

Abstract
In Staphylococcus aureus–caused endocarditis, the pathogen secretes staphylocoagulase (SC), thereby activating human prothrombin (ProT) and evading immune clearance. A previous structural comparison of the SC(1–325) fragment bound to thrombin and its inactive precursor prethrombin 2 has indicated that SC activates ProT by inserting its N-terminal dipeptide Ile1-Val2 into the ProT Ile16 pocket, forming a salt bridge with ProT's Asp194, thereby stabilizing the active conformation. We hypothesized that these N-terminal SC residues modulate ProT binding and activation. Here, we generated labeled SC(1–246) as a probe for competitively defining the affinities of N-terminal SC(1–246) variants preselected by modeling. Using ProT(R155Q,R271Q,R284Q) (ProTQQQ), a variant refractory to prothrombinase- or thrombin-mediated cleavage, we observed variant affinities between ∼1 and 650 nm and activation potencies ranging from 1.8-fold that of WT SC(1–246) to complete loss of function. Substrate binding to ProTQQQ caused allosteric tightening of the affinity of most SC(1–246) variants, consistent with zymogen activation through occupation of the specificity pocket. Conservative changes at positions 1 and 2 were well-tolerated, with Val1-Val2, Ile1-Ala2, and Leu1-Val2 variants exhibiting ProTQQQ affinity and activation potency comparable with WT SC(1–246). Weaker binding variants typically had reduced activation rates, although at near-saturating ProTQQQ levels, several variants exhibited limiting rates similar to or higher than that of WT SC(1–246). The Ile16 pocket in ProTQQQ appears to favor nonpolar, nonaromatic residues at SC positions 1 and 2. Our results suggest that SC variants other than WT Ile1-Val2-Thr3 might emerge with similar ProT-activating efficiency.
Keywords: coagulation factor, Staphylococcus aureus (S. aureus), prothrombin, ligand-binding protein, kinetics, virulence factor, affinity, endocarditis, equilibrium binding, specificity, staphylocoagulase (SC), coagulation, competitive equilibrium binding, clot formation, fibrin
Introduction
Blood clot formation by Staphylococcus aureus can be attributed to the combined effects of pathogen clumping and the generation of fibrin (Fbn).6 The latter is initiated by the secreted virulence factor, staphylocoagulase (SC). Based on the bacteria's ability to promote clot formation in rabbit plasma, S. aureus is divided into coagulase-positive and -negative subgroups. Typing of bacterial isolates for SC is still performed today in clinical diagnosis. Coagulase-positive S. aureus is a potent human pathogen that causes conditions ranging from minor skin infections to life-threatening diseases, such as severe pneumonia, meningitis, and bone, joint, and heart infections. Each year ∼500,000 patients in American hospitals contract staphylococcal infections that lead to ∼30,000 deaths (1, 2).
Turbulent blood flow can cause endothelial damage to heart valves, exposing subendothelium that leads to deposition of platelets and Fbn. The Fbn-platelet matrix deposited on damaged valves serves as a focus for adhering S. aureus bacteria circulating in the blood (3). The S. aureus-platelet interaction is facilitated by fibrinogen (Fbg), fibronectin, thrombospondin, and MSCRAMMs (microbial surface components recognizing adhesive matrix molecules), such as protein A and Fbg-binding ClfA (clumping factor A) (4–11). In acute bacterial endocarditis, Fbn formation is mediated by the thrombin precursor prothrombin (ProT), conformationally activated by bacterial SC. This furthers aggregation of platelets and enlargement of platelet-Fbn-bacteria vegetations on the valves (3). These friable vegetations can break up and cause pulmonary embolism and stroke. The pathogens also utilize these vegetations to disseminate and avert clearance by the host immune system (12). Acute bacterial endocarditis caused by S. aureus leads to 20–40% mortality despite antibiotic therapy (13).
Blood clotting is a highly regulated process, with a delicate balance between clotting and fibrinolysis. SC bypasses the clotting cascade by directly and nonproteolytically activating ProT, thereby shifting the balance to a procoagulant state. SC binds and conformationally activates ProT, forming a complex of catalytically active zymogen and activator, SC·ProT* (with the asterisk denoting a functional catalytic site). The activated complex cleaves Fbg to form Fbn clots involved in enlarging the vegetations. No physiological inhibitors of the SC·ProT* complex have been reported to date, and it is resistant to the plasma serpins, antithrombin-heparin and heparin cofactor II,7 α2-macroglobulin (14), and the leech inhibitor, hirudin (15).
SC is a bifunctional protein with a molecular mass of ∼75,000 Da. Its N-terminal region binds ProT (16), whereas the C-terminal region contains seven 27-amino acid-repeat sequences that bind Fbg (Fig. 1) (17, 18). We previously described that the SC fragment consisting of N-terminal residues 1–325, SC(1–325), binds ProT extremely tightly (∼17–72 pm) and noncovalently in a 1:1 stoichiometry to form the active SC(1–325)·ProT* complex (19). A comparison of the crystal structures of SC(1–325) with thrombin and the inactive zymogen prethrombin 2 (Pre2, prothrombin without the fragments 1 and 2) showed that the first six residues of the SC fragment were fully defined by electron density in the complex with Pre2 but not with thrombin. SC activates the zymogen by inserting its N-terminal Ile1-Val2 (I1V2 amino acid single-letter code) residues into the Ile16 pocket of Pre2, forming a salt bridge with Asp194 and inducing a functional active site in the zymogen (19). Formation of the SC(1–325)·ProT* and SC(1–325)·Pre2* complexes partially blocks exosite 1, the Fbg-binding site, but expresses a new Fbg substrate recognition site that facilitates Fbg binding and cleavage (Fig. 2). SC(1–325) consists of two three-helix-bundle domains (D1 (residues 1–146) and D2 (residues 147–325)) with a boomerang-like structure. The D1 domain interacts with the 148-loop of thrombin or Pre2 and the south rim of the catalytic site, and the D2 domain binds (pro)exosite 1 on Pre2 and thrombin. We report here that SC(1–246), with a partially truncated D2 domain, is capable of ProT activation and binds ProT with a KD of ∼1 nm, which made it a suitable probe for determining the affinities of mutant SC constructs by competitive equilibrium binding. The I1V2 residues are critically involved in SC-mediated ProT activation, and a comparison of the SC N-terminal residues from 12 different S. aureus strains showed strict conservation of I1V2T3. This raised the question of whether S. aureus could possibly make SC mutants that contain N-terminal residues different from I1V2T3 and what the effect of these other N-terminal residues on the affinity and activation of ProT might be. Because 8,000 different combinations are possible, our selection was guided by in silico protein modeling with Rosetta and virtual screening to prioritize combinations with high binding free energy and to identify a range of affinities and activation potencies. In this study, 46 different N-terminal mutants of SC(1–246) were generated through site-directed mutagenesis and characterized for binding and activation of a ProT(R155Q,R271Q,R284Q) mutant (ProTQQQ) that can be proteolytically activated to a meizothrombin form refractory to cleavage by prothrombinase and thrombin (20) but was used in this study to monitor conformational activation of prothrombin instead of proteolytic activation. The panel displayed a wide range of activation potencies and KD values both by equilibrium binding and ProTQQQ activation. The V1V2, I1A2, and L1V2 mutants bound ProTQQQ with affinities similar to that of WT SC(1–246) and with ProTQQQ activation potencies that were similar or up to 1.8-fold greater. Activation potencies of mutants with both weak equilibrium binding and activation-based affinities were typically reduced. Select mutants carrying nonpolar residues at position 1 bound moderately to ProTQQQ when measured by equilibrium binding but exhibited tight (approximately nanomolar) binding when measured by ProTQQQ activation, indicating a difference in affinity when binding to a disordered Ile16 pocket (equilibrium binding) and an Ile16 pocket conformationally stabilized by substrate occupation of the specificity site. These mutants had limiting activation rates similar to or exceeding that of WT SC(1–246). Overall, the Ile16 pocket of ProTQQQ favors nonpolar, nonaromatic residues at positions 1 and 2, however with specific restrictions governed by steric complementarity between the N terminus of SC and the Ile16 pocket of ProT. Our results, including efficient activation of ProT by weaker binding mutants at saturating concentrations, suggest that SC variants might emerge with similar or higher efficiency to activate ProT.
Figure 1.

Full-length staphylocoagulase from S aureus Newman D2 Tager 104 showing different regions of the protein. The full-length protein contains a 26-residue signal sequence, the D1 and D2 domains, a central region, and a C-terminal repeat region consisting of a pseudo-repeat (PR) and seven repeats.
Figure 2.
