

Abstract
OSBPL1 encodes the full-length oxysterol-binding protein-related protein ORP1L, which transports LDL-derived cholesterol at membrane contacts between the late endosomes/lysosomes (LEL) and the endoplasmic reticulum (ER). OSBPL1 also encodes the truncated variant ORP1S that contains only the C-terminal lipid binding domain. HeLa cells in which both variants were knocked out (ORP1-null) were used to determine the functional relationship between ORP1L and ORP1S with respect to cellular cholesterol localization and regulation. ORP1-null cells accumulated cholesterol in LEL and had reduced plasma membrane (PM) cholesterol. PM cholesterol was restored by expression of wild-type ORP1S or a phosphatidylinositol phosphate-binding mutant but not by a sterol-binding mutant. Expression of ORP2, another truncated variant, also restored PM cholesterol in ORP1-null cells. Consistent with a LEL-to-PM cholesterol transport activity, a small fraction of ORP1S was detected on the PM. As a consequence of reduced delivery of cholesterol to the PM in ORP1-null cells, cholesterol was diverted to the ER resulting in normalization of de novo cholesterol synthesis. The deficiency in PM cholesterol also reduced ABCA1-dependent cholesterol efflux and LDL receptor activity in ORP1-null cells. We conclude that ORP1S, which lacks discrete membrane-targeting motifs, transports cholesterol from LEL to the PM.
INTRODUCTION
Oxysterol-binding protein (OSBP) and OSBP-related proteins (ORPs) constitute a large gene family that is widely expressed across eukaryotic phyla (Pietrangelo and Ridgway, 2018). Although originally named for the high affinity binding of 25-hydroxycholesterol by OSBP (Taylor et al., 1984), it is now apparent that OSBP and other family members bind a variety of lipids including cholesterol, phosphatidylinositol polyphosphates (PIPs), and phosphatidylserine (Maeda et al., 2013; Mesmin et al., 2013; Chung et al., 2015; Moser von Filseck et al., 2015). In vitro analysis of several mammalian OSBP/ORPs and yeast Osh proteins indicates their principle function is the transport and exchange of lipids at membrane contact sites (MCS) between organelles (Mesmin et al., 2013; Wang et al., 2019). The activity of OSBP/ORPs at MCS would modify the local lipid environment; however, the pervasiveness of organelle MCS and the size of the gene family suggest the potential to globally modify membrane composition (Mesmin et al., 2017).
Endosomes make numerous contacts with the endoplasmic reticulum (ER) that facilitate organelle maturation, multivesicular body formation, and lipid exchange (Rowland et al., 2014; Raiborg et al., 2015a, b). During the maturation of endosomes, internalized LDL cholesterol esters are hydrolyzed and cholesterol is transported by NPC1 and NPC2 to the limiting membrane of late endosomes/lysosomes (LEL) (Vanier, 2010). ORP1L (Zhao and Ridgway, 2017), ORP5 (Du et al., 2011), and ORP6 (Ouimet et al., 2016) have been implicated in the subsequent transport of cholesterol from the limiting membrane to the ER, a critical step in the utilization and storage of exogenous cholesterol and regulation of de novo cholesterol synthesis. ORP1L is found at contact sites between the ER and the endosomes that are involved in cholesterol transfer (Zhao and Ridgway, 2017; Dong et al., 2019) as well as endosomal maturation (Rocha et al., 2009; van der Kant et al., 2013). The OSBPL1 gene encodes the full-length ORP1L that consists of three N-terminal ankyrin motifs that interact with Rab7 on the surface of LEL, a two phenylalanine in an acid tract (FFAT) motif that binds the ER-resident protein vesicle-associated membrane protein-associated protein (VAP) and a C-terminal OSBP-related domain (ORD) that binds cholesterol and PIPs (Figure 1A) (Johansson et al., 2003, 2005). The protein also has an N-terminal pleckstrin homology (PH) domain that binds PIPs on LEL (Johansson et al., 2003). Association of ORP1L with LEL and VAP-dependent interactions with the ER are required for LDL cholesterol transport from the LEL to ER (Zhao and Ridgway, 2017) and in the reverse direction when LDL cholesterol is absent (Eden et al., 2016). Knockout of ORP1L expression in cultured cells caused cholesterol retention in the LEL and reduced cholesterol delivery to the ER, as measured by decreased acyl-CoA:cholesterol acyltransferase (ACAT) activity and derepression of sterol-regulated genes. The ORP1L ORD also binds and extracts PI(4)P (Zhao and Ridgway, 2017) and other PIPs, but these lipids were not transported in an in vitro assay (Dong et al., 2019). Instead, PI(4,5)P2 and PI(3,5)P2 are allosteric regulators of ORP1 membrane targeting and cholesterol binding at donor membranes (Dong et al., 2019).
FIGURE 1:

CRISPR knockout of ORP1L and ORP1S expression in HeLa cells. (A) Domain organization of ORP1L and ORP1S proteins expressed by the OSBPL1 gene. The position of two gRNAs was used to delete a 1757 base pair region between exons 19 and 20. (B) PCR analysis of genomic DNA from HeLa and ORP1-null cells using primers flanking the deletion site. (C) HeLa, ORP1L-null, and ORP1-null cell lysates were immunoblotted with an ORP1 polyclonal antibody. (D) Cholesterol ester synthesis was measured by [3H]oleate incorporation in cells cultured in FCS (16 h), LPDS (16 h), or FCS plus 25-hydroxycholesterol (6 μM for 2 h, 25OH). Results are mean and SD of five experiments (unpaired t test, *p < 0.01 compared with HeLa cells).
