

Abstract
The dextransucrase DSR-OK from the Gram-positive bacterium Oenococcus kitaharae DSM17330 produces a dextran of the highest molar mass reported to date (∼109 g/mol). In this study, we selected a recombinant form, DSR-OKΔ1, to identify molecular determinants involved in the sugar polymerization mechanism and that confer its ability to produce a very-high-molar-mass polymer. In domain V of DSR-OK, we identified seven putative sugar-binding pockets characteristic of glycoside hydrolase 70 (GH70) glucansucrases that are known to be involved in glucan binding. We investigated their role in polymer synthesis through several approaches, including monitoring of dextran synthesis, affinity assays, sugar binding pocket deletions, site-directed mutagenesis, and construction of chimeric enzymes. Substitution of only two stacking aromatic residues in two consecutive sugar-binding pockets (variant DSR-OKΔ1-Y1162A-F1228A) induced quasi-complete loss of very-high-molar-mass dextran synthesis, resulting in production of only 10–13 kg/mol polymers. Moreover, the double mutation completely switched the semiprocessive mode of DSR-OKΔ1 toward a distributive one, highlighting the strong influence of these pockets on enzyme processivity. Finally, the position of each pocket relative to the active site also appeared to be important for polymer elongation. We propose that sugar-binding pockets spatially closer to the catalytic domain play a major role in the control of processivity. A deep structural characterization, if possible with large-molar-mass sugar ligands, would allow confirming this hypothesis.
Keywords: carbohydrate-binding protein, enzyme catalysis, enzyme mechanism, structure–function, protein engineering, dextransucrase, glucansucrase, polymerization, processivity
Introduction
Glucansucrases (GSs)2 of the family GH70 (1) are bacterial enzymes producing homopolysaccharides of d-glucosyl units from sucrose, a cheap and abundant agroresource (EC 2.4.1.5). Among them, dextransucrases synthesize α-glucans, comprising more than 50% of α-(1→6) osidic linkages called dextrans, which usually display a very high molar mass (HMM) over 106 g/mol. These HMM dextrans can be used as texturing, thickening and emulsifying agents, especially in food and cosmetic fields (2–4). However, most of the applications concern fractions of 1, 10, 40, or 70 × 103 g/mol, traditionally obtained by partial acid hydrolysis and solvent fractionation of a native HMM polymer containing more than 95% of α-(1→6) linkages (5). These clinical dextrans have various pharmaceutical applications (infusion fluids, volume expanders, organ preservatives, iron carriers, anticoagulants, or vaccine adjuvants, for instance (2)).
Considering that dextran applications largely depend on their size, it is thus tempting and would be of great interest to develop enzyme-based processes allowing one-step production of controlled-molar-mass dextrans from sucrose and avoiding the use of acids and solvents. For that purpose, a better understanding of the dextransucrase structural determinants dictating polymer elongation appears to be necessary. In the last decades, sequence analyses and X-ray 3D structure characterizations allowed identifying some amino acid residues involved in linkage specificity (6–11). However, less is known about the enzyme structural determinants controlling the polymer size. To date, five 3D structures of recombinant and truncated glucansucrases have been solved: the dextransucrase GTF180-ΔN from Lactobacillus reuteri 180 (12), which produces a dextran containing around 30% of α-1,3 osidic linkages and 60% of α-1,6 osidic ones; the mutansucrase GTF-SI from Streptococcus mutans (13); the reuteransucrase GTFA-ΔN from L. reuteri 121 (14), the dextransucrase DSR-MΔ2 from Leuconostoc citreum NRRL B-1299 (15), producing a linear α-1,6 dextran; and the alternansucrase ASRΔ2 from L. citreum NRRL B-1355 (16). All of these GSs have in common a five-domain organization with global U-shaped folding. Domains A, B, and C are also found in related GH13 enzymes, whereas domains IV and V are unique to the GH70 family. Among all characterized glucansucrases, DSR-M from L. citreum NRRL B1299 is rather unusual, as it is the first dextransucrase shown to produce only low-molar-mass (LMM) dextrans of around 20–30 kg/mol directly from sucrose and quasi-strictly composed α-1,6 osidic bonds (15). A recent structural analysis of DSR-MΔ2 in complex with isomalto-oligosaccharides, completed by biochemical characterization and mutagenesis studies, allowed us to propose a scenario explaining the distributive mode of LMM dextran synthesis by this enzyme. In particular, the function of specific structural features found in catalytic domain A and domain V were highlighted. Notably, residue Trp-624, located in loop B2 of DSR-MΔ2, has been shown to be crucial for the elongation of gluco-oligosaccharides of a molar mass higher than ∼2 kg/mol and up to ∼16 kg/mol (17). To synthesize dextrans of higher mass (up to 23 kg/mol), the assistance of two sugar-binding pockets, promoting interactions between the growing polymer chain and domain V, was found to be required. The sequences of these sugar-binding pockets are relatively well-conserved; they are found in domain V of every GH70 glucansucrases, and their number varies depending on the enzymes (18). These pockets are defined by a network of amino acids comprising a conserved stacking tyrosine (or phenylalanine) as well as a QXK motif interacting with glucosyl rings. In DSR-MΔ2, the aromatic residue has been proven to be essential for functionality (15). In GTF-SI, combining homology modeling and molecular dynamic studies, Osorio et al. (19) showed the role of domain V in the orientation of the growing glucan chain during elongation because of a higher number of glucan–enzyme contacts compared with the model of GTF-SI devoid of this domain, for which the growing oligosaccharide rapidly protrudes toward the solvent. As these sugar-binding pockets seem to operate during long-chain extension, we suspect that their number and binding affinity could be related to the ability of certain glucansucrases to synthesize HMM polymers by promoting a shift between a distributive (multichain) and a processive (single chain) mode of polymerization. In accordance with this hypothesis, previous work done a decade ago on an HMM dextran–synthesizing glucansucrase, the DSR-S vardel Δ4N from Leuconostoc mesenteroides NRRL B-512F, showed that large truncations in DSR-S domain V led to synthesis of lower-molar-mass dextrans. From these observations, a semiprocessive mechanism of polymerization has been proposed for this enzyme (20). Similar effects were observed for the dextransucrase GTF-180 upon truncation operated in this domain V (20). In all of these studies, the truncations often targeted large sequence fragments of more than 100 amino acids and induced a drastic loss of catalytic efficiency, possibly because of improper protein folding. This weakened the conclusions on the effective impact of the interactions between glucan and domain V in polymerization mechanism and control of polymer molar mass.