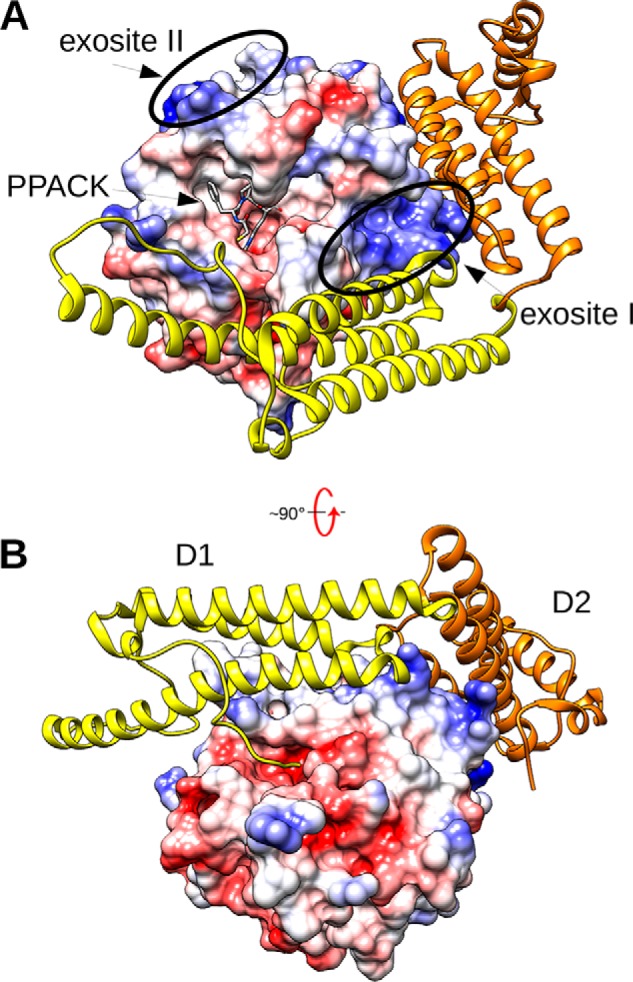
The SC(1–325)·Pre2 complex. A, the molecular surface of Pre2 is shown colored by electrostatic potential, with the ATA-PPACK (N-(sulfanylacetyl)-d-phenylalanyl-N-[(2S,3S)-6-{[amino(iminio)methyl]amino}-1-chloro-2-hydroxyhexan-3-yl]-l-prolinamide) inhibitor displayed as gray sticks in the active site. SC(1–325) is displayed in ribbon mode, with the N-terminal D1 domain colored yellow and the D2 domain colored gold. B, the complex above is rotated ∼90º from the standard orientation to show the insertion of the N-terminal SC peptide Ile1-Val2-Thr3 into the Ile16-binding pocket of Pre2, triggering activation. This figure was constructed with UCSF Chimera using the X-ray crystal structure 1NU9.pdb (16).
Results
Characterization of SC(1–246)-BODIPY and equilibrium binding of labeled and unlabeled SC(1–246) to ProTQQQ
SC(1–246)-BODIPY had a labeling ratio of 0.87 BODIPY-FL thiol-sensitive probe to SC(1–246) with a S7C substitution for covalent probe binding (Fig. 3A). Incubation of ProTQQQ with SC(1–246)-BODIPY showed binding in an approximately 1:1 ratio as observed by native PAGE (Fig. 3B). Competitive equilibrium binding studies were performed to determine the affinity and stoichiometry of four separate ProTQQQ preparations for unlabeled SC(1–246) and SC(1–246)-BODIPY. Unlabeled SC(1–246) bound very tightly to the ProTQQQ preparations, with KD of 0.6–1.0 nm, and a 1:1 stoichiometry (Fig. 4 and Table 1). SC(1–246)-BODIPY bound ProTQQQ with KD of 2.9–5.9 nm and a stoichiometry of 1:1. This weaker affinity is attributed to the BODIPY-FL label. Batch-to-batch variability of ProTQQQ was modest, as reflected by the consistent affinity values for unlabeled and labeled SC(1–246). The maximum fluorescence intensity was 0.6 ± 0.1 for all of the ProTQQQ preparations.
Figure 3.

Characterization of SC(1–246)-BODIPY and its complex with ProTQQQ. A, SDS-PAGE showing the purity of the labeled probe, SC(1–246)-BODIPY. 5 μg of the labeled probe was separated under reduced (lane 2) and nonreduced (lane 4) conditions. Lane 1, protein standards; lane 3, blank. B, native gel electrophoresis was used to show the SC(1–246)-BODIPY·ProTQQQ complex formation. ProTQQQ (2.5 μm, lane 1) was incubated with SC(1–246)-BODIPY (0-fold (lane 2), 0.5-fold (lane 3), 1.0-fold (lane 4), and 1.5-fold (lane 5) excess of ProTQQQ) for 15–30 min at 25 °C, and the complex was separated on native PAGE at 4 °C.
Figure 4.

Equilibrium binding of SC(1–246)-BODIPY and WT SC(1–246) to ProTQQQ. A, C, E, and G, SC(1–246)-BODIPY (29 nm (○) and 502 nm (●)) titrated with ProTQQQ, four separate batches. B, titration of 29 nm SC(1–246)-BODIPY (○); a mixture of 50 nm SC(1–246)-BODIPY and 50 nm WT SC(1–246) (▵); 502 nm SC(1–246)-BODIPY (●); and a mixture of 502 nm SC(1–246)-BODIPY and 500 nm WT SC(1–246) (▴) with ProTQQQ. D, F, and H, titrations of SC(1–246)-BODIPY (29 nm (○) and 502 nm (●)) and a mixture of 502 nm SC(1–246)-BODIPY and 500 nm WT SC(1–246) (▴) with ProTQQQ. Titrations shown in B, D, F, and H were performed with the corresponding, separate ProTQQQ preparations as in A, C, E, and G. The SC(1–246)-BODIPY titration data were analyzed by the quadratic binding equation to obtain the affinity, stoichiometry, and maximum fluorescence intensity (ΔFmax/Fo). Titration data of the probe and competitor were analyzed simultaneously by the cubic binding equation to obtain the affinity and stoichiometry of the competitor, WT SC(1–246) (Table 1).
Table 1.
Parameters for SC(1–246)-BODIPY and unlabeled WT SC(1–246) binding to four separate ProTQQQ preparations
Reference titrations of SC(1–246)-BODIPY with four separate ProTQQQ batches were obtained at two fixed probe concentrations. The competitive binding data for the ProTQQQ preparations were obtained by titrations of fixed concentrations of SC(1–246)-BODIPY probe, and SC(1–246) as competitor, with ProTQQQ. Data were fit simultaneously by the cubic equation to obtain the dissociation constant for ProTQQQ and SC(1–246)-BODIPY (KO, probe) and the competitor SC(1–246) (KC, competitor); the stoichiometric factor for SC(1–246)-BODIPY (n) and SC(1–246) (m); and the maximum fluorescence intensity (ΔFmax/Fo). Experimental error represents ± 2 S.D. Competitive equilibrium binding studies and data analysis were performed as described under “Experimental procedures.” SF, stoichiometric factor.
| SC(1–246) | SF (n or m) | KO or KC | ΔFmax/Fo |
|---|---|---|---|
| nm | |||
| SC(1–246)-BODIPY (n) | 0.90 ± 0.03 | 2.9 ± 0.7 | 0.60 ± 0.01 |
| SC(1–246) (m) | 1.10 ± 0.05 | 0.7 ± 0.2 | |
| SC(1–246)-BODIPY (n) | 0.90 ± 0.02 | 5.9 ± 0.8 | 0.59 ± 0.01 |
| SC(1–246) (m) | 1.10 ± 0.06 | 1.0 ± 0.3 | |
| SC(1–246)-BODIPY (n) | 1.00 ± 0.04 | 4.1 ± 1.1 | 0.57 ± 0.01 |
| SC(1–246) (m) | 1.10 ± 0.05 | 0.7 ± 0.2 | |
| SC(1–246)-BODIPY (n) | 1.01 ± 0.06 | 3.0 ± 1.4 | 0.58 ± 0.01 |
| SC(1–246) (m) | 1.20 ± 0.08 | 0.6 ± 0.4 |
Binding prediction and competitive equilibrium binding of SC(1–246) N-terminal mutants to ProTQQQ
Computational modeling used Rosetta's ΔΔG functionality to calculate the predicted change in binding energy for each mutation (21), sampling all 20 amino acids at positions 1–3. The fixed backbone design strategy allowed discrimination between steric clashing and nonclashing sequences, and based on the resulting energies in Rosetta energy units (REU), a representative double and triple N-terminal mutant panel was selected for experimental binding studies, with expected KD values between 1 and ∼1000 nm. Equilibrium binding showed that the double mutants V1V2, I1A2, and L1V2 bound to ProTQQQ very tightly, with affinities of ∼1 nm, similar to that of WT SC(1–246) (Fig. 5, A–C). Their excellent ProTQQQ activation potency also indicated efficient salt bridge formation with ProTQQQ Asp194 and formation of the ProTQQQ active site. The stoichiometric factor was ∼1, indicating that 1 mol of ProTQQQ binds to 1 mol of SC(1–246) mutant. The KD values from equilibrium binding were measured in the absence of a conformationally stabilizing tripeptide thrombin substrate, and they reflect global binding to ProTQQQ with a disordered Ile16 pocket and specificity subsite. The V1V2, I1A2, and L1V2 combinations of nonpolar residues had comparable equilibrium binding and activation KD values, suggesting a favorable steric complementarity to the Ile16 pocket. Mutants with weaker equilibrium KD values may be governed mainly by the D1 and truncated D2 domain interactions with ProTQQQ, although double mutants with Ile, Val, Leu, and Thr in combination with polar, nonpolar aromatic, bulky, or small residues at position 2 exhibited considerable tightening of the binding to ProTQQQ when an Ile16 pocket-stabilizing substrate was present (Fig. 5 and Tables 2 and 3). The A1S2 and A1T2 mutants exhibited weak equilibrium binding affinity; however, the presence of a chromogenic substrate during ProTQQQ activation caused these mutants to bind with ∼30–100-fold tighter KD, respectively, and exhibit WT-like activation potency. This suggests some conformational flexibility of the ordered Ile16 pocket. Mutants with charged residues (Lys, Arg, Asp, Glu, His) in positions 1 and/or 2; Gly or Trp in position 1; or Pro in position 2 typically bound weakly and were poor activators, with G1G2 the weakest binder (Fig. 6 and Tables 2 and 3). Although disrupted binding for Pro was expected, even some pairs with small, hydrophobic residues in positions 1 and 2 may not optimally fit, illustrating the steric specificity of the Ile16-binding pocket.
