

Abstract
Maternal perturbations or sub-optimal conditions during fetal development can predispose the offspring to diseases in adult life. Animal and human studies show that prenatal androgen excess may be an underlying cause of polycystic ovary syndrome (PCOS) later in life. In women, PCOS is a common fertility disorder with comorbid metabolic dysfunction. Here, using a sheep model of PCOS phenotype, we elucidate the epigenetic changes induced by prenatal (30–90 day) testosterone (T) treatment and its effect on gene expression in fetal day 90 (D90) and adult year 2 (Y2) ovaries. RNA-seq study shows 65 and 99 differentially regulated genes in prenatal T-treated fetal and adult ovaries, respectively. Interestingly, there were no differences in gene inducing histone marks H3K27ac, H3K9ac, and H3K4me3 or in gene silencing marks, H3K27me3 and H3K9me3 in the fetal D90 ovaries of control and excess T-exposed fetuses. In contrast, except for H3K4me3 and H3K27me3, all the other histone marks were upregulated in the prenatal T-treated adult Y2 ovary. Chromatin immunoprecipitation (ChIP) studies in adult Y2 ovaries established a direct relationship between the epigenetic modifications with the upregulated and downregulated genes obtained from RNA-seq. Results show increased gene inducing marks, H3K27ac and H3K9ac, on the promoter region of upregulated genes while gene silencing mark, H3K9me3, was also significantly increased on the downregulated genes. This study provides a mechanistic insight into prenatal T-induced developmental programming and its effect on ovarian gene expression that may contribute to reproductive dysfunction and development of PCOS in adult life.
Keywords: epigenetics, PCOS, prenatal exposure, testosterone, histone modifications, gene expression, sheep ovary
A direct correlation between changes in epigenetic marks and gene expression in adult ovaries from prenatal T-treated sheep establishes epigenetic changes as one of the underlying causes for differential expression of genes in PCOS ovary.
Introduction
In women, polycystic ovary syndrome (PCOS) is a common endocrine disorder, affecting 9–18% reproductive aged women [1]. PCOS is characterized with hyperandrogenism, infertility and comorbid metabolic dysfunctions. Despite years of research, the underlying cause of PCOS is still unknown. During gestation, perturbation or sub-optimal conditions in the maternal environment are now recognized as the contributing factors to the onset of many diseases, manifesting in adulthood [2, 3].
It is well-established that adverse intrauterine condition can permanently reprogram the metabolism and growth of the fetus [4]. Various animal models (mice, rats, primates, and sheep), as well as epidemiological studies in humans now show that prenatal androgen excess can be an underlying cause for the pathophysiology of polycystic ovary syndrome (PCOS), involving oligo/anovulation, polycystic ovaries, hyperandrogenism, and insulin resistance [5–9]. While the concept of fetal programming has been widely accepted to be involved in the “Developmental Origins of Health and Disease” (DOHaD) paradigm, the underlying mechanism is poorly understood.
In this study, we have used a well-established sheep model of PCOS phenotype [9] to understand how prenatal exposure to excess testosterone (T) reprograms the fetus to develop PCOS characteristics later in life. Given that the sheep is a precocial (follicular differentiation occurs before birth) species like humans, it is considered as an excellent animal model to study developmental programing. Moreover, the reproductive and metabolic developmental trajectory of the sheep follows a similar developmental timeline as in humans, with hypothalamus, ovarian follicle formation, and pancreatic functional differentiation occurring during pregnancy [10]. Previous studies show that in the sheep, excess prenatal T exposure leads to neuroendocrine problems [11–13], ovarian disruptions, like abnormal ovarian cycle, anovulation, and multifollicular ovaries [10, 14], and metabolic deficits, like insulin resistance [15] and cardiovascular disease [16], which are very similar to fertility and metabolic disorders in women with PCOS [1, 17]. It is now well-established that prenatal exposure to excess T may cause PCOS and/or PCOS like phenotype later in life, but how this occurs is not known.
Epigenetic changes in response to external stimuli are considered by far the most likely mechanism by which the intrauterine environment affects health and disease of the offspring [18–20]. Early-life insults/adversity may result in stable changes to the epigenotype of germ and somatic cells, having long-term implications [18, 19, 21]. Epigenetic modifications can modulate chromatin compaction and thus gene accessibility through covalent modifications of DNA and histones. In DNA, modifications involve methylation, hydroxymethylation, formylation, and carboxylation of 5’ DNA cytosine residues in CpG sequences. Moreover, histones are subject to a myriad of modifications in their N-terminal tails and to a smaller degree in the C-terminal tails and globular domains, respectively [22]. Modifications of the histone proteins are established either by acetylation, methylation, phosphorylation, or ubiquitination and can be modified by different types of histone-modifying enzymes that can be broadly divided into three categories: writers, readers, and erasers. Epigenetic “writers”, such as histone methyltransferases (HMT), histone acetyltransferases (HAT), DNA methyltransferases (DNMT), and kinases add methyl or acetyl groups to lysine and arginine residues on histone tails and methyl groups to CpG sites on DNA, as well as phosphorylate-specific amino acid residues in histones. These modifications are interpreted by effector proteins called “readers”, like methyl-CpG-binding proteins (MBDs) and proteins containing histone methylation-binding or acetylation-binding domains that further reorganize the histones. Moreover, “erasers”, such as phosphatases, histone demethylases (HDMC), and deacetylases (HDAC) can remove the phosphorylation, methylation, and acetylation marks from DNA and histones, respectively [23, 24]. Essentially, all these epigenetic modifications regulate gene expression by altering the state of chromatin accessibility to transcriptional machinery and/or affecting the ability of transcriptional activators and repressors to bind to specific DNA promotors/regions. Collectively, epigenetic modulation of gene expression can result in a change in cellular phenotype without affecting the actual genotype.
Till date, while there have been numerous studies focused on DNA methylation profiling in understanding PCOS [25–27], studies on histone modifications and its contribution to the development of PCOS are limited. Here, using the sheep as a model, we investigate the level of specific histone modifications and their regulatory role in gene expression in fetal and adult ovaries isolated from prenatal T-treated sheep. This study provides a mechanistic insight into prenatal T-induced developmental programming and its manifestation in ovarian dysfunction and development of PCOS in adult life.
Materials and methods
Ethics statement
Animal care, treatment, and all animal procedures used in this study were performed by Institutional Animal Care and Use Committee of the University of Michigan.