To test our hypothesis and examine the role of the sugar-binding pockets in controlling the chain extension, we focused our study on the recombinant dextransucrase DSR-OK, an enzyme originated from Oenococcus kitaharae DSM 17330, which produces a dextran of the highest molar mass reported to date (>109 g/mol) and with physical properties never observed before (21). Sequence analysis enabled identification of seven putative sugar-binding pockets in domain V of this enzyme, and their role was investigated through different approaches, including pocket deletions, mutagenesis targeting conserved aromatic residues, as well as construction of chimeric enzymes.
Results and discussion
DSR-OKΔ1: design and structural insights into a model enzyme
All attempts to crystallize the full-length DSR-OK, produced and purified according to Ref. 21, were unsuccessful. As the N-terminal extremity of DSR-OK sequence was predicted to be disordered (RONN program), a truncated form of 160 kDa, called DSR-OKΔ1, was constructed (keeping residues 84–1484, Fig. 1A). This truncation did not affect the level of expression in Escherichia coli BL21 (20,000 units/liter of culture for DSR-OKΔ1 and whole-size DSR-OK) but slightly affected its specific activity (90 units/mg for DSR-OKΔ1 versus 120 units/mg for the whole-size enzyme purified under similar conditions). The size exclusion chromatography profiles of the reaction products obtained from 100 g/liter of sucrose were almost equivalent (Fig. 1B). For DSR-OK and DSR-OKΔ1, more than 70% of the transferred glucosyl units were incorporated in a very HMM dextran, 18% in polysaccharides of intermediate size, 8.6% in leucrose, and only 3.5% in free glucose. The size of the HMM polymer was slightly impacted, but DSR-OKΔ1 dextran is of very high molar mass, like DSR-OK dextran, whose molar mass was determined at 1 × 109 g/mol using Asymmetrical flow field-flow fractionation additioned with multi-angle laser light scattering (21). It also surpasses the molar mass value of commercial dextran (2. 106 g/mol) while being of much lower polydispersity. In addition, DSR-OK and DSR-OKΔ1 polymers display equivalent rheological properties (data not shown). As our main objective was to identify determinants differentiating dextransucrases producing HMM polymers (around 108-109 g/mol) from those, like DSR-MΔ2, producing only LMM dextran of around 3 × 104 g/mol, DSR-OKΔ1 was considered a good model for study.
Figure 1.

A, schematic structural organization of DSR-OK and the truncated variant DSR-OKΔ1, based on amino acid alignment with GTF-180-ΔN, and structural analysis. Hatched red lines correspond to nonaligned zones. GH70 enzymes are divided into five structural domains: domain V (red); domain IV (yellow); and the three core domains (domain A (blue), domain B (green), and domain C (purple)). Putative sugar-binding pockets (A–G) are represented by pale yellow stars. B, HPSEC profile of the final medium after depletion of 292 mm sucrose using DSR-OK (blue) and DSR-OKΔ1 (red). Dex2M corresponds to a commercial dextran of 2 × 106 g/mol (dashed line).
Attempts to crystallize DSR-OKΔ1 resulted in formation of poor diffracting crystals whose quality remained insufficient for 3D structure solving. We thus turned toward small-angle X-ray scattering (SAXS) analyses of the protein in solution. SAXS curves revealed that DSR-OKΔ1 is well-behaved in solution without aggregation and shows typical modulations of multidomain proteins resembling those of GTF-180 and DSR-MΔ2 dextransucrases of known structure (Fig. 2A). Compared with DSR-MΔ2, SAXS ab initio envelopes revealed that DSR-OKΔ1 adopts a much more elongated shape in solution (Fig. 2B). This feature is supported by further analyses. P(r) functions of DSR-MΔ2 show an almost bell-shaped curve that nicely tends to zero at Dmax ≈ 125 Å, characteristic of a rather compact particle. In contrast, an asymmetric curve that tends to zero only at very high Dmax ≈ 260 Å is observed for DSR-OKΔ1, characteristic of elongated and flexible proteins (Fig. 3B). A dimensionless Kratky plot of DSR-MΔ2 revealed a well-defined maximum of 1.1 at q*Rg = √3, typical of compact particles (22), whereas the maximum is well over √3 for DSR-OKΔ1, indicating extended and flexible properties. The shoulder observable at high qRg values might suggest interdomain flexibility (Fig. 3C). Overall, we can see that the two enzymes have a similar overall shape for the catalytic domain, but although DSR-MΔ2 has domain V folded back onto the catalytic domain (Fig. 2), DSR-OKΔ1 displays a more extended shape, where domain V (or at least part of it) protrudes away from the catalytic domain. In this respect, DSR-OKΔ1 shows structural features that are in between DSR-MΔ2 and the more extended structure of GTF-180 (23). Unfortunately, the absence of a good-quality model for DSR-OKΔ1 (especially for its domain V) did not allow further modeling into the SAXS data. Therefore, the functional relevance of the different positioning of the domain V remains to be structurally determined.
Figure 2.

A, schematic structural organization of DSR-OKΔ1 compared with GTF-180 and DSR-MΔ2, based on amino acid alignment with both enzyme primary structures and structural analyses. Nonaligned zones are not represented. Domain V is represented in red, domain IV in yellow, domain A in blue, domain B in green, and domain C in purple. For each domain, the percentage of similarity with that of DSR-OK is shown in italics (pairwise alignments with BlastP tool). B, SAXS reconstruction of DSR-OKΔ1 compared with DSR-MΔ2. From left to right: DSR-MΔ2 crystal structure, SAXS ab initio envelope of DSR-MΔ2, SAXS ab initio envelope of DSR-OKΔ1, and GTF-180 crystal structure. Assignment of domains is based on superposition between the DSR-MΔ2 crystal structure and SAXS envelopes. The approximate position of the catalytic cleft is shown with a red circle.
Figure 3.

SAXS data analysis of DSR-OKΔ1 compared with DSR-MΔ2. A, SAXS curves with superimposed fits of the ab initio envelopes (black dots). B, Guinier analysis. C, P(r) functions. D, dimensionless Kratky plot. E, SAXS data summary table. Blue, DSR-MΔ2; red, DSR-OKΔ1.
