

Abstract
Background
Tet methylcytosine dioxygenase 2 (TET2) has been increasingly recognized as an important tumor suppressor involved in tumorigenesis. Here, we aimed to explore the expression pattern of TET2, its clinical significance as well as functional roles in head neck squamous cell carcinoma (HNSCC).
Methods
Both mRNA and protein levels of TET2 in primary HNSCC samples were detected via immunohistochemistry and qRT-PCR, respectively. Correlations between TET2 expression with multiple clinicopathological parameters and patient survival were determined. The biological roles of TET2 in HNSCC were assessed via a gain-of-function approach and in 4-nitroquinoline-1-oxide (4NQO)-induced HNSCC model. Restoration of TET2 by chemicals including 5-Aza-2'-deoxycytidine (5-AZA), metformin or Vitamin C (VC) to inhibit tumor growth was determined in vitro and in a xenograft animal model.
Results
Reduced TET2 expression was found in a large fraction of HNSCC samples. Downregulated TET2 significantly correlated with larger tumor size, advanced clinical stage and inferior prognosis. Reduced TET2 and 5-hydroxymethylcytosine (5hmC) were observed along with disease progression in the 4NQO-induced HNSCC model. Enforced TET2 overexpression significantly inhibited cell proliferation, migration and enhanced the chemosensitivity of cisplatin in HNSCC cells. Restoration of TET2 following 5-AZA, metformin or VC exposure impaired cell proliferation and migration in vitro. Moreover, VC alone or in synergistic with cisplatin potently inhibited tumor growth in vivo.
Conclusions
Our data reveal that reduced TET2 associates with tumor aggressiveness and reduced survival in HNSCC. Genetic or pharmacological restoration of TET2 might be a viable therapeutic strategy for HNSCC patients with TET2 deficiency.
Keywords: Head neck squamous cell carcinoma, tet methylcytosine dioxygenase 2 (TET2), 5-hydroxymethylcytosine (5hmC), DNA demethylation
Introduction
Head neck squamous cell carcinoma (HNSCC) is the sixth common malignancy occurring in oral cavity, oropharynx and hypopharynx, and remains one of the leading causes of cancer-related deaths (1). Smoking, alcohol addiction, betel quid chewing and HPV infection have been established as well-known etiologic factors for this malignancy (2). Although considerable progress has been achieved for HNSCC diagnosis and therapy in the past decades, the 5-year patients’ survival is marginally improved and still less than 60% (3). Local recurrence, cervical node metastasis and advanced clinical stages have been identified as key parameters affecting patient prognosis. Nevertheless, these conventional prognostic biomarkers are far from optimal and quite few druggable targets have been identified for HNSCC thus far (4). These unmet challenges highlight the urgency to develop more valuable biomarkers and effective therapeutics for HNSCC.
Aberrant epigenetic modification is a hallmark of cancer as evidenced that various epigenetics modifiers have been found to be intricately linked to tumor initiation and overgrowth, metastatic dissemination as well as therapeutic resistance (5). In particular, dysregulated DNA methylation such as hypomethylation of promoter in tumor suppressor genes has been linked to cancer development. It was initially believed that DNA methylation in gene repression is an irreversible epigenetic event (5,6). However, the remarkable discovery of the ten-eleven translocation (TET) family proteins (TET1-3) which catalyze the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), indicate that DNA methylation is not at a standstill, but rather dynamic between methylation and demethylation processes (7,8). In addition, these TET enzymes play vital functions in cell fate determination, cell differentiation, development and various diseases including cancer (9). Tet methylcytosine dioxygenase 2 (TET2) was identified as one of the most frequently mutated genes in hematological malignancy (10). Its deficiency promoted hematopoietic stemness and malignant progression of leukemia and lymphomas (11,12). Moreover, reduced TET2 expression and its negative association with patient survival were observed across several solid cancers such as hepatocellular carcinoma, prostate cancer and gastric cancer (13-16). The expression and activity of TET2 itself are modulated by genetic mutation, DNA methylation at its promoter, noncoding RNAs as well as oncometabolites across diverse cancer contexts (8,17,18). We previously found that reduced 5hmC level was observed in a large subset of oral squamous cell carcinoma (the prevalent site for HNSCC), which was presumably due to low TET2 expression (19). In addition, we interrogated transcriptional data from using TCGA-HNSCC datasets and found that only TET2 mRNA was significantly reduced in primary HNSCC samples compared to non-tumor counterparts (19).
Importantly, given the tumor suppressor roles of TET2 in human cancer, several approaches were developed to block tumor growth and progression by restoring TET2 abundance and activity (11,20,21). For instance, VC promoted cell differentiation and chemosensitivity to PARP inhibitors and blocked disease progression presumably via TET2 restoration in leukemia (11). Metformin, an anti-diabetic drug, enhanced TET2 stability through AMPK-mediated TET2 phosphorylation (Ser 99) and in turn robustly inhibited cancer overgrowth (22). In addition, 5-Aza-2'-deoxycytidine (5-AZA), an inhibitor of DNA methylation, had the capacity to increase TET2 expression, global 5hmc levels and enhance platinum chemosensitivity in ovarian cancer (20). Collectively, these findings have established the tumor suppressive roles of TET2 and pharmaceutical restoration of TET2 as a novel and promising approach for cancer therapeutics. However, the expression pattern, biological roles of TET2 and its translational potential as a druggable target in HNSCC remain incompletely defined yet.
In this study, we determined the expression pattern of TET2 in HNSCC specimens and 4NQO-induced HNSCC mouse model as well as its clinicopathological significance. The tumor suppressor roles of TET2 were explored via gain-of-function approach. The therapeutic effects of TET2 restoration by pharmacological agents were measured both in vitro and vivo.
Methods
Cell lines and chemicals
Several HNSCC cell lines including Cal27, FaDu, SCC4, SCC9, SCC25, HN4, HN6 and normal human oral keratinocytes (HOK) cell line were used in this study. SCC4, SCC9, SCC25, Cal27, FaDu and HOK cells were obtained from American Type Culture Collection (ATCC, USA). HN4 and HN6 were generously gifted from Dr. Wantao Chen (Shanghai JiaoTong University). HNSCC cells were cultured in DMEM/F12 (Invitrogen, USA) containing 10% fetal bovine serum (FBS, Gbico, USA) at 37 °C in 5% CO2. Short tandem repeat (STR) profiling was performed to confirm the identities of cell lines and exclude the possibility of cell contamination. 5-AZA (HY-10586) was obtained from MCE (USA). Metformin (A506198-0025) and VC (A610021-0100) were purchased from Sangon Biotech (Shanghai, China). HNSCC cells were treated with different concentrations of 5-AZA, metformin or VC for the indicated times, then trypsinized for further experiments.