Elucidating the role of ORP1L in cholesterol trafficking is complicated by the presence of ORP1S, which has a cytoplasmic/nuclear distribution and is implicated in cholesterol transport from the plasma membrane (PM) to the ER and lipid droplets (Jansen et al., 2011), oxysterol-activation of LXR in the nucleus (Lee et al., 2012), and, more recently, cholesterol transport to the PM (Wang et al., 2019). To determine the function of ORP1S, we deleted a region of OSBPL1 in HeLa cells that ablated expression of ORP1L and ORP1S. These ORP1-null cells displayed reduced PM cholesterol and cholesterol efflux and altered cholesterol homeostasis that was consistent with a role for ORP1S in cholesterol transfer from the LEL to the PM. This is the first evidence that full-length and truncated products of an OSBP gene are involved in opposing lipid transfer pathways, in this case, the delivery of cholesterol to the PM or ER.
RESULTS
Cholesterol delivery to the PM requires ORP1S
We previously characterized ORP1L cholesterol transport activity using cells with a CRISPR/Cas9 deletion that ablated ORP1L but not ORP1S expression (Zhao and Ridgway, 2017). To determine the role of ORP1S in cholesterol transport and homeostasis, CRISPR/Cas9 methodology was used to create a deletion in exons 19 and 20 that knocked out both ORP1L and ORP1S expression in HeLa cells (Figure 1A). The deletion in a clonal cell line (termed ORP1-null) was confirmed by PCR of genomic DNA using primers that flanked the site (Figure 1B) and by immunoblotting of control and ORP1-null cell lysates using an ORP1 antibody (Figure 1C). Cholesterol esterification by ACAT was measured to determine how ORP1S and ORP1L double knockout affected LDL-cholesterol transport to the ER (Figure 1D). As previously reported, ORP1L-null cells had reduced cholesterol esterification by ACAT indicating a defect in LDL-cholesterol transport to the ER (Zhao and Ridgway, 2017). However, cholesterol esterification in ORP1-null cells cultured in fetal calf serum (FCS), LPDS, or 25OH was similar to control HeLa cells (Figure 1D). This lack of effect on ACAT activity was observed in three other HeLa cell-derived ORP1-null cell lines (Supplemental Figure S1, A–C).
The normalization of ER cholesterol esterification in ORP1-null cells relative to ORP1L-null cells indicated a likely role for ORP1S in cholesterol transport from LEL to the ER or other organelles. To elucidate this transport function, the distribution of cellular cholesterol was determined in ORP1-null cells. Only when ORP1-null cells were cultured with human LDL was there a significant increase in unesterified and total cholesterol mass when compared with control HeLa cells (Figure 2A). Filipin staining revealed that both ORP1L- and ORP1-null cells cultured in LDL had increased fluorescent puncta indicative of cholesterol accumulation in the LEL (Figure 2B). Compared with control and ORP1L-null cells, filipin fluorescence in ORP1-null cells was noticeably reduced outside the LEL compartment implying reduced PM cholesterol. This finding was confirmed by coexpression of the cholesterol biosensor mCherry-D4H- and PH-PLCD1-GFP, which detects PI(4,5)P2 in the PM. ORP1-null cells had reduced mCherry-D4H fluorescence at the PM and increased D4H-positive puncta compared with HeLa cells (Figure 2C). The deficiency in PM cholesterol in ORP1-null cells was significant compared with both HeLa and ORP1L-null cells (Figure 2D). To determine whether the cholesterol content of the exofacial leaflet of the PM in ORP-1-null cells was affected, intact cells were incubated with purified mCherry-D4 and GFP-wheat germ agglutinin. A significant reduction in mCherry-D4 fluorescence was detected on ORP1-null cells indicating that both leaflets of the PM are cholesterol deficient (Figure 2, E and F). Also, reduced extraction of [3H]cholesterol from ORP1-null cells by cyclodextrin indicated that the PM is cholesterol deficient relative to HeLa and ORP1L-null cells (Figure 2G). However, this difference was observed only when ORP1-null cells were cultured in human LDL and could represent reduced cholesterol efflux from an intracellular cholesterol pool.
FIGURE 2:

Cholesterol mass and localization in ORP1-null cells. (A) Total and unesterified cholesterol mass was measured in cells cultured in FCS, LPDS, or LPDS plus 50 µg/ml human LDL (mean and SD of four experiments; *p < 0.05 compared with matched HeLa cell controls). (B) Filipin fluorescence was imaged in cells cultured in LPDS (-LDL) or LPDS plus LDL (50 µg/ml) for 16 h. (C) Live cell confocal imaging of HeLa and ORP1-null cells coexpressing Cherry-D4H and PH-PLCD1-GFP-PH for 24 h. (D) Quantification of Cherry-D4H fluorescence in the PM of HeLa, ORP1L-null, and ORP1S-null cells from C. Scatter plots (mean and SD) are shown for three experiments involving 65–100 cells (one-way analysis of variance [ANOVA] with Tukey’s multiple comparison, ***p < 0.001, ****p < 0.0001). (E) Hela and ORP1-null cells were fixed and incubated with Alexa Fluor 488-WGA and recombinant GFP-D4H. Images were captured by confocal microscopy. (F) Quantitation of GFP-D4H fluorescence intensity per cell from the images in E. Results (mean and SD) are from analysis of 50–60 cells (unpaired t test, ***p < 0.001). (G) The percentage of extraction of [3H]cholesterol by 1 mM cyclodextrin from HeLa, ORP1-null, and ORP1L-null cells. Results are the mean SD for a representative experiment repeated two other times with similar results.