Breeding and prenatal treatment
Details of prenatal treatments to generate prenatal T-treated groups have been described previously [28]. Briefly, prenatal T-treated sheep were generated by intramuscular administration of 100 mg of 1.2 mg/kg T propionate (Sigma-Aldrich Corp.) suspended in cottonseed oil to pregnant Suffolk sheep twice weekly from Days 30 to 90 of gestation. The dose of T was chosen based on earlier studies [29, 30] that have been shown to cause PCOS phenotype in adult life. Control breeders were injected with the vehicle for the same duration. Ovaries were collected on fetal Day 90 (D90) and adult year 2 (Y2). For collection of ovaries from fetuses, dams were euthanized by administration of a barbiturate overdose (Fatal Plus; Vortech Pharmaceuticals, Dearborn, MI), and fetuses were removed. As outcomes can be affected by stage of the estrous cycle, the adult year 2 (Y2) females were given two 20 mg injections of Prostaglandin F2α (PGF2α; 5 mg/mL Lutalyse; Pfizer Animal Health, New York, NY) 11 days apart to induce luteolysis and synchronize the initiation of the follicular phase in cycling females. Ewes were euthanized 28 h after the second PGF2α injection, and ovaries were collected during the follicular phase [31]. Fresh flash frozen tissues were briefly minced and sonicated in the lysis buffer (300–450 μL for 50 mg tissue) containing inhibitors to get the protein and DNA for Western blot and ChIP, respectively.
RNA isolation and RNA-seq
Total RNA was extracted from fresh frozen whole ovary samples using Qiagen RNeasy Plus Universal mini kit followed by the manufacturer’s instructions (Qiagen, Hilden, Germany). RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA), and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies, Palo Alto, CA, USA). RNA integrity number (RIN) for all the samples was between 7.3 and 9.2. RNA sequencing libraries were prepared using 500 ng RNA and the NEBNext Ultra RNA Library Prep Kit for Illumina using the manufacturer’s instructions (NEB, Ipswich, MA, USA). Briefly, mRNAs were initially enriched with Oligod (T) beads. Enriched mRNAs were fragmented for 15 min at 94 °C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end-repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing library was validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA) and quantified by using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). The sequencing libraries were clustered on a single lane of a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq instrument (4000 or equivalent) according to the manufacturer’s instructions. The samples were sequenced using a 2x150bp Paired End (PE) configuration. The total number of reads/sample was 45–50 million. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Bioinformatics analysis
After investigating the quality of the raw data (the quality was judged based on Illumina’s Q score, which represents the error rate at each base, built on a log10 score), sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the reference ovine genome available on ENSEMBL (Oaries_v3.1.90) using the STAR aligner v.2.5.2b. The STAR aligner uses a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. BAM files were generated as a result of this step. Unique gene hit counts were calculated by using feature Counts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted. After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis. Using DESeq2 R package, differentially expressed genes (DEGs) were identified between control (n = 3) and T treatment (n = 3) for fetal D90 and control (n = 3) and T (n = 4) for adult Y2, respectively. The heat maps were constructed using rlog-transformed values obtained from RNA-seq data followed by z-normalization for both fetal D90 and adult Y2 ovaries. The Wald test was used to generate P-values and Log2 fold changes. Genes with adjusted P-values < 0.05 and absolute log2 fold changes > 1 were called differentially expressed genes for each comparison (all differentially expressed genes for fetal D90 and adult Y2 are in supplemental figures) [32]. Identification of biological canonical pathways affected by the significant differentially expressed genes was performed with Ingenuity Pathway Analysis (Figure 3A–D) and Function Enrichment (Supplemental Figure S1) software. Gene ontology studies were done using DAVID to identify general biological processes affected by the significant differentially expressed genes (Figure 3E–H) in fetal D90 and adult Y2 ovaries.
Figure 3.
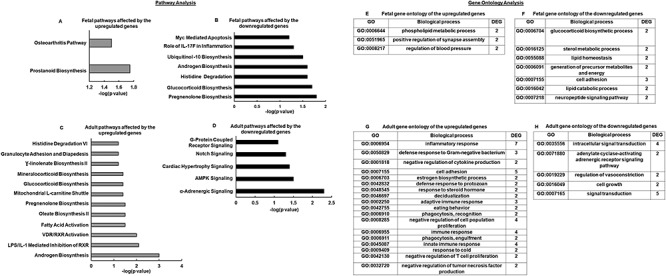
Bioinformatics analysis of the RNA-seq data. Genes differentially expressed were evaluated in silico using Ingenuity Pathway Analysis (IPA) showing upregulated and downregulated canonical pathways in fetal D90 (A, B) and adult Y2 (C, D). The bars represent the P-value at logarithmic scale. Gene ontology of differentially expressed genes analyzing biological process by David showing upregulated and downregulated canonical pathways for fetal D90 ovary (E, F) and adult Y2 ovary (G, H).
Western blot analysis
Using the whole ovary collected on fetal Day 90 (D90) and adult year 2 (Y2) at the end of second breeding season from the control and prenatal T experimental groups, western blot analysis was performed as described previously [33–35]. Primary antibodies (1:1000 dilution) used were: H3K27ac, H3K9ac, H3K4me3, H3, H3K27me3, and H3K9me3 (Supplemental Table S1). Western blot data were quantified by densitometric analysis as described previously [34]. Briefly, protein levels were determined using computer-aided densitometry and expressed as relative increase to H3 protein vs. control. Each experiment consisted of n = 3 ovaries from fetal D90 and adult Y2 animal per treatment group (control and T).
Chromatin immunoprecipitation (ChIP) assay
Using the whole ovary collected from the adult year 2 (Y2) at the end of second breeding season from the control and prenatal T experimental groups, ChIP was performed as described previously [33, 35–37] with MAGnify Chromatin Immunoprecipitation System (Invitrogen), according to the manufacturer’s instructions. Briefly, chromatin fragments were immunoprecipitated with Dynabeads coupled with ChIP grade rabbit polyclonal (Abcam) anti-H3K27ac, anti-H3K9ac, and anti-H3K9me3 (Supplemental Table S1), and quantitative PCR was performed using EXPRESS SYBR GreenER qPCR SuperMixes (Invitrogen). ChIP primers designed for different genes are shown in Supplemental Table S2. IgG was used as non-specific control. N = 3 ovaries from fetal D90 and adult Y2 animals per treatment group (control and T) were used for ChIP studies.