Dextran synthesis with DSR-OKΔ1 becomes rapidly processive
The kinetics of polymer synthesis obtained with DSR-OKΔ1 or DSR-MΔ2 were monitored by HPAEC-PAD analyses, and the two product profiles are clearly different (Fig. 4). During the first 10 min of reaction, DSR-OKΔ1 produced one series of oligosaccharides resulting from successive transfers of glucosyl units onto sucrose (Fig. 4A), as confirmed by invertase digestion of the oligosaccharide mixture (Fig. S1). Similar to DSR-MΔ2 (15) or other glucansucrases, such as GTF-A (24), sucrose is the primer preferentially used at the beginning of the reaction. However, the degree of polymerization (DP) of the oligosaccharides synthesized by DSR-OKΔ1 does not exceed 6, whereas oligosaccharides of DP ranging from 3–30 are produced with DSR-MΔ2 (Fig. 4A). As sucrose consumption is equivalent in both reaction media, this indicates that DSR-OKΔ1 has incorporated glucosyl units in longer chains, which may not be released in the medium or cannot be detected, their size being too high with regard to their quantity. Indeed, at this stage, no HMM polymers can be detected by high performance size exclusion chromatography (HPSEC) chromatography (Fig. 4D). The difference between the two product profiles is even more striking after a 30-min reaction (Fig. 4B) and when total substrate consumption is reached (Fig. 4C). Moreover, HPSEC analyses already revealed the presence of one population of very-HMM dextran after 30 min, which becomes preponderant at the end of the reaction (Fig. 4D). When sucrose is depleted, additional series of oligosaccharides are detected on HPAEC-PAD chromatograms, which probably results from transfer onto glucose, fructose, or leucrose, which accumulate in the medium during the reaction course. Again, the oligosaccharides produced by DSR-OKΔ1 are much shorter in size and present in lower amounts compared with those obtained with DSR-MΔ2. In that respect, DSR-OKΔ1 seems to adopt a semiprocessive mode of dextran synthesis similar to that already proposed for DSR-S-Δ4N (20). At the beginning of the reaction, a distributive mechanism leads to formation of multiple short-chain oligosaccharides, but above DP6, these oligosaccharides are preferentially elongated to yield HMM dextran, suggesting that the chain is retained by the protein for elongation before being released. At the end of the reaction, the oligosaccharide content has increased to represent 18% of the transferred glucosyl units. Their formation is due to the high concentration of fructose, an acceptor becoming a severe competitor of the dextran chains for glucosyl residue capture at this stage.
Figure 4.

A–C, comparison of HPAEC-PAD profiles of DSR-OKΔ1 (red) and DSR-MΔ2 (blue) products from 292 mm sucrose at different reaction times. A, after 10 min. B, after 30 min; C, final reaction medium. Commercial dextran 1500 g/mol is shown as standard (green). D, HPSEC profiles of DSR-OKΔ1 products from 292 mm sucrose at different reaction times.
Functional implications of domain V in processivity
Using a set of sequences encoding structurally characterized sugar-binding pockets from ΔN123-GBD-CD2 and DSR-MΔ2 (15, 18), which are very well structurally conserved despite relatively low sequence conservation (from 25% to 40% identity only), we annotated seven sequences as putative sugar-binding pockets in DSR-OK domain V (Fig. 5). Two of them (A and B) are located at the N-terminal extremity and the other five ones at the C-terminal end (Fig. 1A). All predicted sugar pockets display a conserved aromatic residue (tyrosine or phenylalanine, Fig. 5) identified as an essential stacking platform in structurally characterized pockets (15). The QXK motif is also conserved, except in pockets B and C, which could prevent their functionality (Fig. 5). In total, DSR-OK would possess between five and seven functional sugar-binding pockets. The conserved Tyr-70 of repeat A was truncated in DSR-OKΔ1 (even when the QXK motif was kept), suggesting that this pocket might have lost its functionality in the variant (Fig. 1B).
Figure 5.

Sequences of putative sugar-binding pockets identified in the DSR-OK sequence, also called repeats A–G according to their localization in the primary structure. Sequence alignment was performed against four repeats identified in domains V of ΔN123-GBD-CD2 (from DSR-E) and DSR-MΔ2. A LOGO sequence based on this alignment is also shown. The common aromatic stacking residue and the QXK motif locations are framed in red. Single point mutations were performed on the residues highlighted in gray. rptX, repeat X). DSR-OK repeats (amino acid numbering): A, 66–126; B, 146–218; C, 1093–1160; D, 1661–1222; E, 1223–1287; F, 1347–1412; G, 1413–1484.
Sugar-binding pockets are important for specific activity and dextran binding
Starting from DSR-OKΔ1, several truncated variants were constructed in which sugar-binding pockets were progressively eliminated (Fig. 6). All of these variants were expressed in quasi-equivalent amounts in E. coli, but their specific activity was heavily affected. The DSR-OKΔ2 variant, with two deleted putative sugar-binding pockets at the C-terminal end, lost 93% of activity compared with DSR-OKΔ1. Additional truncations of pockets D and E were even more detrimental, with 99% recorded loss of activity, indicating that these pockets are necessary to maintain activity at a reasonable level. Also, we cannot exclude folding problems, as aggregates were observed during the purification steps of DSR-OKΔ3, DSR-OKΔ4, and DSR-OKΔV. Nevertheless, these findings are strongly contrasting with those obtained previously with DSR-MΔ2, for which complete suppression of domain V had no impact on specific activity. Notably, they get close to those reported for several variants of the DSR-S from L. mesenteroides NRRL B-512F truncated at their C-terminal end (DSR-S vardel Δ3, DSR-S vardel Core), which were also very poorly active (20).
Figure 6.

A, schematic structural organization of DSR-OKΔ1 and its truncated variants. The GTF-180 sequence was used as a template for domain attribution. Hatched red lines correspond to nonaligned zones. Domain V is shown in red, domain IV in yellow, domain A in blue, domain B in green, and domain C in purple. Putative sugar-binding pockets are represented by pale yellow stars. B, HPSEC profile of the products obtained from 292 mm sucrose with DSR-OKΔ1 (red) and DSR-OKΔ2 (green).
