

Abstract
Purpose
Most existing literature describes Lynch syndrome (LS) as a hereditary syndrome leading to high risks of colorectal cancer (CRC) and endometrial cancer mainly as a result of mutations in MLH1 and MSH2. Most of these studies were performed on cohorts with disease suggestive of hereditary CRC and population-based CRC and endometrial cancer cohorts, possibly biasing results. We aimed to describe a large cohort of mismatch repair (MMR) mutation carriers ascertained through multigene panel testing, evaluate their phenotype, and compare the results with those of previous studies.
Methods
We retrospectively reviewed clinical histories of patients who had multigene panel testing, including the MMR and EPCAM genes, between March 2012 and June 2015 (N = 34,981) and performed a series of statistical comparisons.
Results
Overall, MSH6 mutations were most frequent, followed by PMS2, MSH2, MLH1, and EPCAM mutations, respectively. Of 528 patients who had MMR mutations, 63 (11.9%) had breast cancer only and 144 (27.3%) had CRC only. When comparing those with breast cancer only to those with CRC only, MSH6 and PMS2 mutations were more frequent than MLH1 and MSH2 mutations (P = 2.3 × 10−5). Of the 528 patients, 22.2% met BRCA1 and BRCA2 (BRCA1/2) testing criteria and not LS criteria, and 5.1% met neither BRCA1/2 nor LS testing criteria. MSH6 and PMS2 mutations were more frequent than MLH1 and MSH2 mutations among patients who met BRCA1/2 testing criteria but did not meet LS testing criteria (P = 4.3 × 10−7).
Conclusion
These results provide a new perspective on LS and suggest that individuals with MSH6 and PMS2 mutations may present with a hereditary breast and ovarian cancer phenotype. These data also highlight the limitations of current testing criteria in identifying these patients, as well as the need for further investigation of cancer risks in patients with MMR mutations.
INTRODUCTION
Lynch syndrome (LS) is a well-described hereditary cancer syndrome caused by mutations in the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) and large deletions, including the 3′ end of EPCAM. LS has been known as one of the most common hereditary cancer syndromes for some time, and a recent study showed that the population frequency may be as high as 1 in 279, which would make it the most common hereditary cancer syndrome.1 Individuals with LS have high risks for colorectal cancer (CRC) and endometrial cancer (EC), as well as many other cancers. A large body of literature has suggested that most cases of LS are a result of mutations in MLH1 and MSH2 and that the highest cancer risks are for CRC and EC, followed by gastric and ovarian cancers (OCs).2-7 However, many of these studies included individuals with a personal and/or family history of CRC and/or EC or were cohorts of patients suggestive of LS because they met the Amsterdam criteria or Bethesda guidelines. In addition, clinical testing for MSH6, PMS2, and EPCAM became available well after testing for MLH1 and MSH2 became available, so many early studies did not assess for mutations in those genes. An association between breast cancer (BC) and LS has been suggested by a few studies, but it remains controversial, and there are currently no guidelines recommending increased BC screening for women with LS.8-10
Initially, candidates for LS genetic testing were identified on the basis of meeting the Amsterdam criteria or Bethesda guidelines, but more recently, these algorithms have been shown to miss as many as 72% and 27% of individuals with LS, respectively.5 In addition, these criteria are focused on CRC and EC diagnoses. In more recent years, universal screening for LS using microsatellite instability (MSI) and/or immunohistochemistry (IHC) for the MMR proteins has been implemented in a growing number of institutions, but this screening is mostly performed on CRC and, in some cases, EC tumors.2,3,5,11-14 Although they are effective screening tools, MSI and IHC may miss approximately 13% to 23% of patients with LS and may have lower sensitivity for detecting MSH6 and PMS2 mutation carriers in particular.5
The use of multigene panel testing (MGPT) with next-generation sequencing (NGS) for the diagnosis of hereditary cancer has increased dramatically over the past 4 years; however, limited data have been published describing individuals with LS identified through MGPT.15,16 Because more patients are being assessed with MGPT, individuals are being identified who have mutations in what are believed to be well-described, highly penetrant genes, but those patients do not meet established clinical and/or testing criteria for the respective syndrome.15,16
In this study, we aimed to describe the characteristics of individuals with LS identified through MGPT at a single laboratory and compare them to characteristics of patients in the literature with MMR mutations found through traditional means.