DNA constructs, viral production and infection
The human TET2 overexpressing construct (1× FLAG tagged) was generated by inserting the human TET2 full-length cDNA template into pLenti CMV-GFP-Puro plasmid and then verified by direct sequencing. Lentiviral particles were prepared with packaging and envelope plasmids (pCMV-VSV-G and pCMV-Δ8.2) using calcium-phosphate method. These viral supernatants were filtered, concentrated and stored until use. The stable TET2 overexpressing derivative cells were selected by puromycin (2–5 µg/mL, Sigma, USA) for 7 days after infection. Then, ectopic overexpression of TET2 was confirmed via western blot and qPCR assays.
RNA extraction and quantitative RT-PCR
Total RNA was extracted with Trizol (Invitrogen, USA), subjected to reverse transcription and PCR reactions using Prime-ScriptTM RT-PCR kit (Takara, Japan) as we described previously (23,24). All primers used were listed as follows: TET2 (forward: GATAGAACCAACCATGTTGAGGG, reverse: TGGAGCTTTGTAGCCAGAGGT) and GAPDH (forward: AGGTGAAGGTCGGAGTCAAC, reverse: AGTTGAGGTCAATGAAG GGG). Relative mRNA level was quantified via comparative CT method (GAPDH served as the internal control).
Protein extraction and western blot
After trypsin digestion, 1×106 cell/well were pre-seeded six-well plates and exposed to diverse drugs. Following drug exposure for 48 h, these cells were harvested and lysed in ice-cold lysis buffer (Beyotime, China) containing protease inhibitor PMSF (1:100, Beyotime, China). Protein samples were separation through SDS-PAGE gel and transferred to PVDF membranes (Millipore). The PVDF membranes were blocked in 5% fat-free milk and incubated at 4 °C overnight with primary antibodies including TET2 (1:1,000, 21207-1-AP, Proteintech, USA), E-cadherin (1:1,000, #14472, CST, USA), N-cadherin (1:1,000, #13116, CST, USA), Vimentin (1:1,000, GTX100619, GeneTex, USA), Snail1 (1:1,000, #3879, CST, USA), cleaved Caspase-3 (1:1,000, #9664, CST, USA), cleaved PARP (1:1,000, #9541, CST, USA) and GAPDH (1:2,000, sc-47724, Santa Cruz, USA). These membranes were incubated with horseradish HRP-conjugated secondary antibodies (Cell signaling, USA) and detected by ECL chemiluminescence kit (Millipore, Germany).
Dot immunoblot assay
ONE-4-ALL Genomic DNA Mini-Preps Kit (Sangon Biotech, Shanghai, China) was used for genomic DNA extraction. DNA samples were placed on nitrocellulose membranes. Blots were blocked at room temperature and then incubated in anti-5hmC (1:1,000, GTX629765, GeneTex, USA) at room temperature for 3h and imaged by ECL chemiluminescence kit (Millipore, Germany). To ensure equal loading of 5hmC levels, the amount of DNA loaded on the membranes was stained with methylene blue.
Cell proliferation, colony formation and apoptosis assay
Cell proliferation and viability were detected by using CCK-8 Kit (Dojindo, Japan). Briefly, cells were counted and then seeded into 96-well plates with 1×103 cells/ well. Ten µL agents were added into each well at specified time points. After reaction for 4 hours, the absorbance was measured at a wavelength of 450 nm by SpectraMax M2 (Molecular Devices, USA). Colony formation and apoptosis assays were performed as we previously reported (25).
Cell invasion and migration assay
For wound-healing assay, 1×106 cell/well were seeded into six-well plates. Twenty-four hours later, sterile pipette tip was used to create an artificial wound. Wound healing was photographed at indicated times. Cell invasion was determined by transwell invasion assay (Millipore, Germany) as our previous reports (25).
Primary HNSCC patients and tissue specimens
Patients with primary HNSCC who were received surgical resection at our institution between Jan. 2007 to Dec. 2016 were screened and included. Detailed patient inclusion criteria were described as follows: (I) primary HNSCC with no prior chemotherapy or radiotherapy; (II) radical tumor resection and neck lymph node dissection was indicated; (II) detailed demographic, clinical, pathological and follow-up data available. One hundred and one eligible patients were enrolled, and 24 normal adjacent oral mucosa samples obtained from donors during non-tumor surgeries were also included. The archived tissue samples and H&E sections of each patient were retrieved and analyzed to confirm the previous histopathological diagnoses. All enrolled patients signed on written informed consents. This research was reviewed and approved by the Ethic Committees of Nanjing Medical University and carried out under the ethical standards of the 1964 Declaration of Helsinki.
Immunohistochemical staining and scoring
Immunohistochemical staining for TET2 was performed on 4µm-thick paraffin-embedded slides samples as we previously reported (23,24). These sections were incubated with primary TET2 antibody (1:200, 21207-1-AP, Proteintech, USA), 5hmC (1:200, GTX629765, GeneTex, USA), Ki-67 (1:100, Maxin, China) and cleaved Caspase-3 (1:200, #9664, CST, USA). Negative controls (only PBS incubation) were included in each staining run. Immunoreactivity in each slide was semi-quantitatively evaluated according to staining intensity and distribution as we also reported previously (23,25). Intensity score was defined as 0, negative; 1, weak; 2, moderate; 3, strong, while the proportion score was evaluated by two independent pathologists via counting positive nucleus with 0, negative; 1, <10%; 2, 11–50%; 3, 51–80%; 4, >80% positive cells. Immunoreactivity score = intensity score × proportion score. The immunoreactivity of each slide was categorized into three subgroups according to the final score: 0, negative; 1–4, low expression; ≥4, high expression.
Establishment of 4NQO-induced HNSCC models and xenograft models
All the animal protocols were in accordance with institutional animal welfare guidelines of Nanjing Medical University. Six-week-old female C57BL/6 mice were utilized and drunk water containing 50 µg/mL 4-nitroquinoline 1-oxide (4NQO, Cat no. N8141; Sigma, USA) for successive 16 weeks. Mice with normal water were considered as control group. In the 4NQO-induced mouse tongue carcinogenesis model, lesions or tumors of tongue were observed every two weeks. Samples were harvested at the time points (16, 20 and 24 weeks, 10 mice for each time points) after the onset of treatment and used for subsequent histopathological analyses. For xenograft model, FaDu cells with or without stable TET2 overexpression were collected and injected subcutaneously into six-week-old female nu/nu mice with 2×106 cells in 100 µL PBS (6 mice per group). After 12 days, the tumor diameters of these mice bearing tumors were measured by the same caliper every three days until harvested. Tumor volume was assessed by the formula volume (mm3) = [length (mm) × width2 (mm2)]/2. Once tumor samples were harvest, tumor weight was measured. Then, the xenograft tumor samples were processed for further experiments.