Statistical analysis
Data are presented as mean ± SEM. Statistical analysis was performed using GraphPad Prism version 7 (GraphPad Software Inc, California, USA). Statistical comparisons were made by two-tailed unpaired t-test (for comparing two groups), one-way ANOVA followed by Tukey’s post hoc analysis (for comparing multiple groups), or two-way ANOVA followed by Tukey’s post-hoc analysis (for comparing multiple groups and variables), and results with P ≤ 0.05 were considered significant.
Results
Prenatal T treatment causes differential gene expression in fetal and adult ovaries
According to the RNA-seq data, all the quality control parameters were within the acceptable ranges. In fetal D90 ovary, a total of 50 annotated differentially expressed ENSEMBL genes were detected in T vs. control ovaries. Out of these genes, 31 were upregulated and 19 were downregulated genes. Intriguingly, 15 genes were novel genes that were unannotated out of which 10 were upregulated and 5 were downregulated genes. Figure 1A shows the heat map of all the significant differentially expressed genes in the fetal ovary, while Figure 1B and C shows the top 5 upregulated and downregulated genes, respectively. In the adult Y2 ovary, comparing prenatal T vs. control, a total of 74 annotated differentially expressed ENSEMBL genes were detected, out of which 44 were upregulated and 30 were downregulated. There were 25 unannotated novel genes in the Y2 ovaries, out of which 15 were upregulated and 10 were downregulated genes. Figure 2A shows heat map of all the significant differentially expressed genes in the adult ovary, while Figure 2B and C shows the top 5 upregulated and downregulated genes, respectively. The complete list of significant differentially expressed genes from fetal D90 and adult Y2 is shown in Supplementary data S1 and S2, respectively.
Figure 1.
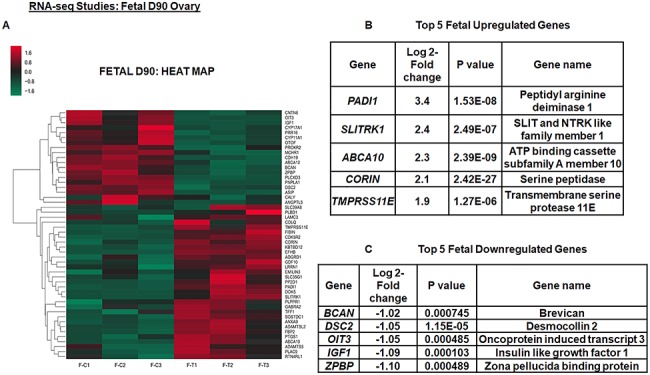
Prenatal T exposure causes differential gene expression in fetal day 90 (D90) ovaries. Top 5 upregulated and downregulated genes for fetal D90 ovary within differentially expressed genes and its corresponding heat map (A–C). The heat map is constructed using rlog-transformed values obtained from RNA-seq data followed by z-normalization for fetal D90 ovaries. The Wald test was used to generate P-values and Log2 fold changes. Genes with adjusted P-values < 0.05 and absolute log2 fold changes > 1 were used as the threshold and called differentially expressed genes for each comparison
Figure 2.
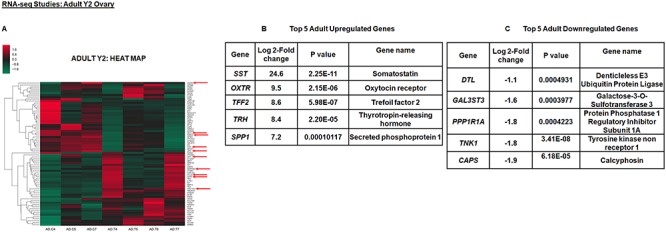
Prenatal T exposure causes differential gene expression in adult year 2 (Y2) ovaries. The top 5 upregulated and downregulated genes for adult Y2 ovary within differentially expressed genes and its corresponding heat map (A–C). The heat map is constructed using rlog-transformed values obtained from RNA-seq data followed by z-normalization for adult Y2 ovaries. The Wald test was used to generate P-values and Log2 fold changes. Genes with adjusted P-values < 0.05 and absolute log2 fold changes > 1 were used as the threshold and called differentially expressed genes for each comparison. Genes chosen for ChIP studies are shown by arrows in the heat map.
Identification of biological canonical pathways affected by the significant differentially expressed genes was performed with Ingenuity Pathway Analysis (Figure 3A–D) and Function Enrichment (Supplemental Figure S1) software. Gene ontology studies were done using DAVID to identify general biological processes affected by the significant differentially expressed genes (Figure 3E–H) in fetal D90 and adult Y2 ovaries. The Ingenuity Pathway Analysis results are presented as negative logarithm of the significance level and shows that downregulated genes in fetal D90 ovary affect pregnenolone, glucocorticoid, and androgen biosynthesis pathways (Figure 3B). In contrast, in the adult Y2 ovary, it is the upregulated genes that affect the same above pathways (Figure 3C). Similarly, functional enrichment analysis in fetal D90 ovary revealed that downregulated genes enriched ovarian steroidogenesis, glucocorticoid, and mineralocorticoid metabolism (FDR ≤ 0.05), while in adult Y2 ovary, it was the upregulated genes that enriched the same pathways (FDR > 0.05) (Supplemental Figure S1). Like Ingenuity Pathway Analysis and Function Enrichment analyses, gene ontology also shows that downregulated genes in fetal D90 ovaries primarily affected the steroid metabolism. Additionally, in the prenatal T-treated adult ovaries, gene ontology analysis indicates inflammatory-immune response and steroid metabolism to be the most affected process by the upregulated genes, while signal transduction the primary process to be affected by downregulated genes.
Prenatal T treatment modifies histone marks in the adult sheep ovary
Given the importance of histone modifications in regulation of gene expression, we checked whether prenatal T treatment modulates key histone marks in the fetal and/or in the adult ovary. For our studies, we choose H3K27me3 and H3K9me3, which are gene repressive marks and H3K27ac, H3K9ac, and H3K4me3, which are gene inducing marks. Results show that prenatal T treatment did not have any effect on both gene repressive (H3K27me3 and H3K9me3) and inducing (H3K27ac, H3K9ac, and H3K4me3) marks compared to control (Figure 4). In contrast, in adult ovaries isolated in Y2, while there was no significant difference in H3K27me3 mark (Figure 5A upper panel and B), ovaries isolated from prenatal T-treated animals had significantly more H3K9me3 (Figure 5A middle panel and C) mark compared to ovaries isolated from control animals. With respect to gene inducing histone marks, H3K27ac (Figure 5D and E) and H3K9ac (Figure 5D and F) were significantly higher in prenatal T-treated ovaries. There was no change in H3K4me3 levels between prenatal T-treated and control animals (Figure 5D and G).