We next determined whether the PM cholesterol deficiency in ORP1-null cells could be rescued by expression of wild-type ORP1S and ligand binding mutants (Figure 3, A–C). Compared to cells expressing empty vector (GFP), GFP-ORP1L expression caused a slight but significant increase in D4H fluorescence on the PM but internal puncta were unaffected (Figure 3, A and B). In contrast, expression of ORP1S-GFP resulted in a significant increase in mCherry-D4H fluorescence on the PM and a reduction in internal fluorescent puncta (Figure 3, A and B). Expression of ORP1S with a ΔELSK sterol-binding defective (SBD) mutation (Vihervaara et al., 2011) did not restore PM cholesterol based on quantitation of mCherry-D4H fluorescence. However, ORP1S with a mutation in the conserved H138H139 motif (HH/AA) required for PIP binding and allosteric regulation (Dong et al., 2019) restored PM D4H fluorescence similar to wild type (Figure 3, A and B). This contrasts with ORP1L cholesterol transfer activity at LEL-ER contacts, which requires both sterol and PIP binding activity (Zhao and Ridgway, 2017; Dong et al., 2019). ORP2 is another ORD-only variant that transfers cholesterol to the PM from endosomal compartments in exchange for PI(4,5)P2 (Wang et al., 2019). ORP2 is expressed in HeLa cells (Koponen et al., 2019) but this is not sufficient to maintain PM cholesterol transport in the absence of both ORP1L and ORP1S (Figure 2D). However, transient overexpression of ORP2 in ORP1-null cells (Figure 3, D and E) significantly increased mCherry-D4H on the PM relative to vector controls (Figure 3F).
FIGURE 3:

Expression of ORP1S restored PM cholesterol in ORP1-null cells. (A, B) ORP1-null cells were transiently cotransfected with vector (GFP) or GFP-tagged ORP1S constructs and mCherry-D4H. Fixed cells were imaged by confocal microscopy (A) and the percentage of D4H fluorescence in the PM of cells was quantified as described in Materials and Methods (B) (bar, 20 μm). Scatter plots (mean and SD) show the results from three experiments involving 50–200 cells per group (one-way ANOVA with Tukey’s multiple comparison; *p < 0.05, **p < 0.01, ***p < 0.0001). (C) Cells transiently expressing GFP vector (lane1), ORP1S-GFP (lane 2), ORP1S-GFP-HH/AA (lane 3), or ORP1S-GFP-SBD (lane 4) were harvested and immunoblotted with a GFP antibody. (D) Cells transiently expressing vector (GFP) or ORP2-GFP and mCherry-D4H were imaged and analyzed as described in A and B (bar, 20 μm). (E) Anti-GFP immunoblot of extracts from ORP1-null cells expressing GFP vector (lane 1) or ORP2-GFP (lane 2). (F) Quantitation of mCherry-D4H fluorescence in the PM. Scatter plots show the results (mean and SD) of analysis of 50–70 cells per group (unpaired t test; **p < 0.01).
If ORP1S transfers cholesterol from the LEL to PM, the protein must transiently interact with these organelles during the binding and release of ligand. A magnified view of the PM of ORP1-null cells expressing ORP1S-GFP and mCherry-D4H indicated regions of colocalization (Figure 4A). Similarly, Flag-tagged ORP1S (Figure 4B) and ORP1S-GFP cultured in lipoprotein deficient medium (Figure 4C) or with LDL (Figure 4D) contained a small fraction of ORP1S that was associated with PM. ORP1S was also variably expressed in the nucleus (Figure 3A) and unaffected by culturing in the presence or absence of LDL (Figure 4, C and D). Since ORP1S lacks the canonical membrane-targeting motifs found in ORP1L, a region of the ORD must interact with the PM during cholesterol transfer.
FIGURE 4:

ORP1S partially localizes on the PM. (A) Colocalization of ORP1S-GFP and mCherry-D4H on the PM of an ORP1-null cell (bar, 5 μm). (B) ORP1-null cells expressing ORP1S-Flag were cultured in 10% FCS and immunostained with an anti-Flag monoclonal. (C, D) ORP1-null cells expressing ORP1S-GFP were cultured in delipidated medium without or with LDL (50 μg/ml) for 16 h (bar, 20 μm). All images are confocal sections (1 μm), and magnified regions are boxed.
Cholesterol homeostasis in ORP1-null cells
Cholesterol accumulated in the LEL of ORP1S-null cells due to defective transport to the PM by ORP1S and to the ER by ORP1L. Despite the retention of cholesterol in LEL, ORP1-null cells had normal ACAT activity (Figure 1D). Similarly, cholesterol, lanosterol, and fatty acid synthesis (measured by [3H]acetate incorporation) in ORP1-null cells cultured in the absence or presence of LDL was similar to HeLa cell controls (Figure 5, A–C). There was a slight reduction in cholesterol synthesis in ORP1-null cells cultured in LDL. Cholesterol and lanosterol synthesis were also unaffected in three other ORP1-null cell lines; however, [3H]acetate incorporation into fatty acids was slightly increased in ORP1-null4 cells (Supplemental Figure S1, D and E). The expression of sterol-regulated genes (SREBP1, LDLR, and HMGCR) in ORP1-null cells was suppressed in the presence of LDL but poorly activated relative to controls when the cells were cultured in LPDS (Figure 5D). However, the defect in transcriptional activation in LDL-free medium had no impact on cholesterol synthesis in ORP1-null cells (Figure 5A). These data suggest that in the absence of ORP1S and ORP1L, an alternate pathway can deliver cholesterol to the ER and reestablish feed-back regulation.
