNA4/TO plasmid carrying an ORF of a destruction-box mutant (DM) Cyclin B1 (Arg-42 and Leu-45 were substituted with Gly and Val, respectively) and a zeocin resistance gene. The ORF of cyclin B1-DM was amplified by PCR using the pCS2-FA-Cyclin B1-DM plasmid (Pe’er et al., 2013) as a template and a primer set flanked with RE sites for BamHI (forward) and XhoI (reverse). Zeocin concentration for selection: 200 µg/ml (BioBasic; # Z706211). NDB cells were stably transfected with a plasmid carrying the ORF of histone H2B-YFP and a neomycin resistance gene. Stably transfected cells were selected by 500 µg/ml G418 (Formedium; #108321-42-2). NDB and NDB-H2B-YFP cell lines were originated from a single cell.
Generation of inducible Cyclin F cell line
Cyclin F was cloned by gateway recombination cloning into pINDUCER20 (Meerbrey et al., 2011). Lentiviral particles were produced in HEK293T cells by transfecting cells with pINDUCER20-Cyclin F, as well as separate plasmids containing TAT, REV, VSVg, and gag-pol. Viral particles were used to transduce MCF7 and T47D cells, which were subsequently selected with geneticin (Life Technologies #10131-035). Cyclin F expression was induced by the addition of doxycycline (5, 25, or 100 ng/ml) to the media for 48 h and cells were analyzed by immunoblot. For in vivo experiments, MG132 (UBP Bio; #F1101) and bortezomib (Sigma-Aldrich; #5043140001) were used at a final concentration of 50 μM and 100 nM, respectively.
Cell synchronization
Synchronization of S3 cells for mitotic extracts.
A 400–500 ml culture of S3 cells was grown in suspension (1-l spinner flask, 85 rpm) until population reached a density of about 2.5 × 105 cells/ml. Cells were then treated with 2 mM thymidine (Sigma-Aldrich; #T9250) for 22 h, washed, and released into prewarmed fresh media. After 3 h, cells were incubated with 50 ng/ml nocodazole (Sigma-Aldrich; #M1404) for 11–12 h and harvested for mitotic extract preparation.
Synchronization of S3 cells for G1 extracts.
Nocodazole-arrested cells (see preceding paragraph) were washed, recultured in 400–500 ml prewarm fresh media, and harvested after 3.5 h for G1 extract preparation. Cells extracts were also generated from cells 6 and 8.5 h after release from nocodazole block.
Synchronization of S3 cells for S-phase extracts.
S3 cells were cultured in suspension until the population reached a density of approximately 5 × 105 cells/ml. Cells were then incubated with 2 mM thymidine for 22 h, washed, and released into prewarmed fresh media for 9 h and blocked again with 2 mM thymidine for 19 h. Cells were either harvested for extract preparation or released from the second thymidine block for 3 or 6 h and then harvested.
Synchronization of NDB cells in late mitosis.
NDB cells were cultured in 15-cm/diameter plates. After reaching a confluency of about 75%, the cells were treated with 1 µg/ml tet (Sigma-Aldrich; #87128) for 22 h and harvested for extract preparation or any other purpose.
Synchronization of NDB cells in prometaphase.
NDB cells at 75% confluency were treated with 100 ng/ml nocodazole for 18 h.
Synchronization of NDB cells in G1.
Asynchronous NDB cells showing the lowest 10% forward scatter width (FSC-W) signal were isolated by FACSAria III (BD). This sorting protocol yields a nearly pure G1 population without presynchronization (Vecsler et al., 2013).
Synchronization of S3 cells for Western blotting.
S3 cells arrested by thymidine-nocodazole block or double thymidine block (see above) were washed and either harvested (t = 0) or recultured for 30 min to 11 h and then harvested.
Live cell imaging
For light phase images, we used a Nikon eclipse TS100 inverted microscope equipped with 20× (NA: 0.4) and 40× (NA: 0.55) LWD lenses, a Nikon Digital-Slight DS-Fi1 camera, and a Nikon C-HGFI Intensilight. Images were processed by ImageJ software.