Figure 4.
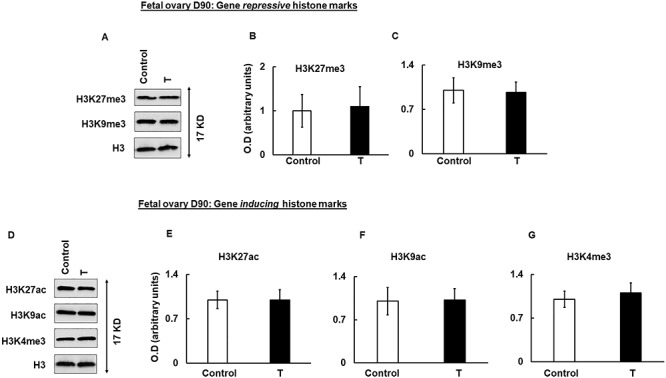
Prenatal T-induced changes in histone marks in fetal day 90 (D90) ovaries. Representative immunoblots demonstrating the level of gene-repressive (H3K27me3 and H3K9me3) (A) and gene-inducing (H3K27ac, H3K9ac, and H3K4me3) histone marks in fetal day 90 (D90) ovaries isolated from control and prenatal T-treated sheep. Densitometric analysis of the gene repressive (B, C) and gene inducing (E–G) immuno blots. Each experiment consisted of n = 3 ovaries from fetal D90 animal per treatment group (control and T). Data are displayed as mean ± SE and normalized to H3.
Figure 5.
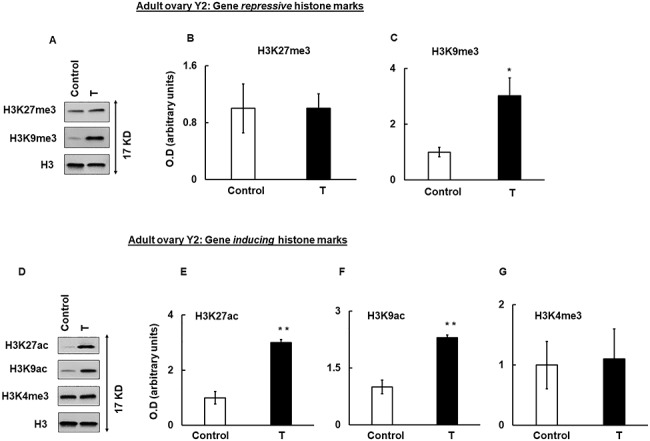
Prenatal T-induced changes in histone marks in adult year 2 (Y2) ovaries. Representative immunoblots demonstrating gene-repressive (H3K27me3 and H3K9me3) (A) and gene-inducing (H3K27ac, H3K9ac, and H3K4me3) histone marks (D) in adult year 2 (Y2) ovaries isolated from control and prenatal T-treated sheep. Densitometric analysis of the gene repressive (B, C) and gene-inducing (E–G) immunoblots. Each experiment consisted of n = 3 ovaries from adult Y2 animal per treatment group (control and T)]. Data are displayed as mean ± SE and normalized to H3. *P ≤ 0.05 vs. control and **P ≤ 0.01 vs. control.
Prenatal T-treatment causes post-translational histone modifications in the promoter of ovarian genes
Next, we determined if changes in the level of epigenetic marks in the ovaries of prenatal T-treated Y2 adult sheep had any effect on gene expression. To correlate the epigenetic modifications with the upregulated and downregulated genes obtained from RNA-seq, we performed ChIP analysis to study post-translational modifications occurring specifically on the ovarian gene promoter. Specific upregulated genes from RNA-seq analysis were selected based on their known ovarian expression/function. For example, SERPINA5 has been shown to be expressed in healthy bovine ovarian follicle [38], CTLA4 is suggested to influence PCOS through regulating obesity [39], HSD3B1 and HSD17B14 [40] genetic variants are responsible in steroid synthesis in ovary, SPP1 regulates cell function in the bovine corpus luteum (CL) [41], and OXTR functions as receptor to the neurotransmitter, oxytocin which is synthesized and secreted in the corpus luteum of the ruminant [42]. Similarly, downregulated genes from RNA-seq analysis were selected based on their ovarian expression/function. TNK1 is a novel tyrosine kinase isolated from human umbilical cord blood found in ovary [43], LIPG is an endothelial lipase (EL) that plays a role in murine reproduction and its expression is high in corpora lutea [44], HEY2 is the target gene for the Notch signaling that regulates ovarian follicle formation and coordinates follicular growth [45], and NDRG4 plays a role in uterine development that occurs postnatally in the ovary of many species [46]. Figure 6 shows ChIP analysis on the promoter region of selected up- and downregulated ovarian genes with anti-H3K9ac (Figure 6A), H3K27ac (Figure 6B), and H3K9me3 (Figure 6C) antibodies in adult Y2 ovaries. Results show that for all the upregulated genes tested, H3K9ac and H3K27ac (gene inducing) marks were significantly higher in the promoter region of these genes in adult Y2 ovaries isolated from prenatal T-treated animals compared to controls. Similarly, for all the downregulated genes tested, the H3K9me3 (gene repressive) mark was significantly more in the promoter region of the selected genes in ovaraies of prenatal T-treated vs. controls (Figure 6C). Epigenetic regulation of gene expression can occur by both increasing and/or decreasing positive (H3K27ac, H3K9ac, and H3K4me3) and/or negative (H3K27me3 and H3K9me3) epigenetic marks. Thus, we also determined H3K9me3 on upregulated genes (Figure 6D) and level of H3K9ac (Figure 6E) and H3K27ac (Figure 6F) on the downregulated genes, respectively. Intriguingly, results show that prenatal T treatment not only increases H3K9ac and H3K27ac gene inducing marks, but also decreases H3K9me3 repressive mark on some of the upregulated genes (Figure 6D). Out of all the upregulated genes tested, only SERPINA5 showed no change in H3K9me3 levels compared to control. In contrast, for the downregulated genes, only TNK1 had lower H3K9ac and NDRG4 had both H3K9ac mark and H3K27ac mark lower in the prenatal T-treated animals compared to control (Figure 6E and F). None of the other genes tested had any change in the H3K9ac and H3K27ac levels (Figure 6E and F). This shows that the epigenetic modulation caused by prenatal T tretment is highly gene-specific. For example, the induction of some genes is caused by modulating both gene expression positive (H3K27ac and H3K9ac) and negative (H3K9me3) marks, while for other upregulated genes, only gene inducing mark (H3K27ac and H3K9ac) is increased. Similarly, for downregulated genes, while some of the genes are epigenetically modulated by increasing gene repressive marks (H3K9me3) as well as lowering gene inducing marks (H3K27ac and H3K9ac), other genes have only increased gene repressive marks.