FIGURE 5:
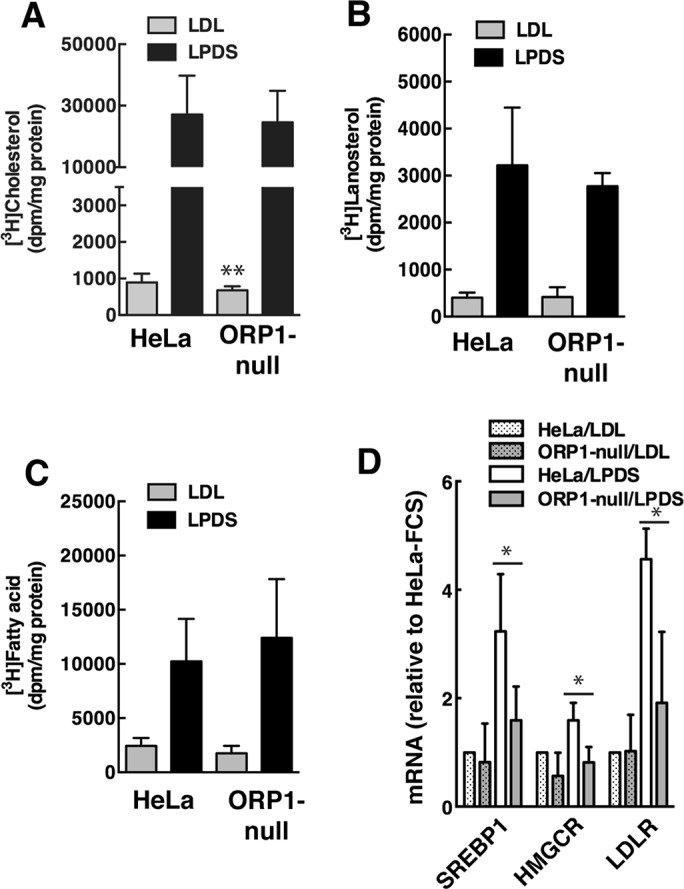
Cholesterol regulation in ORP1-null cells. (A–C) [3H]acetate incorporation into cholesterol (A), lanosterol (B) and fatty acids (C) was measured in HeLa and ORP1-null cells cultured in FBS or LPDS. Results are the mean and SD of 3 experiments (**p < 0.01). (D) qPCR analysis of SREBP1, HMGCR, and LDLR expression in cells cultured in FCS or LPDS. Results are expressed relative to HeLa cells cultured in FBS, and are the mean and SD of 4 experiments (unpaired t test, *p < 0.05 compared with similarly treated HeLa cells).
ORP1L-null cells had a transient delay in the uptake of BODIPY-LDL that was attributed to defects in the endosomal compartment due to loss of ORP1L tethering to the ER (Zhao and Ridgway, 2017). Using the same assay, ORP1-null cells had a 50–60% decrease in BODIPY-LDL uptake at all time points (Figure 6, A and B). Immunostaining in ORP1-null cells revealed a more dispersed distribution of LDLR compared with HeLa cells but cell surface localization was unaffected (Figure 6C). The expression of the LDLR protein in ORP1-null cells cultured in FCS or LPDS was similar to HeLa cells (Figure 6, D and E). When cells were treated with bafilomycin to block lysosomal degradation, LDLR expression in HeLa and ORP1L-null cells was significantly increased severalfold (Figure 6, D and E). In contrast, LDLR expression in bafilomycin-treated ORP1-null cells was unaffected, suggesting reduced LDLR recycling and delivery of LDL to the lysosomes for degradation. Compared to control and ORP1L-null cells, LDLR in ORP1-null cells was dispersed in cytoplasmic endosomes that did not localize with LAMP1 (Supplemental Figure S2A). Treatment of HeLa and ORP1L-null cells with bafilomycin caused increased perinuclear accumulation of LDLR in LAMP1-positive LEL (Supplemental Figure S2B). However, LDLR and LAMP1 in bafilomycin-treated ORP1-null cells remained relatively dispersed in the cytoplasm. Collectively, these results show that ORP1S has an important role in LDLR recycling and cargo delivery to the LEL.
FIGURE 6:

Reduced LDL uptake in ORP1-null cells. (A, B) Spinning disk confocal images of HeLa and ORP1-null cells cultured with BODIPY-LDL. Uptake of BODIPY-LDL was quantified as fluorescence intensity per cell (bar, 20 µm). Scatter plots (mean and SD) are from three experiments (20–27 fields of cells in total, **p < 0.001). (C) Immunofluorescence of total and cell surface LDLR (0.8 μm confocal sections). (D) Immunoblot analysis of LDLR expression in cells cultured in DMEM with FCS, LPDS, or LPDS plus bafilomycin (100 nM for 6 h). (E) From immunoblots in D, LDLR expression was quantified relative to actin. Results are the mean and SD of four to five experiments (unpaired t test, *p < 0.05; ns, not significant).
To determine whether cholesterol delivered to the PM by ORP1S was a substrate for efflux, the expression and activity of ABCA1 was measured in control, ORP1L-null, and ORP1-null cells (Figure 7). ABCA1 protein expression in all three cell lines was increased by the LXR agonists 22-hydroxycholesterol (22OH) and T0901317, with a slightly greater induction of ABCA1 expression in ORP1L-null cells under basal and 22OH-stimulated conditions (Figure 7A). The increase in ABCA1 expression in ORP1L-null cells did not affect cholesterol efflux activity compared with control HeLa cells (Figure 7, B and C). Despite normal induction of ABCA1 protein by 22OH and T0901317 in ORP1-null cells, cholesterol efflux to apoAI remained at the level of unstimulated cells. Thus, the reduction in PM cholesterol caused by ORP1S deficiency negatively impacts ABCA1 activity.
FIGURE 7:

Reduced ABCA1-mediated cholesterol efflux in ORP1-null cells. (A) Cells received no addition (NA, solvent control), 1.0 µM T0901317, and 10 nM RA or 1 μM 22OH and 10 nM RA for 16 h. Cell lysates were immunoblotted for ABCA1 and OSBP. (B) Expression of ABCA1 was quantified relative to OSBP load control. Results are the mean and SD of six experiments (unpaired t test, *p < 0.05, **p < 0.001 compared with similarly treated HeLa cells). (C) The percentage of efflux of [3H]cholesterol to apoAI was measured as described in Materials and Methods. Results are the mean and SD of five experiments (unpaired t test, *p < 0.05, **p < 0.005 compared with similarly treated HeLa cells).