Spread chromosomes were visualized using a Nikon Eclipse Ti-E inverted microscope equipped with 100× oil lens (NA: 1.4) or 40× Oil lens (NA: 1.3), Lumencor illuminator LED light source, and a Zyla sCMOS camera (Andor Technology). Filter sets: excitation 390/18 nm; emission 460/50 nm. Images were processed by Nikon NIS-element and ImageJ software. Time-lapse microscopy of NDB cells was performed with a Leica SP8 inverted scanning confocal microscope equipped with an 63× oil lens (NA: 1.4), and HyD detector. Excitation: 488 nm laser. Emission: 511–552 nm. Images were acquired and processed using LASX and ImageJ software.
Flow cytometry
Overall, cell synchronization was measured by DNA quantification following a standard propidium iodide staining protocol (Sigma-Aldrich; #81845). Gallios (Beckman Coulter) and FACSAria III (Becton Dickinson) flow cytometers were used for analyzing the stained cells. Cell cycle phase distribution was determined by ModFit LT’s Sync Wizard model (Verity Software House). FACSAria III was also used to sort G1 NDB cells for Western blot analysis (see above) and for sorting single NDB and NDB-H2B-YFP cells.
Chromosome spread
Tet-induced and nocodazole-arrested NDB cells (see above) were harvested by gentle pipetting, washed with phosphate-buffered saline (PBS), lysed in hypotonic solution (0.8% KCl, 10 min at room temperature [RT]), and fixed in a methanol/glacial acetic acid solution (3:1 volume ratio). Cell droplets were released from 1-m height onto tilted glass slides. The slides were air-dried and mounted with a mounting solution (Thermo Fisher Scientific; 4112APG) and 4′,6-diamidino-2-phenylindole (DAPI) stain (5 μg/ml [Sigma-Aldrich; #P9542]).
Western blotting
Protein lysis.
Cells were washed twice in cold PBS and lysed in a cold lysis buffer (50 mM Tris, pH 7.6, 150 mM NaCl, 5 mM EDTA, pH 8.0, 0.5% NP-40) supplemented with a protease inhibitor cocktail (Roche; #4693159001), 1 mM phenylmethylsulfonyl fluoride, phosphatase inhibitor cocktail (Sigma-Aldrich; #P5726, #P0044), 10 mM NaF; 20 mM β-glycerophosphate; 1 mM Na3VO4, 20 mM P-nitrophenylphosphate. Protein extracts were incubated on ice for 30 min and the nonsoluble components were pelleted by centrifugation for 45 min at 14,000 × g. Protein concentration was determined by a standard Bradford assay (Bio-Rad #500-0006), a linear bovine serum albumin (BSA) calibration curve, and an Epoch microplate spectrophotometer.
Immunoblotting.
Protein samples were mixed with Laemmli buffer, denatured (5–10 min, 95°C), and resolved on freshly made 8–10% acrylamide gels using a Tris-glycine running buffer. Proteins were then electro-transferred onto a nitrocellulose membrane (Bio-Rad; #162-0115) using a wet transfer or Trans-Blot Turbo transfer system (Bio-Rad). Transfer quality was verified by Ponceau S Solution (Sigma-Aldrich; #81462). Membranes were washed, blocked (5% skimmed milk in TBST), and incubated overnight (4º) with antibody solution (2.5% BSA and 0.05% sodium azide in PBS). The following primary antibodies were used: anti-Tubulin (DSHB; #12G10), anti-Actin (DSHB; #JLA20), anti-HSP70 (Santa Cruz Biotechnology; #SC-24), anti-GAPDH (Santa Cruz Biotechnology; #SC-47724), anti-E2F8 (Abnova; #H00079733-M01 and Abcam; #AB109565), anti-E2F1 (Santa Cruz Biotechnology; #SC-193), anti-Securin (Abcam; #AB3305), anti-Geminin (Abcam; #AB12147), anti-Kifc1 (Bethyl Laboratories; #A300-951A), anti-Cyclin B1 (Santa Cruz Biotechnology; #SC-70898), anti-Cdc20 (Santa Cruz Biotechnology; #SC-8358), anti-Cdh1 (Calibochem; #CC43-100UG), anti-Cdc27 (Santa Cruz Biotechnology #SC-5618 and SC-9972), anti-Cyclin F (Santa Cruz Biotechnology; #SC-952), and anti-HA (Biolegend; #901502); anti-FLAG (Sigma; #A8592). Horseradish peroxidase–conjugated secondary antibodies were purchased from Jackson ImmunoResearch (#115-035-174; #115-035-144; #115-035-003). ECL signal was detected by a SuperSignal West Femtochemiluminescence substrate (Pierce; #34095) or an EZ-ECL (Biological Industries; #20-500-171).