Drug preparation and administration in vivo
For drug treatment in vivo, cisplatin was dissolved into PBS, while VC was dissolved into sterilized pure water and adjusted to pH 7.0 with NaOH solution. VC and cisplatin were administered via intraperitoneal injection at the dose of 4 g/kg/d, 2 mg/kg/3 d, respectively. PBS was delivered as vehicle control. After tumor xenografts were established (the mean tumor volume reached 100 mm3) two weeks after initial cell inoculation, these mice were randomly divided into 4 groups (control, VC, cisplatin and VC +Cis, 6 mice for each group). Tumor sizes were measured with electronic clippers every 3 days. Animals were euthanized at day 14 and tumor samples were harvested, weighted and processed for H&E or IHC staining.
Data mining of TET2, IDH1, IDH2 and L2HGDH in HNSCC via public database
The original data concerning mutational landscape and/or mRNA expression of TET2, IDH1, IDH2 and L2HGDH in primary HNSCC were downloaded from TCGA platform (https://cancergenome.nih.gov/). The log2-transformed mRNA levels of genes of interest in HNSCC as well as normal counterparts were compared.
Statistical analyses
Data were presented as mean ± SD and compared with Student’s t test or ANOVA (Bonferroni post hoc test). Chi-square or Fisher exact test was used for evaluating potential associations between TET2 expression and various clinicopathological parameters. Kaplan-Meier method (Log-rank test), univariate and multivariate Cox regression models was used for estimating patients’ survival. P<0.05 (two-sided) was considered statistically significant. GraphPad Prism 8.0.0 (GraphPad Software Inc., USA) or SPSS 21.0.0 software (IBM, USA) was used for all statistical analyses.
Results
Reduced TET2 expression associates with aggressive clinicopathological features in HNSCC
Our previous findings have revealed that only TET2 mRNA was significantly reduced in primary HNSCC samples compared to non-tumor samples using TCGA-HNSCC datasets (19). To validate this bioinformatics’ results, we measured TET2 expression in 101 archived HNSCC samples by immunohistochemistry (IHC) and 60 pairs of fresh samples by qRT-PCR assay (Figure S1). Patients’ epidemiologic and clinicopathological parameters were listed in Table 1. Briefly, 52 males and 49 females (mean age 62.6 years) with follow-up duration ranging from 4 to 65 months (average 36.6 months) were included. TET2 positive staining was mainly identified in nucleus in cancer cells (Figure 1), which was well consistent with its epigenetic roles in cancer (8). TET2 expression in HNSCC and normal oral mucosa was categorized via IHC scoring regime. As listed in Table 2, TET2 expression were classified as negative (13), low (48) or high expression group (40) in HNSCC samples, while negative (2), low expression (5), or high expression (17) in non-tumor counterparts, respectively. These data indicated significantly reduced TET2 expression in HNSCC samples as compared with its normal counterparts (P=0.0209, Chi-square test). As shown in Table 1, low TET2 expression positively associated with large tumor size (P=0.0278, Fisher exact test) and advanced clinical stage (P=0.0413, Fisher exact test). Moreover, we also collected 60 pairs of primary HNSCC fresh samples and adjacent non-tumor mucosa and measured TET2 mRNA expression by qRT-PCR in these samples. As shown in Figure S1, markedly lower TET2 mRNA levels in tumor as compared to adjacent non-tumor tissues. Collectively, these findings indicate that TET2 is significantly downregulated in HNSCC and its abundance associates with tumor aggressiveness.
Figure S1.
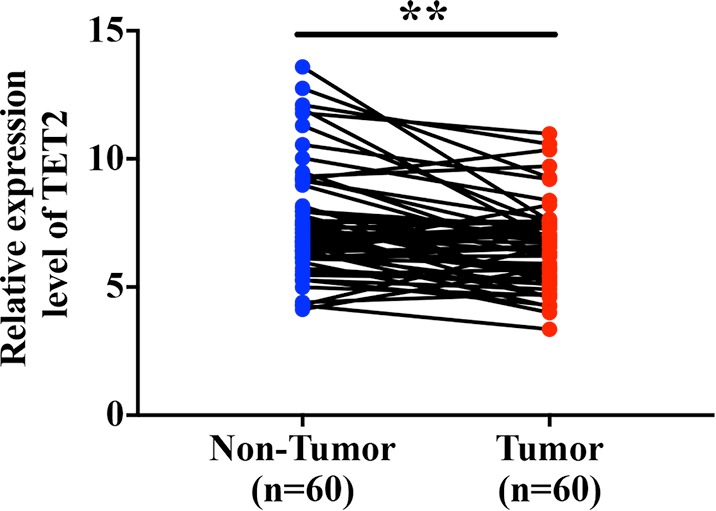
Comparison of TET2 in fresh HNSCC samples and paired non-tumor samples. The mRNA levels of TET2 in 60 pairs of primary HNSCC samples of tumor and adjacent tissues are measured by qRT-PCR assay and compared. Paired t test, **P<0.01. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Table 1. The associations between TET2 expression and multiple clinicopathological parameters in HNSCC samples.
| Clinicopathological parameters | Cases | TET2 | P value | |
|---|---|---|---|---|
| Low* | High | |||
| Gender | 101 | 61 | 40 | |
| Male | 52 | 31 | 21 | >0.9999 |
| Female | 49 | 30 | 19 | |
| Age | ||||
| ≤60 | 40 | 22 | 18 | 0.4098 |
| >60 | 61 | 39 | 22 | |
| Smoking | ||||
| No | 73 | 42 | 31 | 0.3732 |
| Yes | 28 | 19 | 9 | |
| Alcohol use | ||||
| No | 79 | 48 | 31 | >0.9999 |
| Yes | 22 | 13 | 9 | |
| Tumor size | ||||
| T1–T2 | 68 | 38 | 30 | 0.0278# |
| T3–T4 | 43 | 33 | 10 | |
| Pathological grade | ||||
| I | 59 | 35 | 24 | 0.8386 |
| II–III | 42 | 26 | 16 | |
| Cervical node metastasis | ||||
| N0 | 66 | 42 | 24 | 0.3976 |
| N+ | 35 | 19 | 16 | |
| Clinical stage | ||||
| I–II | 52 | 26 | 26 | 0.0413# |
| III–IV | 49 | 35 | 14 | |
*Both of patients with low and negative TET2 staining are stratified into low TET2 category for simplicity. #indicate statistical significance with P values less than 0.05. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Figure 1.

