

Abstract
Cystobactamids belong to the group of arene‐based oligoamides that effectively inhibit bacterial type IIa topoisomerases. Cystobactamid 861‐2 is the most active member of these antibiotics. Most amide bonds present in the cystobactamids link benzoic acids with anilines and it was found that some of these amide bonds undergo chemical and enzymatic hydrolysis, especially the one linking ring C with ring D. This work reports on the chemical synthesis and biological evaluation of thirteen new cystobactamids that still contain the methoxyaspartate hinge. However, we exchanged selected amide bonds either by the urea or the triazole groups and modified ring A in the latter case. While hydrolytic stability could be improved with these structural substitutes, the high antibacterial potency of cystobactamid 861‐2 could only be preserved in selected cases. This includes derivatives, in which the urea group is positioned between rings A and B and where the triazole is found between rings C and D.
Keywords: amides, antibiotics, chemical synthesis, medicinal chemistry, triazoles, urea
Fighting Gram‐negative and Gram‐positive bacteria: Thirteen new cystobactamids, derived from cystobactamid 861‐2 are prepared by chemical synthesis. Main modifications are exchange of the amide group by the chemically more stable urea group and the triazole ring as located in ring A. Antibiotic activity against Gram‐negative and ‐positive bacteria including a multidrug‐resistant strain can be preserved, when the urea function is located between rings A and B and the triazole is positioned between rings C and D.
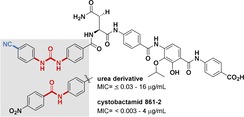
Introduction
Cystobactamids, an unusual group of oligoamides from Cystobacter sp. Cbv34, were first reported in 2014.1 The extracts inhibited the growth of several Gram‐negative and Gram‐positive bacteria and HPLC‐assisted bioactivity‐guided screening provided cystobactamids 919‐1 (1) and 919‐2 (2) as active compounds (Figure 1). These oligoamides contain p‐aminobenzoic acid building blocks and either an iso‐β‐methoxyasparagine or a β‐methoxyasparagine unit. Later, additional derivatives such as the cystobactamids 920‐1 (3), 920‐2 (4) and 862‐1 (5) were reported that structurally differ in the E‐ring and the aspartate hinge region.2a
Figure 1.
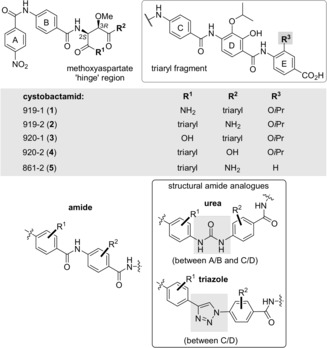
Structures of selected cystobactamids 919‐1 (1), 919‐2 (2), 920‐1 (3), 920‐2 (4) and 861‐2 (5) and the urea as well as triazole groups as structural substitutes for amide group (rings are labelled with letters A–E).
Cystobactamid 862‐1 (5) was found to be the most active natural member. It inhibits several clinically relevant Gram‐positive and Gram‐negative strains (Acinetobacter baumannii: MIC=0.5 μg mL−1, Citrobacter freundii: MIC=0.06 μg mL−1, fluoroquinolone resistant E. coli WT‐III marRΔ74bp: MIC=0.5 μg mL−1, carbapenem resistant P. aeruginosa CRE: MIC=1.0 μg mL−1, and Proteus vulgaris: MIC=0.25 μg mL−1)2 by inhibiting bacterial type IIa topoisomerases. It has to be stressed, that the cystobactamids are structurally related to albicidin with similar antibacterial properties, which has extensively been studied by Süssmuth et al.3
Several total syntheses of cystobactamids 861‐2, 919‐2 and 920‐1 have been published by the Trauner group and by us.2a, 4, 5 These endeavours were essential to revise and prove the constitutions as well as the 2S,3R configurations of cystobactamids.5 Both, the synthetic programmes and the cystobactamid isolation protocols revealed, that the amide bonds between benzoic acids and anilines are prone to hydrolysis with the amide bond that links rings C and D being the most labile one.