Immunoprecipitation
APC/C was immunoprecipitated from NDB mitotic extracts pre- and post–30 min incubation with RO-3306 (see above). To this end, 200 μg cellular extracts were mixed with 300 μl wash buffer (150 mM NaCl, 20 mM Tris-HCI, pH 7.5, 10% glycerol, 0.1% Triton, EDTA 1 mM) and protease inhibitor cocktail (Sigma-Aldrich; #p2714-1BTL). Next, 15 μl agarose–conjugated anti-Cdc27 antibodies (Santa Cruz Biotechnology; SC-9972AC) were incubated with the diluted extracts for 4 h at 4°C. Beads were washed twice with 150 mM NaCl wash buffer and once with 75 mM wash buffer, resuspended with 4× Laemmli sample buffer, and denatured 10 min at 95°C. Protein samples were resolved by SDS–PAGE.
HA/FLAG immunoprecipitation.
HEK293T cells were transfected with the indicated plasmids using PolyJet DNA in vitro transfection reagent (SignaGen #SL1000688). At 24 h posttransfection, cells were harvested, lysed in NETN buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 0.5 mM EDTA, 0.5% NP40, 2 µg/ml pepstatin, 2 µg/ml apoprotinin, 10 µg/ml leupeptin, 1.0 mM 4-(2 aminoethyl) benzenesulfonyl fluoride, 1.0 mM Na3VO4) and immunoprecipitated with EZview Red anti-HA or EZview Red anti-FLAG M2 affinity gel (Sigma #E6779; #F2426). IPs were performed for 2 h at 4°, washed 5× in cold NETN buffer by rotating for 5 min each time, and eluted in Laemmli buffer prior to analysis by immunoblot.
Preparation of cell extracts
HeLa S3 extracts.
Synchronous S3 cells were washed with cold PBS and lysed in a swelling buffer (20 mM HEPES, pH 7.5, 2 mM MgCl2, 5 mM KCl, 1 mM dithiothreitol [DTT], and protease inhibitor cocktail [Roche; #11836170001]) supplemented with energy regenerating mixture (1 mM ATP, 0.1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid [EGTA], 1 mM MgCl2, 7.5 mM creatine phosphate, 50 μg/ml creatine phosphokinase). Cells were swelled on ice for 30 min and homogenized by freeze–thawing in liquid nitrogen and passed through a 21-G needle 10 times. Extracts were cleared by subsequent centrifugations (14,000 RPM; 10 and 40 min), and stored at –80º.
NDB mitotic extracts.
Tet-induced NDB cells were harvested from 20 150-mm plates. Cells were washed gently with cold PBS and lysed for extract preparation (see preceding paragraph).
NDB G1-like extracts.
NDB mitotic extracts (see above) were preactivated with RO-3306 for 15 to 30 min. This treatment overrides the blocking effect of nondegradable Cyclin B1 and induces mitotic exit into an APC/CCdh1-active state in tube. Only then were in vitro translated (IVT) substrates added to the reaction mixture for degradation assays.
