

Abstract
Tumor-associated macrophages (TAMs) have been shown to both aid and hinder tumor growth, with patient outcomes potentially hinging on the proportion of M1, pro-inflammatory/growth-inhibiting, to M2, growth-supporting, phenotypes. Strategies to stimulate tumor regression by promoting polarization to M1 are a novel approach that harnesses the immune system to enhance therapeutic outcomes, including chemotherapy. We recently found that nanotherapy with mesoporous particles loaded with albumin-bound paclitaxel (MSV-nab-PTX) promotes macrophage polarization towards M1 in breast cancer liver metastases (BCLM). However, it remains unclear to what extent tumor regression can be maximized based on modulation of the macrophage phenotype, especially for poorly perfused tumors such as BCLM. Here, for the first time, a CRISPR system is employed to permanently modulate macrophage polarization in a controlled in vitro setting. This enables the design of 3D co-culture experiments mimicking the BCLM hypovascularized environment with various ratios of polarized macrophages. We implement a mathematical framework to evaluate nanoparticle-mediated chemotherapy in conjunction with TAM polarization. The response is predicted to be not linearly dependent on the M1:M2 ratio. To investigate this phenomenon, the response is simulated via the model for a variety of M1:M2 ratios. The modeling indicates that polarization to an all-M1 population may be less effective than a combination of both M1 and M2. Experimental results with the CRISPR system confirm this model-driven hypothesis. Altogether, this study indicates that response to nanoparticle-mediated chemotherapy targeting poorly perfused tumors may benefit from a fine-tuned M1:M2 ratio that maintains both phenotypes in the tumor microenvironment during treatment.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02504-z) contains supplementary material, which is available to authorized users.
Keywords: Cancer immunotherapy, Macrophage polarization, Nanotherapy, Breast cancer liver metastases, Mathematical modeling, computational simulation
Introduction
Breast cancer disseminates to the liver in approximately 30–50% of patients suffering from metastatic disease [1]. Unfortunately, breast cancer liver metastasis (BCLM) median survival is 4.23 months, which compares unfavorably with metastases at other sites (e.g., lung 6–15 months, bone 33–48 months, and isolated soft tissue metastases median survival > 50 months) [1]. Despite recent developments in radiation, surgical techniques, and chemo- and target-specific immune/hormone therapies, BCLM remains a leading cause of mortality. In particular, the complexity of the BCLM microenvironment has hindered the development of efficacious chemotherapeutic strategies [2]. As the liver has a dense network of capillaries reaching inner cells and efficiently providing oxygen and soluble nutrients, BCLM do not initially rely on angiogenesis for survival but rather on the existing vasculature in the surrounding parenchyma [3]. At later stages, these metastases can also change the surrounding microenvironment via angiogenesis [4], as has been observed clinically [5]. Hypo-perfusion limits diffusive transport into BCLM, as is clinically observed via MRI (magnetic resonance imaging) by the lack of contrast agent permeation yielding hypo-attenuating lesions [6]. Experimental evidence supports the notion that hypovascularization makes BCLM less susceptible to chemotherapeutic agents [7]. We have previously observed that impaired vascularity in BCLM prevents macromolecules from fully penetrating these lesions [8]. Inadequate transport is especially acute with high molecular weight (HMW) molecules and particles, as has been shown for m99Tc microaggregated albumin [9].
The complexity of BCLM further leads to dynamic changes in the cells of the tumor microenvironment (TME), especially in tumor-associated macrophages (TAM) [10, 11]. TAM can be locally polarized to pro- or anti-inflammatory phenotypes, based on stimuli in the TME [12, 13]. The M1 phenotype favors an anti-tumor immune response [14], characterized by release of pro-inflammatory cytokines, such as IL-1, -6 and -12, TNFα, and reactive oxygen species (ROS), and expression of inducible nitric oxide synthase [15]. The M2 phenotype suppresses inflammation, favoring the formation of tumor stroma and neovasculature in a wound healing-type of response [16, 17], and thus promotes tumor development [18, 19]. Therapeutically induced macrophage polarization to M1 has been shown to inhibit cancer progression and metastasis [20], and has become a goal for immunotherapeutic strategies targeting macrophage populations. Recent examples include carboxyl- and amino-functionalized polystyrene nanoparticles [21] and immunostimulatory agents such as RRx-001 (ABDNAZ) [22]. For breast cancer, immunotherapy involving checkpoint inhibitors or cancer vaccines in combination with established treatment strategies is undergoing promising evaluation [23].
As TAM tend to accumulate near hypoxic tissue, which is difficult to reach via vascular-borne molecules [24], we have recently evaluated taking advantage of their presence in the TME to overcome the limitations of therapeutics targeting hypo-perfused lesions. We have shown that shifting the transport of a chemotherapeutic drug from circulation towards TAM in BCLM can significantly improve outcomes and survival benefits [8, 25]. As professional phagocytes, macrophages recognize circulating solid particles, and have been shown to be a suitable target for intravenously administered nanotherapeutics. As terminally differentiated cells, macrophages are unaffected by most anti-cancer therapeutics, and, thus, can act as sources of drug in the vicinity of hypo-perfused tumor tissue, especially for HMW therapeutics.
In this study, we develop an interdisciplinary framework to facilitate effective analysis of immunotherapy aiming to affect macrophage polarization in BCLM to maximize the cytotoxic effect of HMW-based therapeutics. As a purely empirical approach would be insurmountable due to the complex interaction between nanotherapies, drugs, cells, and the TME, we employ both experimental and computational approaches to evaluate therapeutic response by shifting the transport of therapeutics towards macrophages in the TME while inducing polarization towards the M1 phenotype. In particular, we employ for the first time a CRISPR (clustered regularly interspaced short palindromic repeats) system to permanently modulate macrophage polarization. This modification allows study with varying ratio of M1 and M2 macrophages in a controlled in vitro environment. The experimental component is performed in a 3D co-culture that mimics the hypovascularized TME of BCLM, as previously reported [8, 25]. To enable enrichment of the M1 phenotype, we use liposomes loaded with CRISPR complex targeting RICTOR, rapamycin-insensitive companion of mTOR (mammalian target of rapamycin). Although several factors and pathways are involved in macrophage polarization, in our studies we have discovered that CRISPR-RICTOR-Liposomes are one of the most efficient systems to prevent polarization to the M2 phenotype [26]. RICTOR is an adapter protein in the mTORC2 complex, and has been shown to influence differentiation of immunosuppressive M2 macrophages [27, 28]. We have found that knockdown of the RICTOR gene can block M2 differentiation and redirect the polarization towards the M1 subtype, even when subjected to pro- M2 stimulatory influences in the TME.