Despite its very low activity, the DSR-OKΔ2 variant was able to consume 292 mm sucrose. The HPSEC profiles show that DSR-OKΔ2 kept its ability to synthesize HMM dextran of slightly reduced size compared with DSR-OKΔ1 but also produced a new population of LMM dextran around 13 kg/mol (Fig. 6). Similar behavior was observed for variant DSR-S vardel Δ3 and DSR-S vardel Core (20). This reveals that not all dextran chains are elongated to the same extent, which could be attributed to the simultaneous suppression of sugar-binding pockets F and G. Deletion of pocket A in DSR-OKΔ1 also induced a slight decrease in HMM polymer size compared with that produced by DSR-OK (Fig. 1B).
To evaluate the strength of the dextran–enzyme interaction, affinity gel electrophoreses of pure DSR-OKΔ1, DSR-OKΔV, and DSR-MΔ2 (as a positive control) were carried out using gels prepared with dextran of 6000 g/mol (Dex6000) or 68,400 g/mol (Dex68,400). As shown in Fig. 7, a marked migration delay was observed for DSR-OKΔ1 and a lower one for DSR-MΔ2 in gels containing dextran. In contrast, there was no delay for DSR-OKΔV. This confirms that the affinity for dextran is mainly conferred by domain V and decreases when its size is reduced. Moreover, the migration delay is higher in the gel containing Dex68,400 than in the gel prepared with Dex6000, meaning that affinity is stronger for longer dextran. Furthermore, the delay did not significantly vary when lowering dextran concentrations, preventing any Kd determination because of too-strong binding interactions.
Figure 7.
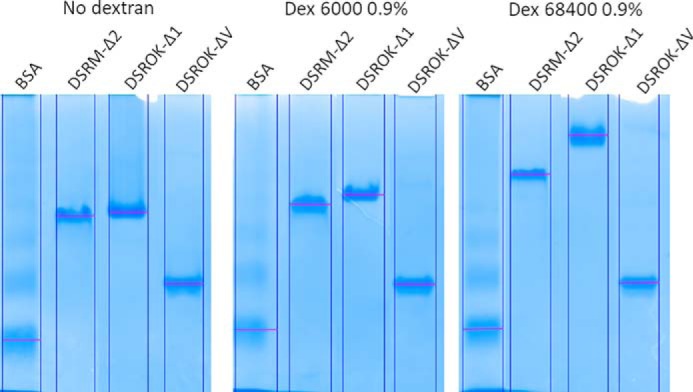
Affinity gel electrophoresis of DSR-OK variants in gels containing dextran of 6000 g/mol or 68,400 g/mol. BSA, negative control; DSR-MΔ2, positive control.
Sugar-biding pockets contribute to enzyme processivity
Because truncations can cause profound changes in 3D structure, we turned to mutagenesis to investigate the role of the identified putative sugar-binding pockets. The conserved stacking residues (Phe or Tyr) identified in the different pockets were individually replaced with alanine. All mutants were well expressed in E. coli and active (Table S1). NMR analyses and dextranase digestion of the reaction products confirmed that the mutations did not affect linkage specificity (data not shown).
In pockets B, C, F, and G, single mutation of Tyr-153, Phe-1098, Phe-1354, or Tyr-1420 into alanine did not affect the product profile compared with the nonmutated form (Fig. 8). The ability to synthesize HMM dextran was fully maintained, and the population of LMM products remained unchanged. Mutation of only one stacking residue was not sufficient to alter the pocket functionality or prevent binding in other pockets, considering that different pockets could act in synergy. A double mutant, F1354A/Y1420A (targeting pockets F and G), was constructed. This mutant produced a HMM dextran population of slightly lower size compared with DSR-OKΔ1, suggesting that these two pockets may not play a major role in elongation (Fig. 8). Notably, variant DSR-OKΔ2 entirely devoid of pockets F and G, produced a higher amount of LMM dextran, showing that deletion and point mutation are not strictly equivalent (Fig. 6).
Figure 8.

HPSEC analysis of the products synthesized by DSR-OK variants from 292 mm sucrose. Blue, DSR-OK; red, DSR-OKΔ1; light purple, DSR-OKΔ2; green, DSR-OKΔ1-Y1420A (pocket G); turquoise, DSR-OKΔ1-Y1354A (pocket F); light blue, DSR-OKΔ1-F1228A (pocket E); light red, DSR-OKΔ1-Y1162A (pocket D); dark purple, DSR-OKΔ1-F1098A (pocket C); dark orange, DSR-OKΔ1-Y153A (pocket B); light turquoise, DSR-OKΔ1-Y1354A-Y1420A (pockets F and G); light green, DSR-OKΔ1-Y1162A-F1228A (pockets D and E).
In pockets D and E, the single mutations of Y1162A and F1228A, respectively, impacted the product profile as well as the enzyme activity more severely, which was estimated to be 49% for DSR-OKΔ1-Y1162A (pocket D) and 73% for DSR-OKΔ1-F1228A (pocket E) relative to that of the WT. Both single mutants produced a significant amount of LMM dextran in addition to the HMM one (Fig. 8). The effect was even more pronounced for the double mutant DSR-OK1-Y1162A/F1228A, which retained only 20% of activity and almost totally lost its ability to synthesize HMM dextran while producing a LMM dextran population of around 10–13 kg/mol (estimated at the peak apex of the HPSEC chromatogram). In addition, the kinetics of dextran synthesis with this double mutant were very similar to that obtained with DSR-MΔ2 (Fig. 9), revealing that the double mutations caused a switch from a semiprocessive to a totally distributive mode of elongation. These two mutations probably impaired two anchoring points of the elongated dextran chain, lowering the strength of enzyme–product interaction, which appears to be clearly related to enzyme processivity. This demonstrates that binding to domain V, mediated by the succession of certain sugar-binding pockets, is the main determinant that governs the enzyme processivity and elongation efficiency of GSs synthesizing HMM dextran.
Figure 9.

Comparison of HPAEC-PAD product profiles of DSR-MΔ2 (blue) and DSR-OKΔ1-Y1162A/F1228A (orange) at different reaction times from 292 mm sucrose after 10 min, 30 min, and final reaction medium (tf).