METHODS
Study Population
Deidentified clinical histories and molecular results were reviewed for all patients who underwent MGPT, including the MMR and EPCAM genes between March 2012 and June 2015 at Ambry Genetics (Aliso Viejo, CA). The study was reviewed by Solutions Institutional Review Board and found to be exempt.
MGPT
Patients underwent comprehensive analysis of nine to 49 hereditary cancer predisposition genes, depending on which test was ordered (Appendix Table A1, online only). The test ordered was determined by the ordering clinician. The specific indication for testing was not available in our data set but may be inferred in some cases from the personal and/or family history of cancer and the testing criteria met. Gene analysis is described in detail in the Appendix (online only).
All variants, with the exception of previously characterized benign alterations, underwent thorough assessment and review of available evidence (eg, population frequency information, published case reports, case/control and functional studies, locus-specific databases [such as the InSIGHT database from International Society of Gastrointestinal Hereditary Tumors],17 internal co-occurrence and cosegregation data, evolutionary conservation, and in silico predictions). Variants were classified per the Ambry five-tier variant classification protocol (pathogenic mutation; variant, likely pathogenic; variant of unknown significance [VUS]; variant, likely benign; and benign) which is based on guidelines published by the American College of Medical Genetics and Genomics, the Association for Molecular Pathology, and the International Agency for Research on Cancer.18-20
Data Analysis
Information on demographic characteristics and personal and family cancer history was extracted from clinician-completed test requisition forms (TRFs) and, when available, clinic notes and pedigrees. Individuals known to be family members of a proband previously tested at our laboratory, as indicated by the ordering provider, and those who were likely to be related because of a shared mutation and similar personal and family histories, were excluded. Patients were eligible for inclusion in phenotype comparisons if the personal and family history sections were completed on the TRF and they had one MMR or EPCAM pathogenic mutation or likely pathogenic variant. Individuals with two or more mutations or likely pathogenic variants were excluded. Tumor-specific cohorts used for genotype-phenotype comparisons were designed to reduce bias by focusing on isolated tumor types as follows: patients with BC only who had BC but not OC, CRC, or EC; patients with CRC only who had CRC, but not BC, EC, or OC; patients with OC only who had OC, but not BC, CRC, or EC; and patients with EC only who had EC but not CRC, BC, or OC. Comparisons of gene-specific mutation frequencies were performed between these groups by using multivariable logistic regression analysis and controlling for age at testing, sex, race/ethnicity, and family history of CRC and/or EC among first- and second-degree relatives. Clinical histories were further evaluated to determine whether National Comprehensive Cancer Network (NCCN) LS and/or BRCA1 and BRCA2 (BRCA1/2) testing criteria, two of the most commonly used testing criteria in the United States, were met.21,22 Gene-specific mutation frequencies among those who met each set of criteria were compared by using Fisher’s exact test. P values from the multivariable analysis and Fisher’s exact test were adjusted for multiple comparisons via false discovery rate with a cutoff of 5%. The χ2 and binomial proportion tests were used to compare our mutation frequencies with those from previous studies.