As part of a medicinal chemistry programme on cystobactamids, we therefore envisaged to prepare a library of methoxyaspartate containing cystobactamids. One aspect of this programme covers the substitution of the amide group by supposedly chemically more stable functional elements such as urea and triazole (Figure 1). Besides 1,2,4‐triazoles, a cis amide bond surrogate,6 the 1,2,3‐triazole ring has recently been found to be a prominent amide bond bioisostere.7, 8
Here, we report on our recent total synthetic endeavours towards new cystobactamid derivatives with improved chemical stability and the determination of their antibacterial properties. This includes the direct comparison with ciprofloxacin (CP).
Results and Discussion
Chemical syntheses
General considerations: Cystobactamids are natural products with a modular architecture. The key structural elements are amide bonds, composed of combinations of benzoic acids, anilines and carboxylate and amino group in the methoxyaspartate unit. In addition, protecting groups for the carboxylates, amines and phenols have to be chosen. Substantial search and optimisation led to a toolbox of reactions listed in Scheme 1 (cases I–VI), that also repeatedly served to prepare the new cystobactamids reported here. Three principal types of amide forming processes have been developed, that combine two aromatic building blocks (case I), a benzoic acid with an amine (case II), and an aniline with a carboxylate (case III).
Scheme 1.
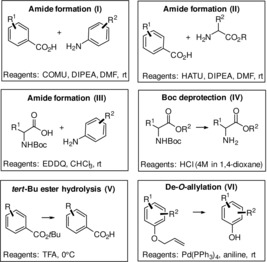
Reagents employed for principal reaction for constructing cystobactamids (COMU=(1‐cyano‐2‐ethoxy‐2‐oxoethylidene‐aminooxy)dimethylaminomorpholinocarbenium‐hexa‐fluorophosphate; EEDQ=N‐ethoxycarbonyl‐2‐ethoxy‐1,2‐dihydroquinoline; HATU=[O‐(7‐azabenzotriazole‐1‐yl)‐N,N,N′,N′‐tetramethyluronium‐hexa‐fluorophosphate]; DIPEA=diisopropyl‐ethylamine, DMF=dimethylformamide, TFA=trifluoroacetic acid).
Boc‐deprotection of the methoxyaspartate hinge region was achieved under mildly acidic conditions (case IV), avoiding the hydrolysis of the tert‐butyl ester in ring E. This ester was commonly cleaved under stronger acidic conditions in neat TFA, routinely the final step of our chemical syntheses (case V). Finally, the allyl deprotection of the phenol function was achieved under Pd0‐catalysed conditions (case VI).
Syntheses of urea‐modified cystobactamids: For exchanging the amide group by urea one of the coupling partners was chosen to contain the isocyanate group as in 4‐isocyanato‐benzonitrile 6 and 1‐isocyanato‐4‐nitrobenzene 10 (Scheme 2). These building blocks were flexibly used to prepare cystobactamid derivatives containing an urea bridge between rings A and B as well as rings C and D. Several coupling partners 9, 19 and 20 were at hand from our previously published syntheses of cystobactamids 861‐2 (5) and 920‐1 (3).2a, 5
Scheme 2.
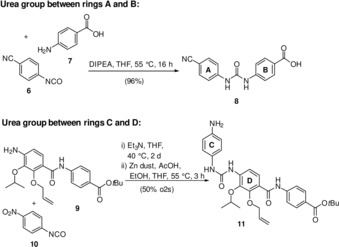
Synthesis of urea‐containing building blocks 8 and 11.
4‐Isocyanato‐benzonitrile (6) was efficiently coupled with 4‐aminobenzoic acid 7 to yield disubstituted urea 8 resembling the linkages between rings A and B.
Diamide 9 was coupled with 1‐isocyanato‐4‐nitrobenzene 10 followed by reduction of the nitro group yielding urea derivative 11 in which the urea group is located between rings C and D. We first prepared cyano derivative 16 in which the urea group is located between the A and B rings (Scheme 3). The cyano derivative was chosen because it has been established that exchange of the nitro group by cyanide leads to improved antibacterial properties.8
Scheme 3.