Moreover, the role and functionality of each sugar-binding pocket are clearly not equivalent. Indeed, the single mutations in pockets D and E had a major impact on the dextran distribution profile, in contrast to those targeting pockets B, C, F, and G. Nonconservation of the QXK motif in pockets B and C could explain the limited role of these pockets. Their position (more or less distant from the active site) may also be important. In DSR-MΔ2, mutation of the stacking residue of pocket A had a higher impact on polymer size than that in pocket B. Pocket A is farther from the active site in the primary sequence than pocket B, but considering the compact form of DSR-MΔ2, this pocket is spatially closer to the catalytic gorge (15). Based on the SAXS envelope of DSR-OKΔ1, pockets F and G would be further away from the active site than all other putative pockets, which could explain their less important role. However, these interpretations should be treated with caution, as domain V is known to be mobile and could move around domain IV, as seen for GTF-180 (23). Of course, the 3D structure of the enzyme, especially the enzyme in complex with a long dextran chain, would provide interesting information, and efforts will have to be pursued in this direction.
Domain swapping between distributive and processive enzymes influences the size of the HMM polymer
We envisioned exchanging domain V between DSR-OKΔ1 and DSR-MΔ2 to examine the effect on polymer size. Swapping of domain IV was also considered to limit misfolding and conserve a native connection between domains IV and V. To determine at which position the domains should be swapped, we used sequence alignments and structural superimposition with GH70 glucansucrase structures solved to date (12–14). We noticed the presence of a helix in domain IV (amino acids 424–429, DSR-M numbering), which corresponds to the hinge region between domains IV and V described previously for GTF-180ΔN (23). This helix was swapped together with domain V, and the most conserved positions at the borders of each domain were selected as swapping points (Fig. S2), yielding chimeras 1 and 3 (Fig. 10). In addition, to limit possible problems of chimera folding because of the spatial proximity of domains coming from different enzymes, concomitant swapping of domains IV and V was also performed, yielding chimeras 2 and 4 (Fig. 10).
Figure 10.

Schematic representation of the chimeric enzymes and HPSEC profiles of the final reaction medium of the different enzyme forms. Parts of DSR-OKΔ1 are represented in orange and parts of DSR-MΔ2 in blue. Molar mass at the apex is indicated in grams per mole.
The levels of expression of chimeras 1 and 2 (with DSR-OK core) and their activities were lower than those obtained for DSR-OKΔ1 (Fig. S3). In contrast, chimeras 3 and 4 (with DSR-M core) were produced in equivalent amounts as the parental enzyme. In all cases, the production yields were sufficient to incubate each enzyme extract with 292 mm of sucrose until total substrate consumption.
As shown on HPSEC chromatograms (Fig. 10), when DSR-OK domain V (or IV plus V) is replaced with those of DSR-MΔ2, the resulting enzymes (chimeras 1 and 2) synthesize two main populations of dextran: one of molar mass superior to 106 g/mol (but inferior to that produced by the parental enzyme) and a major population of molar mass again estimated around 13 kg/mol. The concomitant swap of domain IV and V did not influence the dextran profile, suggesting that the two chimeras have similar catalytic and perhaps structural properties. Hence, the nature of domain IV does not influence the elongation process, acting as a “spacer” between the catalytic domain and domain V. The product profiles look like those obtained with DSR-OKΔ2, DSR-OKΔ1-Y1162A, DSR-OKΔ1-F1228A, and DSR-OKΔ1-Y1162A/F1228A (mutation of stacking residues of pockets D and/or E), with a bimodal dextran population. In fact, the interchange with DSR-M domain V, known to be of low binding efficiency (15), has the same effect on the DSR-OK polymerization process as alteration of its own domain V ability to interact with growing dextran chains.
Concerning chimeras 3 and 4, constructed in a DSR-MΔ2 template, they both produced a population of dextran between 10 and 40 kg/mol with a molar mass at the apex of around 35 kg/mol. The synthesized products are higher in size than those produced by DSR-MΔ2. However, grafting DSR-OKΔ1 domain V in place of its own domain and introducing two additional sugar-binding pockets did not result in production of an HMM dextran superior to 106 g/mol. Again, stability and folding issues also need to be considered, and swapping of entire domains at both enzyme extremities is certainly very damaging.
Domain V exchanges between the semiprocessive and distributive enzymes impacted product size in the expected directions but was not sufficient to completely change the mode of dextran elongation. We have shown previously for DSR-MΔ2 that the interplay between the catalytic domain and domain V governs the size of the polymer. Specific determinants in the catalytic domain of DSR-OK and interplay with domain V may also participate in the high processivity of this enzyme, as shown for other enzymes (Leu-940 in GTF180-ΔN (10) and Trp-624 in DSR-MΔ2 (17)). However, these residues are not conserved at other GH70 catalytic sites (or their role is not equivalent in other enzymes), and so the identification of such interacting residues is not predictable in the absence of structural data.
Conclusion
The recombinant enzyme DSR-OK, originated from O. kitaharae DSM17330, was demonstrated to synthesize a HMM and quasi-linear dextran. This polymer presents exceptional rheological properties (viscosity, yield stress, and gel-like behavior) never before observed for other dextrans. For these reasons, DSR-OK was selected as a model to identify the determinants involved in the control of polymer size. More specifically, we focused on the role of the numerous putative sugar-binding pockets covering domain V of the protein.
Monitoring dextran synthesis revealed that DSR-OKΔ1, a model of study, adopts a semiprocessive mode of elongation comparable with that of DSR-S vardel Δ4N. The first phase of chain initiation, producing oligosaccharides, is quickly followed by synthesis of an HMM dextran (>108 g/mol). This contrasts with the mode of dextran elongation described previously for the DSR-MΔ2 enzyme (15), which displays an original distributive mode of elongation responsible for synthesis of LMM dextran of around 20–30 kg/mol. On the other hand, affinity assays revealed that the binding ability of DSR-OKΔ1 toward glucan chains is very high and mainly conferred by its domain V. In contrast, the binding affinity of DSR-MΔ2 domain V has been shown previously to be low (15). Such differences revealed that the polymerization mechanism (distributive versus semiprocessive) could be determinant for the ability of dextransucrases to synthesize HMM dextran. We propose here that some sugar-binding pockets of domain V are important promotors of processivity because of strong interaction with the growing dextran chain.