Materials and methods
Experimental system for cancer cell and macrophage co-culture
Cell culture
4T1 mouse breast cancer cells were cultured in MEM (minimum essential medium) with the addition of 10% FBS (fetal bovine serum), 1% antibiotic/antimycotic, 1% GlutaMAX, 1% NEAA (non-essential amino acids), 1% MEM vitamin, and 1% sodium pyruvate supplements and maintained in humidified atmosphere at 37 °C and 5% CO2.
Mouse macrophages were obtained by isolation from hind leg bone marrow from Balb/c mice (6–8 weeks, females), as previously described [25]. Briefly, mouse bone marrow was extracted by flushing with syringe, washed twice with PBS (phosphate-buffered saline), and erythrocytes were lysed by red blood cell lysis buffer (Sigma, USA). Cells were filtered with a 70-µm filter (BD Lifesciences, USA). To initiate differentiation to resting macrophages (M0), cells were incubated with macrophage medium, containing 10% FBS and 1% penicillin/streptomycin in RPMI 1640 medium. Further differentiation of macrophages was initiated by incubation in relevant media with the addition of the following factors: 50 ng/mL IFN-γ and 20 ng/mL LPS for M1 macrophage differentiation, and 50 ng/mL IL-4 and 50 ng/mL M-CSF for M2 macrophage differentiation.
crRNA production by in vitro transcription
CRISPR crRNA sequences targeting RICTOR were designed for the use with Cas12a using Benchling tool (benchling.com/crispr). Template oligonucleotides in reverse complement sequence which included T7 promoter were ordered from Eurofins Genomics (Louisville, KY). gRNA (guide RNA) were obtained by transcribing template oligonucleotides using MEGAscript™ T7 Transcription Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Produced crRNA was purified using Oligo Clean & Concentrator™ (Zymo Research, Irvine, CA, USA). crRNA concentration was measured by assessing the absorption at 260 nm using Take3 plates and Synergy H4 Hybrid Reader (Biotek, Winooski, VT, USA).
CRISPR liposome design and characterization
CRISPR liposomes were designed to enrich the population of M1 macrophages through RICTOR molecular pathway targeting (CRISPR-RICTOR-Liposomes), which prevents macrophage differentiation into M2 phenotype. Liposomes were prepared using lipid hydration–extrusion method as previously described [29]. Briefly, 3.62 mg soybean phosphatidylcholine (Lipoid S100, Lipoid, Germany), and 0.88 mg 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) (Avanti Polar Lipids, Alabama, USA) were dissolved in 5 mL ethanol. Solvent was evaporated for 30 min at 41 °C and 150 rpm using rotary evaporator (Rotavapor, Buchi, Switzerland). Resulting thin film was rehydrated with 0.5 mL PBS, pH 7.2, and further sonicated intermittently for 15 min using Branson 1510 bath sonicator (Branson, Danbury, CT, USA) to create a homogeneous dispersion. CRISPR systems were prepared by mixing 1 μg Cas12a nuclease (Integrated DNA Technologies, Coralville, IA, USA) to 400 ng sgRNA (single-guide RNA) in 50 μL serum-free MEM medium for 5 min and adding this mixture to 2 μL of liposome dispersion. CRISPR-RICTOR-Liposome size and zeta potential were assessed by dynamic light scattering using Zetasizer instrument (Malvern, Worcestershire, UK). Analysis was conducted in triplicates.
Characterization of CRISPR effect on macrophages
Undifferentiated primary macrophages were seeded on 16-well chamber slides (Nunc™ Lab-Tek™) with 30,000 cells/cm2. CRISPR-RICTOR-Liposomes were added to each well at 5 μL liposome/mL, and cells were kept overnight under incubation. After 24 h, the medium was changed and samples were stimulated with IL-4 and M-CSF to induce M2 differentiation. After 48 h of incubation, cells were fixed with 4% paraformaldehyde for 30 min at 4 °C. Macrophages were stained with 2.5 μg/mL rat anti-mouse CD80 primary antibody (Thermo Fisher Scientific, Waltham, MA), Alexa Fluor 568-goat anti-rat IgG secondary antibody, and FITC-rat anti-mouse CD204 antibody (Abcam, Cambridge, UK) for macrophage phenotype analysis, and counterstained with DAPI (4′,6-diamidino-2-phenylindole) for nucleus stain. Cells were analyzed using Nikon A1 confocal microscope (Nikon Inc., Melville, NY, USA) and macrophage phenotypes were assessed with NIS-Elements software (Nikon Inc.).
Western blot analysis
Protein expression of CRISPR target RICTOR was analyzed in bone marrow-derived macrophages via Western blot. Isolated mouse macrophage cells were seeded in six-well plates and incubated with CRISPR-Liposome targeting RICTOR for 48 h. At the end of incubation, semi-adherent and adherent cells were collected, centrifuged (200×g for 5 min), and the resulting cell pellet was washed with ice-cold PBS before lysing with radioimmunoprecipitation assay buffer with Halt™ protease and phosphatase cocktail (Thermo Scientific, USA) and left on ice for 20 min. Combined cell lysates were briefly sonicated at 20% amplitude for 10 s using a probe sonicator (QSonica, LLC, Newton, CT, USA). Sonicated lysates were centrifuged (15,000×g for 20 min). Supernatants (cell lysates) were collected and used for protein determination using a Pierce™ BCA (bicinchoninic acid) kit (Thermo Scientific, USA), following the manufacturer’s instructions. Aliquots of cell lysates were denatured at 95 °C for 5 min after the addition of 6 × Laemmli sample buffer (Alfa Aesar, MA, USA). Proteins in cell lysates were resolved by electrophoresis on a 3–8% Tris–acetate gels (NuPage™, Invitrogen, USA) using a constant voltage of 150 V for 1 h. Resolved proteins were blotted onto PVDF (polyvinylidene fluoride) membrane using iBlot™ stacks at 20 V for 10 min using a semi-dry iBlot™ system (Invitrogen, USA). Blots were then blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline solution containing 0.1% Tween-20 (TBST) for 1 h at room temperature. Probing with primary antibodies was then started under gentle shaking overnight at 4 °C (RICTOR rabbit monoclonal antibody, Cell Signaling Technology, 1:1000 dilution in 5% BSA in TBST and mouse monoclonal β-actin antibody (Invitrogen), 1:10,000 in 5% BSA). Blots were then washed using TBST (3 × 5 min, room temp) and probed with appropriate horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibodies for RICTOR (Cell Signaling Technology) and HRP-conjugated antimouse β-actin antibody (Invitrogen). Membranes were then washed as above in TBST. Protein bands were visualized using Forte™ ECL (enhanced chemiluminescence) kit (EMD Millipore, MA, USA) using manufacturer instructions. Blots were imaged using a ChemiDoc XRS + CCD (charge-coupled device) Imager (BioRad, USA). Densitometric analysis of images were performed using VisionWorks LS™ analysis software V8.20 (UVP, LLC) for RICTOR bands and normalized to corresponding β-actin intensity. Data were analyzed for significance (P < 0.05) using Student’s t test on GraphPad Prism software V8.0 (San Diego, USA).