RESULTS
Demographic Characteristics
Of 34,980 individuals who, to the best of our knowledge, were unrelated and who underwent MGPT for the MMR genes and EPCAM, 618 MMR and EPCAM mutations were identified in 612 (1.7%) individuals. Thirty-three individuals had a second pathogenic mutation in an LS (n = 6) or another cancer predisposition gene (n = 27) and were excluded from further analyses, leaving a cohort of 579 probands. Of these, 120 (20.7%) also had a VUS in one of the genes analyzed. This cohort was mostly female (73.4%), white (66.7%), and affected with cancer (85.8%, excluding nonmelanoma skin cancer; Table 1). Mutations in MSH6 were most frequent (29.3%), followed by PMS2 (24.2%), MSH2 (23.7%), MLH1 (21.6%), and EPCAM (1.2%; Fig 1). This distribution is statistically significantly different from those in the largest previous studies (Fig 2).23 EPCAM mutation carriers were combined with MSH2 carriers for further analyses. MLH1 carriers were diagnosed with their first primary cancer at the youngest average age (42.2 years), followed by MSH2/EPCAM, PMS2, and MSH6 carriers (Table 1).
Table 1.
Demographic and Clinical Characteristics

Fig 1.

Overall mismatch repair gene and EPCAM mutation distribution (n = 579).
Fig 2.

Percentage of mutation carriers per mismatch repair gene compared with previous studies. The χ2 test was used to compare mutation frequencies from this study with those from previous studies. Hampel et al, A2,14 is a population-based endometrial cancer cohort. Hampel et al, B3,13 is a population-based colorectal cancer cohort. Palomaki et al5 calculated a weighted proportion from four previous studies on colorectal cancer cohorts. Bonadona et al4 is a cohort of patients who were suggestive of Lynch syndrome (LS). Moreira et al6 includes patients from four large registries of population-based and/or high-risk families with colorectal cancer. Yurgelun et al16 is a cohort of patients with histories suggestive of LS. Sjursen et al23 is a cohort of individuals with a clinical diagnosis of hereditary nonpolyposis colorectal cancer and/or a molecular diagnosis of LS. The cohort in Møller et al7 is a pooled prospective database of individuals with LS from several collaborative groups. (*) EPCAM mutations were excluded for all comparisons except those with Møller et al. Most previous studies did not analyze EPCAM, but in Møller et al, EPCAM mutations were combined with MSH2 and could not be excluded. As a result, the number of patients for the comparison with Møller et al in this study is 579, and the MSH2 percentage for this study is 24.9 with EPCAM mutations included.
Mutation Spectrum
All unique variants classified as pathogenic or likely pathogenic (n = 346) are listed in Appendix Table A2 (online only). Five mutations were seen 10 or more times each. PMS2 c.137G>T p.S46I (n = 21) is reported as a European founder mutation, and MSH2 c.1906G>C p.A636P (n = 13) is a known Ashkenazi Jewish founder mutation.24,25 MSH2 c.942+3A>T (n = 18) and MSH6 c.3261dupC p.F1088Lfs*5 (n = 16) are recurrent mutations occurring in microsatellite tracts,26-28 and PMS2 c.1831dupA (n = 10) was previously reported in three patients.24
Phenotype
Individuals for whom personal and/or family history of cancer was not provided (n = 51) were excluded from phenotype analyses, leaving a cohort of 528. The most common cancers in MMR mutation carriers were CRC (n = 186 [35.2%]), EC (n = 136 [25.8%]), BC (n = 124 [23.5%]), and OC (n = 74 [14.0%]; Table 1). In the remainder of the cohort tested for MMR gene mutations for whom personal and family cancer history information was available (n = 31,545), BC was most common (n = 12,173 [38.6%]), followed by OC (n = 4,416 [14.0%]), CRC (n = 3,378 [10.7%]), and EC (n = 1,831 [5.8%]). The average age of first primary cancer diagnosis was 46.6 years. In addition, 19.3% of LS probands reported one or more adenomatous polyps and 4.5% had 10 or more polyps (Table 1).
BC was the first primary cancer for 84 (15.9%) of the MMR mutation carriers, and OC was the first primary cancer for 61 (11.6%) probands. Overall, 145 patients (27.5%) presented with BC or OC, 73.1% of whom had not been diagnosed with CRC or EC at the time of genetic testing. Nine MMR mutation carriers had both BC and OC without CRC or EC, seven of whom had MSH6 (n = 4) or PMS2 (n = 3) mutations.