By mutating only two stacking aromatic residues of two consecutive binding pockets in the variant DSR-OKΔ1-Y1162A/F1228A, we switched the semiprocessive mode of DSR-OKΔ1 toward a distributive one, showing the influence of these pockets on enzyme processivity. Elimination of the sugar-binding pockets in DSR-OKΔ1 (this study) and DSR-S vardelΔ4N (20) also led to LMM dextran formation but with a much higher decrease in specific activity. Targeting residues responsible for functionality of the sugar-binding pockets is thus a very good means to modulate glucansucrase dextran size. The position of the pockets relative to the active site also seems to be important, and it is clear that all binding pockets do not play exactly the same role. Our assumption is that binding pockets spatially closer to the catalytic domain would be more involved in control of processivity, but this will have to be confirmed by structural data. In addition, the avidity for dextran increases with the number of pockets interacting with the polymer. Short oligosaccharides bind with lower affinity than dextran, and one can imagine cooperativity between the pockets when the glucan chain is long enough to interact with several binding pockets, a cooperativity that may extend to the catalytic site.
Indeed, as revealed by the behaviors of the chimeric enzymes constructed from DSR-OKΔ1 and DSR-MΔ2, in which domains IV and V were interchanged, transformation of an HMM-synthesizing enzyme into one dedicated to LMM dextran synthesis and vice versa is not trivial, especially without losing too much catalytic efficiency. Activity loss also arises when domain V becomes less efficient, after mutation of binding pockets, for instance, and can be attributed to a decrease in the apparent affinity for the dextran substrate and/or perturbation of the concerted action between the catalytic domain and domain V. This highlights the fact that HMM-synthesizing GSs have optimally evolved to ensure perfect tuning between the catalytic domain and domain V. Their interplay may be due to continuity between the acceptor binding subsites located in the catalytic domain and the sugar-binding pockets. However, to have an idea of the position of the dextran during chain extension and start touching on the dynamics of this process, efforts to solve the structure of GH70 polymerases in complex with long oligosaccharides must be pursued.
Materials and methods
Construction of DSR-OKΔ1
The primary sequence of DSR-OK (GenBank accession number EHN59583) was analyzed with the RONN program (https://academic.oup.com/bioinformatics/article/21/16/3369/215577)3 to identify regions that will possibly lack a well-defined 3D structure under native conditions. Construction of DSR-OKΔ1, consisting of deletion of the first 107 amino acids from the entire form pET-53/DsrOK plasmid DNA template (21), including the His tag, was performed following the method described by Wang and Malcolm (25, primers in Table S1). Briefly, this method consists of a two-stage procedure based on the QuikChange site-directed mutagenesis protocol (Stratagene, La Jolla, CA), in which a single-primer extension stage before the standard PCR protocol allows deletion of a sequence of interest.
Protein expression and purification
Proteins cloned in a pET55-DEST vector were expressed and purified as described previously (15). For the proteins cloned in a pET53-DEST vector, freshly transformed BL21 star (DE3) E. coli cells (Thermo Fisher Scientific) were grown overnight in Lysogeny Broth medium supplemented with 100 μg/ml of ampicillin at 37 °C. Then 1 liter of modified ZYM5052 medium (30) (with the following changes: 1% lactose, 1% glycerol, and no glucose added) was inoculated from the starter culture at an A600 nm of 0.05 and incubated at 23 °C under agitation. After 23 h of culture, cells were harvested by centrifugation, resuspended at a final A600 nm of 200 in purification buffer (1× PBS and 280 mm NaCl (pH 7.4)), and disrupted by sonication. Enzymes were recovered in soluble supernatant extract after centrifugation (20,000 rpm, 30 min). Protein purification was performed using ÄKTAxpress (GE Healthcare) at 12 °C, with a first step consisting of Strep-tag® affinity chromatography. Proteins were injected at 1 ml/min into a 5-ml Strep Trap HPTM column (GE Healthcare) pre-equilibrated with purification buffer. After binding, Strep-tagged enzymes were eluted at 4 ml/min using a gradient of d-desthiobiotin (from 0.05–2.5 mm) in 20 column volumes. Affinity purification was followed by a size exclusion step on a Superose 12 column, 16 × 60 (GE Healthcare), from which the protein preparation was eluted with buffer for SAXS analysis (30 mm MES (pH 6.5), 100 mm NaCl, and 0.05 g/liter CaCl2) or with 50 mm sodium acetate buffer (100 mm NaCl and 0.05 g/liter CaCl2 (pH 5.75)) for biochemical characterization. Protein purity was verified by SDS-PAGE. Protein concentration was determined by spectroscopy at 280 nm on a Nanodrop ND-1000 (Thermo Fisher Scientific) using the protein theoretical molar extinction coefficient and the molecular weight calculated by the ExPASy ProtParam tool (RRID: SCR_018087).
Activity assays
Activity was assayed using the dinitrosalicylic method (26). One unit of DSR-OK variant is defined as the amount of enzyme that catalyzes production of 1 μmol of fructose/min from 292 mm sucrose in 50 mm sodium acetate buffer (pH 5.75) at 30 °C.
Enzymatic reaction
All dextran syntheses were carried out with 292 mm sucrose at 30 °C using 1 unit/ml of pure enzyme in 50 mm sodium acetate buffer. For kinetics studies, samples were taken at regular intervals until total sucrose depletion, reactions were stopped by 10 min incubation at 95 °C, and samples were stored at −20 °C until further analyses.
Product characterization
High-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) analyses were performed using a CarboPacTM PA100 analytical column (4 × 250 mm) coupled with a CarboPacTM PA100 guard (4 × 50 mm). Product separation was performed using a sodium acetate gradient (6–500 mm) in 150 mm NaOH over 45 min (1 ml/min).
HPSEC analyses were performed using two Shodex OH-Pak SB-802.5 and SB-805 columns (Showa Denko, Tokyo, Japan) in series coupled with a Shodex OH-Pak SB-G guard column and placed in a 70 °C oven. The samples were diluted to a maximum of 10 g/kg of total sugars in the eluent (0.45 m NaNO3 and 1% ethylene glycol). Elution was performed at a flow rate of 0.3 ml/min. When possible, the weight-average molar masses of synthesized dextrans were determined using a calibration curve with standards (concentration of 10 g/kg) of fructose, sucrose, and dextrans of 11 300, 39 100, 68 400, 503 × 103, and 2 × 106 g/mol (Sigma-Aldrich). Dextrans produced by DSR-OK are very-HMM polymers, and precise molar mass determination would necessitate more expensive methods, such as AF4-MALS (21). Consequently, for dextrans of more than 2 × 106 g/mol, HPSEC is used as a comparative method but is limited to discriminate slight differences.