Degradation and mobility shift assays
Target proteins were in vitro transcribed and translated in rabbit reticulocyte lysate (TNT-coupled reticulocyte system; Promega; #L4600, #L4610) supplemented with 35S-methionine (IsoLabel L-35S Steady Blue, Izotop; #TSM-01). Degradation/mobility-shift assays were performed in 20-μl cell extracts supplemented with 1 μl of ×20 energy regenerating mixture (see above), 1 μl of 10 mg/ml Ub solution (Boston Biochem; #U-100H), and 1 µl radiolabeled IVT product of interest. As indicated, reaction mixture was supplemented with one or more of the following reagents: 1) recombinant His-tagged UbcH10 or UbcH10DN (5 μg), 2) recombinant GST-tagged Emi1 C-terminus (1 μg), 3) Cdk1 inhibitor RO-3306 (15-30 μM; Enzo Life Sciences; #ALX-270-463-M001), 4) MG132 (20 μM; Boston Biochem; #I-130), and 5) dimethyl sulfoxide (DMSO). Reaction mixtures were incubated at 28°C, unless otherwise is indicated. Aliquots of 3–5 μl were taken every 10 to 20 min, mixed with Laemmli buffer, denatured (10 min, 95°C), and quickly frozen in liquid nitrogen. Protein samples were resolved by SDS–PAGE. Gels were soaked in a methanol/acetic acid (10%/7.5%) solution for 20 min, dried in vacuum and heat, and exposed to phosphor screen. IVT proteins were visualized by autoradiography using Typhoon FLA 9500 phosphorimager (GE Healthcare Life Sciences). Signal intensity was measured by ImageJ software after background subtraction and normalized to 35S signal at time 0 min (t0). All plots were created in Microsoft Excel software, version 16.20. Mean and SE were calculated from three or four independent degradation assays.
On-chip ubiquitination and Ub-chain preference assays
Mold fabrication.
The device was designed using AutoCAD 2011 (Autodesk) and each layer was reproduced as a chrome mask at 40,000 dpi (Fineline-Imaging). Flow molds were fabricated on 4″ silicon wafers (Silicon Quest International) with pretreatment of O2 plasma 34% for 5 min. The wafers were spin coated with SPR 220-7 (Shipley; 1500 rpm, 60 s) yielding a substrate height of around 13–15 μm. The molds were baked at 105°C for 6 min followed by a 120-s I-line exposure on a MA6 contact mask aligner (Karl Suss). Next, molds were incubated for 2 h in RT, baked in 110°C for 5 min, incubated for an additional 45 min at RT, and developed with AZ 726 MIF Developer followed by DW H2O wash. Finally, molds were annealed at ramping temp (70–200°C; 10°C/h) for 15 h. Control molds were fabricated on 4″ silicon wafers by spin coating SU-8 2025/3025 (MicroChem) at 500 rpm for 5 s followed by 3000 rpm for 70 s yielding a substrate height of around 15–18 μm. The molds were baked at 65°C for 2 min and 95°C for 7 min. Next, the wafers were exposed for 15 s on the mask aligner, followed by a postexposure baking series of 65°C for 1 min and 95°C for 3 min. The wafers were developed in AZ EBR Solvent for 4.5 min followed by an isopropanol wash. At the end of the fabrication step, control and flow molds were Teflon coated to promote elastomer release during subsequent use.
Device fabrication.
The microfluidic devices were fabricated on silicone molds casting silicone elastomer polydimethylsiloxane (PDMS, SYLGARD 184, Dow Corning). Each device consists of two aligned PDMS layers: the flow layer and the control layer. A mixture of a silicone-based elastomer and curing agent was prepared in two different ratios 5:1 and 20:1 for the control and flow molds, respectively. The control layer was degassed and baked for 30 min at 80°C/60 min at 90°C. The flow layer was initially spin coated (Laurell Technologies) at 1500–2000 rpm for 60 s and then baked at 80°C for 30 min. The control layer was separated from its mold and then control channel access holes were punched. The flow and control layers were aligned manually and baked for 2 h at 80°C. The two-layer device (chip) was peeled from the flow mold and flow channels access holes were punched.