Immunohistochemical staining of TET2 in primary HNSCC samples. (A,B) Representative positive staining of TET2 in normal oral epithelial; (C,D) representative low expression of TET2 in primary human HNSCC sample; (E,F) representative high expression of TET2 in primary human HNSCC sample; nuclei are counterstained with hematoxylin. The areas marked by black box in the A, C, E images (upper panel) are shown in larger magnification as B, D, F images (lower panel), respectively. Scale bar: 50 µm. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Table 2. TET2 protein expression in HNSCC and normal oral mucosa.
| TET2 expression | P value | |||
|---|---|---|---|---|
| Negative | Low | High | ||
| Normal oral mucosa | 2 | 5 | 17 | 0.0209 |
| HNSCC | 13 | 48 | 40 | |
TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Reduced TET2 expression in HNSCC patients correlates with unfavorable survival
Next, we sought to discern the prognostic values of TET2 expression in HNSCC. Until the last follow-up, 52 (51.5%) patients were disease-free, 5 (4.9%) alive but with local recurrences and/or cervical nodal metastases, whereas 44 (43.6%) died due to post-surgical relapse, metastases, or other reasons. As shown in Figure 2, low TET2 expression had an adverse prognostic impact on patient survival. Patients with reduced TET2 significantly associated with reduced overall (Figure 2A) and disease-free (Figure 2B) survival (P=0.0305, 0.0404, respectively, log-rank test). Furthermore, as shown in Table 3, low TET2 expression was identified as an independent prognostic factor for HNSCC via both univariate and multivariate Cox regression analyses.
Figure 2.

Decreased TET2 expression positively associates with reduced survival in HNSCC patients. Overall survival (A) and disease-free survival (B) analyses of patients stratified with high or low expression of TET2 were estimated by Kaplan-Meier method and compared with Log-rank test. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Table 3. Univariate and multivariate survival analyses (proportional hazards method) for patients with primary HNSCC.
| Variable | Univariate survival analysis | Multivariate survival analysis | |||||
|---|---|---|---|---|---|---|---|
| Hazard ratio | 95% CI | P value | Hazard ratio | 95% CI | P value | ||
| Gender (male, female) | 1.133 | 0.622–2.062 | 0.683 | N/A | |||
| Smoking (no, yes) | 0.922 | 0.538–1.830 | 0.980 | N/A | |||
| Alcohol use (no, yes) | 0.920 | 0.472–1.793 | 0.807 | N/A | |||
| Age (>60, ≤60) | 1.333 | 0.671–2.646 | 0.412 | 0.961 | 0.517–1.787 | 0.901 | |
| Tumor size (T3–T4, T1–T2) | 1.555 | 0.847–2.853 | 0.154 | 1.197 | 0.574–2.497 | 0.632 | |
| Pathological grade (II–III, I) | 1.182 | 0.667–2.094 | 0.567 | 1.164 | 0.601–2.220 | 0.645 | |
| Cervical nodal metastasis (N+, N0) | 1.096 | 1.590–2.035 | 0.773 | 0.856 | 0.283–2.588 | 0.783 | |
| Clinical stage (III–IV, I–II) | 1.658 | 0.903–3.044 | 0.103 | 0.751 | 0.375–1.579 | 0.450 | |
| TET2 expression (high, low) | 0.514 | 0.268–0.987 | 0.045# | 0.509 | 0.265–0.982 | 0.044# | |
#Indicate statistical significance with P values less than 0.05. HNSCC, head neck squamous cell carcinoma; TET2, tet methylcytosine dioxygenase 2.
Reduced TET2 and 5hmc expression along with disease progression in 4NQO-induced HNSCC tumorigenesis
Having established that reduced TET2 associated with aggressiveness and poor prognosis in HNSCC, we next utilized the 4NQO-induced HNSCC mouse model to reveal its expression along with disease progression. The 4NQO-induced HNSCC largely mimics the multi-stage tumorigenic events found in patients (26). Lesions were detected in the tongue after 4NQO administration at 16th, 20th and 24th week. Histological evaluations of these lesions revealed hyperkeratosis, dysplasia and invasive SCC, which were well corresponded to the main stages during HNSCC tumorigenesis. As shown in Figure 3 and Table 4, we performed immunohistological staining of TET2 and found prevalent nuclear staining of TET2 in normal tongue mucosa (8/10, 80%) while negative or low staining in SCC. In parallel with TET2 expression, the abundance of 5hmc, the enzymatic product and a surrogate of TET2 activity, as evaluated by immunohistochemistry, was found lower in SCC as compared to other types of lesions (Figure S2 and Table S1). Collectively, these data support that reduced TET2 and 5hmc likely contribute to HNSCC tumorigenesis.
Figure 3.

TET2 expression during tumorigenesis in 4NQO-induced HNSCC animal model. Immunohistochemical staining of TET2 in samples from diverse stages in 4NQO-induced animal model. Images in the upper panel (A,C,E,G) are representative staining of TET2 in normal, epithelial with hyperplasia, epithelial with severe dysplasia/carcinoma in situ and squamous cell carcinoma, respectively. Images in the lower panel (B,D,F,H) are magnified from the black box area in the A, C, E, G images in the upper panel, respectively. The number of animals in each stage during 4NQO-induced HNSCC model was 10. Scale bar: 100 µm. TET2, tet methylcytosine dioxygenase 2; 4NQO, 4-nitroquinoline-1-oxide; HNSCC, head neck squamous cell carcinoma.
Table 4. TET2 expression pattern in samples harvested from different stages in 4NQO-induced HNSCC model.
| TET2 expression | P value | ||
|---|---|---|---|
| Negative/low | High | ||
| Healthy mucosa | 2 | 8 | 0.0370 |
| Hyperplasia | 3 | 7 | |
| Dysplasia/carcinoma in situ | 5 | 5 | |
| Invasive carcinoma | 8 | 2 | |
TET2, tet methylcytosine dioxygenase 2; 4NQO, 4-nitroquinoline-1-oxide; HNSCC, head neck squamous cell carcinoma.
Figure S2.

5hmC expression pattern during HNSCC tumorigenesis in 4NQO-induced animal model. Immunohistochemical staining of 5hmC in samples from diverse stages in 4NQO-induced animal model. Images in the upper panel (A,C,E,G) are representative staining of 5hmC in normal, epithelial with hyperplasia, epithelial with severe dysplasia/carcinoma in situ and squamous cell carcinoma, respectively. Images in the lower panel (B,D,F,H) are magnified from the black box area in the A, C, E, G images in the upper panel, respectively. Scale bar: 100 µm. HNSCC, head neck squamous cell carcinoma; 4NQO, 4-nitroquinoline-1-oxide.