When comparing those with BC only (n = 63; Fig 3A) with those who had CRC only (n = 144; Fig 3B), MSH6 and PMS2 mutations were more frequent than MLH1 and MSH2 mutations (P = 2.3 × 10−5). When comparing those with OC only (n = 40; Fig 3C) with those who had CRC only, there was a trend toward MSH6 mutations being more frequent than MLH1 and MSH2 mutations, but it did not reach statistical significance. When comparing probands with EC only (n = 71; Fig 3D) with those who had CRC only, MSH6 mutations were more frequent than MLH1 and PMS2 mutations (P = 1.1 × 10−3 and P = 1.7 × 10−2, respectively), and MSH2 mutations were more frequent than MLH1 mutations (P = 1.2 × 10−2). Detailed results of statistical comparisons are listed in Table 2.
Fig 3.

Mismatch repair gene mutation distributions among tumor-specific subgroups. (A) Patients with breast cancer (BC) without ovarian cancer (OC), colorectal cancer (CRC), or endometrial cancer (EC; n = 63). (B) Patients with CRC without BC, EC, or OC (n = 144). (C) Patients with OC without BC, CRC, or EC (n = 40). (D) Patients with EC without CRC, BC, or OC (n = 71).
Table 2.
Comparison of Genotypes and Phenotypes

In our study, mutation frequencies for patients with CRC only and EC only were significantly higher than recently published population frequencies for all four MMR genes (Appendix Table A3, online only).1 For patients with BC only, the frequency of PMS2 mutations was significantly higher than the population estimates, but there was no significant difference in MLH1, MSH2, or MSH6 mutations. Conversely, among patients with OC only, the frequencies of MLH1, MSH2, and MSH6 mutations were significantly higher than the population estimates, but there was no significant increase in the frequency of PMS2 mutations (Appendix Table A3).
Testing Criteria
In our cohort, 18.2% of MMR mutation carriers met the Amsterdam criteria and 37.3% met the revised Bethesda guidelines, although 68.6% had a PREMM1,2,6 score ≥ 5% and 72.7% met the NCCN guidelines for LS testing (not mutually exclusive).21,29 Interestingly, 58.9% met the NCCN guidelines for BRCA1/2 testing and 22.2% met only these guidelines, although 36.7% met both guidelines. An additional 5.1% did not meet any testing criteria for LS or BRCA1/2. Altogether, 27.3% of the patients in this cohort who had MMR mutations would not have been identified by current LS testing criteria. MSH6 and PMS2 carriers were more likely to meet only the NCCN BRCA1/2 testing criteria than MLH1 and MSH2 carriers (P = 4.3 × 10−7). Conversely, MLH1 and MSH2 mutation carriers were more likely to meet the NCCN guidelines for LS testing than MSH6 and PMS2 carriers (P = 7.5 × 10−8; Table 3).
Table 3.
Genetic Testing Criteria Gene by Gene Comparisons

Tumor Testing
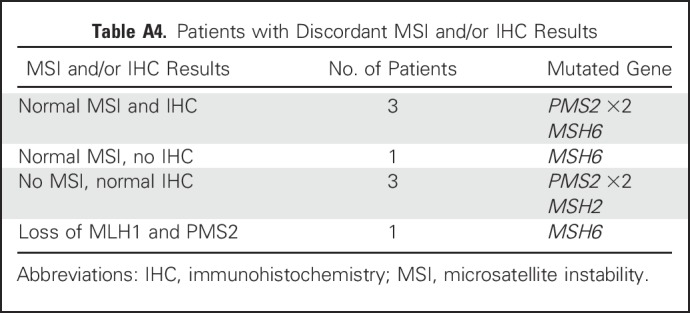
Results of MSI and/or IHC analysis (tumor testing) were provided on the TRF for 99 MMR mutation carriers (18.8%); 10 (1.9%) had MSI only, 65 (12.3%) had IHC only, and 24 (4.5%) had both MSI and IHC results. Of those with tumor testing results, eight (8.1%) were discordant with the mutated gene (Appendix Table A4, online only). Seven of the eight had normal tumor testing and one had loss of proteins, which indicated a mutation in a different gene. Of MLH1 and MSH2/EPCAM mutation carriers who had tumor testing results (n = 23 and n = 30, respectively), the results were discordant for one MSH2 carrier (3.3%). Of MSH6 and PMS2 mutation carriers who had MSI and/or IHC results (n = 24 and n = 22, respectively), results were discordant for three MSH6 carriers (12.5%) and four PMS2 carriers (18.2%).