Surface chemistry.
To bind the expressed protein, we first cover the epoxy slides with biotinylated BSA (1 µg/µl; Thermo Scientific). Streptavidin (0.5 µg/µl Neutravidin; Pierce) was then introduced to interact with the biotinylated BSA layer. Next, a designated set of pneomatic valves, also known as “button valves” (Noach-Hirsh et al., 2015), were closed and a layer of biotinylated PEG (1 µg/µl; Nanocs) was used to block the protein chamber periphery, leaving the center itself exposed to avidin to which antibodies and eventually target proteins can be attached. Then, the button valves were opened and anti-GFP biotinylated antibodies (0.2 µg/µl; Abcam; #ab6658) were flowed through the device to interact with the exposed avidin at the protein chamber center. This creates an array of protein chamber with anti GFP antibodies to which EGFP-tagged E2F8 IVT product can bind. PBS buffer or PBS buffer with 1% BSA was used to the wash between steps.
On-chip ubiquitination and Ub-chain preference assays.
E2F8-EGFP IVT product was flowed into the chip and immobilized on the surface at the protein chamber via its EGFP tag, followed by a wash with a PBS buffer. S3 G1 extracts were mixed with 1) 0.04 mg/ml Rd-Ub (Boston Biochem; #U-600); 2) mock or 0.4 mg/ml one of the following unlabled Ubiqutin variants: WT (Boston Biochem; #U100-H), Lys11 to Arg mutant (Boston Biochem; #UM-K11R), Lys-48 to Arg mutant (Boston Biochem;#UM-K48R), Lys-63 to Arg mutant (Boston Biochem; #UM-K63R); and 3) mock or 0.3 mg/ml His-tagged UbcH10DN (recombinant) or 0.48 mg/ml GST-tagged Emi1 C-terminus (recombinant). Next, reaction mixture was flowed to protein chambers for 10 min (RT). Unbound material was washed by HEPES buffer (50 mM). Rd-Ub levels were determined by 535 nm excitation (emission filters: 575/50). Rd-Ub signal was normalized to the immobilized protein level, as measured by 488 nm-excited GFP (emission filter: 535/25). This normalization provides net ubiquitnation signal per microcompartment. Ub-chain preference of E2F8 was validated in tube using degradation assays in S3 G1 extracts to which 8 µg UbcH10 (recombinant) and 0.4 µg WT- or mutant K-to-R Ub (see above) were added.
Image and data analysis.
LS reloaded microarray scanner (LS Reloaded; Tecan, Männedorf, Switzarland) and GenePix7.0 (Molecular Devices) image analysis software were used for analysis and presentation of the images for the figures.
For more details, see Noach-Hirsh et al. (2015).
Supplementary Material
Acknowledgments
We thank Tamar Listovsky-Yulzary, Ofir Hakim, Doron Ginsberg, Itay Koren, Vamsi Mootha, and Yaron Shav-Tal for sharing reagents. The Tzur lab is supported by the Israel Cancer Research Fund, Grant no. RCDA00102, and the Israel Science Foundation Grant nos. 659/16 and 2038/19. The Emanuele lab is supported by the University of North Carolina University Cancer Research Fund, National Institutes of Health (R01GM120309), American Cancer Society (RSG-18-220-01-TBG), and donations from the Brookside Foundation.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- BSA
bovine serum albumin
- CB
Cy box
- DBD
DNA-binding domain
- IVT
in vitro translated
- KM
KEN mutant
- KO
knockout
- NBD
nondegradable allele of Cyclin B1
- ORF
open reading frame
- PBS
phosphate-buffered saline
- PDMS
polydimethylsiloxane
- Rd-Ub
rhodamine-labeled ubiquitin
- RE
restriction enzyme
- tet
tetracycline
- Ub
ubiquitin
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-12-0725) on January 29, 2020.