Figure 6.
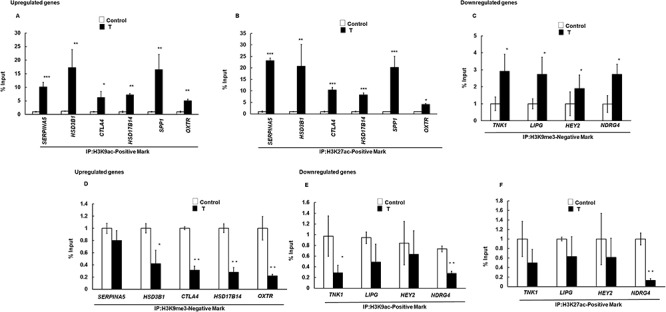
Prenatal T exposure changes the level of gene-inducing and gene-repressive histone marks on differentially expressed genes in adult year 2 (Y2) ovaries. Epigenetic marks were determined by ChIP assay in the whole ovary isolated from prenatal T-treated adult Y2. A, B: Levels of H3K9ac and H3K27ac positive mark on the promoter region of upregulated ovarian genes. C: Level of H3K9me3 negative mark on the promoter region of downregulated ovarian genes. D: Level of H3K9me3 on the promoter region of upregulated ovarian genes. E, F: Levels of H3K9ac and H3K27ac positive mark on the promoter region of downregulated ovarian genes. Values represent percentage input (mean ± SE, each experiment consisted of n = 3 ovaries from adult Y2 animal per treatment group (control and T). *P ≤ 0.05 vs. control, **P ≤ 0.01 vs. control and ***P ≤ 0.005. IP-immunoprecipitation.
Discussion
This study provides an insight into prenatal T-induced epigenetic changes in the ovary and its effect on gene expression. Previous studies [10, 14, 31] have clearly established that prenatal T treatment in the sheep changes ovarian morphology and causes PCOS phenotype in adult life. This is the first RNA-seq study in the prenatal T-treated sheep model to determine global changes in ovarian gene expression both in fetal life and adult life. Moreover, our studies demonstrate a direct relationship between changes in epigenetic marks and gene expression.
Our studies show that genes primarily in steroid biosynthesis, inflammation, and regulatory/cell signaling categories were affected by prenatal T treatment. For example, hyperandrogenism is a key characteristic feature of PCOS condition. Notably, HSD3B1, HSD17B14, CYP11A1, and CYP17A1 genes that we find to be differentially regulated in T vs. control fetal and adult ovaries are known to be involved in androgen biosynthesis [40]. Previous studies in prostate cancer have shown that androgens can upregulate HSD3B1 [47], while studies in sheep have reported that prenatal androgen exposure leads to downregulation of CYP11A1 [48, 49] and CYP17A1 [49, 50]. Similar to these studies, here we also find that HSD3B1and HSD17B14 are upregulated in the prenatal T adult ovaries, while CYP11A1 and CYP17A1 are downregulated in the excess T-exposed fetal D90 ovaries compared to control. This suggests that while excess T exposure to fetuses initially may lower androgen production, in adult stage, the androgen biosynthetic pathway gets upregulated in ovaries that were exposed to excess prenatal T. This might explain the increased expression of AR reported in prenatal T adult ovaries [51], since androgens in a positive feedback loop increases androgen receptor (AR) expression [52, 53]. Moreover, in our study, pathway and function enrichment analyses also show that the androgen biosynthesis pathway is the most significantly affected pathway in the prenatal T-exposed fetus and adult ovaries. With respect to inflammation, gene ontology studies show that about 22 upregulated genes in prenatal T adult Y2 ovary are involved in the inflammation/immune response. Inflammation has been shown to be extensively associated with PCOS and hypothesized to be one of the underlying causes of PCOS [54]. Intriguingly, previous studies [55] in sheep have reported that prenatal T can promote low grade chronic inflammation in placenta. Interestingly, our studies (Figure 3B) find that the IL-17F-induced inflammatory pathway is affected in fetal ovary. Moreover, this prenatal T-induced chronic inflammation seen in the fetal ovary persists in the adult ovary. Therefore, it is possible that prenatal T induced chronic inflammation in the ovary can be a driving force of PCOS development and progression in adult life. In case of cell signaling, we find that HEY2 gene, involved in Notch Signaling is downregulated in prenatal T adult Y2 ovary (RNA-seq). Notch Signaling plays an important role in follicular growth [45] and has been shown to be associated with PCOS [56]. Another signaling protein that is downregulated in prenatal T-treated Y2 ovary is diacylglycerol kinase (DGKG). DGKG is important in insulin signaling, lipid metabolism, and a regulator of diacylglycerol and phosphatidic acid, which are important mediators of signal transduction. Pathway and function enrichment analyses demonstrate, in general, that signal transduction pathways and specifically the inositol pathway are affected in the prenatal T-treated ovaries. Previous studies have also reported that key proteins in the insulin signaling pathway are downregulated in the adult prenatal T-treated ovaries [57]. Additionally, GPCR signaling is affected by the downregulated genes in the prenatal T-treated adult ovary. One of the main genes associated with this pathway that is downregulated in our RNA-seq data is adrenergic receptor (ADRA1A and 2A). Adrenergic receptors are known to play an important role in follicular development and normal ovarian function [58]. Thus, our studies suggest that prenatal T treatment modulates the expression of ovarian genes involved in key biological processes that have been implicated to be either an underlying cause and/or progression of PCOS.
Notably, all previous studies in the ovary of prenatal T-treated sheep model reported differentially expressed protein expression by immunohistochemical studies in specific cells using ovarian sections [51, 57, 59]. Other than the expressions of HSD3B1 and HSD17B that are upregulated in adult ovaries and CYP11A1 and CYP17A1 that are downregulated in fetal ovaries, we did not find any differentially regulated genes that were common with the proteins reported in previous studies in the prenatal T-treated fetal and adult ovaries. This may be due to the fact that our RNA-seq study was done with total RNA isolated from fetal and adult whole ovaries, which may have masked any significant cell-specific changes in the expression of these genes. Whole ovaries from fetal and adult sheep enabled us to perform RNA-seq and ChIP studies as cell number is a major limiting factor for these studies. Moreover, it is well established [60] that in addition to the genomic effects of androgens, the later can also increase protein levels in a transcription-independent non-genomic fashion [37].