Table S1. 5hmC expression pattern in samples harvested from different stages in 4NQO-induced HNSCC model.
| 5hmC expression | P value | ||
|---|---|---|---|
| Negative/low | High | ||
| Healthy mucosa | 2 | 8 | 0.0133 |
| Hyperplasia | 4 | 6 | |
| Dysplasia/carcinoma in situ | 6 | 4 | |
| Invasive carcinoma | 9 | 1 | |
4NQO, 4-nitroquinoline-1-oxide; HNSCC, head neck squamous cell carcinoma.
TET2 downregulation might be partially due to genetic deletion in HNSCC
To explore the possible mechanisms responsible for reduced TET2 expression and activity in HNSCC, we firstly interrogated the genetic alterations and transcriptional profiles of TET2 and several well-known modulators of TET2 in TCGA-HNSCC dataset. Consistent with mutational status of TET2 in other solid malignancies, total frequency of TET2 genetic alteration in HNSCC was relatively rare (<5%), thus precluding the possibility that TET2 mutation was primarily responsible for its low expression (Figure S3A,B). However, copy number variations such as deletion (30.47%) and amplification (9.61%) were detected in approximately 40% samples (Figure S3C). Noticeably, samples with deletion had significantly lower abundance of TET2 mRNA as compared to those without deletion, implying that genetic deletion might contribute to reduced TET2 expression in HNSCC (Figure S3D). Moreover, we examined three key metabolic enzymes associated with TET2 activity including IDH1,2 and L2HGDH in TCGA-HNSCC dataset (27). The mutational frequencies of these three enzymes in HNSCC were fewer than 10% (Figure S3A). In addition, as shown in Figure S3E,F,G, only IDH2 was found to be differentially expressed between HNSCC samples and their normal counterparts. Unexpected, IDH2 mRNA abundance was significantly lower in HNSCC than that in healthy samples. Given the negative regulatory effects of IDH1/2 on TET2 activity, we reasoned that TET2 activity in HNSCC might be not directly affected by these oncometabolites like IDH1/2 and 2-hydroxyglutarate. Collectively, our data reveal that TET2 deficiency with low expression and activity in HNSCC is likely linked to its genetic deletion.
Enforced TET2 overexpression inhibits phenotypic changes in HNSCC cells
Having revealed reduced TET2 expression in HNSCC, we next aimed to characterize the roles of TET2 via gain-of-function approach. Both TET2 mRNA and protein levels in a panel of HNSCC cell lines were measured and it was significantly decreased in all HNSCC cell lines detected compared to HOK cells (Figure 4A and Figure S4A). Cal27 and FaDu cells with relatively low endogenous TET2 were selected for lentiviral vector-mediated ectopic TET2 overexpression. As displayed in Figure 4B and Figure S4B, marked overexpression of TET2 was observed in FaDu and Cal27 cells following lentiviral infection. Subsequently, the global abundance of 5hmC, a well-established product of TET2, was significantly increased following TET2 enforced overexpression as determined by dot immunoblot assay (Figure 4C). As shown in Figure 4D,E, cell proliferation was significantly inhibited in cells upon TET2 overexpression as evidenced by the data from CCK-8 and colony formation assays. In addition, as shown in the Figure S5, enforced TET2 overexpression markedly enhanced sensitivity of cells to the common chemotherapeutic agent cisplatin in vitro. The migratory and invasive potentials of TET2-overexpressing cells were measured using wound healing and transwell assays, respectively. As shown in Figure 4F,G, both migratory and invasive properties of FaDu and Cal27 cells were significantly reduced after TET2 overexpression. In agreement with this, upregulation of E-cadherin and downregulation of Vimentin, N-cadherin and Snail were observed upon TET2 overexpression (Figure 4H).
Figure 4.

Enforced TET2 overexpression inhibits cell proliferation, invasion and migration in HNSCC cells. (A) Endogenous TET2 protein expression in a panel of HNSCC cell lines is measured via western blot assay (left panel). Quantification of endogenous TET2 protein expression (right panel) is shown; (B) enforced TET2 overexpression is confirmed after lentivirus infection in Cal27 and FaDu cells; (C) 5hmC expression levels in Cal27 and FaDu cells with TET2 overexpression are detected by dot bolt assay. Methylene blue staining is utilized as loading control; (D,E) cell proliferation is markedly suppressed upon TET2 overexpression as measured by CCK-8 viability assay (D). The potentials of colony formation are significantly inhibited in TET2 overexpression cells as compared to control (E); (F,G) the invasive (F) and migratory (G) abilities are significantly reduced in TET2-overexpressing cells compared to control as detected via transwell and wound healing assays; (H) the abundance of invasion/metastasis markers E-cadherin, N-cadherin, Vimentin and Snail following TET2 overexpression is assayed by western blot. Representative images are shown. Data are mean ± SD from three independent experiments. **P<0.01, *P<0.05, Student t-test, two-way ANOVA. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Figure S5.

Enforced TET2 overexpression enhanced chemosensitivity in HNSCC cells. (A,B) Cell viability is significantly impaired in TET2 overexpressing cells treated with cisplatin (2.5 µg/mL, B) alone than those treated with vehicle. Student’s t-test, *P<0.05. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Enforced TET2 overexpression inhibits tumor growth in a HNSCC xenograft model
To further confirm the tumor-inhibitory ability of TET2 in vivo, we next developed a HNSCC xenograft model. TET2-overexpressing cells were subcutaneously inoculated into nude mice. As showed in Figure 5A,B,C, TET2 overexpression impaired tumor overgrowth in vivo as evidenced by much smaller tumor burden formed in cells with TET2 overexpression than those from controls. Moreover, IHC staining of samples revealed significantly increased TET2 as well as 5hmc, and reduced Ki67+ cells in TET2-overexpressing cell derived samples (Figure 5D,E,F,G). Together, these findings revealed that ectopic TET2 overexpression impaired tumor outgrowth of HNSCC in vivo, further supporting its tumor suppressor roles in HNSCC.
Figure 5.