Figure 5.

Competitive binding of SC(1–246) N-terminal double mutants that bind tightly to ProTQQQ. Titrations of the probe, SC(1–246)-BODIPY (29 nm (○) and 502 nm (●)), with ProTQQQ from Fig. 4 served as reference curves for competitive titrations of SC(1–246) mutants. The probe concentration for all of the competitive titrations (▵, ▴) in A–H was 50 nm. Concentrations of competing SC(1–246) mutants were as follows: V1V2 0.90 μm (A), I1A2 0.74 μm (B), L1V2 0.75 μm (C), I1T2 2.24 μm (D), I1W2 0.52 μm (▵) and 9.97 μm (▴) (E), T1V2 6.16 μm (F), L1T2 9.55 μm (G), and L1Q2 3.95 μm (H). Titrations of probe and competitor were simultaneously analyzed by the cubic binding equation to obtain KC, stoichiometry, and maximum fluorescence intensity (ΔFmax/Fo) of the SC(1–246) mutants (Tables 2 and 3).
Table 2.
Characteristics of N-terminal residues in relation to mutant SC(1–246) affinity and ProTQQQ activation potency
| N-terminal mutants | Residue 1 characteristics | Residue 2 characteristics | Affinity range (KD) | Activation potency normalized (Vlim) | Free energy (ΔG) |
|---|---|---|---|---|---|
| nm | |||||
| I1V2 WT | Nonpolar | Nonpolar | 0.7 ± 0.2 | 1.0 | −12.48 |
| V1V2, I1A2, and L1V2 | Similar nonpolar | Similar nonpolar | ∼0.9–1.3 | ∼1.0–1.80 | − 12.33 to − 12.11 |
| I1T2, I1W2, I1L2, and V1G2 | Similar nonpolar | Polar, nonpolar, indole | ∼5–100 | ∼0.9–1.60 | − 11.29 to − 9.55 |
| T1V2, T1P2, and T1A2 | Polar | Nonpolar, pyrrolidine | ∼11–100 | ∼0.4–1.70 | − 10.88 to − 9.55 |
| L1T2, L1Q2, L1K2, and L1P2 | Similar nonpolar | Nonpolar, pyrrolidine, charged | ∼19–220 | ∼0.3–1.6 | − 10.54 to − 9.08 |
| W1A2 and W1E2 | Indole | Nonpolar, charged | ∼117–191 | NDa | − 9.45 to − 9.17 |
| M1L2, M1W2, M1K2, and M1E2 | Bulky nonpolar | Nonpolar, indole, charged | ∼127–514 | ∼0.06–1.1 | − 9.40 to − 8.58 |
| G1H2, G1A2, G1P2, G1D2, G1G2, A1W2, A1K2, A1S2, and A1T2 | Small nonpolar | Small nonpolar, polar, charged, aromatic | ∼153–503 | ∼0.02–1.0 | − 9.29 to − 8.62 |
| Q1K2, T1D2, S1K2, Q1L2, and N1D2 | Polar | Charged, nonpolar | ∼169–493 | ∼0.02–0.24 | − 9.24 to − 8.58 |
| R1R2, K1A2, E1T2, R1Q2, and E1S2 | Charged | Charged, nonpolar, polar | ∼379–650 | ∼0.004–0.1 | − 8.76 to − 8.44 |
a ND, not determined.
Table 3.
ProTQQQ binding and activation by SC(1–246) N-terminal double mutants
Reference titrations of SC(1–246)-BODIPY with ProTQQQ were obtained at two fixed probe concentrations. Competitive binding data were obtained by titrations of fixed concentrations of SC(1–246)-BODIPY probe, and mutant SC(1–246) as competitor, with ProTQQQ. Data were fit simultaneously by the cubic equation to obtain the dissociation constant for ProTQQQ and SC(1–246)-BODIPY (KO, probe) and mutant SC(1–246) (KC, competitor); the stoichiometric factor for SC(1–246)-BODIPY (n) and mutant SC(1–246) (m); and the maximum fluorescence intensity (ΔFmax/Fo). Experimental error represents ± 2 S.D. Competitive equilibrium binding studies and data analysis were performed as described under “Experimental procedures.” ND, not determined; SF, stoichiometric factor.
| SC(1–246) mutants | SF (m) | KC | ΔFmax/Fo | ΔG | KD from ProTQQQ activation | Vlim | REU complex (predicted) |
|---|---|---|---|---|---|---|---|
| mol ProTQQQ/mol SC (1–246) | nm | kcal/mol | nm | REU | |||
| I1V2 | 1.10 ± 0.05 | 0.7 ± 0.2 | 0.60 ± 0.01 | −12.48 | 1.5 ± 0.2 | 1.0 ± 0.01 | −130 |
| V1V2 | 1.17 ± 0.04 | 0.9 ± 0.3 | 0.60 ± 0.05 | −12.33 | 2.1 ± 0.5 | 1.0 ± 0.01 | −128 |
| I1A2 | 1.13 ± 0.02 | 1.1 ± 0.3 | 0.57 ± 0.01 | −12.25 | 4.2 ± 0.5 | 1.8 ± 0.01 | −126 |
| L1V2 | 1.14 ± 0.04 | 1.3 ± 0.4 | 0.57 ± 0.004 | −12.11 | 4.1 ± 1.0 | 1.4 ± 0.1 | −117 |
| I1T2 | 1.01 ± 0.09 | 5.3 ± 1.7 | 0.56 ± 0.01 | −11.29 | 5.0 ± 1.4 | 0.90 ± 0.08 | −125 |
| I1W2 | 1.13 ± 0.12 | 7.1 ± 2.4 | 0.59 ± 0.01 | −11.11 | 1.8 ± 0.2 | 0.92 ± 0.01 | 481 |
| T1V2 | 1.02 ± 0.16 | 11 ± 4 | 0.56 ± 0.01 | −10.88 | 8 ± 2 | 1.7 ± 0.1 | −124 |
| L1T2 | Fixed to 1 | 19 ± 6 | 0.60 ± 0.01 | −10.54 | 3.0 ± 0.4 | 1.4 ± 0.1 | −112 |
| L1Q2 | Fixed to 1 | 27 ± 7 | 0.57 ± 0.01 | −10.33 | 1.2 ± 0.4 | 1.3 ± 0.1 | −108 |
| T1P2 | Fixed to 1 | 61 ± 40 | 0.57 ± 0.01 | −9.84 | 169 ± 34 | 0.38 ± 0.03 | −57 |
| I1L2 | Fixed to 1 | 64 ± 17 | 0.57 ± 0.01 | −9.82 | 7 ± 1 | 1.3 ± 0.1 | −119 |
| L1K2 | Fixed to 1 | 76 ± 12 | 0.60 ± 0.01 | −9.71 | 3.0 ± 0.3 | 1.6 ± 0.1 | −110 |
| V1G2 | Fixed to 1 | 100 ± 27 | 0.60 ± 0.01 | −9.55 | 3.1 ± 0.4 | 1.6 ± 0.1 | −122 |
| T1A2 | Fixed to 1 | 100 ± 27 | 0.60 ± 0.01 | −9.55 | 5.2 ± 0.2 | 1.3 ± 0.1 | −120 |
| W1A2 | Fixed to 1 | 117 ± 36 | 0.60 ± 0.01 | −9.45 | ND | ND | 1,298 |
| M1L2 | Fixed to 1 | 127 ± 38 | 0.58 ± 0.01 | −9.41 | 16 ± 2 | 0.90 ± 0.02 | −107 |
| M1W2 | Fixed to 1 | 148 ± 27 | 0.60 ± 0.01 | −9.32 | 8 ± 1 | 0.25 ± 0.01 | 493 |
| G1H2 | Fixed to 1 | 153 ± 43 | 0.61 ± 0.01 | −9.29 | 346 ± 134 | 0.02 ± 0.003 | −115 |
| G1A2 | Fixed to 1 | 166 ± 42 | 0.60 ± 0.01 | −9.25 | 46 ± 5 | 0.17 ± 0.01 | −118 |
| Q1K2 | Fixed to 1 | 169 ± 49 | 0.60 ± 0.01 | −9.24 | 59 ± 8 | 0.20 ± 0.01 | −116 |
| A1W2 | Fixed to 1 | 170 ± 60 | 0.58 ± 0.01 | −9.23 | 44 ± 5 | 0.14 ± 0.01 | 487 |
| M1K2 | Fixed to 1 | 174 ± 54 | 0.58 ± 0.01 | −9.22 | 9 ± 1 | 1.1 ± 0.1 | −111 |
| W1E2 | Fixed to 1 | 191 ± 54 | 0.60 ± 0.01 | −9.17 | 5,700 ± 4,200 | 0.13 ± 0.06 | 1,307 |
| A1K2 | Fixed to 1 | 191 ± 62 | 0.58 ± 0.01 | −9.16 | 27 ± 2 | 0.55 ± 0.11 | −116 |
| L1P2 | Fixed to 1 | 220 ± 64 | 0.58 ± 0.01 | −9.08 | 22 ± 3 | 0.31 ± 0.01 | −50 |
| T1D2 | Fixed to 1 | 248 ± 72 | 0.58 ± 0.01 | −9.01 | 100 ± 18 | ∼0.2 | −115 |
| S1K2 | Fixed to 1 | 251 ± 76 | 0.58 ± 0.01 | −9 | 44 ± 2 | 0.16 ± 0.01 | −115 |
| Q1L2 | Fixed to 1 | 277 ± 83 | 0.58 ± 0.01 | −8.94 | 188 ± 21 | 0.024 ± 0.001 | −111 |
| A1S2 | Fixed to 1 | 295 ± 69 | 0.60 ± 0.01 | −8.91 | 11 ± 0.4 | 1.0 ± 0.1 | −118 |
| R1R2 | Fixed to 1 | 379 ± 88 | 0.61 ± 0.01 | −8.76 | 26 ± 5 | 0.1 ± 0.01 | −104 |
| G1P2 | Fixed to 1 | 446 ± 81 | 0.61 ± 0.01 | −8.66 | 64 ± 6 | 0.020 ± 0.001 | −54 |
| K1A2 | Fixed to 1 | 455 ± 80 | 0.60 ± 0.01 | −8.65 | 69 ± 20 | 0.010 ± 0.001 | −111 |
| G1D2 | Fixed to 1 | 468 ± 80 | 0.61 ± 0.01 | −8.63 | 76 ± 7 | 0.020 ± 0.001 | −113 |
| A1T2 | Fixed to 1 | 480 ± 95 | 0.60 ± 0.01 | −8.62 | 5 ± 0.3 | 1.0 ± 0.1 | −118 |
| E1T2 | Fixed to 1 | 486 ± 80 | 0.60 ± 0.01 | −8.61 | 105 ± 11 | 0.030 ± 0.001 | −113 |
| N1D2 | Fixed to 1 | 493 ± 99 | 0.60 ± 0.01 | −8.6 | 89 ± 7 | 0.020 ± 0.001 | −113 |
| G1G2 | Fixed to 1 | 503 ± 90 | 0.61 ± 0.01 | −8.59 | 26 ± 2 | 0.26 ± 0.01 | −115 |
| R1Q2 | Fixed to 1 | 503 ± 94 | 0.60 ± 0.01 | −8.59 | 35 ± 26 | 0.004 ± 0.010 | −104 |
| M1E2 | Fixed to 1 | 514 ± 96 | 0.60 ± 0.01 | −8.58 | 56 ± 8 | 0.060 ± 0.001 | −105 |
| E1S2 | Fixed to 1 | 650 ± 120 | 0.61 ± 0.01 | −8.44 | 205 ± 14 | 0.010 ± 0.002 | −112 |
Figure 6.