DISCUSSION
Traditionally, LS has been described as a hereditary CRC and EC syndrome caused primarily by mutations in MLH1 and MSH24-6,29,30; however, our data suggest that this may not be a complete description of the syndrome. Although CRC and EC were the most common cancers among MMR mutation carriers in this cohort, affecting 35.2% and 25.8% of probands, respectively, BC was nearly as common (23.5% of probands) and 14.0% had OC. In fact, 27.5% (n = 145) of MMR mutation carriers presented with BC (15.9%) or OC (11.6%) as their first primary cancer, the majority of which (n = 106 [73.1%]) had not been diagnosed with CRC or EC at the time of genetic testing. In addition, there was an approximately equal distribution of mutations in MLH1, MSH2, MSH6, and PMS2 in this study, with the greatest percentages of mutations in MSH6 and PMS2.
Several previous studies have suggested a higher risk for EC in MSH6 mutation carriers than in carriers of other MMR mutations.2,30-32 More recently, MSH6 and PMS2 mutations have been reported at higher frequencies than MLH1 and MSH2 in hereditary breast and/or ovarian cancer (HBOC) MGPT cohorts, but in small numbers.33-36 Another recent MGPT study assessing patients with disease suggestive of LS found that of 114 patients with LS, 35% had a mutation in MSH2, 27% in MLH1, 23% in MSH6, 12% in PMS2, and 3% in EPCAM. This is more consistent with previous LS studies and significantly different from the distribution in our cohort (P = 8.7 × 10−4; Fig 2).16 To the best of our knowledge, this study reports on the largest cohort of individuals tested for and with mutations in the MMR genes identified through MGPT to date.
Previous studies have shown that the Amsterdam criteria will identify approximately 13% to 58% of families with LS, and the revised Bethesda criteria will identify approximately 65% to 91% of patients with LS compared with 18.2% and 37.3%, respectively, in this study.2,5,12,23 We found that 27.3% of patients did not meet any current criteria for LS testing, and MSH6 and PMS2 mutation carriers were significantly more likely to meet only BRCA1/2 testing criteria (and not LS criteria) than MLH1 and MSH2 mutation carriers. In addition, 15.2% of available MSI and/or IHC results for patients with MSH6 or PMS2 mutations were discordant, which could have led to the mutation being missed in the absence of MGPT. It is known that MSI and IHC do not have 100% sensitivity for LS, and many of the discordant MSH6 mutations may be explained by known issues with IHC for the MSH6 protein.5,37,38
Most recently, data from a population-based cohort of patients with CRC who were screened for MMR gene mutations were used to estimate population frequencies of MMR mutations.1 Results of that study demonstrated that of the MMR genes, mutations in PMS2 are the most frequent in the general population, followed by MSH6, MLH1, and MSH2. It is not surprising that the mutation frequencies for all MMR genes are significantly higher in our CRC and EC only probands because these are the cancers with the highest risks in LS. The fact that PMS2 mutations were significantly more frequent in probands with BC only suggests an excess of BC in our cohort of MMR mutation carriers, particularly among PMS2 carriers, beyond what would be expected in the general population. Although the study by Win et al1 is population based, our cohort represents a high-risk cohort, with most probands submitted for testing as a result of disease suggestive of some kind of hereditary cancer but not necessarily LS. This may explain why MSH6 mutations, which have been published in association with higher risk for gynecologic cancers and more moderate risk for CRC,7,32 were most frequent in our cohort, while PMS2 mutations, which are believed to have the lowest penetrance of MMR mutations, were most frequent in the population-based estimates.1
Although this cohort did not have the bias of being selected for patients with a personal and/or family history suggestive of LS, the majority of patients submitted for MGPT at this laboratory were females with disease suggestive of HBOC, which may have biased our results and explain some of the BC and OC in our patients with MMR mutations. We do not know whether the BCs seen in this cohort were sporadic or if they were caused by LS, so it is possible at least in part, that MSH6 and PMS2 carriers were more likely to have an HBOC phenotype because they had a lower risk for CRC and EC, while BC and OC were common in our testing population. MSH6 and PMS2 mutations are not commonly seen in HBOC overall. Among all 32,103 patients tested who had data on personal and family history, MSH6 and PMS2 mutations were 4.4% (49 of 1,120) of mutations found in patients with BC only and in 5.5% (25 of 453) of mutations in patients with OC only.