REFERENCES
- Ayad NG, Rankin S, Murakami M, Jebanathirajah J, Gygi S, Kirschner MW. (2003). Tome-1, a trigger of mitotic entry, is degraded during G1 via the APC. Cell , 101–113. [DOI] [PubMed] [Google Scholar]
- Ayad NG, Rankin S, Ooi D, Rape M, Kirschner MW. (2005). Identification of ubiquitin ligase substrates by in vitro expression cloning. Methods Enzymol , 404–414. [DOI] [PubMed] [Google Scholar]
- Boekhout M, Yuan R, Wondergem AP, Segeren HA, van Liere EA, Awol N, Jansen I, Wolthuis RM, de Bruin A, Westendorp B. (2016). Feedback regulation between atypical E2Fs and APC/CCdh1 coordinates cell cycle progression. EMBO Rep , 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhavarapu VN, White ED, Mahanic CS, Chen L, Lin FT, Lin WC. (2012). Regulation of E2F1 by APC/C Cdh1 via K11 linkage-specific ubiquitin chain formation. Cell Cycle , 2030–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdova K, Yang H, Faedda R, Hume S, Chauhan J, Ebner D, Kessler BM, Vendrell I, Drewry DH, Wells CI, et al (2019). E2F1 proteolysis via SCF-cyclin F underlies synthetic lethality between cyclin F loss and Chk1 inhibition. EMBO J e101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. (1999). SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol , 193–199. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. (2009). Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer , 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, Bonacci T, Arceci A, Lahiri D, Mills CA, Kernan JL, Branigan TB, DeCaprio JA, Burke DJ, Emanuele MJ. (2016). APC/C and SCF(cyclin F) constitute a reciprocal feedback circuit controlling S-phase entry. Cell Rep , 3359–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Cloos P, Toftegaard U, Klinkenberg D, Bracken AP, Trinh E, Heeran M, Di Stefano L, Helin K. (2005). Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res , 5458–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clijsters L, Hoencamp C, Calis JJA, Marzio A, Handgraaf SM, Cuitino MC, Rosenberg BR, Leone G, Pagano M. (2019). Cyclin F controls cell-cycle transcriptional outputs by directing the degradation of the three activator E2Fs. Mol Cell , 1264–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Vecsler M, Liberzon A, Noach M, Zlotorynski E, Tzur A. (2013). Unbiased transcriptome signature of in vivo cell proliferation reveals pro- and antiproliferative gene networks. Cell Cycle , 2992–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, Pagano M. (2010). SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature , 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Esencay M, Pagano M. (2013). A cyclin without cyclin-dependent kinases: cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol , 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G. (2003). Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem , 42041–42049. [DOI] [PubMed] [Google Scholar]
- Di Stefano L, Jensen MR, Helin K. (2003). E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J , 6289–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Murray AW. (2000). The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell , 411–424. [DOI] [PubMed] [Google Scholar]
- Galper J, Rayner SL, Hogan AL, Fifita JA, Lee A, Chung RS, Blair IP, Yang S. (2017). Cyclin F: A component of an E3 ubiquitin ligase complex with roles in neurodegeneration and cancer. Int J Biochem Cell Biol , 216–220. [DOI] [PubMed] [Google Scholar]
- Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, Smith DL, Pines J, Hagan IM. (2015). A PP1-PP2A phosphatase relay controls mitotic progression. Nature , 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Den Elzen N, Vodermaier HC, Waizenegger IC, Peters JM, Pines J. (2002). Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J Cell Biol , 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR. (2009). Balancing the decision of cell proliferation and cell fate. Cell Cycle , 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Krutchinsky AN, Morgan DO. (2008). Positive feedback sharpens the anaphase switch. Nature , 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JN, Park I, Ellingson E, Littlepage LE, Pellman D. (2001). Activity of the APC(Cdh1) form of the anaphase-promoting complex persists until S phase and prevents the premature expression of Cdc20p. J Cell Biol , 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO. (1999). Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol , 227–236. [DOI] [PubMed] [Google Scholar]