Competitive binding titrations of SC(1–246) N-terminal double mutants that bind weakly to ProTQQQ. Titrations of the probe, SC(1–246)-BODIPY (29 nm (○) and 502 nm (●)) with ProTQQQ from Fig. 4 served as reference curves for competitive titrations of SC(1–246) mutants. The probe concentration for all of the competitive titrations (▴) in A–F was 50 nm. Concentrations of competing SC(1–246) mutants were as follows: G1G2 8.12 μm (A), R1R2 9.06 μm (B), K1A2 9.50 μm (C), E1T2 9.04 μm (D), R1Q2 10.30 μm (E), and E1S2 11.38 μm (F). Titrations of probe and competitor were simultaneously analyzed by the cubic binding equation to obtain KC, stoichiometry, and maximum fluorescence intensity (ΔFmax/Fo) of the SC(1–246) mutants (Tables 2 and 3).
Equilibrium binding KD values of triple mutants varied from ∼4 to ∼55 nm (Table 4). The mutant I1I2V3 bound ProTQQQ with a KD of 4 ± 3 nm (Fig. 7A) and activated ProTQQQ at an appreciable rate, suggesting that the ProTQQQ Ile16 pocket accommodates these nonpolar residues with reasonable fit, conducive to forming a salt bridge with ProTQQQ Asp194. The mutants R1H2W3, F1L2Q3, E1S2W3, D1D2Y3, G1G2G3, and E1L2K3 had KD values of 36–55 nm (Fig. 7, B–G) but activated ProTQQQ poorly. Interestingly, substituting Gly3 for WT Thr3 rescued equilibrium binding ∼10-fold, compared with G1G2, but with no improvement in activation potency.
Table 4.
ProTQQQ binding and activation by SC(1–246) N-terminal triple mutants
Reference titrations of SC(1–246)-BODIPY with ProTQQQ were obtained at two fixed probe concentrations. The competitive binding data were obtained by titrations of fixed concentrations of SC(1–246)-BODIPY probe, and triple mutant SC(1–246) as competitor, with ProTQQQ. Data were fit simultaneously by the cubic equation to obtain the dissociation constant for ProTQQQ and SC(1–246)-BODIPY (KO, probe) and mutant SC(1–246) (KC, competitor); the stoichiometric factor for SC(1–246)-BODIPY (n) and mutant SC(1–246) (m); and the maximum fluorescence intensity (ΔFmax/Fo). Experimental error represents ± 2 S.D. Competitive equilibrium binding studies and data analysis were performed as described under “Experimental procedures.”
| SC(1–246) mutants | KC | KD from ProTQQQ activation | Vlim | ΔFmax/Fo | ΔG |
|---|---|---|---|---|---|
| nm | nm | kcal/mol | |||
| I1I2V3 | 4 ± 3 | 3 ± 1 | 0.80 ± 0.03 | 0.57 ± 0.01 | −11.51 |
| R1H2W3 | 35 ± 23 | 124 ± 48 | 0.31 ± 0.03 | 0.58 ± 0.01 | −10.16 |
| F1L2Q3 | 39 ± 25 | > 500 | 0.06 ± 0.05 | 0.58 ± 0.01 | −10.11 |
| E1L2K3 | 42 ± 27 | 309 ± 54 | 0.26 ± 0.01 | 0.58 ± 0.01 | −10.07 |
| E1S2W3 | 48 ± 32 | 28 ± 42 | 0.03 ± 0.01 | 0.58 ± 0.01 | −9.98 |
| G1G2G3 | 48 ± 33 | 29 ± 20 | 0.11 ± 0.01 | 0.58 ± 0.01 | −9.98 |
| D1D2Y3 | 55 ± 36 | 545 ± 145 | 0.19 ± 0.03 | 0.58 ± 0.01 | −9.90 |
Figure 7.

Competitive binding titrations of SC(1–246) N-terminal triple mutants with ProTQQQ. Titrations of the probe, SC(1–246)-BODIPY (29 nm (○) and 502 nm (●)) with ProTQQQ from Fig. 4 served as reference curves for competitive titrations of SC(1–246) mutants. The probe concentration for all of the competitive titrations (▴) in A–G was 50 nm. Concentrations of competing SC(1–246) mutants were as follows: I1I2V3 2.84 μm (A), R1H2W3 7.59 μm (B), F1L2Q3 5.49 μm (C), E1S2W3 5.74 μm (D), D1D2Y3 7.65 μm (E), E1L2K3 5.77 μm (F), and G1G2G3 6.44 μm (G). Titrations of probe and competitor were simultaneously analyzed by the cubic binding equation to obtain KC, stoichiometry, and maximum fluorescence intensity (ΔFmax/Fo) of the SC(1–246) mutants (Table 4).
The Gibbs free energy ΔG for binding of the mutants varied from −12.45 to −8.44 kcal/mol (Tables 2–4 and Fig. 8), with the lowest value for WT SC(1–246), calculated from the averaged KD for binding to the four ProTQQQ batches. V1V2, I1A2, L1V2, I1T2, I1W2, and I1I2V3 had ΔG values similar to the WT protein, consistent with KD values in the nanomolar range. A good correlation was observed between the predicted Rosetta energies and the ΔG values calculated from equilibrium binding (Fig. 9), except for a few outliers (T1P2, W1A2, M1W2, A1W2, W1E2, L1P2, and G1P2) that gave inconsistent Rosetta energies but also exhibited reduced activation potency (Fig. S1). These outliers occur in the presence of Pro2 and of Trp1 or Trp2 and display an off-scale energetic prediction because Rosetta is unable to fit them into the structure of the complex. This is not unexpected behavior; the introduction of a Pro or Trp residue may require such major conformational rearrangement, due to steric clash or backbone geometry restriction, that the score penalty increases beyond the ability of the sampling protocol to incorporate it. Additional sampling might be necessary to create accurate models for these large or conformationally restricted amino acids. The only unexpected discrepancy was I1W2 with an REU score of 481 but exhibiting WT-like binding and ProTQQQ activation properties. The behavior of I1W2 is difficult to rationalize; the high Rosetta score reflects our expectation that inserting the steric bulk of a Trp residue at position 2 would be unfavorable. This prediction is consistent with the predictions and measurements of other mutants containing Trp at position 2. We can only conjecture that the I1W2 combination permits binding and activation via an unknown mechanism.
Figure 8.

Gibbs free energy of the SC(1–246) N-terminal double and triple mutants for binding to ProTQQQ. ΔG values were calculated using KD values obtained from equilibrium binding, as described under “Experimental Procedures.”
Figure 9.

Correlation between REU and ΔG values calculated from equilibrium binding.