RNA isolation and qPCR analysis
Macrophages were stimulated to differentiate into M2 phenotype with IL-4 and M-CSF for 5 days before the experiment. Macrophages were then treated with CRISPR-RICTOR-Liposomes and cultured in M2 medium to maintain an environment favorable for differentiation to M2. After 4 h of treatment, the total RNA was isolated from macrophages using RNeasy Mini Kit (Qiagen, Hilden, Germany), followed by reverse transcription using QuantiTect® Reverse Transcription Kit (Qiagen). Resulting cDNA (complementary DNA) was diluted in nuclease-free water before real-time polymerase chain reaction (PCR) step. mRNA (messenger RNA) levels were measured using QuantiTect® SYBR® Green PCR Kit (Qiagen) and StepOnePlus™ Real-Time PCR System (Applied Biosystems™). Gene expressions were normalized to the expression of corresponding β-actin housekeeping gene.
Efficacy studies in 3D TME co-culture model of breast cancer spheres and macrophages
4T1 breast cancer cell tumor spheres were generated using Bio-Assembler™ system based on protocols we reported previously [8, 25]. For cytotoxicity studies, spheroids were grown to ~ 450–500 μm diameters in 96-well plate. Macrophages were treated with CRISPR-RICTOR-Liposomes one day before co-culture for blocking differentiation to M2. Another batch of macrophages was differentiated to M2 according to the above protocol. On the day of co-culture, both macrophages were tested for their phenotype with rat anti-mouse CD80 antibody (Thermo Fisher Scientific, Waltham, MA) confirming M1 phenotype and rabbit anti-mouse CD204 antibody (Abcam, Cambridge, UK) confirming M2 phenotype. After phenotype confirmation, macrophages were harvested and co-cultured with 4T1 spheroids with different ratio of CRISPR-RICTOR- liposome-treated macrophages (M1) and M2 macrophages, with 2 × 103 macrophages in total.
In the efficacy study with mesoporous particles loaded with albumin-bound paclitaxel (MSV-nab-PTX), M1 and M2 macrophages were seeded in co-culture with 4T1 spheroids at different ratios of M1 and M2, similar to the previous setup. Further, MSV-nab-PTX was added to the co-culture at a dose equivalent to ~ 30 ng of PTX (paclitaxel). After 48 or 72 h, the 4T1 spheres were harvested and analyzed for viability using CellTiter-Glo 3D Cell Viability Assay (Promega, Madison, Wisconsin) according to the manufacturer’s protocol.
Evaluation of CRISPR-RICTOR-Liposomes effect in vivo in breast cancer model
Breast cancer model was generated by orthotopic injection of 105 4T1 cells/100 μL PBS in Balb/c mice (6–8 weeks, females). Tumors were grown for 10 days prior to injection of CRISPR-RICTOR-Liposome systems. Animals were randomized into treatment and control groups (n = 6), and 100 µL of CRISPR-RICTOR-Liposomes or 100 µL PBS (control) were injected intratumorally to the lesion. Mice were sacrificed after 24 h. Tumors were harvested and frozen in optimal cutting temperature (OCT) compound followed by cryo-sectioning and staining for CD80 (M1) and CD206 (M2) macrophage markers. Slides were stained with Alexa Fluor647-anti mouse CD80 and Alexa Fluor488-anti mouse CD206 antibodies (Abcam, Cambridge, UK), and counterstained with DAPI (Thermo Fisher, Waltham, MA, USA). Colocalizations of CD80 in macrophages were confirmed by co-staining of CD80 and Alexa Fluor488-anti mouse CD11b antibodies (Abcam, Cambridge, UK).
Mathematical model of BCLM response to therapy as a function of macrophage polarization
The model presented in [8, 25] simulated the tumor response to MSV-nab-PTX nanoparticles taken up by macrophages. This model was extended here to simulate the effect of a hypothetical agent affecting macrophage polarization (AAMP) towards M1 or M2 subtypes. The main model parameters are summarized in Supplementary Table 1, with values previously calibrated in [8, 25, 30, 31] to achieve biologically meaningful results. The model includes the vasculature in the extratumoral space because it represents the in vivo condition of the metastatic lesions in the liver, for which the extratumoral space is vascularized. The angiogenesis model component simulates the model by [32] and is based on [30, 31]. Oxygen and vasculature-related parameters are as in [2, 3]. A simplified liver vascular organization composed of square elements is simulated, acknowledging that in biological reality, these elements are heterogeneously delineated by the sinusoids between the portal tracts and central veins at high density.
The model was calibrated following [8]. Details of the numerical implementation are in [31] and references therein, including [30].
Simulation of macrophages
As in [33], undifferentiated macrophages extravasate from the vasculature in proportion to local concentration of macrophage chemoattractants (e.g., pro-angiogenic factors released by tumor cells). They migrate through the interstitium following gradients of oxygen, chemoattractants, and pressure, as described in [33]. Polarization into M1 or M2 subtypes occurs in the vicinity of the tumor microenvironment based on the ratio of pro-M1 and pro-M2 macrophage factors released by viable tumor cells [8, 25]. The number of macrophages and their localization in the simulations is stochastic, and thus variability is introduced in the M1:M2 ratios. Simulations were run n = 5 to obtain statistically significant results.
M1 macrophages are simulated to penetrate deeper than M2 subtypes into tumor tissue, as shown in Supplementary Fig. 1a (showing an average macrophage number), to replicate this effect observed in our recently published experiments [25]. This effect is modeled via an additional chemoattractant with increasing concentration towards the center of the lesion selectively influencing M1 movement [25].
Further, we have experimentally observed [25] that the presence of MSV-nab-PTX shifts the ratio of M1 to M2 macrophages to be 1.2:1.0. This effect is calibrated in the model by simulating a one-time bolus injection of MSV-nab-PTX into the system. Supplementary Fig. 1b shows the accumulation of MSV-nab-PTX-loaded M1 and M2 macrophages in the simulated tumor over time. The number of drug-loaded macrophages peaks in the tumor within 24 h post-MSV-nab-PTX injection, and decreases to zero by 36 h. Although macrophages in vivo have longer and more variable lifespans in the TME, this timeframe provides a consistent period for the evaluation of macrophage polarization and its effects on the growing tumor.