Intriguingly, our RNA-seq data identified several significant differentially expressed unannotated (novel) genes. Furthermore, some of the annotated genes like PADI1 (peptidyl arginine deiminase, type I,), SLITRK1 (SLIT and NTRK like family member 1), CORIN (corin, serine peptidase), TMPRSS11E (transmembrane serine protease 11E), EFHB (EF-hand domain family member B) and TFF2 (trefoil factor 2), MMP12 (matrix metallopeptidase 12), SERPINA5 (serpin family A member 5), BPI (bactericidal permeability increasing protein), and TREM2 (triggering receptor expressed on myeloid cells 2) that were significantly differentially expressed in the prenatal T-treated fetal and adult ovaries have not been studied in the ovary with respect to PCOS. Thus, further studies are needed to not only identify the novel genes but also determine the role of these unannotated and annotated genes in the ovary that might play a crucial role in the development of PCOS.
Previously, we have shown that androgens may influence gene expression through modifying epigenetic marks [33]. We and others [18, 20, 61] have also shown that adverse intra-uterine environment may cause epigenetic changes in the fetus that persist in adult life contributing to reprograming of gene expression. However, surprisingly in this study we did not find any prenatal T-induced epigenetic changes in the D90 fetal ovary. This may be because prenatal T might not have an immediate effect in the fetal ovary (at D90 when treatment was stopped), but epigenetic changes may occur at latter gestational time points. However, our results clearly show that when the fetus develops into adult, several histone marks appeared to be modified in the prenatal T-treated ovary. Further studies are needed to identify the key developmental window where changes in epigenetic marks become more pronounced. Another question that needs to be answered is how these epigenetic changes are occurring and whether this is a direct effect of prenatal T treatment or is a secondary effect of the prenatal T (hyperandrogenism) that is seen in the adult stage. Importantly, this study provides direct evidence towards establishing epigenetic changes as one of the underlying causes for differential expression of genes in PCOS ovary. In future, ChIP-seq studies are required to determine the global effect of these epigenetic changes and identify the genes regulated though epigenetic modulation in prenatal T-treated ovaries.
Laser capture micro-dissection and immunofluorescence in prenatal T-treated adult Y2 sheep ovary show [61] ovarian cell-specific upregulation of H3K9me3 and H3K4me2 marks and differential expression of specific histone and DNA epigenetic modifying enzymes at the mRNA level. Using the whole ovary, the current study also finds increase in H3K9me3 (gene repressive) mark in adult Y2 prenatal T-treated ovaries. We also see increased H3K27ac and H3K9ac (gene activating) marks but no change in H3K27me3 (gene repressive) and H3K4me3 (gene activating) marks. Interestingly, no change in the mRNA expression of p300, the enzyme responsible for acetylation of H3 at K27, and increased mRNA expression of EZH2 and SYMD3, the enzymes responsible for trimethylation of K27 and K4 on H3, respectively, has also been reported [61] in prenatal T-treated adult Y2 sheep ovary. Our RNA-seq data do not show any changes in the expression levels of the histone modifying enzymes. Moreover, we have done western blot analysis (data not shown) of EZH2, JMJD3, JMJD2B, and p300 and we did not see any change in the levels of these enzymes in adult year 2 (Y2) ovaries isolated from control and prenatal T-treated sheep. Changes in mRNA or protein levels may not be enough to explain the downstream effects of these enzymes as the activity of these enzymes may be regulated at a post-translational level. For example, phosphorylation of EZH2 at serine 21 [62] is known to block the methyltransferase activity of EZH2 by disrupting its association with histone proteins, and we have shown [33] that androgens through the PI3K/Akt pathway can phosphorylate EZH2 at serine 21. Therefore, future studies should focus towards correlating the expression and activity of epigenetic enzymes with its specific downstream epigenetic marks with respect to prenatal T treatment in the ovary. Intriguingly, in human PCOS patients, global increase in H3K9ac mark and a decrease of H3K9me2 mark were reported in cumulus cells of PCOS patients compared to control groups [63]. Our study also shows increase of H3K9ac mark which can be an important mark that can be targeted for future studies.
In summary, while it is well-established that prenatal T treatment may lead to PCOS phenotype in adult life, this study establishes prenatal T-induced epigenetic modulations as one of the underlying causes for manifestation of PCOS. Here we provide direct evidence that changes in epigenetic marks lead to differential expression of genes in adult ovaries from prenatal T-treated sheep. Future studies are needed to understand how prenatal T modulates epigenetic enzymes, which changes specific epigenetic marks and its effect on downstream gene expression and ovarian physiology.
Supplementary Material
Acknowledgments
We acknowledge the assistance of Douglas Doop and Gary McCalla for animal care and Dr. Almudena Veiga-Lopez, Dr. Teresa Stecker, Mr. James Lee, Ms. Carol Herkimer, and students in the University of Michigan Undergraduate Research Opportunity Program for assistance with administration of treatments and tissue collection.
Footnotes
† Grant Support: This work was supported by NIH R01HD086062-03 grant, USDA-HATCH project MICL02383, Michigan State University-General Funds and MSU AgBioResearch to AS, R01GM131398 to JW and HD PO1HD44232 to VP.