- Jin L, Williamson A, Banerjee S, Philipp I, Rape M. (2008). Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell , 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Ohtani K, Nevins JR. (1994). Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev , 1514–1525. [DOI] [PubMed] [Google Scholar]
- Kernan J, Bonacci T, Emanuele MJ. (2018). Who guards the guardian? Mechanisms that restrain APC/C during the cell cycle. Biochim Biophys Acta , 1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. (1995). A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell , 279–288. [DOI] [PubMed] [Google Scholar]
- Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM. (2000). Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol Biol Cell , 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravtsova-Ivantsiv Y, Sommer T, Ciechanover A. (2013). The lysine48-based polyubiquitin chain proteasomal signal: not a single child anymore. Angew Chem Int Ed Engl , 192–198. [DOI] [PubMed] [Google Scholar]
- Lammens T, Li J, Leone G, De Veylder L. (2009). Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol , 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, et al (2008). Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell , 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listovsky T, Zor A, Laronne A, Brandeis M. (2000). Cdk1 is essential for mammalian cyclosome/APC regulation. Exp Cell Res , 184–191. [DOI] [PubMed] [Google Scholar]
- Liu B, Shats I, Angus SP, Gatza ML, Nevins JR. (2013). Interaction of E2F7 transcription factor with E2F1 and C-terminal-binding protein (CtBP) provides a mechanism for E2F7-dependent transcription repression. J Biol Chem , 24581–24589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, Patil K, Tuttle J, Cleghorn W, Leone G. (2005). Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem , 18211–18220. [DOI] [PubMed] [Google Scholar]
- Manning AL, Longworth MS, Dyson NJ. (2010). Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev , 1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrommati I, Faedda R, Galasso G, Li J, Burdova K, Fischer R, Kessler BM, Carrero ZI, Guardavaccaro D, Pagano M, D’Angiolella V. (2018). beta-TrCP- and casein kinase II-mediated degradation of cyclin F controls timely mitotic progression. Cell Rep , 3404–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry TJ, Kirschner MW. (1998). Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell , 1043–1053. [DOI] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci USA , 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, Rape M. (2011). Processive ubiquitin chain formation by the anaphase-promoting complex. Semin Cell Dev Biol , 544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AW, Desai AB, Salmon ED. (1996). Real time observation of anaphase in vitro. Proc Natl Acad Sci USA , 12327–12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AW, Kirschner MW. (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature , 275–280. [DOI] [PubMed] [Google Scholar]
- Nguyen PA, Groen AC, Loose M, Ishihara K, Wuhr M, Field CM, Mitchison TJ. (2014). Spatial organization of cytokinesis signaling reconstituted in a cell-free system. Science , 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noach-Hirsh M, Nevenzal H, Glick Y, Chorni E, Avrahami D, Barbiro-Michaely E, Gerber D, Tzur A. (2015). Integrated microfluidics for protein modification discovery. Mol Cell Proteomics , 2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orian A, Gonen H, Bercovich B, Fajerman I, Eytan E, Israel A, Mercurio F, Iwai K, Schwartz AL, Ciechanover A. (2000). SCF(beta)(-TrCP) ubiquitin ligase-mediated processing of NF-kappaB p105 requires phosphorylation of its C-terminus by IkappaB kinase. EMBO J , 2580–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouseph MM, Li J, Chen HZ, Pecot T, Wenzel P, Thompson JC, Comstock G, Chokshi V, Byrne M, Forde B, et al (2012). Atypical E2F repressors and activators coordinate placental development. Dev Cell , 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pe’er T, Lahmi R, Sharaby Y, Chorni E, Noach M, Vecsler M, Zlotorynski E, Steen H, Steen JA, Tzur A. (2013). Gas2l3, a novel constriction site-associated protein whose regulation is mediated by the APC/C Cdh1 complex. PLoS One , e57532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister K, Pipka JL, Chiang C, Liu Y, Clark RA, Keller R, Skoglund P, Guertin MJ, Hall IM, Stukenberg PT. (2018). Identification of drivers of aneuploidy in breast tumors. Cell Rep , 2758–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger CM, Kirschner MW. (2000). The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev , 655–665. [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D. (2008). E2F - at the crossroads of life and death. Trends Cell Biol , 528–535. [DOI] [PubMed] [Google Scholar]