TET2 overexpression inhibits tumor growth in a HNSCC xenograft model. (A) FaDu cells with or without TET2 overexpression are inoculated subcutaneously in nude mice. Images of samples from TET2-overexpressing and control groups (n=6 for each group) are shown; (B) the volume curves show tumor growth from TET2-overexpressing and control cells; (C) tumor weights at the endpoint are shown as the mean ± SD; (D) H&E staining and immunohistochemical staining of TET2, Ki-67 and 5hmC in tumors harvested from xenograft mice. Scale bar: 50 μm; (E, F, G) The abundance of TET2 (E), Ki-67 (F) and 5hmc (G) positive staining was quantified. Representative images are shown. **P<0.01, *P<0.05, Student t-test, two-way ANOVA.
Pharmacological restoration of TET2 inhibits cell proliferation, migration and induced apoptosis in HNSCC cells
Several lines of evidence have suggested that some chemical compounds with anti-cancer effects such as 5-AZA, metformin and VC could reverse TET2 loss and potently restrained tumor growth in selected cancer contexts (11,20). Inspired by these findings, we set out to examine whether these drugs can restore TET2 expression/activity and have therapeutic effects in HNSCC. Significantly increased expression of TET2 was observed at both mRNA and protein levels in Cal27 and FaDu cells after 5-AZA, metformin or VC treatment (Figure 6 and Figure S6). As expected, the abundance of 5hmC was also concordantly upregulated in both cells (Figure 6B,D,F). Subsequently, cell proliferation, migration and apoptosis were investigated following chemical treatments. As shown in Figure 6G,H, cell proliferation and migration of Cal27 and FaDu cells were significantly impaired following individual treatments of 5-AZA, metformin and VC. In parallel, cells undergoing apoptosis were markedly increased upon exposure of these chemicals (Figure 6I,J). Collectively, these findings suggest that 5-AZA, metformin and VC have the capacities to restore TET2 expression/activity and exert therapeutic effects in vitro.
Figure 6.

Treatments with 5-AZA, metformin or VC inhibit cell proliferation, migration and induces apoptosis via increasing TET2 expression and activity in HNSCC cells. (A,C,E) TET2 protein expression in Cal27 and FaDu cells is measured after 5-AZA (5, 10 µM), metformin (5, 10 mM) or VC (5, 10 mM) treatment for 48 h via western blot assay; (B,D,F) 5hmC abundance in Cal27 and FaDu cells is measured after 5-AZA (5, 10 µM), metformin (5, 10 mM) or VC (5, 10 mM) treatment for 48h via dot blot assay; (G) cell proliferation is remarkably suppressed after chemical treatments as measured by CCK-8 viability assay; (H) the migration abilities are significantly reduced in cells by wound healing assay (12, 24 h after cell scratching); (I) increased percentages of cell undergoing apoptosis were evident following 5-AZA (10 µM), metformin (10 mM) or VC (10 mM) treatment for 48 h as assayed by Annexin V-PI staining; (J) the expression of cleaved PARP and Caspase-3 protein in Cal27 cells is measured after 5-AZA (10 µM), metformin (10 mM) or VC (10 mM) treatment for 48 h via western blot assay. Representative images were shown. Data are mean ± SD from three independent experiments. **P<0.01, *P<0.05, Student t-test, two-way ANOVA. 5-AZA, 5-Aza-2'-deoxycytidine; VC, Vitamin C; TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Figure S6.

Treatments of 5-AZA, metformin or VC increased TET2 mRNA expression. The mRNA levels of TET2 are significantly upregulated following treatments with 5-AZA (5, 10 µM) (A), metformin (5, 10 mM) (B) or VC (5, 10 mM) (C) in Cal27 and FaDu cells. Two-way ANOVA, *P<0.05, **P<0.01. 5-AZA, 5-Aza-2'-deoxycytidine; TET2, tet methylcytosine dioxygenase 2; VC, Vitamin C.
Vitamin C alone or in synergistic with cisplatin inhibits tumor growth by TET2 restoration in vivo
The potent therapeutic effects of VC against HNSCC attracted our attention and prompted us to further validate the anti-cancer effects of VC in vivo. Previous studies have revealed that therapeutic effects are achieved by high-dose systematic administration of VC in solid cancers (21). We hypothesized that when VC was delivered in combination with the common therapeutic agents like cisplatin, more potent or synergistic effects can be achieved for HNSCC. As shown in Figure S7, synergistic effects were detected with VC plus cisplatin as compared to single agent in both Cal27 and Fadu cells. Next, we developed a HNSCC xenograft model to explore the therapeutic effects of VC alone or in combination with cisplatin in vivo. After subcutaneous xenografts were established, VC, cisplatin alone or their combination were delivered via intraperitoneal injection for consecutive 14 days. The detailed schedule for drug administration was illustrated in Figure 7A. As shown in Figure 7B,C,D, the volume and weight of tumor in mice treated with VC or cisplatin alone were significantly lower as compared to those in vehicle-treated animal. Moreover, combined treatment with VC and cisplatin induced the most potent growth-inhibitory effects as evidenced by the trajectory of tumor volume and final weight. Finally, we conducted IHC staining of TET2, Ki67 and cleaved Caspase 3 in specimens from four animal subgroups. As displayed in Figure 7E,F, the densities of TET2 positive staining were significantly increased in samples from animals treated by VC or VC plus cisplatin, while cisplatin treatment had undetectable effects on TET2 expression. Moreover, samples from VC or cisplatin-treated animals had less amount of Ki67+ staining and more cleaved Caspase 3+ staining in relative to those from vehicle-treated mice. Notably, samples from combinational treatment showed the least Ki67+ staining and the most cleaved Caspase 3+ staining among four subgroups (Figure 7G,H).
Figure S7.

Sensitivities of Cal27 and FaDu to VC, cisplatin or in combination. (A,C) Survival fractions for Cal27 (A) and FaDu (C) cells are shown. (B,D) Combination index of Cal27 (B) and FaDu (D) cells are shown. Fa, fraction affected. Error bars represent means ± SD. VC, Vitamin C.
Figure 7.

VC alone or in synergistic with cisplatin inhibit HNSCC tumor growth in vivo. (A) A schematic chart of experimental design to determine the therapeutic effects of VC and cisplatin in vivo. Dosages of drug administration are shown respectively. N=6 mice in each group of drug treatments. (B) Samples harvested from each treatment group are photographed. (C) Tumor volume is monitored every 3 days. (D) Final tumor weights are recorded and compared. (E,F,G,H) H&E staining and immunohistochemical staining of TET2, Ki67 and cleaved Caspase3 in xenograft samples are shown and evaluated. Scale bar: 50 μm. Quantitative data are presented in F, G and H as mean ± SD. Representative images were shown. **P<0.01, *P<0.05, Student t-test, two-way ANOVA.