Although we do not know the indication for genetic testing, we found that 72.7% of individuals with MMR mutations met NCCN guidelines for LS testing, 58.9% met NCCN guidelines for BRCA1/2 testing, and 36.7% met both criteria, so it is likely that these syndromes were high on the ordering clinician's differential diagnosis for these patients. Regarding the possibility of other genetic causes for the BC and OC in our MMR mutation carriers, the BRCA1/2 genes were analyzed in 92.3% of patients with BC and/or OC (167 of 181), and no pathogenic or likely pathogenic variants were found. Of the 14 patients who had a panel that did not include BRCA1/2 and did not indicate previously completed and negative BRCA1/2 testing, only five met current NCCN guidelines for this testing.22 In addition, 76.8% of patients with BC and/or OC (139 of 181) had a panel that included several other genes associated with increased risk for BC and/or OC (Appendix Table A1), and no pathogenic or likely pathogenic variants were found. Regarding the exclusion of hereditary polyposis genes, a panel that included APC, BMPR1A, MUTYH, PTEN, SMAD4, and STK11 was performed for all patients with 10 or more polyps, and no pathogenic or likely pathogenic variants were found in those genes. Finally, most of the clinical information analyzed in this study was ascertained from TRFs and some from clinical notes and pedigrees submitted with the TRFs. Additional studies are needed to confirm our results, assess the causative role of MMR mutations in BC, and clarify gene-specific cancer risks for individuals with LS.
In summary, these data from an MGPT cohort provide a different perspective of LS than previous reports and add to the literature describing genotype-phenotype correlations within LS. Results from this study suggest that carriers of MSH6 and PMS2 mutations may present with an HBOC phenotype and are more likely to be missed by current LS screening methods and testing guidelines. This may explain why MSH6 and PMS2 mutations have been underrepresented in previous cohorts and suggests they may be underidentified in general.
Appendix
Methods
Genomic DNA (gDNA) was isolated from blood or saliva by using a QIAsymphony DNA kit (Qiagen, Valencia, CA) and quantified. Sequence enrichment was performed by incorporating the gDNA onto a microfluidics chip or into microdroplets with primer pairs or by a bait-capture methodology by using long biotinylated oligonucleotide probes (RainDance Technologies, Billerica, MA or Integrated DNA Technologies, San Diego, CA), followed by polymerase chain reaction (PCR) and next-generation sequencing (NGS; Illumina, San Diego, CA) of all coding regions plus at least five bases into the 5′ and 3′ ends of all introns and untranslated regions (5′UTR and 3′UTR). NGS of the promoter region was also performed for PTEN (c.-1300 to c.-745), MLH1 (c.-337 to c.-194), and MSH2 (c.-318 to c.-65). Sanger sequencing was performed for regions with insufficient depth of coverage and for verification of all variant calls other than known nonpathogenic alterations. Analysis for the MSH2 inversion of coding exons 1-7 was performed on select patients per the ordering clinician, by NGS and/or PCR and agarose gel electrophoresis.