Prothrombin activation by SC(1–246) N-terminal double and triple mutants
Initial rates v0 of p-nitroanilide formation upon chromogenic substrate cleavage by the ProTQQQ·SC complexes were linear for WT SC(1–246) and tight-binding mutants, and the titrations showed saturation around 20 nm SC(1–246) variant. A few mutants caused hysteresis-like lag phases in substrate hydrolysis by their complexes with ProTQQQ, and post-lag, linear v0 rates were used for analysis of these mutants. The limiting velocity (Vlim) of WT SC(1–246) was 20 ± 5 mAbs/min, in good agreement for all four ProTQQQ batches, and used as the 100% value, or 1.00, for normalizing assays of slow-activating mutants to 1 nm ProTQQQ. KD and Vlim values derived from the ProTQQQ activation analysis are given in Table 3. The V1V2, I1A2, and L1V2 mutants with affinities similar to WT SC(1–246) activated ProTQQQ with similar or higher potency than WT SC(1–246). I1A2 and L1V2 showed ∼81 and ∼41% increase in ProTQQQ activity, suggesting that Ala2 and Leu1 nonpolar residues bound tightly and fit optimally in the Ile16 pocket of ProTQQQ for Ile1 and Leu1 bonding with Asp194, resulting in increased ProTQQQ activity. Apparent KD values for these tight-binding mutants, derived from the activation kinetics, were in good agreement with those measured by equilibrium binding (Fig. 10). Most mutants with a weaker equilibrium binding KD for ProTQQQ also activated ProTQQQ weakly, with relative Vlim ≪ 1, although some, like M1L2, A1T2, A1S2, M1K2, T1A2, L1Q2, I1L2, L1T2, L1K2, V1G2, and T1V2 activated ProTQQQ similarly or up to 1.7-fold better than WT SC(1–246) at mutant concentrations approaching ProTQQQ saturation with regard to KD calculated from the activation profiles (Tables 2–4). The affinities of these mutants, defined by ProTQQQ activation, were typically higher than their counterparts defined by equilibrium binding, due to allosteric modulation of the binding by the presence of the chromogenic substrate occupying the specificity pocket of the zymogen. This was previously also reported for vWbp binding to FPR derivatives of prothrombin, prethrombin 1 and 2 (22), and the binding of oligopeptides to trypsinogen with the specificity site occupied by a covalent ligand or a tight-binding inhibitor (23). Affinities defined for triple mutants that activated ProTQQQ poorly were not well-defined due to large experimental error. Overall, nonpolar hydrophobic residues were well-tolerated in position 1, whereas polar, aromatic, or charged residues generally diminished the ProTQQQ activation potency. Due to its conformational rigidity and unusual configuration, Pro in position 2 is thought to hamper efficient salt bridge formation of any residue at position 1, with a greatly diminished activation potency as a result.
Figure 10.
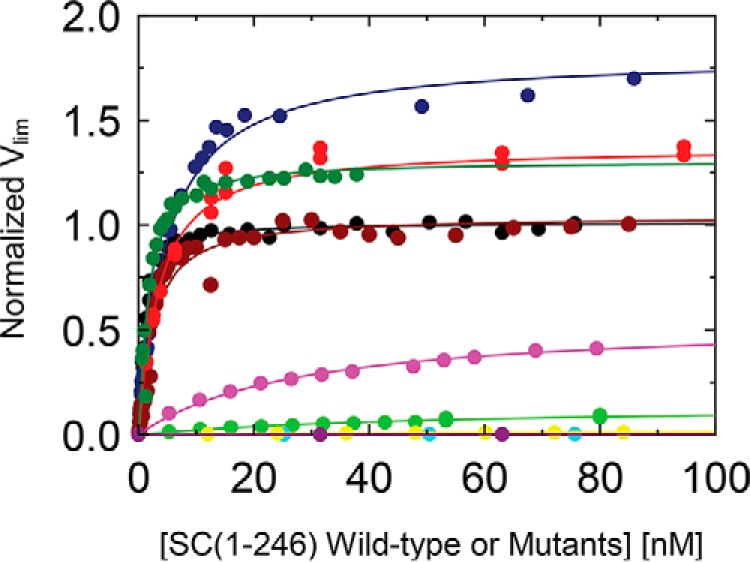
Activation of ProTQQQ by SC(1–246) WT or double mutants. ProTQQQ (1, 10, or 20 nm) and SC(1–246) WT (black circles) and a representative selection of mutants (navy blue, I1A2; red, L1T2; dark green, L1Q2; brown, V1V2; purple, A1K2; green, A1W2; cyan, K1A2; yellow, Q1K2; eggplant, E1S2) were incubated for 10 min at 25 °C, and the reaction was initiated by adding S-2238. Activation of ProTQQQ was measured by the relative rates of increase in absorbance at 405 nm, and weak-binding mutants with low activation potency were titrated up to 1300 nm. The data were analyzed as described under “Experimental Procedures.”
Discussion
In physiological blood coagulation, ProT is proteolytically cleaved in a multistep process to form the central clotting protease, thrombin (24). Proteolytic activation of ProT and Pre2 is initiated by cleavage of the peptide bond between Arg15 and Ile16 (chymotrypsinogen numbering). The newly formed I1V2 N terminus inserts into the Ile16 pocket of the zymogen, triggering the folding of zymogen activation domain residues 142–152, 186–194, and 216–226, and the α-ammonium of group of Ile1 forms a salt bridge with the carboxylate of Asp194. This generates the substrate recognition subsites and the oxyanion hole (25, 26). In contrast, SC, a virulence factor secreted by S. aureus, is a potent nonproteolytic ProT activator. Our structure-function studies on SC constructs in complex with ProT, Pre2, and thrombin demonstrate that the SC I1V2 residues are critical for ProT and Pre2 activation. As shown in the crystal structure of the SC(1–325)·Pre2 complex, these residues insert into the Ile16 pocket of Pre2, with SC Ile1 forming the salt bridge with Asp194, and conformational changes resulting in the generation of the Pre2 active site in the complex (19). Based on the ability to clot plasma, SC variants from different S. aureus strains are classified into 12 different serotypes (27, 28). The SC D1 domain (except for the first seven residues) and the D2 domain of 10 different S. aureus serotypes showed identities ranging from 53 to 89% (D1) and from 57 to 92% (D2), with the conservation of predicted ProT residues (29). A new classification scheme, based on the D1 domain of SC from 103 S. aureus strains, showed an average of 67.1% D1 domain identity among the 12 SC serotypes, with the first seven N-terminal residues being highly conserved (28). Our studies showed that the conserved I1V2 residues in the natural N terminus of SC(1–325) were required for ProT activation, with SC(2–325) being 6-fold less efficient and SC(3–325) only exhibiting <2% activity. However, a Met-SC(1–325) construct still containing the initiating Met residue (Met1-Ile2-Val3) also showed ProT activation potency and binding with KD = 17 ± 2 nm, suggesting some degree of SC promiscuity and also flexibility of the ProT Ile16 pocket in accommodating different residues (19). The first six N-terminal SC residues were fully resolved in the structure of the SC(1–325)·Pre2 complex, and modeling showed that extending or shortening this hexapeptide by one residue still allowed for interaction with the prothrombin 2 activation pocket. Also, von Willebrand factor–binding protein (vWbp), a nonproteolytic ProT activator from S. aureus Tager 104 Newman D2, and streptokinase, a nonproteolytic plasminogen activator from Streptococcus equisimilis and Streptococcus pyogenes, have N-terminal I1V2 and I1A2 sequences, respectively, suggesting that the Ile16 pocket of serine protease zymogens is highly suited to accommodate small nonpolar residues. Hence, we wanted to examine the tolerance of the Ile16 pocket of ProT for a panel of N-terminal residues. This could help decipher why SC has this unique N-terminal conservation and predict whether under selective pressure, S. aureus may be capable of producing SC variants with other N-terminal residues that could have affinity and potency similar or superior to that of SC with the canonical I1V2 residues.
Our previous studies reported that SC(1–325) binds to ProT with KD 0.3 ± 0.2 nm (19). The extremely high affinity makes this construct less suitable for use as a probe in measuring competitive binding of SC mutants with weaker affinity. Therefore, we used truncated SC(1–246) with a S7C substitution for BODIPY labeling, which we characterized to have a KD of ∼3 nm for ProTQQQ binding, a minimally weaker interaction than what we measured for competitive binding of WT SC(1–246) with KD of ∼1 nm, due to introduction of the fluorescence label. This probe allowed measurement of affinities of competitive N-terminal mutants up to KD ∼650 nm. SC(1–246) contains an intact D1 domain but lacks residues 247–282 of the (pro)thrombin (pro)exosite I-binding D2 domain, which is thought to result in weakened affinity compared with a construct with an intact D2 domain.
The mutants in this study can be categorized in four groups with regard to ProTQQQ binding and activation, compared with WT SC(1–246): (a) similar affinity, and similar or increased activation potency, in constructs with conserved or homologous nonpolar N-terminal residues; (b) modestly to significantly weaker equilibrium binding affinity, but substrate-induced, tight KD and induced fit to the Ile16 pocket, and similar or increased activation potency; (c) modestly weaker affinity but significantly reduced activation potency due to poor fit of the N-terminal residues and mainly governed by partial D2 binding; and (d) significantly weaker affinity and significantly reduced or abolished activation potency in constructs with polar or proline-containing N termini, perhaps by triggering unfavorable long-range binding interactions through electrostatic or steric conformational changes. The overall affinity of our constructs for ProTQQQ is proposed to result from the combined effects of binding of N-terminal SC(1–246) residues to the ProTQQQ Ile16 pocket, contact of the D1 domain with the ProTQQQ 148-loop, and binding of the truncated D2 domain to ProT proexosite I, whereas the activation potency is critically defined by the capacity of SC(1–246) residue 1 forming a salt bridge with ProTQQQ Asp194 and an adequate and, if necessary, substrate-inducible fit of residue 2 in the ordered Ile16 pocket. Our previous studies indicated that isolated D1 bound to ProT with modest affinity, KD ∼780 nm when measured by ProT activation and KD ∼3.5 μm when measured by fluorescence equilibrium binding in the absence of a substrate. Isolated D2 binding to ProT proexosite I did not cause ProT activation due to the absence of the critical N-terminal residues; however, its affinity by equilibrium binding was ∼30 nm (19, 30).