DISCUSSION
There are numerous examples of alternative splicing or promoter usage in OSBP genes that result in the expression of “long” ORPs that are targeted to organelle MCS and “short” ORD-only ORPs (Pietrangelo and Ridgway, 2018). OSBPL1 encodes ORP1L, which localizes to LEL-ER contact sites and transfers cholesterol, and a cytosolic ORP1S variant that lacks the ankyrin, FFAT, and PH domains in the N-terminus of ORP1L (Johansson et al., 2005; Zhao and Ridgway, 2017). To understand the functional significance of this differential expression pattern, we used cells with a double knockout of OSBPL1 to show that its two protein products are involved in opposing cholesterol transport pathways from the LEL to the PM (ORP1S) or ER (ORP1L).
A striking phenotype of ORP1-null cells was a reduction in PM cholesterol and accumulation in the endolysosomal system. D4H and D4 cholesterol sensors were used to quantify depletion of cholesterol in the cytoplasmic and exofacial leaflets of the PM in ORP1-null cells. As well, filipin staining and extraction of radiolabeled cholesterol with cyclodextrin revealed that the PM was cholesterol depleted. The shift in cholesterol distribution between the PM and the LEL in ORP1-null cells caused only minor changes in total cholesterol mass. However, two functional consequences of this redistribution of cholesterol in ORP1-null cells were identified. First, ORP1-null cells had a significant deficiency in ABCA1-dependent efflux to apoA1 in the medium despite normal levels of ABCA1 protein expression. ABCA1 efflux was normal in ORP1L-null cells consistent with the proposal that ORP1S maintains a pool of cholesterol at the PM that is accessible to the D4H probe and ABCA1. Second, cholesterol deficiency in the PM or excess cholesterol in the endolysosomal compartment also negatively affected LDLR activity. ORP1-nulls had reduced BODIPY-LDL uptake but normal expression and surface localization of the LDLR. However, internalized LDLR was localized to a dispersed endosomal compartment in ORP1-null cells and did not significantly accumulate in response to a lysosomal inhibitor, suggesting that endocytic recycling and LDL delivery to the lysosomes was partially compromised. Despite the apparent inhibition of LDL uptake, ORP1-null cells still accumulated cholesterol in LEL pointing to a primary role for ORP1S and ORP1L in cholesterol egress from that compartment.
Coupled with the abnormal cholesterol distribution and PM dysregulation in ORP1-null cells was a paradoxical normalization of ER cholesterol homeostasis. To explain this, a model is proposed wherein ORP1L and ORP1S work in opposing pathways that export LDL-cholesterol to the ER and PM, respectively (Figure 8). In wild-type cells, ORP1L transfers cholesterol to the ER, where it is esterified by ACAT and negatively regulates SREBP processing and cholesterol synthesis (Zhao and Ridgway, 2017), and ORP1S transfers cholesterol to the PM. In cells lacking ORP1L, cholesterol transfer from the LEL is reduced by 60–70% but ORP1S continues to transfer cholesterol to the PM. The unabated removal of cholesterol from the LEL by ORP1S could further deplete the ER of cholesterol in ORP1L-null cells by competition with other LEL-to-ER cholesterol transfer pathways. In cells lacking both ORP1 variants, a new homeostatic balance is established due to inhibition of PM cholesterol transport and expansion of the LEL cholesterol pool, resulting in enhanced delivery to the ER. There are two potential mechanisms to explain this phenotype. First, ORP1L-independent cholesterol transfer to the ER could be in competition with ORP1S cholesterol transfer to the PM. In the absence of ORP1S, this alternate pathway would have access to an expanded cholesterol pool and enhance sterol delivery to the ER. Second, abnormal LDL uptake and cycling of the LDLR in ORP1-null cells could result in delivery of LDL-cholesterol to other endosomal compartments with ER cholesterol transport activities. For instance, a distinct population of STARD3-positive late endosomes receives LDL-cholesterol prior to those that are ORP1L-positive (van der Kant et al., 2013). The cholesterol esterification phenotype of ORPL-null and ORP1-null cells differs from a recent report that showed a 20–40% reduction in cholesterol esterification in single and double knockout HeLa cells, respectively (Dong et al., 2019). This discrepancy could be related to the shorter exposure to LDL-containing medium prior to measure radioactive oleate incorporation into cholesterol ester. Importantly, we observed that knockout of ORP1S in previously characterized ORP1L-null cells (Zhao and Ridgway, 2017) normalized their low cholesterol esterification activity (see ORP1-null4 cells, Supplemental Figure S1B).
FIGURE 8:

ORP1L and ORP1S have opposing cholesterol transfer activities at the LEL. The distribution of cholesterol and ER cholesterol regulatory responses are shown in wild-type, ORP1L-null, and double knockout ORP1-null cells. Broken arrows in ORP1L- and ORP1-null cells indicate alternative ORP1L-independent cholesterol transfer pathways. The relative distribution of cholesterol in the ER, LEL, and PM is indicated by the grayscale. ACAT activity and cholesterol synthesis (Chol Syn) is indicated relative to wild-type conditions.
Expression of GFP-ORP1S in ORP1-null cells was sufficient to restore a normal distribution of cholesterol between the PM and the LEL. As expected, an ORP1S sterol-binding mutant was ineffective in this regard. However, the wild-type activity of ORP1S-HH/AA was unexpected since the same mutation in ORP1L prevented LEL-to-ER cholesterol transfer activity in cells (Zhao and Ridgway, 2017; Dong et al., 2019) and, in ORP1-ORD, inhibited sterol transfer in a liposomal assay (Dong et al., 2019). The ORP1 ORD binds to all PIP species in vitro but does not transfer them between liposomes. Rather, PI(4,5)P2 and PI(3,4)P2 were identified as allosteric activators of ORP1S that promote membrane targeting and sterol extraction from LEL (Dong et al., 2019). Although the LEL appears to be the donor membrane for ORP1S transfer, the ability of the ORP1S-HH/AA to restore activity suggests a PIP-independent mechanism for LEL-to-PM transfer. ORP1S-GFP, which lacks canonical membrane-targeting motifs, was detected in the cytoplasm and occasionally on the PM with mCherry-D4H but not on LEL in HeLa cells (Figure 4A). Thus, ORP1S is not regulated by PIPs at MCS like its full-length counterpart ORP1L and could primarily recognize and transfer cholesterol to membranes based on their sterol content.