A targeted chromosomal microarray was used for the detection of gross deletions and duplications for all genes except PMS2 (Agilent, Santa Clara, CA). Gross deletion and duplication analysis of PMS2 was performed by using MLPA kit P008-B1 (MRC-Holland, Amsterdam, Netherlands). If a deletion was detected in exons 13, 14, or 15 of PMS2, double-stranded sequencing of the appropriate exon(s) of the pseudogene PMS2CL was performed to determine whether the deletion was located in the PMS2 gene or the pseudogene.
Initial data processing and base calling, including extraction of cluster intensities, was performed by using RTA 1.12.4 (HiSeq Control Software 1.4.5; Illumina, Hayward, CA). Sequence quality filtering was executed with the CASAVA software (version 1.8.2; Illumina). Sequence fragments were aligned to the reference human genome (GRCh37), and variant calls were generated by using CASAVA. A minimum quality threshold of Q30 was applied, translating to an accuracy of > 99.9% for called bases.
Table A1.
Genes Tested for MMR Mutation Carriers

Table A2.
Mutation Data

Table A3.
Gene-Specific Mutation Frequencies by Proband Tumor Type Compared with Population Frequencies

Table A4.
Patients with Discordant MSI and/or IHC Results
Footnotes
Portions of this study have been presented at the Annual Meeting of the Western Association of Gynecologic Oncologists, June 8-11, 2016, Sedona, AZ; 35th Annual Conference of the National Society of Genetic Counselors, September 28-October 1, 2016, Seattle, WA; and the 20th Annual Meeting of the Collaborative Group of the Americas on Inherited Colorectal Cancer, October 2-3, 2016, Seattle, WA.
AUTHOR CONTRIBUTIONS
Conception and design: Carin R. Espenschied, Holly LaDuca, Rachel McFarland, Chia-Ling Gau, Heather Hampel
Collection and assembly of data: Carin R. Espenschied, Holly LaDuca, Rachel McFarland
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Multigene Panel Testing Provides a New Perspective on Lynch Syndrome
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Carin R. Espenschied
Employment: Ambry Genetics
Holly LaDuca
Employment: Ambry Genetics
Stock or Other Ownership: Ambry Genetics
Shuwei Li
Employment: Ambry Genetics
Rachel McFarland
Employment: Ambry Genetics
Chia-Ling Gau
Employment: Ambry Genetics
Stock or Other Ownership: Ambry Genetics
Heather Hampel
Stock or Other Ownership: Genome Medical
Honoraria: Beijing Genomics Institute
Consulting or Advisory Role: Beacon Laboratory Benefit Solutions, InVitae, Genome Medical
Research Funding: Myriad Genetics (Inst)
REFERENCES
- 1.Win AK, Jenkins MA, Dowty JG, et al. : Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev 26:404-412, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hampel H, Frankel W, Panescu J, et al. : Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 66:7810-7817, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Hampel H, Frankel WL, Martin E, et al. : Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 26:5783-5788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonadona V, Bonaïti B, Olschwang S, et al. : Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305:2304-2310, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Palomaki GE, McClain MR, Melillo S, et al. : EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 11:42-65, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moreira L, Balaguer F, Lindor N, et al. : Identification of Lynch syndrome among patients with colorectal cancer. JAMA 308:1555-1565, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Møller P, Seppälä T, Bernstein I, et al. : Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 66:464-472, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lotsari JE, Gylling A, Abdel-Rahman WM, et al. : Breast carcinoma and Lynch syndrome: Molecular analysis of tumors arising in mutation carriers, non-carriers, and sporadic cases. Breast Cancer Res 14:R90, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wasielewski M, Riaz M, Vermeulen J, et al. : Association of rare MSH6 variants with familial breast cancer. Breast Cancer Res Treat 123:315-320, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Walsh MD, Buchanan DD, Cummings MC, et al. : Lynch syndrome-associated breast cancers: Clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res 16:2214-2224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beamer LC, Grant ML, Espenschied CR, et al. : Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol 30:1058-1063, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heald B, Plesec T, Liu X, et al. : Implementation of universal microsatellite instability and immunohistochemistry screening for diagnosing Lynch syndrome in a large academic medical center. J Clin Oncol 31:1336-1340, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampel H, Frankel WL, Martin E, et al. : Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 352:1851-1860, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Hampel H, Panescu J, Lockman J, et al. : Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) Among Endometrial Cancer Patients. Cancer Res 67:9603, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Cragun D, Radford C, Dolinsky JS, et al. : Panel-based testing for inherited colorectal cancer: A descriptive study of clinical testing performed by a US laboratory. Clin Genet 86:510-520, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yurgelun MB, Allen B, Kaldate RR, et al: Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 149:604-613.e20, 2015. [DOI] [PMC free article] [PubMed]