Macrophages act as point sources of drug to simulate the release of paclitaxel from the MSV-nab-PTX system, as described in [8]. This drug is diffused in the TME to induce local cytotoxicity [8, 25]. For simplicity, drug uptake by tumor tissue, death effect, and washout from the interstitium are simulated to take effect immediately [34]. Additionally, M1 macrophages induce cytotoxicity while M2 promote tumor cell proliferation [25, 33].
Simulation of agent inducing polarization to M1 phenotype
Macrophage polarization to the M1 phenotype is driven by simulating a bolus infusion of a hypothetical “agent affecting macrophage polarization” (AAMP), here generically named N:
AAMP with diffusivity DN is released at rate from vessels at location 1vessel (of value 1 if a vessel is present and 0 otherwise), and decays at rate , acting locally on undifferentiated macrophages to promote their conversion to the M1 phenotype. The likelihood of conversion is proportional to the local concentration of N and its strength, [35]. This strength starts with a value of 0 to attain a M1:M2 ratio of 1.2:1 (as experimentally observed when MSV-nab-PTX are present [25]), and is incremented in steps of 80 to achieve an increasing magnitude of these ratios, as depicted in Supplementary Fig. 2.
Statistical analysis
Data were statistically analyzed using Student’s t test on GraphPadPrism software. p values below 0.05 were considered significant, and p < 0.01 as very significant.
Results
Since macrophage polarization is a dynamic process influenced by the TME, to achieve a stable phenotype we used here for the first time a CRISPR system targeting RICTOR that allows to block M2 and to induce M1 macrophage differentiation [27, 28, 36]. As this modulation of M1 to M2 ratio can only be achieved in a controlled in vitro environment, a 3D co-culture system that mimics the hypovascularized TME of BCLM was employed, as previously reported [8].
Treatment with CRISPR-RICTOR-Liposome was able to knock down RICTOR expression, as shown in western blot in Fig. 1a and quantified in Fig. 1b. Fluorescence images in Fig. 1c show that a stable M1 phenotype was achievable even when stimulated to differentiate towards M2. Further, the reprogramming was assessed by measurement of mRNA expression of inflammatory-related genes. Treatment of M2-differentiated macrophages with CRISPR-RICTOR-Liposomes caused a switch to an M1-like subtype, as indicated by the increase in the expression of pro-inflammatory genes. As shown in Fig. 1d, expression of MCP-1, MIP-1α, MIP-1β, RANTES, KC, and IL-1β were significantly elevated (up to 100-fold induction). On the other hand, expression of anti-inflammatory cytokine IL-10 was reduced, as expected when macrophage polarization shifts to a more pro-inflammatory M1 subtype. Differentiation of CRISPR-treated macrophages was evaluated following cell stimulation with IL-4 and M-CSF, which normally induces M2 differentiation. Cells not treated with IL-4 and M-CSF showed preferential differentiation towards the M1 macrophage phenotype, with ~ 75% M1 macrophages in both CRISPR-treated and -untreated control. CRISPR treatment was very effective in blocking M2 differentiation upon stimulation with IL-4 and M-CSF, with the cells retaining ~ 85% of the M1 phenotype, while all cells that did not undergo genetic editing differentiated towards the M2 phenotype (Fig. 1e).
Fig. 1.

Evaluation of effects of CRISPR-RICTOR-Liposome on M2 polarized macrophages in vitro. a Immunoblotting analysis of RICTOR protein expression in untreated (MΦ) versus macrophages treated to CRISPR-CRISPR-Liposome (MΦ CRISPR). Isolated mouse macrophages were cultured, treated, and analyzed by Western blot. b Densitometric analysis of RICTOR band intensities normalized to β-actin. n = 3, *significant to untreated macrophage control (MΦ) (p < 0.05). c Effect on macrophage differentiation of CRISPR treatment targeting RICTOR, coupled with macrophage differentiation stimulated towards M1 (IFNγ + LPS) or M2 (IL-4 + M-CSF). Cells were stained with CD163 (green- M2 marker) and CD80 (red-M1 marker). d mRNA expression from M2 macrophages with and without treatment with CRISPR-RICTOR-Liposome, measured via qPCR (n = 4). e Quantitative analysis of cell phenotype. Scale bar = 100 μm, mean ± SD, biological replicates n = 5, *p < 0.05, **p < 0.01 vs. control
The ability of the CRISPR-RICTOR-Liposome treatment to shift the macrophage phenotype was evaluated in an in vivo model of breast cancer. The treatment caused a more than 12-fold increase in fluorescence intensity of the M1 macrophage marker CD80 in the tumor tissue as compared to the tumor injected with PBS (Supplementary Fig. 3). CD80 has been reported as a robust phenotypic marker for human MΦIFN-γ (M1 phenotype) macrophages [37]; however, as it can also be expressed by other immune cells (e.g., B and T cells), we confirmed that the signal originated with M1 macrophages by co-staining with the CD11b pan-macrophage marker (Supplementary Fig. 4). The co-localization of CD80 and CD11b fluorescent signals shows that the vast majority of CD80+ cells were M1 macrophages in the CRISPR-Lip-treated tumor. In the untreated control, while CD11b staining intensity was similar to the treated tumor, there was a very weak signal of CD80+, as expected since most of the macrophages in the tumor were M2-like phenotype. The figure further shows that the level of cells expressing the M2 marker CD206 was not significantly affected by the treatment. CD206 is a well-accepted (classical) and very specific marker for M2 differentiation [38, 39], which has been used as a benchmark to evaluate new M2 candidate markers [40].
Next, we evaluated the response to MSV-nab-PTX at different M1:M2 ratios. We confirmed that the macrophage phenotypes as well as the M1:M2 ratios in the presence of cancer spheroids remained stable for the duration of the experiments, equaling the initial ratios introduced at t = 0 (Supplementary Fig. 5). The ratios in the figure refer to macrophages differentiated towards the M2 phenotype vs. macrophages arrested in the M1 phenotype using CRISPR. In Fig. 2, varying strengths of a hypothetical “agent affecting macrophage polarization” (AAMP) that shifts macrophage polarization towards the M1 subtype were mimicked by varying the ratio of M1:M2 subtypes in the spheroid co-cultures. When the cells were not treated with MSV-nab-PTX, cancer cell viability increased as this ratio decreased, plateauing for values lower than 1:1 for both 48 and 72 h treatment durations. In contrast, the MSV-nab-PTX case was non-uniform with respect to this ratio. The lowest cell viability for 48 h exposure was for the M1-only case and increased with higher numbers of M2, with M1:M2 ratios of 1500:500 and 1000:1000 showing statistically similar results. For 72 h, an M1:M2 ratio of 1500:500 achieved lower viability than 2000:0 (all M1), indicating that the presence of the M2 subtype augments the therapeutic efficacy. Likewise, the ratio of 0:2000 (all M2) yielded lower viability than the 500:1500 ratio.