ORP2 is another ORD-only protein that binds and transfers cholesterol to the PM in exchange for PI(4,5)P2 (Wang et al., 2019). HeLa cells express ORP2 but overexpression was required to restore PM cholesterol in ORP1-null cells, indicating that ORP2 and ORP1S function in the same pathway but are regulated by different mechanisms. ORP2 interacts with ORP1L on LEL as part of a concerted pathway to deliver LDL-cholesterol to the PM in HeLa cells (Koponen et al., 2019). In our ORP1-null HeLa cells, ORP2 might not be fully active with respect to PM cholesterol transport due to the absence of ORP1L. Thus, ORP2 and ORP1S deliver cholesterol to the PM but ORP2 may require the presence of ORP1L and PI(4,5)P2 for optimal activity.
MATERIALS AND METHODS
Cell culture
HeLa cells were cultured in DMEM with 10% FCS at 37°C in a 5% CO2 atmosphere. CRISPR/Cas9 gene editing (Ran et al., 2013) in HeLa cells was used to introduce a deletion in OSBPL1 using two guide RNAs (gRNAs) targeting exons 19 (TGTATTCAGTTAGGCGCTGT) and 20 (GCTTCTCAGTGGGAACGGAC) that disrupted the expression of ORP1L and ORP1S (Figure 1A). Immunoblotting indicated that these cells, referred to hereafter as ORP1-null (clone 16-5), had no detectable expression of ORP1L and ORP1S (Figure 1C). Additional double knockout HeLa cell lines were generated as follows: ORP1-null2 cells (clone 89-S3) were derived by targeting HeLa cells with the exon 19 gRNA, ORP-null3 (clone 16-10) were isolated during screening for ORP1-null cells (see above), and ORP1-null4 cells (clone 81-3) were derived by targeting ORP1L-null cells (Zhao and Ridgway, 2017) with exon 19 and 20 gRNAs. All ORP1-null cells were isolated by limiting dilution and verified by PCR (forward primer; TCCATGGTATTAGGTGCTC: reverse primer; TTCTCCTCTTCATCTGCAACGC), genomic sequencing, and immunoblotting for ORP1 isoforms. Cells were transiently transfected for 24 or 48 h with ORP1L and ORP1S expression vectors using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
Analysis of cholesterol ester and cholesterol synthesis
Cholesterol esterification was measured by incubating cells with 100 μM [3H]oleate/bovine serum albumin (BSA) (6:1 mol/mol) for 4 h (Goldstein et al., 1983), extraction of the oleate-labeled lipids from dishes with hexane:isopropanol (3:2, vol/vol), and separation by thin-layer chromatography in hexane:diethylether:acetic acid (90:30:1, vol/vol). Radioactivity in cholesterol esters and triglycerides was quantified by liquid scintillation counting and normalized to cell protein.
Sterol and fatty acid synthesis was measured by incubating HeLa cells with 2.5 μCi/ml [14C]acetate for 4 h (Brown et al., 1978). After extraction, lipids were saponified in ethanol and 50% (wt/vol) KOH for 1 h at 60°C, extracted with hexane, and cholesterol and lanosterol were resolved by thin-layer chromatography in petroleum ether/diethyl ether/acetic acid (60:40:1, vol/vol) and radioactivity was measured by liquid scintillation counting. [3H]fatty acids were extracted from the hydrolysate with hexane after acidifying with HCl (pH < 3). Radioactivity was measured by liquid scintillation counting and normalized to cell protein.
Immunoblotting and immunofluorescence
Cells were lysed in SDS sample buffer (12.5% SDS, 30 mM Tris-HCl, 12.5% glycerol, and 0.01% bromophenol blue, pH 6.8), heated at 90°C for 5 min, separated by SDS–PAGE, and transferred to nitrocellulose. The following primary antibodies were used: ORP1L (1:2000, Abcam, ab131165), LAMP1 and LAMP2 (1:1000, Developmental Studies Hybridoma Bank, 1D4B and H4B4), LDLR (1:2000, rabbit polyclonal Ab3143 targeting the C-terminus) (Russell et al., 1984), or actin (1:10,000, Sigma AC15). Proteins were visualized with IRDye-800– or -680–conjugated secondary antibodies using an Odyssey Imaging System and application software v3.0 (LI-COR Biosciences).
Cells cultured on glass coverslips were fixed in 4% (wt/vol) paraformaldehyde and permeabilized with 0.05% (wt/vol) Triton X-100 at 4°C. Cells were probed with LAMP1 or LDLR antibodies and appropriate secondary Alexa Fluor–conjugated antibodies (1:5000, ThermoFisher) in phosphate-buffered saline (PBS) containing 1% (wt/vol) BSA. Confocal imaging was performed using a Zeiss LSM510/Axiovert 200M microscope with a Plan-Apochromat 100×/1.40 NA oil immersion objective.