SAXS measurements and processing
SAXS experiments were performed on the SWING beamline at the SOLEIL synchrotron (Gif-sur-Yvette, France) as described previously (15). Briefly, a 50-μl volume of protein sample (10 mg/ml) was injected onto a size-exclusion column (Bio SEC3 300, Agilent, Santa Clara, CA) equilibrated in MES buffer (30 mm MES (pH 6.5), 100 mm NaCl, and 0.05 g/liter CaCl2) and eluted directly into the SAXS capillary cell at a flow rate of 200 μl/min at a temperature of 10 °C. Using a wavelength of 1.033 Å, SAXS data were collected online throughout the elution time, and the recorded curves were normalized to the transmitted intensity, buffer-subtracted, and averaged using FOXTROT as described previously (27). Radius of gyration values were determined by a Guinier fit of the one-dimensional curves using the ATSAS package (28). The p(r) function was calculated using the GNOM program and the corresponding ab initio envelopes were calculated using the GASBOR program.
Mutagenesis study
Construction of deletion mutants
For construction of DSR-OKΔ2 to V, the corresponding genes were amplified by PCR from the pET-53/DsrOK plasmid DNA template (21) using the primers described in Table S2; addition of the CACC sequence (underlined) to the 5′ forward primers allowed correct insertion of genes into the pENTR/d-TOPO® vector (Life Technologies). From a positive entry clone, LR recombination (Gateway® LR Clonase® II Enzyme Mix, Life Technologies) was performed with the pET-55-DEST and pET53-DEST destination vectors (Novagen). Expression clones were selected on LB agar plates supplemented with 100 μg/ml ampicillin. Plasmids were extracted with the Sigma-Aldrich GenElute HP Plasmid Miniprep Kit, verified by restriction analyses, and the genes of interest were sequenced (GATC Biotech). E. coli TOP10-competent cells (Life Technologies) were used for all cloning experiments. Finally, deletion of the sugar-binding pockets in DSR-OKΔ1ΔFG and DSR-OKΔ1ΔDEFG were cloned in the pENTR/d-TOPO® vector (Life Technologies) using the In-Fusion® HD Cloning Kit from Clontech. As for previous constructions, LR recombination of positive entry clones was then realized with the pET-55-DEST and pET53-DEST destination vectors (Novagen).
Site-directed mutagenesis
Mutants Y1662A, F1228A, F1354A, and Y1420A were constructed by inverse PCR (oligo-mediated introduction of site-specific mutations) as described in Ref. 15 using the pET53-dsrok-Δ1 plasmid as a template and the primers described in Table S1. Double mutants were constructed by novel mutation onto a previous single mutant.
Chimera construction
Two DNA fragments corresponding to gene regions to be swapped were amplified by PCR using the primers and DNA templates detailed in Table S3 and were then assembled into a single vector using Gibson assembly (New England Biolabs) (29). All chimera genes were cloned in the pET55/DEST vector comprising a Strep and a His6 tag at the N- and C-terminal ends, respectively. Every construction was verified by sequencing (GATC Biotech, Constance, Germany).
Author contributions
M. C., G. C., and C. M. formal analysis; M. C., M. R.-S., and C. M. validation; M. C., M. V., P. B., M. R.-S., and C. M. investigation; M. C., G. C., M. V., P. B., M. R.-S., and C. M. methodology; M. C. and C. M. writing-original draft; M. C., G. C., M. V., M. R.-S., and C. M. writing-review and editing; G. C. and C. M. supervision; M. R.-S. funding acquisition; M. R.-S. and C. M. project administration; C. M. conceptualization.
Supplementary Material
Acknowledgments
We are grateful to the ICEO facility, which is part of the Integrated Screening Platform of Toulouse, for providing access to HPLC and protein purification equipment. We acknowledge the staff of the SWING beamline of synchrotron SOLEIL (Gif-Sur-Yvette, France) for SAXS data acquisition and especially Pierre Roblin for advice regarding SAXS interpretation.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3 and Tables S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- GS
- glucansucrase
- HMM
- high molar mass
- LMM
- low molar mass
- DP
- degree of polymerization
- SAXS
- small-angle X-ray scattering
- HPAEC-PAD
- high-performance anion exchange chromatography with pulsed amperometric detection
- Rg
- Radius of gyration
- HPSEC
- high performance size exclusion chromatography.
References
- 1. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Belder A. (2003). In Dextran A. A. (Ed.). Handbook (Amersham biosciences). Uppsala: Amersham Biosciences AB. [Google Scholar]
- 3. Kothari D., Das D., Patel S., and Goyal A. (2014). Dextran and food application. In Polysaccharides (Ramawat K. G., and Mérillon J.-M., eds.), pp. 1–16, Springer International Publishing AG, Cham, Switzerland. [Google Scholar]
- 4. Vettori M. H. P. B., Blanco K. C., Cortezi M., Lima C. J. B., and Contiero J. (2012) Dextran: effect of process parameters on production, purification and molecular weight and recent applications. Diálogos & Ciência 2012, 171–186 [Google Scholar]
- 5. Naessens M., Cerdobbel A., Soetaert W., and Vandamme E. J. (2005) Leuconostoc dextransucrase and dextran: production, properties and applications. Journal of Chemical Technology & Biotechnology 80, 845–860 10.1002/jctb.1322 [DOI] [Google Scholar]
- 6. Kralj S., Stripling E., Sanders P., van Geel-Schutten G. H., and Dijkhuizen L. (2005) Highly hydrolytic reuteransucrase from probiotic Lactobacillus reuteri strain ATCC 55730. Appl. Environ. Microbiol. 71, 3942–3950 10.1128/AEM.71.7.3942-3950.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moulis C., Joucla G., Harrison D., Fabre E., Potocki-Veronese G., Monsan P., and Remaud-Simeon M. (2006) Understanding the polymerization mechanism of glycoside-hydrolase family 70 glucansucrases. J. Biol. Chem. 281, 31254–31267 10.1074/jbc.M604850200 [DOI] [PubMed] [Google Scholar]