References
- 1. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, Lizneva D, Natterson-Horowtiz B, Teede HJ, Yildiz BO. Polycystic ovary syndrome. Nat Rev Dis Primers 2016; 2:16057. [DOI] [PubMed] [Google Scholar]
- 2. Barker DJ. In utero programming of chronic disease. Clin Sci (Lond) 1998; 95:115–128. [PubMed] [Google Scholar]
- 3. Nijland MJ, Ford SP, Nathanielsz PW. Prenatal origins of adult disease. Curr Opin Obstet Gynecol 2008; 20:132–138. [DOI] [PubMed] [Google Scholar]
- 4. Barker D. Mothers, Babies, and Disease in Later Life. London: United Kingdom: Programming the baby: BMJ Publishing Group; 1994: 14–36. [Google Scholar]
- 5. Abbott DH, Levine JE, Dumesic DA. Translational insight into polycystic ovary syndrome (PCOS) from female monkeys with PCOS-like traits. Curr Pharm Des 2016; 22:5625–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walters KA, Bertoldo MJ, Handelsman DJ. Evidence from animal models on the pathogenesis of PCOS. Best Pract Res Clin Endocrinol Metab 2018; 32:271–281. [DOI] [PubMed] [Google Scholar]
- 7. Huang-Doran I, Franks S. Genetic rodent models of obesity-associated ovarian dysfunction and subfertility: insights into polycystic ovary syndrome. Front Endocrinol (Lausanne) 2016; 7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Houten EL, Visser JA. Mouse models to study polycystic ovary syndrome: a possible link between metabolism and ovarian function? Reprod Biol 2014; 14:32–43. [DOI] [PubMed] [Google Scholar]
- 9. Cardoso RC, Padmanabhan V. Developmental programming of PCOS traits: insights from the sheep. Med Sci (Basel) 2019; 7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol 2013; 373:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foster DL, Jackson LM, Padmanabhan V. Programming of GnRH feedback controls timing puberty and adult reproductive activity. Mol Cell Endocrinol 2006; 254-255:109–119. [DOI] [PubMed] [Google Scholar]
- 12. Cardoso RC, Puttabyatappa M, Padmanabhan V. Steroidogenic versus metabolic programming of reproductive neuroendocrine, ovarian and metabolic dysfunctions. Neuroendocrinology 2015; 102:226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson JE, Birch RA, Foster DL, Padmanabhan V. Prenatal exposure of the ovine fetus to androgens sexually differentiates the steroid feedback mechanisms that control gonadotropin releasing hormone secretion and disrupts ovarian cycles. Arch Sex Behav 2002; 31:35–41. [DOI] [PubMed] [Google Scholar]
- 14. Puttabyatappa M, Padmanabhan V. Developmental programming of ovarian functions and dysfunctions. Vitam Horm 2018; 107:377–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puttabyatappa M, Padmanabhan V. Prenatal testosterone programming of insulin resistance in the female sheep. Adv Exp Med Biol 2017; 1043:575–596. [DOI] [PubMed] [Google Scholar]
- 16. King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab 2007; 292:E1837–E1841. [DOI] [PubMed] [Google Scholar]
- 17. Pasquali R, Stener-Victorin E, Yildiz BO, Duleba AJ, Hoeger K, Mason H, Homburg R, Hickey T, Franks S, Tapanainen JS, Balen A, Abbott DH et al. PCOS forum: research in polycystic ovary syndrome today and tomorrow. Clin Endocrinol 2011; 74:424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sinha N, Biswas A, Nave O, Seger C, Sen A. Gestational diabetes epigenetically reprograms the cart promoter in fetal ovary, causing subfertility in adult life. Endocrinology 2019; 160:1684–1700. [DOI] [PubMed] [Google Scholar]
- 19. Xu N, Kwon S, Abbott DH, Geller DH, Dumesic DA, Azziz R, Guo X, Goodarzi MO. Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS One 2011; 6:e27286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 2007; 104:13056–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abbott DH, Dumesic DA, Franks S. Developmental origin of polycystic ovary syndrome-a hypothesis. J Endocrinol 2002; 174:1–5. [DOI] [PubMed] [Google Scholar]
- 22. Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693–705. [DOI] [PubMed] [Google Scholar]
- 23. Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41–45. [DOI] [PubMed] [Google Scholar]
- 24. Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 25. Lambertini L, Saul SR, Copperman AB, Hammerstad SS, Yi Z, Zhang W, Tomer Y, Kase N. Intrauterine reprogramming of the polycystic ovary syndrome: Evidence from a pilot study of cord blood global methylation analysis. Front Endocrinol (Lausanne) 2017; 8:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sagvekar P, Kumar P, Mangoli V, Desai S, Mukherjee S. DNA methylome profiling of granulosa cells reveals altered methylation in genes regulating vital ovarian functions in polycystic ovary syndrome. Clin Epigenetics 2019; 11:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. VA-m ER, Gomez-Viais YI, Garcia-Gomez E, Reyes-Mayoral C, Reyes-Munoz E, Camacho-Arroyo I, Cerbon MA. DNA methylation in the pathogenesis of polycystic ovary syndrome. Reproduction 2019; 155:R27–R40. [DOI] [PubMed] [Google Scholar]
- 28. Manikkam M, Crespi EJ, Doop DD, Herkimer C, Lee JS, Yu S, Brown MB, Foster DL, Padmanabhan V. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology 2004; 145:790–798. [DOI] [PubMed] [Google Scholar]
- 29. Sarma HN, Manikkam M, Herkimer C, Dell'Orco J, Welch KB, Foster DL, Padmanabhan V. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology 2005; 146:4281–4291. [DOI] [PubMed] [Google Scholar]
- 30. Veiga-Lopez A, Ye W, Phillips DJ, Herkimer C, Knight PG, Padmanabhan V. Developmental programming: deficits in reproductive hormone dynamics and ovulatory outcomes in prenatal, testosterone-treated sheep. Biol Reprod 2008; 78:636–647. [DOI] [PubMed] [Google Scholar]
- 31. Smith P, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programming: differential effects of prenatal testosterone and dihydrotestosterone on follicular recruitment, depletion of follicular reserve, and ovarian morphology in sheep. Biol Reprod 2009; 80:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma X, Hayes E, Biswas A, Seger C, Prizant H, Hammes SR, Sen A. Androgens regulate ovarian gene expression through modulation of Ezh2 expression and activity. Endocrinology 2017; 158:2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma X, Hayes E, Prizant H, Srivastava RK, Hammes SR, Sen A, Leptin-Induced CART. Cocaine- and amphetamine-regulated transcript is a novel intraovarian mediator of obesity-related infertility in females. Endocrinology 2016; 157:1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roy S, Gandra D, Seger C, Biswas A, Kushnir VA, Gleicher N, Kumar TR, Sen A. Oocyte-derived factors (GDF9 and BMP15) and FSH regulate AMH expression via modulation of H3K27AC in granulosa cells. Endocrinology 