Cholesterol efflux to apoA1 and cyclodextrin
ABCA1-mediated efflux was assayed as previously described (Bowden and Ridgway, 2008). Cells were cultured on 12-well plates in DMEM containing 10% lipoprotein deficient serum (LPDS) and 10 µg/ml [3H]cholesterol (0.3 µCi/ml). At the same time, cells received the LXR activators T0901317 (1 µM) plus retinoic acid (10 nM) or 22OH (1 µM) plus retinoic acid (10 nM) to induce ABCA1 expression After 16–18 h, medium was removed and cells were incubated for 1 h in DMEM containing 5% LPDS and rinsed twice with 0.5 ml of warm serum-free DMEM. Cells were then incubated for 4 h in 0.5 ml of serum-free DMEM with or without 20 µg/ml human apoAI (controls received an equivalent amount of BSA). Medium was collected on ice and centrifuged at 10,000 × g for 1 min at 4°C to remove cellular debris, and radioactivity in the supernatant was quantified by liquid scintillation counting. Cells were rinsed in cold PBS and lysed in 0.5 N NaOH for 30 min, and radioactivity was measured by liquid scintillation. The percentage of cholesterol efflux to apoAI (minus BSA control) was calculated from dpmmedium/ dpmtotal (medium plus cell lysate)*100.
For experiments involving cyclodextrin extraction, cells were cultured in 12-well plates in DMEM containing 50 mg/ml human LDL, 0.3 µCi/ml [3H]cholesterol for 24 h at 37°C. The medium was replaced with DMEM containing 5% FCS for 1 h, and cells were then rinsed twice with 0.5 ml of warm serum-free DMEM, followed by the addition of 0.5 ml of serum-free DMEM containing 1 mM of cyclodextrin. Over a period of 30 min, medium was collected on ice and subjected to centrifugation at 10,000 rpm for 1 min a 4°C to remove cellular debris, and the supernatant was collected and radioactivity was measured by liquid scintillation counting. Radioactivity in the cells was measured after lysis in 0.5 N NaOH for 30 min. The percentage of cholesterol extraction was calculated from dpmmedium/dpmtotal (medium plus cell lysate)*100.
BODIPY-LDL uptake by cultured cells
Uptake of BODIPY-LDL (515ex/520em, ThermoFisher) by HeLa and ORP1L-null cells was quantified as described (Zhao and Ridgway, 2017). Briefly, cells were cultured on 35-mm Cellview dishes (Greiner Bio-One) with DMEM containing 5% LPDS for 16 h and then incubated on ice for 15 min with DMEM containing BODIPY-LDL (10 μg/ml) (Molecular Probes, ThermoFisher). Dishes were then mounted on the 37°C environmentally controlled stage of a Zeiss Cell Observer spinning disk confocal microscope equipped with an Axiocam Hrm CCD and a 63× (1.4 NA) oil immersion objective. Fluorescence intensity was quantified in Z-stack images collected over 0.24-μm intervals.
Analysis of cellular cholesterol distribution and mass
Cellular cholesterol was visualized with filipin (0.25 µg/ml for 30 min) after cells were fixed in 4% paraformaldehyde. Total and unesterified cholesterol mass was measured using the Amplex Red method (ThermoFisher Scientific) and expressed relative to cell protein.
mCherry-D4H (Maekawa and Fairn, 2015) and PH-PLCD1-GFP (Varnai and Balla, 1998) were used to visualize cholesterol and PI(4,5)P2, respectively, after transient coexpression in HeLa and ORP1L-null cells for 24 h. Cells were mounted on an environmentally controlled stage of a Zeiss Cell Observer spinning disk confocal microscope at 37°C and imaged as described above. Cellular mCherry D4H fluorescence intensity was quantified from confocal images split into PLCD-PH (PM and cell outline) and mCherry-D4H channels using ImageJ (v1.46). The total area of the cell and the cytosol was chosen based on the PLCD-PH channel and added to the ROI manager list. In the mCherry-D4H channel, the fluorescence intensity of respective areas was measured based on the ROI manager list. The fluorescence intensity of the total cell area is designated as Itotal while the cytosol is Icytosol. The mCherry-D4H fluorescence intensity on the PM is IPM = (Itotal – Icytosol) and was expressed as IPM/Icytosol*100. In cells transfected with ORP1-GFP or ORP-GFP and mCherry-D4H, the total cell area was selected based on the cytoplasmic GFP signal. A 0.1- to 0.3-μm perimeter corresponding to the PM was manually selected based on GFP fluorescence. Both areas were added to the ROI manager and then applied to the mCherry-D4H channel, and fluorescence intensity was quantified as described above.
The cholesterol content of the exofacial leaflet of the PM was quantified using recombinant mCherry-D4 expressed in Escherichia coli (provided by Barbara Karten, Dalhousie University). Cells cultured on glass coverslips were incubated in PBS containing mCherry-D4 (10 μg/ml) and Alexa Fluor 488-wheat germ agglutinin (WGA; 0.8 μg/ml) for 20 min on ice. Cells were washed twice with cold PBS and fixed in 3% PFA for 10 min. Cells were mounted and imaged by confocal microscopy, and the fluorescence intensity of the PM was quantified in individual cells that were WGA positive.
PCR analysis
Total RNA was isolated from cells (RNA Easy, Qiagen) and 1 µg was reverse transcribed (QuantiTect Reverse Transcription Kit, Qiagen). Quantitative PCR (qPCR) was performed on a CFX96 Real-Time System (Bio-Rad) using the 2-ΔCt method with normalization to GAPDH. The primer sets used to SREBP1, LDLR, HMGCR, and GAPDH were previously described (Zhao and Ridgway, 2017). Reverse transcriptase-PCR analysis of OSBPL1 mRNA expression was determined using primers flanking the deletion site.
Supplementary Material
Acknowledgments
We thank Mark Charman and Robert Douglas for technical assistance. Funding was received from the Canadian Institutes of Health Research (MOP-15284) and the Bernard and Winnifred Lockwood Endowment for Research. J.F. was supported by a John and Elizabeth Beattie studentship award.