Fig. 2.

Viability of breast cancer cells growing in tumor spheres representing hypo-vascularized BCLM in 3D co-culture with M1 and M2 polarized macrophages. M1 macrophages were CRISPR-RICTOR-Liposomes treated and differentiated in the presence of IFN-gamma/LPS, while M2 macrophages were polarized in vitro in the presence of IL-4/M-CSF. The viability is shown as a function of varying ratios of M1:M2 macrophages under conditions of no treatment or exposure to MSV-nab-PTX for a 48 h and b 72 h. The varying ratios mimic the varying strength of a hypothetical polarization regimen that shifts this ratio. Mean ± SD, biological replicates n = 5, *p < 0.05, **p < 0.01 vs control
To more systematically explore the effect of macrophage polarization on the response of hypovascularized cancer lesions to MSV-nab-PTX, we applied mathematical modeling to simulate BCLM growth and treatment response. With the model parameters calibrated as described in “Materials and methods”, a representative BCLM lesion was first grown by the model to a diameter ~ 400 μm (Supplementary Fig. 6). As the lesion grows, tissue regions with adequate access to oxygen and nutrients are able to proliferate while regions with inadequate access become hypoxic. The dense liver capillary network is modeled by the rectangular grid, with irregular sprouts generated through angiogenesis during the lesion progression.
During the tumor growth process, individual macrophages are recruited to the vicinity of the lesion based on attraction to chemoattractants released by the hypoxic tumor cells. Based on the local TME conditions, these macrophages differentiate into M1 or M2 subtypes, which, respectively, either hinder or aid the tumor progression. The M1 macrophages release cytotoxins, e.g., nitric oxide, which affect the viability of tumor cells in their immediate vicinity. This is simulated to alter the tumor tissue proportionally to the concentration of the toxins released. Figure 3a shows the anti-proliferative effect of M1 macrophages. As the released nitric oxide from M1 macrophages is quickly degraded, the model simulates a cytotoxic effect in the locations of M1 macrophages to achieve a local effect. The effect of the nitric oxide is reflected by the tumor shrinkage compared to the initial condition (Supplementary Fig. 6). Figure 3b shows the same situation but including MSV-nab-PTX uptake by the M1 macrophages 24 h after therapy initiation, which accentuates the tumor shrinkage. The M2 macrophages release growth factors (e.g., TGF-β) that stimulate the survival and proliferation of tumor cells. This effect was simulated in the model by having the M2 subtypes release a generic growth factor that adds to the overall proliferation term. In the model, these growth factors up-regulate the proliferation of both proliferating and hypoxic tumor tissues, which increases the overall tumor growth rate. Figure 3c shows the M2 macrophage influence on the tumor growth. In this snapshot, the macrophages are in peak concentration and predominantly distributed within the tumor or at the boundary. Figure 3d shows the same situation but includes MSV-nab-PTX uptake by the M2 macrophages 24 h after therapy initiation, which now leads to substantial tumor shrinkage.
Fig. 3.

Simulation of polarized macrophage activity on a growing BCLM lesion at 24 h post-initiation. aM1-only, without MSV-nab-PTX treatment; bM1-only, with MSV-nab-PTX shown. cM2-only, without MSV-nab-PTX treatment; dM2-only, with MSV-nab-PTX. As the lesion shrinks during treatment [with viable tumor tissue (red) enclosing a hypoxic region (blue) without necrosis], the oncotic pressure (non-dimensional units) due to cell proliferation correspondingly decreases. The dense liver capillary network is modeled by the rectangular grid (brown), with irregular sprouts generated through angiogenesis during the lesion progression. The M2-derived growth factor (non-dimensional units) is only present for the M2 case. Bar = 200 μm
Next, we evaluated the delivery of a hypothetical AAMP to the tumor microenvironment to dynamically affect the M1:M2 polarization, which in turn alters the tumor growth characteristics. An increasing strength of AAMP was simulated to have a corresponding steady increase in the ratio of M1 to M2 macrophages. Supplementary Fig. 2 summarizes the average M1:M2 ratio for different cases of the AAMP strength. As is increased, the probability of AAMP causing conversion to an M2 macrophage is increased. With five simulation samples for each AAMP strength (n = 5), there is variability in the M1:M2 ratio. This is due to the stochastic nature of macrophage production and movement through the simulated TME. Macrophages near the vasculature may have more exposure to AAMP and, thus, locally higher M1:M2 ratios. Despite this variability, an overall trend of increasing M1:M2 ratios with increasing AAMP strength is attained (Supplementary Fig. 2).
The strength of AAMP increases the overall M1 macrophage population and decreases the M2 population, while both of these subtypes are also releasing albumin-bound paclitaxel (nab-PTX) in the tumor vicinity from the MSV-nab-PTX that they have taken up in the circulation. Figure 4 qualitatively displays the tumor course over 36 h under exposure to AAMP therapy of medium strength (, for which M1:M2 is 3.0:1), while simultaneously simulating a bolus injection of MSV-nab-PTX drug-loaded nanotherapy at 0 h. The MSV-nab-PTX uptaken by the macrophages are retained near and within the lesion by the macrophage infiltration, while the nab-PTX is released from them in the tumor proximity. The hypothetical AAMP administered intravenously as a bolus injection to polarize the macrophages has its maximum concentration at t = 0 h, and is progressively washed out from the tissue. Both PTX and the macrophages reach maximal numbers later in the simulation, as shown at 12 h. By 36 h, the immunotherapy and PTX effects have waned, and the tumor begins to regrow.
Fig. 4.

Simulated progression over 36 h (a through d) of a representative tumor lesion after exposure to a medium strength () of a hypothetical “agent affecting macrophage polarization” (AAMP), simulating an immune therapeutic (as a fraction of the maximum in the vasculature) shifting the M1:M2 ratio to 3.0:1, in conjunction with a bolus injection of MSV-nab-PTX drug-loaded nanoparticles. Colors as in Fig. 3. Bar = 200 μm
The tumor response over time when treated with MSV-nab-PTX loaded macrophages is shown in Fig. 5. A general trend of increased M1:M2 ratio leading to decreased tumor size can be seen (Fig. 5a). When simulating the effect of M1 only (i.e., inactivated M2, Fig. 5b) while maintaining the same proportion of macrophages, i.e., the same number of activated M1, the tumor response is significantly less than when the M2 are active (Fig. 5a), even in the case of a high ratio of M1:M2 of 3.8:1. Thus, a dual effect of the M2 macrophages is predicted by the model. Since PTX is a cell-cycle inhibitor, M2 macrophages potentiate the effect of AAMP during treatment while also accelerating tumor recovery post-treatment (Fig. 5a). To evaluate this effect further, we simulated repeated treatment cycles with MSV-nab-PTX. For treatments every 2d (Fig. 5c or every 3d (Fig. 5d), the presence of both M1 and M2 subtypes yielded significantly higher tumor regression than when M2 macrophages were inactive.