Active-site ligands with high affinity for the proteinase are known to induce a proteinase-like conformation in the zymogen. Occupation of the ProTQQQ specificity site by a thrombin substrate caused a ∼5-fold allosteric tightening of the D1 binding, which is facilitated by favorable steric complementarity of the first two N-terminal residues of SC and the Ile16-binding pocket (Fig. 11). A similar allosteric modulation is caused by binding of the S. aureus-secreted vWbp to prothrombin derivatives, with tighter binding to prothrombin forms that have their active site labeled with d-Phe-Pro-Arg-chloromethylketone (22). This tightening effect was previously also observed for binding of small peptides in the Ile16 pocket of trypsinogen with its specificity site occupied by pancreatic trypsin inhibitor (PTI) or covalently bound p-guanidinobenzoate (pGB) (23, 31). In the presence of PTI or pGB, the Ile16 pocket is fully formed, in contrast with free trypsinogen, which shows a disordered specificity pocket and Ile16-binding pocket in the crystal structure. We found that the V1V2, I1A2, and L1V2 double mutants of SC(1–246) bind ProTQQQ with similar affinities and show similar or higher potency in activating ProTQQQ, and the I1I2V3 triple mutant binds and activates ProTQQQ only slightly more weakly. In a study with isolated di- and oligopeptides, Bode (23) reported that the more effective peptides I1V2 and I1V2G3 used in activation studies of trypsinogen carrying a ligand in the specificity pocket (pGB or pancreatic trypsin inhibitor) were identical to the newly formed N-terminal sequence after cleavage of the activation peptide. However, the peptides V1V2, I1A2, and L1V2 bound to pGB-trypsinogen with affinities 30-, 160-, and 190-fold weaker than I1V2, respectively. In our studies, additional interactions of the D1 and the truncated D2 domains were shown to contribute to enhanced binding affinity and ProTQQQ activation potential by SC(1–246) mutants with these N-terminal sequences. In the I1I2V3 triple mutant, Ile2 is similar in size and hydrophobicity to Val2 of the WT construct. Consequently, there was not much reduction in ProTQQQ affinity and activation potency of this mutant. The equilibrium binding and kinetic data presented here indicate that small and nonpolar residues are preferred over bulky and charged ones for sufficient ProT binding and activation (Table 2), due to a better fit in the Ile16 pocket, even in the disordered state. The Val1, Leu1, and Ala2 residues of the double mutants are as functional as Ile1 and Val2 of WT SC(1–246). The presence of these residues in the pocket favors the packing and alignment of the side chains triggering conformational activation in a similar fashion as seen in the SC(1–325)·Pre2 complex, with the α-ammonium group of Val1 possibly connecting through a salt bridge with Asp194. Ala2 may be stabilized through the formation of a hydrogen bond with Asp189 in the ProTQQQ specificity pocket, as observed for Val2 in the SC(1–325)·Pre2 crystal structure. In the crystal structure of SC(1–325)·Pre2, Ile1 is completely buried in the hydrophobic Ile16 pocket, whereas Val2 partially contacts the outer solvent. Replacement of valine with the bulky amino acid leucine showed a 15-fold decrease in affinity for pGB-trypsinogen (23). Even though binding and insertion of the first two N-terminal are additive, the Ile16 pocket can accommodate a less favorable residue at the second position.
Figure 11.
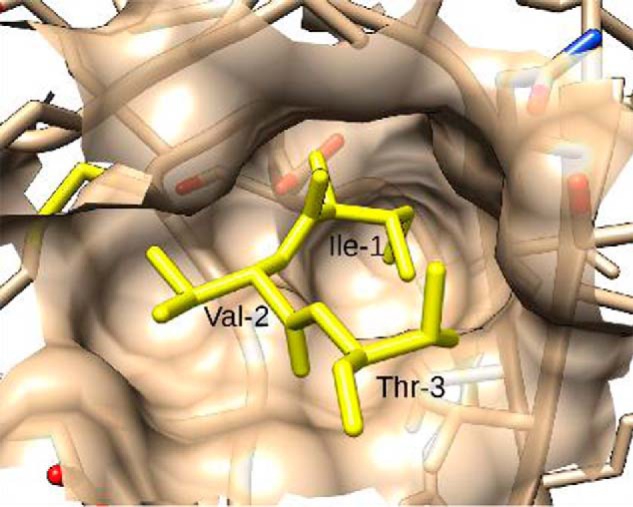
Binding site complementarity between SC(1–325) and prethrombin 2. Native residues I1V2T3 of SC are shown in the Ile16 pocket of prothrombin 2. The steric complementarity excludes the possibility of adding additional bulk to the N terminus of SC without generating energetically unfavorable clashes with neighboring residues.
Interestingly, the T1A2 mutant has the same T1A2T3 residues as those proteolytically generated in a ProT mutant upon cleavage at Arg320 (32). Substitution of IVE to TAT following ProT Arg320 did not prevent cleavage but ultimately generated a thrombin mutant IIaTAT with zymogen-like properties that bound the active site probe DAPA with ∼32,000-fold weaker affinity than WT thrombin (KD ∼1 nm) and only had 0.2% specific activity toward the thrombin-specific chromogenic substrate S2238. Our T1A2 mutant bound ProTQQQ with equilibrium KD of 100 ± 27 nm, and 5.2 ± 0.2 nm from ProT activation, in the presence of a chromogenic substrate and an activation potential 1.3 times that of WT SC(1–246). This functional mutant may attribute its potential of salt bridge formation with ProTQQQ Asp194 and zymogen activation to cumulative N-terminal, D1 and D2 conformational interactions that shift the zymogen–protease equilibrium in IIaTAT toward the protease conformation.
Equilibrium binding of various mutants involving Pro and Trp was moderate to weak (T1P2, M1W2, A1W2, and L1P2, ∼60–200 nm), and their dramatically reduced activation potency suggests nonproductive interaction with the Ile16 pocket and binding through the partial D2 domain that may be perturbed by electrostatic or steric effects introduced by the mutations. Variations in the structural orientation of the partial D2 domain may alter binding and lead to weaker overall mutant affinity for ProTQQQ. Typically, these mutants were outliers in the correlation of predicted REU scores and measured ΔG values of equilibrium binding (Fig. S1), suggesting that the sampling protocol employed was not sufficient to overcome the major structural perturbation required to insert the steric bulk of a Trp residue or to accommodate the backbone angle restriction imposed by a Pro residue. The high energy penalty can be interpreted as a clear signal that the new sequence is incompatible with the native structure. Triple mutants containing bulky aromatic or charged residues as well as G1G2G3 bound ∼10-fold tighter than the G1G2 double mutant with KD of 48 ± 33 nm, respectively, suggesting that Gly at position 3 instead of native Thr is more conducive to steric complementarity. However, the low activation potencies of both the double and triple mutant indicated impaired salt bridge formation with ProTQQQ Asp194.
In conclusion, we have determined the affinities of a panel of 46 different SC N-terminal mutants for ProTQQQ and showed that the Ile16 pocket is specific for accommodating residues that are similar in size to I1V2, but improved fit for a variety of preferably noncharged position 2 residues except for proline can be induced by small substrate binding. This characterization of the SC N-terminal residues in the SC-ProT complex provides further information to better design antibodies as drugs to target the SC N terminus. Our results suggest the distinct possibility that S. aureus may be capable of adapting to continuous use of antibiotics and selection pressure to escape the human immune response, by generating SC variants with similar or higher efficiency to activate ProT.
Experimental procedures
Expression, purification, and labeling of proteins
SC(1–246) was cloned into a modified pET30b(+) vector (Novagen) containing an N-terminal His6 tag followed by a tobacco etch virus cleavage site (19, 30). The SC(1–246) N-terminal mutants were prepared through site-directed mutagenesis using degenerate and specific primers (Table S1), and mutations were confirmed by DNA sequencing. The mutants were expressed in Rosetta 2 (DE3) pLysS Escherichia coli in the presence of 100 μg/ml kanamycin, and expression was induced by 10 mg/ml lactose for 4 h. Mutants were purified from inclusion bodies, and the His6 tag was removed as described (33, 34). The proteins were stored in 50 mm HEPES, 125 mm NaCl, pH 7.4, at −80 °C until use. The mutant concentrations were determined using the extinction coefficients and molecular mass calculated by the Expasy tool, RRID:SCR_018087 (Table S2). HEK293 cells expressing ProTQQQ, in which the prothrombinase cleavage site Arg271 and the thrombin cleavage sites Arg155 and Arg284 were replaced by glutamine to prevent degradation, were a gift from Dr. Sriram Krishnaswamy (University of Pennsylvania School of Medicine) (20). ProTQQQ was expressed, purified, and stored as described (35, 36). Four separate ProTQQQ batches were prepared, and the concentrations were determined using E280 nm,0.1% 1.47 ml mg−1 cm−1 and Mr 72,000.