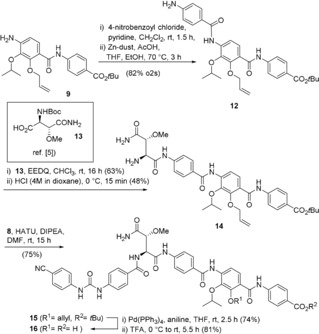
Synthesis of cystobactamid derivative 16 bearing an urea group between rings A and B.
The synthesis commenced with the coupling of amide 9 with 4‐nitrobenzoic acid and reduction of the resulting nitroarene to yield the aniline 12. Next, the aniline was linked to the (2S,3R)‐methoxyaspartate derivative 13.2a Then, the Boc group was hydrolysed under mildly acidic conditions circumventing cleavage of the tert‐butyl ester. Next, the resulting tetramide 14 was coupled with the urea derivative 8 and the resulting product 15 was transformed into the cystobactamid derivative 16 after O‐deallylation and ester hydrolysis.
Next, we installed the urea group between rings C and D (Scheme 4). For that, urea derivative 11 was coupled with methoxyaspartate derivative 13 to yield Boc‐amide 17, which was transformed into amine 18 under mildly acidic conditions. Amide formation with diamides 19 and 20, respectively, yielded cystobactamid precursors 21 and 22, that underwent the common two‐step protocol for removing the allyl‐ and tert‐butyl groups. The two urea‐bearing cystobactamid derivatives 23 and 24 were purified by RP‐HPLC. The truncated derivative 25 was prepared from Boc‐protected tetramide 17 under harsher acidic conditions that led to hydrolysis of both, the Boc‐group as well as the tert‐butyl ester. This was followed by cleavage of the allylether group. In order to remove any traces of the transition metal the crude product was also purified by RP‐HPLC.
Scheme 4.
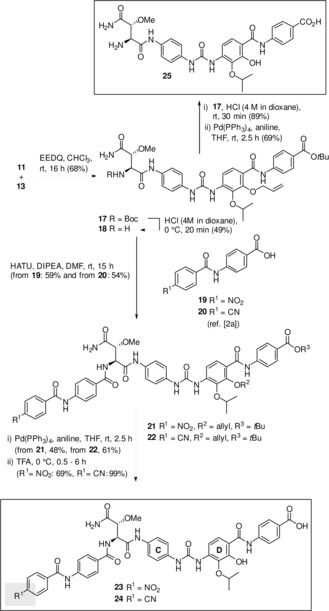
Synthesis of cystobactamid derivatives 23–25 bearing an urea group between rings C and D.
Finally, urea derivative 18 also served as starting point to prepare cystobactamid 27 with two urea groups between rings A/B and C/D (Scheme 5). Coupling with urea derivative 8 yielded compound 26 which was transformed into the cystobactamid derivative 27 in two steps under our standard conditions.
Scheme 5.
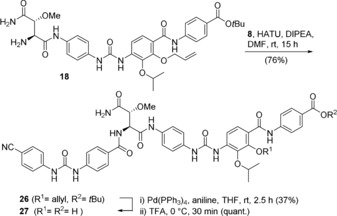
Synthesis of cystobactamid derivative 27 that contains two urea linkages between rings A/B and C/D.
With a set of new urea‐modified cystobactamids in hand we tested the chemical stability of derivative 24 representing the group of urea cystobactamids prepared here. It was dissolved in [D6]DMSO, triethyl amine (12 equiv.) was added and a series of 1H NMR spectra were recorded over a period of 27 h. Careful inspection revealed no degradation products derived from urea derivative 24. As expected, exchange of the amide group by urea leads to enhanced stability under basic conditions.