Fig. 5.

Simulated average tumor radius (n = 5, mean ± SD) over time when treated with MSV-nab-PTX-loaded macrophages. a Single treatment with both M1 and M2 subtypes active for three different M1:M2 ratios; b single treatment with only M1 active for three different M1:M2 ratios; c treated every 2d with M1:M2 of 3.0:1; d treated every 3d with M1:M2 of 3.0:1
Figure 6 summarizes the minimum tumor radius achieved by the MSV-nab-PTX bolus injection for the three treatment protocols in Fig. 5 in the case of an M1:M2 ratio of 3.0:1, comparing the case when both M1 and M2 are active vs. when M2 is inactive. The model results indicate that the presence of the M2 can provide a significantly higher tumor regression, whether the MSV-nab-PTX is administered as a single dose or in multiple doses over a number of days.
Fig. 6.
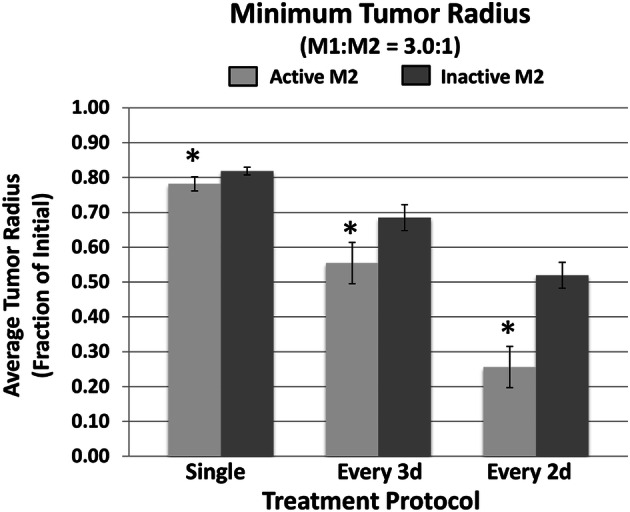
Simulated minimum tumor radius achieved by the MSV-nab-PTX bolus injection for three treatment protocols in the case of an M1:M2 ratio of 3.0:1. Simulation results (n = 5, mean ± SD) are shown for single dose, administration every 3d, and administration every 2d. (*p < 0.05)
Altogether, these results indicate that there may exist tumor-specific conditions for which a certain number of MSV-nab-PTX-loaded M2 may help to amplify the tumor response due to the M2 phenotype promoting tumor proliferation in the presence of cell-cycling drugs, whereas a predominantly M1 population would be less effective.
Discussion
Targeting macrophages in the TME is gaining recognition as a promising strategy for tumor therapy [41], with the critical role of macrophages in cancer growth, progression and immunotherapy recently reviewed in [42], highlighting the potential of innate immunity/macrophage modulation to restrain tumor growth. Macrophages are a key cell population of the innate immune system, with the M1 type triggering naïve T cells to have a Th1/cytotoxic response and the M2 type triggering T cells to have a Th2-type response associated with antibody production [43]. Both types serve multi-faceted purposes to maintain tissue homeostasis, with the M1 upholding immunity against foreign threats and the M2 modulating tissue repair and healing. Consequently, an imbalance in either one can potentially lead to severe illness; strong M1 activity has been associated with auto-immune diseases and organ rejection, while predominance of M2 activity has been linked to tumor progression.
Multiple studies focusing on finding agents that can shift macrophage polarization from an anti-inflammatory and tumorigenic M2 phenotype to a pro-inflammatory and anti-cancerous M1 phenotype are underway [25, 44–46]. While modulating macrophage polarization as a solo therapy has shown some promise [44], pronounced clinical benefits are expected mainly in combination with standard therapy. Recently, we reported that MSV-nab-PTX nanotherapy shifts the transport of therapeutics in BCLM from circulating in the bloodstream and, thus, unable to penetrate hypo-perfused metastatic lesions, to therapeutics specifically taken up and retained/transported by macrophages into the BCLM TME to be released there [8]. Integrating both experiments and mathematical modeling, we demonstrated that the proposed nanotherapy targeting cancer cells can additionally influence macrophage polarization from M2 to M1 [25].
In the current study, we evaluated the response of breast cancer cells to MSV-nab-PTX in 3D co-culture mimicking the TME of hypovascularized lesions in the liver with various ratios of polarized macrophages. To enable a stable polarization of macrophages and avoid their repolarization under the dynamic biochemical stresses in the TME, CRISPR technology was utilized. Consistent with the computational simulation predictions from our previous work [25], our experimental results here confirmed that the response to the cytotoxic agent MSV-nab-PTX depends non-linearly on the M1:M2 ratio. To explore this phenomenon further, we employed the mathematical modeling to analyze the effects of therapy while simulating manipulation of the macrophage phenotype via a hypothetical “agent affecting macrophage polarization” (AAMP). Although the role of macrophages in cancer therapy has been previously investigated via mathematical modeling, as recently reviewed in [47], the influence of varying macrophage phenotypes on the response to nanotherapy has had limited evaluation. The model-based finding that the M2–tumor interaction may have a dual role in the response to MSV-nab-PTX, initially promoting tumor death and subsequently aiding tumor recovery, highlights the nonlinear effect of the macrophage polarization in the TME during treatment. This interaction is expected to depend on nanotherapy and tumor tissue-specific conditions, including vascularization, hypoxia, and other microenvironment characteristics affecting macrophage behavior, which require further elucidation. Further, the model results suggest that immunotherapy strategies solely based on maximizing the M1:M2 ratio may be less effective than protocols which establish an M1:M2 proportion that first maximizes tumor regression during chemotherapeutic exposure, and then maximizes this ratio in favor of the M1 phenotype during the tumor recovery phase.