Discussion
Aberrant DNA methylation and demethylation has been increasingly recognized as essential molecular events underlying tumorigenesis and promising diagnostic biomarkers and therapeutic targets (5,28). In particular, TET2, the key DNA-demethylation enzyme, serves as a novel tumor suppressor gene critically involved in cancer initiation and progression (9). Importantly, restoration of TET2 expression and activity in cancer by genetic or pharmacological approaches have shown promising therapeutic effects in multiple preclinical models (11,21). Here, we interrogated the expression and functional roles of TET2 behind HNSCC pathogenesis and assessed whether TET2 restoration had anti-cancer effects in HNSCC. Our data revealed that TET2 deficiency serves as a novel prognostic biomarker for HNSCC. VC exposure alone or in synergistic with cisplatin had robust anti-cancer effects via enhancing TET2 expression and activity, thus suggesting a novel and alternative therapeutic strategy for those HNSCC with TET2 deficiency.
Previous reports have established that TET2 is frequently downregulated in a large fraction of human cancer and its expression correlates with clinicopathological features and therapeutic outcomes (13,16,29). For instance, reduced TET2 expression was detected in gastric cancer and its low expression significantly associated with inferior overall and disease-free survival (13). Our previous study has revealed reduced expression of TET2 mRNA and 5hmc in OSCC samples (19). In agreement with these abovementioned reports, our data from HNSCC cell lines and specimens further revealed that TET2 protein was significantly downregulated in HNSCC as compared to its normal counterparts. These findings are well consistent with the previous study regarding TET2 protein expression in OSCC, although sample size in their report was relatively small (30). Moreover, reduced TET2 abundance associated with larger tumor size, advanced clinical stage and unfavorable prognosis in our HNSCC cohort. Complementarily, univariate and multivariate Cox hazard regression analyses identified the expression status of TET2 as an independent factor affecting patient prognosis. Collectively, our results indicate that TET2 abundance is a novel prognostic biomarker for HNSCC. Obviously, more patients from multiple centers are still required to convincingly establish its prognostic utility in HNSCC before it can translate into routine clinical practice.
Accumulating evidence have delineated several layers of regulatory mechanisms concerning TET2 expression and activity across several cancer contexts (8,9). For example, TET2 was found to be frequently mutated or deleted in hematological malignancy, but rare in solid cancers (31). In agreement with this, our data mining and interrogation of TET2 mutation in TCGA-HNSCC dataset revealed that few mutations but 30.4% deletions were detected in HNSCC samples and samples with TET2 deletion had lower expression. In addition, we also screened the mutational and transcriptional profiles of enzymes affecting TET2 activity in other malignancies, but failed to find direct evidence to support their roles in modulating TET2 expression and activity in HNSCC. Altogether, these data suggest that genetic deletion might be one of mechanisms responsible for reduced TET2 expression in HNSCC. However, the detailed and accurate mechanisms underlying TET2 deficiency in HNSCC are warranted further investigations.
Mounting evidence has revealed that TET2 plays critical roles underlying cell proliferation, migration, invasion as well as epithelial mesenchymal transition (EMT) in various human cancers (11,16,32,33). For example, the proliferative, migratory and invasive properties were significantly enhanced following TET2 inhibition via siRNA in prostate cancer cells, while cell proliferation and invasion were potently inhibited upon TET2 overexpression in melanoma cells (16,32). Additionally, increased TET2 expression in malignant melanoma could rescue the TGF-β1-induced EMT-like process in vitro and block tumor outgrowth and metastasis in vivo (33). Our data from gain-of-function experimental approach clearly indicated that enforced TET2 overexpression inhibited cell proliferation, migration, invasion and enhanced the chemosensitivity of cisplatin in vitro. Importantly, tumor growth was impaired when cells with TET2 overexpression were subcutaneously inoculated in a xenograft model. Consistently, reduced TET2 and its enzymatic product 5hmc were found along with disease progression in the 4NQO-induced HNSCC mice. Together, these findings reveal that TET2 serves as a novel tumor suppressor via inhibiting cell proliferation, aggressiveness during HNSCC progression. Detailed downstream targets responsible for TET2’s roles in HNSCC remain to be identified.
Several reports revealed that DNA methyltransferase inhibitor 5-AZA, antidiabetic drug metformin as well as VC inhibited tumor growth and progression in part via modulating TET2 and its related demethylation process (11,20-22). For example, 5-AZA treatment increased 5hmC abundance through TET2 and TET3 upregulation and restored the chemosensitivity in ovarian cancer (20). Wu and his colleagues have reported that metformin inhibited cancer outgrowth through protecting AMPK-mediated phosphorylation of TET2 and the resultant increased TET2 as well as 5hmC (22). Moreover, VC potently restored TET2 activity and enhanced the sensitivity to PARP inhibitors in leukemia and renal cell carcinoma (11,21). Consistent with these previous studies, our results indicated that these three chemicals robustly restored the expression and activity of TET2 in HNSCC cells, inhibited cell proliferation and migration as well as triggered cell apoptosis. These data support that TET2 deficiency in human cancer can be pharmacologically restored to inhibit cancer growth and progression, irrespective of cancer origin. Noticeably, our data revealed that VC exposure induced significant upregulation of TET2 mRNA in vitro, although previous studies rarely reported this phenomenon in other contexts whereby their findings indicated increased activity of TET2 upon VC treatment (21). In particular, administration of VC alone had therapeutic effects in the xenograft model and even better and synergistic effects when it was delivered in combination with cisplatin. These treatments were well-tolerated in preclinical models, thus supporting the feasibility to translate these findings into clinical practice. Collectively, our data together with previous findings reveal that blocking tumor growth via pharmacological restoration of TET2 is feasible and effective in HNSCC.
In conclusion, our data reveal that reduced TET2 expression and activity contribute to HNSCC tumorigenesis and serve as a novel prognostic biomarker for HNSCC. Therapeutic targeting TET2 via genetic or pharmacologic approaches might be a viable and alternative strategy against HNSCC with TET2 deficiency.
Figure S3.

Genetic alternations and expression levels of TET2, IDH1, IDH2 and L2HGDH in TCGA-HNSCC dataset. (A) Genetic alternations of TET2, IDH1, IDH2 and L2HGDH in TCGA-HNSCC dataset via cBioPortal platform. (B) Relative mRNA expression level of TET2 (log2-transformed) in TCGA-HNSCC dataset is shown. (C) The distribution of TET2 copy number variations in TCGA-HNSCC dataset is shown. (D) Relative mRNA expression level of TET2 (log2-transformed) in patients accompanied with or without TET2 genetic deletion in TCGA-HNSCC dataset is shown. (E,F,G) Relative mRNA expression level of IDH1, IDH2 and L2HGDH (log2-transformed) in TCGA-HNSCC dataset is shown. Y-axis represents the median intensity, 25th and 75th percentile data. Student’s t test or Mann-Whitney U test as appropriate. ns, not significant, *P<0.05, **P<0.01. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Figure S4.