Preparation and characterization of SC(1–246)-BODIPY
To create a labeled SC construct, Ser7 of SC(1–246) was converted to cysteine through site-directed mutagenesis (Agilent Technologies) and confirmed by DNA sequencing. Purified SC(1–246)-S7C was reduced with 2 mm DTT and dialyzed against 5 mm MES, 125 mm NaCl, 2 mm DTT, pH 6.0. The reduced protein was run on Sephadex G-25 (1 × 25 cm) in 50 mm HEPES, 125 mm NaCl, 1 mg/ml PEG, 10 mm EDTA, pH 7.4, buffer to remove free DTT. Approximately 5–10 mg of protein was incubated for 1 h at 25 °C with a 10-fold molar excess of BODIPY-FL-iodoacetamide (Thermo Fisher Scientific) to label the free S7C thiol. The excess probe was removed by Sephadex G-25 chromatography in 50 mm HEPES, 125 mm NaCl, 0.1 mm EDTA, pH 7.4, buffer. Labeled SC(1–246)-S7C-BODIPY-FL (SC(1–246)-BODIPY) was dialyzed against storage buffer (50 mm HEPES, 125 mm NaCl, pH 7.4) and stored at −80 °C. The concentration and labeling ratio were determined using E280 nm, 0.1% 0.936 ml mg−1 cm−1 and Mr 29,150 for WT SC(1–246) and SC(1–246)-S7C, and E505 nm of 63,771 cm−1 m−1 for BODIPY-FL-iodoacetamide. An absorbance ratio (A280 nm/A505 nm) of 0.03 was used to correct for the probe contribution to absorbance at 280 nm. The purity of SC(1–246)-BODIPY was established by 4–20% polyacrylamide SDS-PAGE under reduced and nonreduced conditions. The fluorescence of the labeled protein was imaged under UV light, and proteins were then stained with colloidal Coomassie Blue G-250. To determine whether SC(1–246)-BODIPY forms a binary complex with ProTQQQ, a fixed concentration (2.5 μm) of ProTQQQ was incubated with different concentrations of SC(1–246)-BODIPY (0, 0.5, 1.0, and 1.5 μm) at 25 °C for 15–30 min. The samples were run on a 6% polyacrylamide gel under native conditions (Tris-glycine buffer, pH 8.3, no SDS) at 4 °C. The fluorescence was imaged, and the proteins were stained as described above.
Prothrombin activation assay
Activity titrations of ProT complexes with WT or mutant SC(1–246) were performed in 50 mm HEPES, 110 mm NaCl, 5 mm CaCl2, 1 mg/ml PEG 8000, pH 7.4, buffer, in PEG 20,000-coated 96-well plates (Nunc). Varying concentrations of WT or mutant SC(1–246) were incubated with 1, 10, or 20 nm ProT for 10 min at 25 °C. The reaction was initiated by the addition of 600 μm chromogenic substrate S-2238 (Diapharma), and the rate was measured in a ThermoMax plate reader (Thermo Fisher Scientific) for 10 min at 405 nm until the absorbance reached 0.1. Initial rates (mAbs/min) for the mutants were normalized to that of WT SC(1–246). The normalized rate dependences as a function of the SC(1–246) concentration were fitted by the quadratic binding equation using SCIENTIST (MicroMath) to obtain the Vlim and KD (19).
Direct and competitive fluorescence equilibrium binding
Fluorescence measurements were performed with a PTI QuantaMaster 30 spectrofluorometer at 25 °C using acrylic cuvettes coated with PEG 20,000. Titrations were performed in 50 mm HEPES, 110 mm NaCl, 5 mm CaCl2, 1 mg/ml PEG 8000, pH 7.4, buffer with 1 mg/ml ovalbumin, and fluorescence was measured at λex = 496 nm (3–6-nm band pass) and λem = 535 nm (4–6-nm band pass). Two fixed concentrations of SC(1–246)-BODIPY were titrated with the ligand, ProTQQQ. The competitive binding assays were performed with one fixed concentration of SC(1–246)-BODIPY in the presence of a single fixed concentration of unlabeled WT or mutant SC(1–246) competitor, titrated with the ligand, ProTQQQ. The SC(1–246)-BODIPY control titrations with ProTQQQ were performed to obtain the stoichiometry and KO for SC(1–246)-BODIPY. The titrations of SC(1–246)-BODIPY in the presence of competing WT or mutant SC(1–246) were performed to obtain the stoichiometry and dissociation constant KC for the competitors. The fractional change in fluorescence was calculated as (Fobs − Fo)/Fo = ΔF/Fo, and the data were fit by the quadratic binding equation (37) using SCIENTIST (MicroMath) software. For the competition experiments, titrations in the absence and presence of competitor were fit simultaneously by the cubic binding equation (37). Nonlinear least-squares fitting was performed using SCIENTIST (MicroMath) either with or without fixed stoichiometry for the competitive data to obtain the dissociation constants KO and KC, maximum fluorescence intensities ((Fmax − Fo)/Fo = ΔFmax/Fo), and stoichiometric factors n for SC(1–246)-BODIPY and m for unlabeled WT and mutant SC(1–246). The error estimates represent the 95% confidence interval. The Gibbs free energy (ΔG) values for WT and mutant SC(1–246) binding to ProTQQQ were calculated using the equation, ΔG = RT ln KD, where R = 1.987 × 10−3 kcal mol−1 degree−1 and T = 298.15 K (25 °C), and KD is expressed in m.
Computational modeling
To analyze the energetic effects of the N-terminal mutations of SC(1–246) on the binding with ProTQQQ, the mutations were performed in silico using the Rosetta software suite (RRID:SCR_015701) (38). The X-ray crystal structure of the complex 1nu9 (19) was relaxed using Rosetta3 (August 2016 build 58479), using constraints to maintain atomic positions close to the experimental input structure. The relaxation was performed 100 times, and the lowest-energy structure was selected for mutation (/rosetta-3.9/main/source/bin/relax.default.linuxgccrelease -s 1nu9_cleaned.pdb -in:file:fullatom -nstruct 100 -relax:fast -relax:constrain_relax_to_start_coords). Mutations were introduced into the structure using Rosetta's fixed backbone design (fixbb) application (39) and a resfile specifying the identities of residues at positions 1, 2, and 3. All 20 amino acids were tested at each of the three positions, resulting in 8,000 output structures, representing putative single, double, and triple mutants (/rosetta-3.9/main/source/bin/fixbb.default.linuxgccrelease -s relaxed_with_constraints.pdb -resfile resfiles/$key -in:file:fullatom -use_input_sc -ex1 -ex2 -out:path:pdb pdbfiles -out:path:score scorefiles). The predicted change in the interaction energy between the mutated SC(1–246) and Pre-2 was calculated using Rosetta's interface analyzer application, to evaluate the resulting changes in the energetic binding contribution of the N-terminal segment of SC(1–246). (/rosetta-3.9/main/source/bin/InterfaceAnalyzer.default.linuxgccrelease -in:path ../pdbfiles -in:file:l structure_batch.list -add_regular_scores_to_scorefile -out:path:pdb pdbfiles -out:path:score scorefiles -out:file:scorefile structure_batch.fasc), and the sixth column “dG_separated” was extracted using awk. (% cat ../scorefiles/*.fasc | awk '{print $NF “ ” $6}' | grep -v SEQUENCE | grep -v description | sort -nrk2 > sorted_interface_energies.list).
Data availability
The structure of the SC(1–325)·Pre2 complex 1NU9 is available in the Protein Data Bank. All remaining data are contained within the article and supporting information.
Author contributions
A. A. M., H. K. K., M. E. A., B. H. G., J. H. S., J. M., P. E. B., and I. M. V. data curation; A. A. M., H. K. K., M. E. A., B. H. G., and J. M. formal analysis; A. A. M., H. K. K., M. E. A., and P. P. investigation; A. A. M., H. K. K., M. E. A., B. H. G., P. P., J. H. S., and P. E. B. methodology; A. A. M. and H. K. K. writing-original draft; H. K. K., P. P., P. E. B., and I. M. V. conceptualization; P. P. and I. M. V. writing-review and editing; J. H. S. and J. M. software; J. H. S. and J. M. validation; P. E. B. and I. M. V. supervision; P. E. B. and I. M. V. funding acquisition; P. E. B. and I. M. V. project administration.
Supplementary Material
This research was supported in part by National Institutes of Health Grants R01 HL071544 (to P. E. B. and I. M. V.), R01 HL114477 (to P. P.), and R01 HL122010 (to Alfred George, providing support for J. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Fig. S1.
I. M. Verhamme, P. Panizzi, and P. E. Bock, unpublished observations.
- Fbn
- fibrin
- SC
- staphylocoagulase
- SC(1–246)-BODIPY
- SC(1–246)-S7C-BODIPY-FL
- Fbg
- fibrinogen
- ProT
- prothrombin
- ProTQQQ
- prothrombin mutant with mutations at arginines 155, 271, and 284 substituted to glutamine
- Pre2
- prethrombin 2
- vWbp
- von Willebrand factor–binding protein
- REU
- Rosetta energy unit(s)
- PTI
- pancreatic trypsin inhibitor
- pGB
- p-guanidinobenzoate
- Vlim
- limiting velocity.