It is to be noted that there are multiple factors in the liver metastatic TME affecting tumor growth and therapy response. These factors include, among others, liver fibrosis and activation of stellate cells, T-cell exhaustion, and enrichment of myeloid-derived suppressor cells (MDSCs). Here, we focused on the polarization of tumor-associated macrophages, which in itself may be insufficient to restrain metastatic growth. However, it is considered valuable to study each of these factors to understand the associated mechanisms and to explore therapeutic combinations. The results here show that the polarization of macrophages may play an important role in the planning of combinatorial therapeutic regimens. We further note that PTX is a chemotherapeutic that targets proliferating cells; thus, differentiated cells such as macrophages are typically unaffected by PTX. We have previously shown that macrophage viability was not impacted by PTX up to 50 µg/mL in vitro, and that macrophages in uninvolved liver were not affected by MSV-nAb-PTX treatment in vivo [48]. However, PTX in high concentration may induce intracellular signals that mimic lipopolysaccharides in murine macrophages [49].
The goal for effective therapy would be to deliver and maintain a therapeutic drug dose to a target site while minimizing systemic toxicity. Numerous macromolecule-based therapeutic strategies have been proposed and clinically approved in recent years to treat advanced breast cancer, including albumin-bound drug conjugates (e.g., nab-PTX or Abraxane®), various antibodies [e.g., anti-HER2 mAb (monoclonal antibody) or Trastuzumab] and genetic materials [such as siRNA (small interfering RNA), miRNA (micro-RNA) and aptamers]. In hypo-vascularized BCLM, these potent therapeutics are unable to be transported in cytotoxic concentrations into tumor tissue prior to their clearance from circulation. Thus, new approaches to enhance therapeutic macromolecule accumulation in hypo-perfused tumor tissue are necessary. The results in this study show that effectiveness of a cytotoxic regimen with MSV-nab-PTX, which has shown promise to overcome these transport barriers in vitro [8, 25] and in vivo [45] by leveraging phagocytic uptake by TAM in the TME, could potentially be accentuated with immunotherapy that adjusts the M1:M2 ratio to first boost tumor death during drug exposure and then to hinder tumor recovery post-chemotherapy. Further work is necessary to elucidate the therapy and TME parameters that define the conditions to maximize response. The interdisciplinary framework presented here lays a first step towards the design of therapies customized to specific TME and immunological conditions.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- AAMP
Agent affecting macrophage polarization
- BCA
Bicinchoninic acid
- BCLM
Breast cancer liver metastasis
- BSA
Bovine serum albumin
- CCD
Charge-coupled device
- cDNA
Complementary DNA
- CRISPR
Clustered regularly interspaced short palindromic repeats
- crRNA
CRISPR RNA
- DAPI
4′,6-Diamidino-2-phenylindole
- DNA
Deoxyribonucleic acid
- ECL
Enhanced chemiluminescence
- FBS
Fetal bovine serum
- gRNA
Guide RNA
- HMW
High molecular weight
- HRP
Horseradish peroxidase
- mAb
Monoclonal antibody
- MEM
Minimum essential medium
- miRNA
Micro-RNA
- mRNA
Messenger RNA
- MRI
Magnetic resonance imaging
- MSV-nab-PTX
Mesoporous particles loaded with nab-PTX
- mTOR
Mammalian target of rapamycin
- nab-PTX
Albumin-bound paclitaxel
- NEAA
Non-essential amino acids
- OCT
Optimal cutting temperature
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PTX
Paclitaxel
- PVDF
Polyvinylidene fluoride
- qPCR
Quantitative PCR
- RICTOR
Rapamycin-insensitive companion of mTOR
- RIPA
Radioimmunoprecipitation assay
- RNA
Ribonucleic acid
- sgRNA
Single-guide RNA
- siRNA
Small interfering RNA
- TAM
Tumor-associated macrophage
- TME
Tumor microenvironment
- TBST
Tris-buffered saline with Tween-20
Author contributions
Study conception and design: FL, LC, BG, and HF; experiments: FL, AH, BG, DS, EC, and CZ; CRISPR liposome system development: FL; mathematical model implementation and testing: LC and HF; data collection and analysis: FL, LC, AH, BG, HF, DS, EC, and CZ; manuscript preparation and revision: FL, LC, BG, and HF.
Funding
Leonard acknowledges Houston Methodist Research Institute Department of Nanomedicine Innovative Grant Award and METAvivor Foundation Early Career Investigator Award. Leonard and Godin gratefully acknowledge funding from George and Angelina Kostas Research Center for Cardiovascular Nanomedicine Grant. Frieboes acknowledges partial support by the National Institutes of Health/National Cancer Institute Grant R15CA203605.
Compliance with ethical standards
Conflict of interest
The authors have no conflicts to disclose.
Ethical approval and ethical standards
In vivo mouse studies were performed in accordance with the Houston Methodist Research Institute Institutional Animal Care and Use Committee (IACUC—approval number: AUP-0617–0020). The animal research was conducted in full compliance with federal, state, and local regulations and institutional policies.
Animal source
Balb/c mice (6–8 weeks, females) were purchased from Jackson laboratory for all of the animal experiments in this study.
Cell line authentication
4T1 mouse breast cancer cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA), which tests and authenticates the cells in its collection.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Fransisca Leonard and Louis T. Curtis: equal contribution.
Contributor Information
Biana Godin, Email: bgodin@houstonmethodist.org, Email: bianagodinv@gmail.com.