- 8. Hellmuth H., Wittrock S., Kralj S., Dijkhuizen L., Hofer B., and Seibel J. (2008) Engineering the glucansucrase GTFR enzyme reaction and glycosidic bond specificity: toward tailor-made polymer and oligosaccharide products. Biochemistry 47, 6678–6684 10.1021/bi800563r [DOI] [PubMed] [Google Scholar]
- 9. van Leeuwen S. S., Kralj S., Eeuwema W., Gerwig G. J., Dijkhuizen L., and Kamerling J. P. (2009) Structural characterization of bioengineered α-d-glucans produced by mutant glucansucrase GTF180 enzymes of Lactobacillus reuteri strain 180. Biomacromolecules 10, 580–588 10.1021/bm801240r [DOI] [PubMed] [Google Scholar]
- 10. Meng X., Dobruchowska J. M., Pijning T., López C. A., Kamerling J. P., and Dijkhuizen L. (2014) Residue Leu940 has a crucial role in the linkage and reaction specificity of the glucansucrase GTF180 of the probiotic bacterium Lactobacillus reuteri 180. J. Biol. Chem. 289, 32773–32782 10.1074/jbc.M114.602524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meng X., Pijning T., Dobruchowska J. M., Gerwig G. J., and Dijkhuizen L. (2015) Characterization of the functional roles of amino acid residues in acceptor-binding subsite +1 in the active site of the glucansucrase GTF180 from Lactobacillus reuteri 180. J. Biol. Chem. 290, 30131–30141 10.1074/jbc.M115.687558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vujicic-Zagar A., Pijning T., Kralj S., López C. A., Eeuwema W., Dijkhuizen L., and Dijkstra B. W. (2010) Crystal structure of a 117 kDa glucansucrase fragment provides insight into evolution and product specificity of GH70 enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 21406–21411 10.1073/pnas.1007531107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ito K., Ito S., Shimamura T., Weyand S., Kawarasaki Y., Misaka T., Abe K., Kobayashi T., Cameron A. D., and Iwata S. (2011) Crystal structure of glucansucrase from the dental caries pathogen Streptococcus mutans. J. Mol. Biol. 408, 177–186 10.1016/j.jmb.2011.02.028 [DOI] [PubMed] [Google Scholar]
- 14. Pijning T., Vujičić-Žagar A., Kralj S., Dijkhuizen L., and Dijkstra B. W. (2012) Structure of the α-1,6/α-1,4-specific glucansucrase GTFA from Lactobacillus reuteri 121. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1448–1454 10.1107/S1744309112044168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Claverie M., Cioci G., Vuillemin M., Monties N., Roblin P., Lippens G., Remaud-Siméon M., and Moulis C. (2017) Investigations on the determinants responsible for low molar mass dextran formation by DSR-M dextransucrase. ACS Catalysis 7, 7106–7119 10.1021/acscatal.7b02182 [DOI] [Google Scholar]
- 16. Molina M., Moulis C., Monties N., Pizzut-Serin S., Guieysse D., Morel S., Cioci G., and Remaud-Siméon M. (2019) Deciphering an undecided enzyme: investigations of the structural determinants involved in the linkage specificity of alternansucrase. ACS Catalysis 9, 2222–2237 10.1021/acscatal.8b04510 [DOI] [Google Scholar]
- 17. Claverie M., Cioci G., Guionnet M., Schörghuber J., Lichtenecker R., Moulis C., Remaud-Simeon M., and Lippens G. (2019) Futile encounter engineering of the DSR-M dextransucrase modifies the resulting polymer length. Biochemistry 58, 2853–2859 10.1021/acs.biochem.9b00373 [DOI] [PubMed] [Google Scholar]
- 18. Brison Y., Malbert Y., Czaplicki G., Mourey L., Remaud-Simeon M., and Tranier S. (2016) Structural insights into the carbohydrate binding ability of an α-(1→2) branching sucrase from glycoside hydrolase family 70. J. Biol. Chem. 291, 7527–7540 10.1074/jbc.M115.688796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Osorio M. I., Zúñiga M. A., Mendoza F., Jaña G. A., and Jiménez V. A. (2019) Modulation of glucan-enzyme interactions by domain V in GTF-SI from Streptococcus mutans. Proteins 87, 74–80 10.1002/prot.25624 [DOI] [PubMed] [Google Scholar]
- 20. Meng X., Dobruchowska J. M., Pijning T., Gerwig G. J., Kamerling J. P., and Dijkhuizen L. (2015) Truncation of domain V of the multidomain glucansucrase GTF180 of Lactobacillus reuteri 180 heavily impairs its polysaccharide-synthesizing ability. Appl. Microbiol. Biotechnol. 99, 5885–5894 10.1007/s00253-014-6361-8 [DOI] [PubMed] [Google Scholar]
- 21. Vuillemin M., Grimaud F., Claverie M., Rolland-Sabaté A., Garnier C., Lucas P., Monsan P., Dols-Lafargue M., Remaud-Siméon M., and Moulis C. (2018) A dextran with unique rheological properties produced by the dextransucrase from Oenococcus kitaharae DSM 17330. Carbohydrate Polymers 179, 10–18 [DOI] [PubMed] [Google Scholar]
- 22. Receveur-Brechot V., and Durand D. (2012) How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 13, 55–75 10.2174/138920312799277901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pijning T., Vujičić-Žagar A., Kralj S., Dijkhuizen L., and Dijkstra B. W. (2014) Flexibility of truncated and full-length glucansucrase GTF180 enzymes from Lactobacillus reuteri 180. FEBS J. 281, 2159–2171 10.1111/febs.12769 [DOI] [PubMed] [Google Scholar]
- 24. Dobruchowska J. M., Meng X., Leemhuis H., Gerwig G. J., Dijkhuizen L., and Kamerling J. P. (2013) Gluco-oligomers initially formed by the reuteransucrase enzyme of Lactobacillus reuteri 121 incubated with sucrose and malto-oligosaccharides. Glycobiology 23, 1084–1096 10.1093/glycob/cwt048 [DOI] [PubMed] [Google Scholar]
- 25. Wang W., and Malcolm B. A. (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange site-directed mutagenesis. BioTechniques 26, 680–682 10.2144/99264st03 [DOI] [PubMed] [Google Scholar]
- 26. Miller G. L. (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428 10.1021/ac60147a030 [DOI] [Google Scholar]
- 27. David G., and Pérez J. (2009) Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 42, 892–900 10.1107/S0021889809029288 [DOI] [Google Scholar]
- 28. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gibson D. G. (2009) Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 37, 6984–6990 10.1093/nar/gkp687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Studier F.W. (2005) Protein production by auto-induction in high-density shaking cultures. Prot. Exp. Pur. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.