References
- 1. Klein E., Smith D. L., and Laxminarayan R. (2007) Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg. Infect. Dis. 13, 1840–1846 10.3201/eid1312.070629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McGavin M. J., and Heinrichs D. E. (2012) The staphylococci and staphylococcal pathogenesis. Front. Cell Infect. Microbiol. 2, 66 10.3389/fcimb.2012.00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lowy F. D. (1998) Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- 4. Hartleib J., Köhler N., Dickinson R. B., Chhatwal G. S., Sixma J. J., Hartford O. M., Foster T. J., Peters G., Kehrel B. E., and Herrmann M. (2000) Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood 96, 2149–2156 [PubMed] [Google Scholar]
- 5. Herrmann M., Suchard S. J., Boxer L. A., Waldvogel F. A., and Lew P. D. (1991) Thrombospondin binds to Staphylococcus aureus and promotes staphylococcal adherence to surfaces. Infect. Immun. 59, 279–288 10.1128/IAI.59.1.279-288.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herrmann M., Lai Q. J., Albrecht R. M., Mosher D. F., and Proctor R. A. (1993) Adhesion of Staphylococcus aureus to surface-bound platelets: role of fibrinogen/fibrin and platelet integrins. J. Infect. Dis. 167, 312–322 10.1093/infdis/167.2.312 [DOI] [PubMed] [Google Scholar]
- 7. Kuypers J. M., and Proctor R. A. (1989) Reduced adherence to traumatized rat heart valves by a low-fibronectin-binding mutant of Staphylococcus aureus. Infect. Immun. 57, 2306–2312 10.1128/IAI.57.8.2306-2312.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moreillon P., Entenza J. M., Francioli P., McDevitt D., Foster T. J., François P., and Vaudaux P. (1995) Role of Staphylococcus aureus coagulase and clumping factor in pathogenesis of experimental endocarditis. Infect. Immun. 63, 4738–4743 10.1128/IAI.63.12.4738-4743.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patti J. M., Allen B. L., McGavin M. J., and Höök M. (1994) MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48, 585–617 10.1146/annurev.mi.48.100194.003101 [DOI] [PubMed] [Google Scholar]
- 10. Que Y. A., François P., Haefliger J. A., Entenza J. M., Vaudaux P., and Moreillon P. (2001) Reassessing the role of Staphylococcus aureus clumping factor and fibronectin-binding protein by expression in Lactococcus lactis. Infect. Immun. 69, 6296–6302 10.1128/IAI.69.10.6296-6302.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siboo I. R., Cheung A. L., Bayer A. S., and Sullam P. M. (2001) Clumping factor A mediates binding of Staphylococcus aureus to human platelets. Infect. Immun. 69, 3120–3127 10.1128/IAI.69.5.3120-3127.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McAdow M., Missiakas D. M., and Schneewind O. (2012) Staphylococcus aureus secretes coagulase and von Willebrand factor binding protein to modify the coagulation cascade and establish host infections. J. Innate Immun. 4, 141–148 10.1159/000333447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cabell C. H., Jollis J. G., Peterson G. E., Corey G. R., Anderson D. J., Sexton D. J., Woods C. W., Reller L. B., Ryan T., and Fowler V. G. Jr. (2002) Changing patient characteristics and the effect on mortality in endocarditis. Arch. Intern. Med. 162, 90–94 10.1001/archinte.162.1.90 [DOI] [PubMed] [Google Scholar]
- 14. Soulier J. P., and Prou-Wartelle O. (1967) Study of thrombin-coagulase. Thromb. Diath. Haemorrh. 17, 321–334 [PubMed] [Google Scholar]
- 15. Kawabata S., Morita T., Iwanaga S., and Igarashi H. (1985) Difference in enzymatic properties between α-thrombin-staphylocoagulase complex and free α-thrombin. J. Biochem. 97, 1073–1078 10.1093/oxfordjournals.jbchem.a135150 [DOI] [PubMed] [Google Scholar]
- 16. Kawabata S., Miyata T., Morita T., Miyata T., Iwanaga S., and Igarashi H. (1986) The amino acid sequence of the procoagulant- and prothrombin-binding domain isolated from staphylocoagulase. J. Biol. Chem. 261, 527–531 [PubMed] [Google Scholar]
- 17. Kawabata S., and Iwanaga S. (1994) Structure and function of staphylothrombin. Semin. Thromb. Hemost. 20, 345–350 10.1055/s-2007-1001925 [DOI] [PubMed] [Google Scholar]
- 18. Panizzi P., Friedrich R., Fuentes-Prior P., Bode W., and Bock P. E. (2004) The staphylocoagulase family of zymogen activator and adhesion proteins. Cell Mol. Life Sci. 61, 2793–2798 10.1007/s00018-004-4285-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedrich R., Panizzi P., Fuentes-Prior P., Richter K., Verhamme I., Anderson P. J., Kawabata S., Huber R., Bode W., and Bock P. E. (2003) Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature 425, 535–539 10.1038/nature01962 [DOI] [PubMed] [Google Scholar]
- 20. Kamath P., and Krishnaswamy S. (2008) Fate of membrane-bound reactants and products during the activation of human prothrombin by prothrombinase. J. Biol. Chem. 283, 30164–30173 10.1074/jbc.M806158200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kellogg E. H., Leaver-Fay A., and Baker D. (2011) Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79, 830–838 10.1002/prot.22921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kroh H. K., and Bock P. E. (2012) Effect of zymogen domains and active site occupation on activation of prothrombin by von Willebrand factor-binding protein. J. Biol. Chem. 287, 39149–39157 10.1074/jbc.M112.415562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bode W. (1979) The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. II. The binding of the pancreatic trypsin inhibitor and of isoleucine-valine and of sequentially related peptides to trypsinogen and to p-guanidinobenzoate-trypsinogen. J. Mol. Biol. 127, 357–374 10.1016/0022-2836(79)90227-4 [DOI] [PubMed] [Google Scholar]
- 24. Krishnaswamy S. (2013) The transition of prothrombin to thrombin. J. Thromb. Haemost. 11, 265–276 10.1111/jth.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bode W., and Huber R. (1978) Crystal structure analysis and refinement of two variants of trigonal trypsinogen: trigonal trypsin and PEG (polyethylene glycol) trypsinogen and their comparison with orthorhombic trypsin and trigonal trypsinogen. FEBS Lett. 90, 265–269 10.1016/0014-5793(78)80382-2 [DOI] [PubMed] [Google Scholar]
- 26. Khan A. R., and James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 10.1002/pro.5560070401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Watanabe S., Ito T., Sasaki T., Li S., Uchiyama I., Kishii K., Kikuchi K., Skov R. L., and Hiramatsu K. (2009) Genetic diversity of staphylocoagulase genes (coa): insight into the evolution of variable chromosomal virulence factors in Staphylococcus aureus. PLoS ONE 4, e5714 10.1371/journal.pone.0005714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanemitsu K., Yamamoto H., Takemura H., Kaku M., and Shimada J. (2001) Relatedness between the coagulase gene 3′-end region and coagulase serotypes among Staphylococcus aureus strains. Microbiol. Immunol. 45, 23–27 10.1111/j.1348-0421.2001.tb01270.x [DOI] [PubMed] [Google Scholar]
- 29. Watanabe S., Ito T., Takeuchi F., Endo M., Okuno E., and Hiramatsu K. (2005) Structural comparison of ten serotypes of staphylocoagulases in Staphylococcus aureus. J. Bacteriol. 187, 3698–3707 10.1128/JB.187.11.3698-3707.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Panizzi P., Friedrich R., Fuentes-Prior P., Kroh H. K., Briggs J., Tans G., Bode W., and Bock P. E. (2006) Novel fluorescent prothrombin analogs as probes of staphylocoagulase-prothrombin interactions. J. Biol. Chem. 281, 1169–1178 10.1074/jbc.M507955200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bode W., and Huber R. (1976) Induction of the bovine trypsinogen-trypsin transition by peptides sequentially similar to the N-terminus of trypsin. FEBS Lett. 68, 231–236 10.1016/0014-5793(76)80443-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bianchini E. P., Orcutt S. J., Panizzi P., Bock P. E., and Krishnaswamy S. (2005) Ratcheting of the substrate from the zymogen to proteinase conformations directs the sequential cleavage of prothrombin by prothrombinase. Proc. Natl. Acad. Sci. U.S.A. 102, 10099–10104 10.1073/pnas.0504704102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kroh H. K., Panizzi P., and Bock P. E. (2009) Von Willebrand factor-binding protein is a hysteretic conformational activator of prothrombin. Proc. Natl. Acad. Sci. U.S.A. 106, 7786–7791 10.1073/pnas.0811750106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panizzi P., Boxrud P. D., Verhamme I. M., and Bock P. E. (2006) Binding of the COOH-terminal lysine residue of streptokinase to plasmin(ogen) kringles enhances formation of the streptokinase.plasmin(ogen) catalytic complexes. J. Biol. Chem. 281, 26774–26778 10.1074/jbc.C600171200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orcutt S. J., and Krishnaswamy S. (2004) Binding of substrate in two conformations to human prothrombinase drives consecutive cleavage at two sites in prothrombin. J. Biol. Chem. 279, 54927–54936 10.1074/jbc.M410866200 [DOI] [PubMed] [Google Scholar]
- 36. Newell-Caito J. L., Laha M., Tharp A. C., Creamer J. I., Xu H., Maddur A. A., Tans G., and Bock P. E. (2011) Notecarin D binds human factor V and factor Va with high affinity in the absence of membranes. J. Biol. Chem. 286, 38286–38297 10.1074/jbc.M111.247122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bock P. E., Olson S. T., and Björk I. (1997) Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite I. J. Biol. Chem. 272, 19837–19845 10.1074/jbc.272.32.19837 [DOI] [PubMed] [Google Scholar]
- 38. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 10.1016/B978-0-12-381270-4.00019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuhlman B., Dantas G., Ireton G. C., Varani G., Stoddard B. L., and Baker D. (2003) Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368 10.1126/science.1089427 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structure of the SC(1–325)·Pre2 complex 1NU9 is available in the Protein Data Bank. All remaining data are contained within the article and supporting information.