- Powers BL, Hall MC. (2017). Re-examining the role of Cdc14 phosphatase in reversal of Cdk phosphorylation during mitotic exit. J Cell Sci , 2673–2681. [DOI] [PubMed] [Google Scholar]
- Rape M, Kirschner MW. (2004). Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature , 588–595. [DOI] [PubMed] [Google Scholar]
- Rape M, Reddy SK, Kirschner MW. (2006). The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell , 89–103. [DOI] [PubMed] [Google Scholar]
- Silverman JS, Skaar JR, Pagano M. (2012). SCF ubiquitin ligases in the maintenance of genome stability. Trends Biochem Sci , 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SA, Winter D, Kirchner M, Chauhan R, Ahmed S, Ozlu N, Tzur A, Steen JA, Steen H. (2014). Co-regulation proteomics reveals substrates and mechanisms of APC/C-dependent degradation. EMBO J , 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. (1995). The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell , 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley IA, Gaspar I, Ephrussi A, Surrey T. (2012). Aster migration determines the length scale of nuclear separation in the Drosophila syncytial embryo. J Cell Biol , 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlings I, de Bruin A. (2016). E2F transcription factors control the roller coaster ride of cell cycle gene expression. Methods Mol Biol , 71–88. [DOI] [PubMed] [Google Scholar]
- Vecsler M, Lazar I, Tzur A. (2013). Using standard optical flow cytometry for synchronizing proliferating cells in the G1 phase. PLoS One , e83935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman D, Nachum S, Cohen M, Enrico TP, Noach-Hirsh M, Parasol J, Zomer-Polak S, Auerbach N, Sheinberger-ChornI E, Nevenzal H, Levi-Dadon N, Wang X, Lahmi R, Michaely E, et al (2019). Cell cycle oscillators underlying orderly proteolysis of E2F8. BioRxiv 10.1101/672964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ER, Brown NG, Peters JM, Stark H, Schulman BA. (2019). Posing the APC/C E3 ubiquitin ligase to orchestrate cell division. Trends Cell Biol , 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley SP, Hinchcliffe EH, Glotzer M, Hyman AA, Sluder G, Wang Y. (1997). CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J Cell Biol , 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Merbl Y, Huo Y, Gallop JL, Tzur A, Kirschner MW. (2010). UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc Natl Acad Sci USA , 1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzenberger C, Gerlich DW. (2011). Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol , 469–482. [DOI] [PubMed] [Google Scholar]
- Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y. (1998). Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature , 590–594. [DOI] [PubMed] [Google Scholar]
- Yuan R, Liu Q, Segeren HA, Yuniati L, Guardavaccaro D, Lebbink RJ, Westendorp B, de Bruin A. (2019). Cyclin F-dependent degradation of E2F7 is critical for DNA repair and G2-phase progression. EMBO J e101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. (1998). Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science , 1721–1724. [DOI] [PubMed] [Google Scholar]
- Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS, La Thangue NB. (2008). DNA-damage response control of E2F7 and E2F8. EMBO Rep , 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, McGarry TJ, Bernal T, Kirschner MW. (1999). Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science , 418–422. [DOI] [PubMed] [Google Scholar]
- Zur A, Brandeis M. (2001). Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis. EMBO J , 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur A, Brandeis M. (2002). Timing of APC/C substrate degradation is determined by fzy/fzr specificity of destruction boxes. EMBO J , 4500–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.