- 17.Thompson BA, Spurdle AB, Plazzer JP, et al. : Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 46:107-115, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. doi: 10.1155/2016/2469523. Pesaran T, Karam R, Huether R, et al: Beyond DNA: An integrated and functional approach for classifying germline variants in breast cancer genes. Int J Breast Cancer 2016:1-10, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plon SE, Eccles DM, Easton D, et al. : Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29:1282-1291, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405-424, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Provenzale D, Gupta S, Ahnen DJ, et al. : Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 14:1010-1030, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Daly MB, Pilarski R, Axilbund JE, et al. : Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J Natl Compr Canc Netw 14:153-162, 2016 [DOI] [PubMed] [Google Scholar]
- 23.Sjursen W, McPhillips M, Scott RJ, et al. : Lynch syndrome mutation spectrum in New South Wales, Australia, including 55 novel mutations. Mol Genet Genomic Med 4:223-231, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomsic J, Senter L, Liyanarachchi S, et al. : Recurrent and founder mutations in the PMS2 gene. Clin Genet 83:238-243, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foulkes WD, Thiffault I, Gruber SB, et al. : The founder mutation MSH2*1906G-->C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population. Am J Hum Genet 71:1395-1412, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green J, O’Driscoll M, Barnes A, et al. : Impact of gender and parent of origin on the phenotypic expression of hereditary nonpolyposis colorectal cancer in a large Newfoundland kindred with a common MSH2 mutation. Dis Colon Rectum 45:1223-1232, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Desai DC, Lockman JC, Chadwick RB, et al. : Recurrent germline mutation in MSH2 arises frequently de novo. J Med Genet 37:646-652, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pritchard CC, Morrissey C, Kumar A, et al. : Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun 5:4988, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kastrinos F, Steyerberg EW, Mercado R, et al. : The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 140:73-81, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talseth-Palmer BA, McPhillips M, Groombridge C, et al. : MSH6 and PMS2 mutation positive Australian Lynch syndrome families: Novel mutations, cancer risk and age of diagnosis of colorectal cancer. Hered Cancer Clin Pract 8:5, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramsoekh D, Wagner A, van Leerdam ME, et al. : A high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic setting. Gut 57:1539-1544, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Baglietto L, Lindor NM, Dowty JG, et al. : Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 102:193-201, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desmond A, Kurian AW, Gabree M, et al. : Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol 1:943-951, 2015 [DOI] [PubMed] [Google Scholar]
- 34.Minion LE, Dolinsky JS, Chase DM, et al. : Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol Oncol 137:86-92, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Norquist BM, Harrell MI, Brady MF, et al. : Inherited mutations in women with ovarian carcinoma. JAMA Oncol 2:482-490, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tung N, Lin NU, Kidd J, et al. : Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol 34:1460-1468, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao F, Panarelli NC, Rennert H, et al. : Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am J Surg Pathol 34:1798-1804, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Okkels H, Lindorff-Larsen K, Thorlasius-Ussing O, et al. : MSH6 mutations are frequent in hereditary nonpolyposis colorectal cancer families with normal pMSH6 expression as detected by immunohistochemistry. Appl Immunohistochem Mol Morphol 20:470-477, 2012 [DOI] [PubMed] [Google Scholar]