The mRNA expression of TET2 in HNSCC cell lines and TET2 overexpression in HNSCC cells. (A) The mRNA levels of TET2 in a panel of HNSCC cell lines are determined by qRT-PCR assay; (B) the mRNA levels of TET2 via overexpression was confirmed after lentivirus infection in Cal27 and FaDu cells. Data are mean ± SD. Two-way ANOVA or Student’s t test, *P<0.05, **P<0.01. TET2, tet methylcytosine dioxygenase 2; HNSCC, head neck squamous cell carcinoma.
Acknowledgments
Funding: This work was supported, in whole or in part, by National Natural Science Foundation of China [81572669], Natural Science Foundation of Jiangsu Province [BK20161564, BK20161024], A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions [2018-87], Research grant from Nanjing Medical University and Southeast University [2017DN20] and Project from Nanjing Municipal Committee of Science and Technology [201803044].
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All enrolled patients signed on written informed consents. This research was reviewed and approved by the Ethic Committees of Nanjing Medical University and carried out under the ethical standards of the 1964 Declaration of Helsinki.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7-30. 10.3322/caac.21442 [DOI] [PubMed] [Google Scholar]
- 2.Gillison ML, Chaturvedi AK, Anderson WF, et al. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J Clin Oncol 2015;33:3235-42. 10.1200/JCO.2015.61.6995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271-89. 10.3322/caac.21349 [DOI] [PubMed] [Google Scholar]
- 4.Budach V, Tinhofer I. Novel prognostic clinical factors and biomarkers for outcome prediction in head and neck cancer: a systematic review. Lancet Oncol 2019;20:e313-26. 10.1016/S1470-2045(19)30177-9 [DOI] [PubMed] [Google Scholar]
- 5.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012;150:12-27. 10.1016/j.cell.2012.06.013 [DOI] [PubMed] [Google Scholar]
- 6.Koch A, Joosten SC, Feng Z, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol 2018;15:459-66. 10.1038/s41571-018-0004-4 [DOI] [PubMed] [Google Scholar]
- 7.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930-5. 10.1126/science.1170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013;502:472-9. 10.1038/nature12750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016;30:733-50. 10.1101/gad.276568.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko M, An J, Pastor WA, et al. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev 2015;263:6-21. 10.1111/imr.12239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cimmino L, Dolgalev I, Wang YB, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017;170:1079-1195.e20. 10.1016/j.cell.2017.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominguez PM, Ghamlouch H, Rosikiewicz W, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov 2018;8:1632-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng W, Wang J, Zhang J, et al. TET2 regulates LncRNA-ANRIL expression and inhibits the growth of human gastric cancer cells. IUBMB Life 2016;68:355-64. 10.1002/iub.1490 [DOI] [PubMed] [Google Scholar]
- 14.García MG, Carella A, Urdinguio RG, et al. Epigenetic dysregulation of TET2 in human glioblastoma. Oncotarget 2018;9:25922-34. 10.18632/oncotarget.25406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Jiang J, Mo J, et al. Global DNA 5-Hydroxymethylcytosine and 5-Formylcytosine Contents Are Decreased in the Early Stage of Hepatocellular Carcinoma. Hepatology 2019;69:196-208. 10.1002/hep.30146 [DOI] [PubMed] [Google Scholar]
- 16.Nickerson ML, Das S, Im KM, et al. TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 2017;36:2172-83. 10.1038/onc.2016.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet 2014;30:464-74. 10.1016/j.tig.2014.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song SJ, Poliseno L, Song MS, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013;154:311-24. 10.1016/j.cell.2013.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Hu H, Wang Q, et al. The level and clinical significance of 5-hydroxymethylcytosine in oral squamous cell carcinoma: An immunohistochemical study in 95 patients. Pathol Res Pract 2017;213:969-74. 10.1016/j.prp.2017.04.016 [DOI] [PubMed] [Google Scholar]
- 20.Tucker DW, Getchell CR, McCarthy ET, et al. Epigenetic Reprogramming Strategies to Reverse Global Loss of 5-Hydroxymethylcytosine, a Prognostic Factor for Poor Survival in High-grade Serous Ovarian Cancer. Clin Cancer Res 2018;24:1389-401. 10.1158/1078-0432.CCR-17-1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shenoy N, Bhagat TD, Cheville J, et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J Clin Invest 2019;130:1612-25. 10.1172/JCI98747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu D, Hu D, Chen H, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018;559:637-41. 10.1038/s41586-018-0350-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Wang Y, Zhu Y, et al. The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol Oncol 2015;9:1091-105. 10.1016/j.molonc.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Zhu Y, Wang Q, et al. The histone demethylase LSD1 is a novel oncogene and therapeutic target in oral cancer. Cancer Lett 2016;374:12-21. 10.1016/j.canlet.2016.02.004 [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, Wang YL, Diao PF, et al. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics 2019;9:1777-93. 10.7150/thno.31581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol 2006;42:655-67. 10.1016/j.oraloncology.2005.10.013 [DOI] [PubMed] [Google Scholar]
- 27.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553-67. 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630-41. 10.1038/nrg.2016.93 [DOI] [PubMed] [Google Scholar]
- 29.Zhang TJ, Zhou JD, Yang DQ, et al. TET2 expression is a potential prognostic and predictive biomarker in cytogenetically normal acute myeloid leukemia. J Cell Physiol 2018;233:5838-46. 10.1002/jcp.26373 [DOI] [PubMed] [Google Scholar]
- 30.Jäwert F, Hasseus B, Kjeller G, et al. Loss of 5-hydroxymethylcytosine and TET2 in oral squamous cell carcinoma. Anticancer Res 2013;33:4325-8. [PubMed] [Google Scholar]
- 31.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009;114:144-7. 10.1182/blood-2009-03-210039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonvin E, Radaelli E, Bizet M, et al. TET2-Dependent Hydroxymethylome Plasticity Reduces Melanoma Initiation and Progression. Cancer Res 2019;79:482-94. 10.1158/0008-5472.CAN-18-1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong F, Guo Y, Niu Y, et al. Epigenetic silencing of TET2 and TET3 induces an EMT-like process in melanoma. Oncotarget 2017;8:315-28. [DOI] [PMC free article] [PubMed] [Google Scholar]