Hermann B. Frieboes, Email: hbfrie01@louisville.edu
References
- 1.Wyld L, et al. Prognostic factors for patients with hepatic metastases from breast cancer. Br J Cancer. 2003;89(2):284–290. doi: 10.1038/sj.bjc.6601038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Eynden GG, et al. The multifaceted role of the microenvironment in liver metastasis: biology and clinical implications. Can Res. 2013;73(7):2031–2043. doi: 10.1158/0008-5472.CAN-12-3931. [DOI] [PubMed] [Google Scholar]
- 3.Stessels F, et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer. 2004;90(7):1429–1436. doi: 10.1038/sj.bjc.6601727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma R, et al. Mechanisms involved in breast cancer liver metastasis. J Transl Med. 2015;13:64. doi: 10.1186/s12967-015-0425-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braga L, et al. Does hypervascularity of liver metastases as detected on MRI predict disease progression in breast cancer patients? AJR Am J Roentgenol. 2004;182(5):1207–1213. doi: 10.2214/ajr.182.5.1821207. [DOI] [PubMed] [Google Scholar]
- 6.Liu LX, Zhang WH, Jiang HC. Current treatment for liver metastases from colorectal cancer. World J Gastroenterol. 2003;9(2):193–200. doi: 10.3748/wjg.v9.i2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pezzella F, Gatter KC. Evidence showing that tumors can grow without angiogenesis and can switch between angiogenic and nonangiogenicphenotypes. J Natl Cancer Inst. 2016;108(8):djw032. doi: 10.1093/jnci/djw032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonard F, et al. Enhanced performance of macrophage-encapsulated nanoparticle albumin-bound-paclitaxel in hypo-perfused cancer lesions. Nanoscale. 2016;8(25):12544–12552. doi: 10.1039/c5nr07796f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daly JM, et al. Predicting tumor response in patients with colorectal hepatic metastases. Ann Surg. 1985;202(3):384–393. doi: 10.1097/00000658-198509000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Martinez FO. Regulators of macrophage activation. Eur J Immunol. 2011;41(6):1531–1534. doi: 10.1002/eji.201141670. [DOI] [PubMed] [Google Scholar]
- 13.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Investig. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- 15.Galdiero MR, et al. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–1410. doi: 10.1016/j.imbio.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Mills CD. Anatomy of a discovery: m1 and m2 macrophages. Front Immunol. 2015;6:212. doi: 10.3389/fimmu.2015.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sica A, et al. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42(6):717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Cao W, et al. Macrophage subtype predicts lymph node metastasis in oesophageal adenocarcinoma and promotes cancer cell invasion in vitro. Br J Cancer. 2015;113(5):738–746. doi: 10.1038/bjc.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pantano F, et al. The role of macrophages polarization in predicting prognosis of radically resected gastric cancer patients. J Cell Mol Med. 2013;17(11):1415–1421. doi: 10.1111/jcmm.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Georgoudaki A-M, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15(9):2000–2011. doi: 10.1016/j.celrep.2016.04.084. [DOI] [PubMed] [Google Scholar]
- 21.Fuchs AK, et al. Carboxyl- and amino-functionalized polystyrene nanoparticles differentially affect the polarization profile of M1 and M2 macrophage subsets. Biomaterials. 2016;85:78–87. doi: 10.1016/j.biomaterials.2016.01.064. [DOI] [PubMed] [Google Scholar]
- 22.Oronsky B, et al. RRx-001: a systemically non-toxic M2-to-M1 macrophage stimulating and prosensitizing agent in Phase II clinical trials. Expert Opin Investig Drugs. 2017;26(1):109–119. doi: 10.1080/13543784.2017.1268600. [DOI] [PubMed] [Google Scholar]
- 23.Nathan MR, Schmid P. The emerging world of breast cancer immunotherapy. Breast. 2017;37:200–206. doi: 10.1016/j.breast.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 24.Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol. 2005;167(3):627–635. doi: 10.1016/S0002-9440(10)62038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard F, et al. Macrophage polarization contributes to the anti-tumoral efficacy of mesoporous nanovectors loaded with albumin-bound paclitaxel. Front Immunol. 2017;8:693. doi: 10.3389/fimmu.2017.00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leonard F, Godin B. Agents for macrophage polarization. Houston: Houston Methodist; 2018. [Google Scholar]
- 27.Babaev VR, et al. Loss of rictor in monocyte/macrophages suppresses their proliferation and viability reducing atherosclerosis in LDLR null mice. Front Immunol. 2018;9:215. doi: 10.3389/fimmu.2018.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Festuccia WT, et al. Myeloid-specific Rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS ONE. 2014;9(4):e95432. doi: 10.1371/journal.pone.0095432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Refuerzo JS, et al. Liposomes: a nanoscale drug carrying system to prevent indomethacin passage to the fetus in a pregnant mouse model. Am J Obstet Gynecol. 2015;212(4):508 e1–7. doi: 10.1016/j.ajog.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Macklin P, et al. Multiscale modelling and nonlinear simulation of vascular tumour growth. J Math Biol. 2009;58(4–5):765–798. doi: 10.1007/s00285-008-0216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu M, et al. The effect of interstitial pressure on tumor growth: coupling with the blood and lymphatic vascular systems. J Theor Biol. 2013;320:131–151. doi: 10.1016/j.jtbi.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDougall SR, Anderson ARA, Chaplain MAJ. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J Theor Biol. 2006;241(3):564–589. doi: 10.1016/j.jtbi.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 33.Mahlbacher G, et al. Mathematical modeling of tumor-associated macrophage interactions with the cancer microenvironment. J Immunother Cancer. 2018;6(1):10. doi: 10.1186/s40425-017-0313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Ven AL, et al. Integrated intravital microscopy and mathematical modeling to optimize nanotherapeutics delivery to tumors. AIP Adv. 2012;2(1):11208. doi: 10.1063/1.3699060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curtis LT, Frieboes HB (HB) Modeling of combination chemotherapy and immunotherapy for lung cancer. In: 41st Annual international conference of the IEEE engineering in medicine and biology society (EMBC). IEEE, Berlin, Germany, pp 273–276 [DOI] [PMC free article] [PubMed]
- 36.Hallowell RW, et al. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat Commun. 2017;8:14208. doi: 10.1038/ncomms14208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambarus CA, et al. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J Immunol Methods. 2012;375(1–2):196–206. doi: 10.1016/j.jim.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Porcheray F, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142(3):481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda A, et al. Poly(I:C) stimulation is superior than Imiquimod to induce the antitumoral functional profile of tumor-conditioned macrophages. Eur J Immunol. 2019;49(5):801–811. doi: 10.1002/eji.201847888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jablonski KA, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. 2015;10(12):e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown JM, Recht L, Strober S. The promise of targeting macrophages in cancer therapy. Clin Cancer Res. 2017;23(13):3241–3250. doi: 10.1158/1078-0432.CCR-16-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mills CD, Lenz LL, Harris RA. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res. 2016;76(3):513–516. doi: 10.1158/0008-5472.CAN-15-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mills CD, et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 44.Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tariq M, et al. Macrophage polarization: anti-cancer strategies to target tumor-associated macrophage in breast cancer. J Cell Biochem. 2017;118(9):2484–2501. doi: 10.1002/jcb.25895. [DOI] [PubMed] [Google Scholar]
- 46.Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. 2018;8:49. doi: 10.3389/fonc.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mahlbacher GE, Reihmer KC, Frieboes HB. Mathematical modeling of tumor-immune cell interactions. J Theor Biol. 2019;469:47–60. doi: 10.1016/j.jtbi.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanei T, et al. Redirecting transport of nanoparticle albumin-bound paclitaxel to macrophages enhances therapeutic efficacy against liver metastases. Can Res. 2016;76(2):429–439. doi: 10.1158/0008-5472.CAN-15-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogel SN, Carboni JM, Manthey CL (1994) Paclitaxel, a mimetic of bacterial lipopolysaccharide (LPS) in murine macrophages, in taxane anticancer agents. In: Georg GI, Chen TT, Ojima I (eds) American Chemical Society, Washington, DC, pp 162–172
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.