Abbreviations used:
- ACAT
acyl-CoA:cholesterol acyltransferase
- BSA
bovine serum albumin
- ER
endoplasmic reticulum
- FCS
fetal calf serum
- FFAT
two phenylalanine in an acid tract
- gRNA
guide RNA
- LEL
late endosomes/lysosome
- LPDS
lipoprotein-deficient serum
- MCS
membrane contact sites
- 22OH
22-hydroxycholesterol
- ORD
OSBP-related domain
- ORP
OSBP-related protein
- OSBP
oxysterol binding protein
- PBS
phosphate-buffered saline
- PH
pleckstrin homology
- PIP
phosphatidylinositol polyphosphates
- PM
plasma membrane
- SBD
sterol-binding defective
- VAP
vesicle-associated membrane protein-associated protein
- WGA
wheat germ agglutinin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-12-0697) on February 5, 2020.
REFERENCES
- Bowden K, Ridgway ND. (2008). OSBP negatively regulates ABCA1 protein stability. J Biol Chem , 18210–18217. [DOI] [PubMed] [Google Scholar]
- Brown MS, Faust JR, Goldstein JL. (1978). Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem , 1121–1128. [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P. (2015). PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science , 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Du X, Wang H, Wang J, Lu C, Chen X, Zhu Z, Luo Z, Yu L, Brown AJ, et al (2019). Allosteric enhancement of ORP1-mediated cholesterol transport by PI(4,5)P2/PI(3,4)P2. Nature Comm , 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H. (2011). A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol , 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden ER, Sanchez-Heras E, Tsapara A, Sobota A, Levine TP, Futter CE. (2016). Annexin a1 tethers membrane contact sites that mediate ER to endosome cholesterol transport. Dev Cell , 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, Brown MS. (1983). Receptor mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol , 241–260. [DOI] [PubMed] [Google Scholar]
- Jansen M, Ohsaki Y, Rita Rega L, Bittman R, Olkkonen VM, Ikonen E. (2011). Role of ORPs in sterol transport from plasma membrane to ER and lipid droplets in mammalian cells. Traffic , 218–231. [DOI] [PubMed] [Google Scholar]
- Johansson M, Bocher V, Lehto M, Chinetti G, Kuismanen E, Ehnholm C, Staels B, Olkkonen VM. (2003). The two variants of oxysterol binding protein-related protein-1 display different tissue expression patterns, have different intracellular localization, and are functionally distinct. Mol Biol Cell , 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Lehto M, Tanhuanpaa K, Cover TL, Olkkonen VM. (2005). The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell , 5480–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen A, Arora A, Takahashi K, Kentala H, Kivela AM, Jaaskelainen E, Peranen J, Somerharju P, Ikonen E, Viitala T, Olkkonen VM. (2019). ORP2 interacts with phosphoinositides and controls the subcellular distribution of cholesterol. Biochimie , 90–101. [DOI] [PubMed] [Google Scholar]
- Lee S, Wang PY, Jeong Y, Mangelsdorf DJ, Anderson RG, Michaely P. (2012). Sterol-dependent nuclear import of ORP1S promotes LXR regulated trans-activation of apoE. Exp Cell Res , 2128–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, Gavin AC. (2013). Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature , 257–261. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Fairn GD. (2015). Complementary probes reveal that phosphatidylserine is required for the proper transbilayer distribution of cholesterol. J Cell Sci , 1422–1433. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. (2013). A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell , 830–843. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Polidori J, Jamecna D, Lacas-Gervais S, Antonny B. (2017). Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J , 3156–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser von Filseck J, Copic A, Delfosse V, Vanni S, Jackson CL, Bourguet W, Drin G. (2015). Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science , 432–436. [DOI] [PubMed] [Google Scholar]
- Ouimet M, Hennessy EJ, van Solingen C, Koelwyn GJ, Hussein MA, Ramkhelawon B, Rayner KJ, Temel RE, Perisic L, Hedin U, et al (2016). miRNA targeting of oxysterol-binding protein-like 6 regulates cholesterol trafficking and efflux. Arterioscler Thromb Vasc Biol , 942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrangelo A, Ridgway ND. (2018). Bridging the molecular and biological functions of the oxysterol-binding protein family. Cell Mol Life Sci , 3079–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, et al (2015a). Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature , 234–238. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Wenzel EM, Stenmark H. (2015b). ER-endosome contact sites: molecular compositions and functions. EMBO J , 1848–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc , 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J. (2009). Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol , 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK. (2014). ER contact sites define the position and timing of endosome fission. Cell , 1027–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, Schneider WJ, Yamamoto T, Luskey KL, Brown MS, Goldstein JL. (1984). Domain map of the LDL receptor: sequence homology with the epidermal growth factor precursor. Cell , 577–585. [DOI] [PubMed] [Google Scholar]
- Taylor FR, Saucier SE, Shown EP, Parish EJ, Kandutsch AA. (1984). Correlation between oxysterol binding to a cytosolic binding protein and potency in the repression of hydroxymethylglutaryl coenzyme A reductase. J Biol Chem , 12382–12387. [PubMed] [Google Scholar]
- van der Kant R, Zondervan I, Janssen L, Neefjes J. (2013). Cholesterol-binding molecules MLN64 and ORP1L mark distinct late endosomes with transporters ABCA3 and NPC1. J Lipid Res , 2153–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT. (2010). Niemann-Pick disease type C. Orphanet J Rare Dis , 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Balla T. (1998). Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol , 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara T, Uronen RL, Wohlfahrt G, Bjorkhem I, Ikonen E, Olkkonen VM. (2011). Sterol binding by OSBP-related protein 1L regulates late endosome motility and function. Cell Mol Life Sci , 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ma Q, Qi Y, Dong J, Du X, Rae J, Wang J, Wu WF, Brown AJ, Parton RG, et al (2019). ORP2 delivers cholesterol to the plasma membrane in exchange for phosphatidylinositol 4, 5-bisphosphate (PI(4,5)P2). Mol Cell , 1–16. [DOI] [PubMed] [Google Scholar]
- Zhao K, Ridgway ND. (2017). Oxysterol-binding protein-related protein 1L regulates cholesterol egress from the endo-lysosomal system. Cell Rep , 1807–1818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.