2018; 159:3433–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sen A, De Castro I, Defranco DB, Deng FM, Melamed J, Kapur P, Raj GV, Rossi R, Hammes SR. Paxillin mediates extranuclear and intranuclear signaling in prostate cancer proliferation. J Clin Invest 2012; 122:2469–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sen A, Prizant H, Light A, Biswas A, Hayes E, Lee HJ, Barad D, Gleicher N, Hammes SR. Androgens regulate ovarian follicular development by increasing follicle stimulating hormone receptor and microRNA-125b expression. Proc Natl Acad Sci U S A 2014; 111:3008–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayashi KG, Ushizawa K, Hosoe M, Takahashi T. Differential gene expression of serine protease inhibitors in bovine ovarian follicle: possible involvement in follicular growth and atresia. Reprod Biol Endocrinol 2011; 9:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Su J, Li Y, Su G, Wang J, Qiu T, Ma R, Zhao L. Genetic association of CTLA4 gene with polycystic ovary syndrome in the Chinese Han population. Medicine (Baltimore) 2018; 97:e11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quirke LD, Juengel JL, Tisdall DJ, Lun S, Heath DA, McNatty KP. Ontogeny of steroidogenesis in the fetal sheep gonad. Biol Reprod 2001; 65:216–228. [DOI] [PubMed] [Google Scholar]
- 41. Poole DH, Ndiaye K, Pate JL. Expression and regulation of secreted phosphoprotein 1 in the bovine corpus luteum and effects on T lymphocyte chemotaxis. Reproduction 2013; 146:527–537. [DOI] [PubMed] [Google Scholar]
- 42. McArdle CA, Holtorf AP. Oxytocin and progesterone release from bovine corpus luteal cells in culture: Effects of insulin-like growth factor I, insulin, and prostaglandins. Endocrinology 1989; 124:1278–1286. [DOI] [PubMed] [Google Scholar]
- 43. Hoehn GT, Stokland T, Amin S, Ramirez M, Hawkins AL, Griffin CA, Small D, Civin CI. Tnk1: a novel intracellular tyrosine kinase gene isolated from human umbilical cord blood CD34+/Lin-/CD38- stem/progenitor cells. Oncogene 1996; 12:903–913. [PubMed] [Google Scholar]
- 44. Lindegaard ML, Nielsen JE, Hannibal J, Nielsen LB. Expression of the endothelial lipase gene in murine embryos and reproductive organs. J Lipid Res 2005; 46:439–444. [DOI] [PubMed] [Google Scholar]
- 45. Vanorny DA, Prasasya RD, Chalpe AJ, Kilen SM, Mayo KE. Notch signaling regulates ovarian follicle formation and coordinates follicular growth. Mol Endocrinol 2014; 28:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Q, Gu Y, Zhang X, Wang JM, He YP, Shi Y, Sun ZG, Shi HJ, Wang J. Uterine expression of NDRG4 is induced by estrogen and up-regulated during embryo implantation process in mice. PLoS One 2016; 11:e0155491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hettel D, Zhang A, Alyamani M, Berk M, Sharifi NAR. Signaling in prostate cancer regulates a feed-forward mechanism of androgen synthesis by way of HSD3B1 upregulation. Endocrinology 2018; 159:2884–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luense LJ, Veiga-Lopez A, Padmanabhan V, Christenson LK. Developmental programming: gestational testosterone treatment alters fetal ovarian gene expression. Endocrinology 2011; 152:4974–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hogg K, McNeilly AS, Duncan WC. Prenatal androgen exposure leads to alterations in gene and protein expression in the ovine fetal ovary. Endocrinology 2011; 152:2048–2059. [DOI] [PubMed] [Google Scholar]
- 50. Padmanabhan V, Salvetti NR, Matiller V, Ortega HH. Developmental programming: prenatal steroid excess disrupts key members of intraovarian steroidogenic pathway in sheep. Endocrinology 2014; 155:3649–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ortega HH, Salvetti NR, Padmanabhan V. Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction 2009; 137:865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eriksen MB, Glintborg D, Nielsen MF, Jakobsen MA, Brusgaard K, Tan Q, Gaster M. Testosterone treatment increases androgen receptor and aromatase gene expression in myotubes from patients with PCOS and controls, but does not induce insulin resistance. Biochem Biophys Res Commun 2014; 451:622–626. [DOI] [PubMed] [Google Scholar]
- 53. Weil SJ, Vendola K, Zhou J, Adesanya OO, Wang J, Okafor J, Bondy CA. Androgen receptor gene expression in the primate ovary: cellular localization, regulation, and functional correlations. J Clin Endocrinol Metab 1998; 83:2479–2485. [DOI] [PubMed] [Google Scholar]
- 54. Gonzalez F. Inflammation in polycystic ovary syndrome: underpinning of insulin resistance and ovarian dysfunction. Steroids 2012; 77:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelley AS, Puttabyatappa M, Ciarelli J, Zeng L, Smith YR, Lieberman R, Subramaniam P, Padmanabhan V. Prenatal testosterone excess disrupts placental function in a sheep model of polycystic ovary syndrome. Endocrinology 2019; 160:2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu B, Zhang YW, Tong XH, Liu YS. Characterization of microRNA profile in human cumulus granulosa cells: identification of microRNAs that regulate notch signaling and are associated with PCOS. Mol Cell Endocrinol 2015; 404:26–36. [DOI] [PubMed] [Google Scholar]
- 57. Ortega HH, Rey F, Velazquez MM, Padmanabhan V. Developmental programming: effect of prenatal steroid excess on intraovarian components of insulin signaling pathway and related proteins in sheep. Biol Reprod 2010; 82:1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Merz C, Saller S, Kunz L, Xu J, Yeoman RR, Ting AY, Lawson MS, Stouffer RL, Hennebold JD, Pau F, Dissen GA, Ojeda SR et al. Expression of the beta-2 adrenergic receptor (ADRB-2) in human and monkey ovarian follicles: a marker of growing follicles? J Ovarian Res 2015; 8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ortega HH, Veiga-Lopez A, Sreedharan S, del Lujan Velazquez MM, Salvetti NR, Padmanabhan V. Developmental programming: does prenatal steroid excess disrupt the ovarian VEGF system in sheep? Biol Reprod 2015; 93:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol 2016; 17:783–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guo X, Puttabyatappa M, Thompson RC, Padmanabhan V. Developmental programming: contribution of epigenetic enzymes to antral follicular defects in the sheep model of PCOS. Endocrinology 2019; 160:2471–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005; 310:306–310. [DOI] [PubMed] [Google Scholar]
- 63. Hosseini E, Shahhoseini M, Afsharian P, Karimian L, Ashrafi M, Mehraein F, Afatoonian R. Role of epigenetic modifications in the aberrant CYP19A1 gene expression in polycystic ovary syndrome. Arch Med Sci 2019; 15:887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.