Syntheses of triazole‐modified cystobactamids: A second structural element for amide substitution is the triazole ring that is straightforwardly prepared from an alkyne and an azide.7, 8 We pursued to substitute the most labile amide group between rings C and D. Thus, we chose 4‐ethynylaniline 29 as alkyne building block and consequently arylazide 28 as second component. The latter was prepared from aniline 9 and reacted under classical Sharpless conditions to yield triazole 30. Amide coupling with methoxyaspartate 13 provided amine 31 after Boc‐deprotection under mildly acidic conditions (Scheme 6).
Scheme 6.
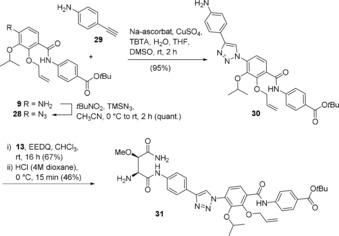
Synthesis of the central triazole derivative 31.
Having installed the triazole ring between rings C and D, we turned our attention to prepare several A/B‐ring fragments that vary in ring A (Table 1).
Table 1.
Preparation of diamides 39–44 (COMU = 1‐cyano‐2‐ethoxy‐2‐oxoethylideneaminooxy) dimethylamino‐morpholino‐carbenium‐hexafluorophosphate).
|
| |||
|---|---|---|---|
|
Building block ring A |
Building block ring B |
Conditions |
Yields [%] |
|
33 |
7 |
Na2CO3, THF, H2O, rt, 3 h |
39 (98) |
|
34 |
32 |
for 34–38: COMU, DIPEA, DMF, rt, 16 h, then TFA, rt, 30 min |
40 (75) |
|
35 |
32 |
41 (66) |
|
|
36 |
32 |
42 (94) |
|
|
37 |
32 |
43 (83) |
|
|
38 |
32 |
44 (79) |
|
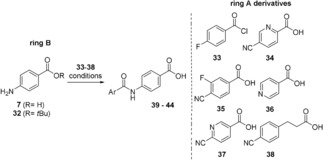
We mainly focused on modifications in the 4‐position and exchange of the benzene by the pyridine ring (Table 1; building blocks 33–37). In one case, we used a benzoyl chloride (33) which was directly reacted with the aminobenzoic acid (7). In most cases, however, the tert‐butyl ester 32 which was coupled with A‐ring derivatives 34–37 by in situ activation of their carboxylic function. Finally, also 3‐(4‐isocyanophenyl)propanoic acid (38) was chosen as elongated bishomo analogue of 4‐cyanobenzoic acid. Consequently, we had a small library of A/B‐ring fragments (39–44) at hand that was used to access triazole‐bearing cystobactamids 45–52 in a three step sequence (coupling of A/B fragments to triazole‐containing aspartate building block 31, de‐O‐allylation and tert‐butyl ester hydrolysis) (Scheme 7).
Scheme 7.
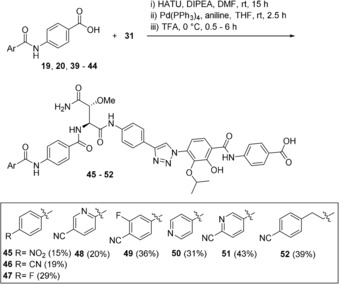
Synthesis of triazole‐modified cystobactamids 45–52 (yields refer to the three step sequence).
Evaluation of antibacterial properties
The new urea‐modified cystobactamids 16, 23–25 and 27 were tested against a panel of different Gram‐negative and Gram‐positive pathogens, including intrinsically multidrug‐resistant P. aeruginosa (Table 2) and directly compared with cystobactamid 861‐2 (5). Incorporation of the urea group between rings C and D as in derivatives 23–25 and 27 leads to substantial and even complete loss of antibacterial activity. This also includes truncation that is represented by cystobactamid derivative 25. In essence, although chemical stabilisation of the cystobactamids can be achieved by exchanging the amide group with urea, this approach fails for connecting rings C and D due to loss of antibacterial activity. However, when the urea group is positioned between rings A and B as in cystobactamid derivative 16, biological activity is almost completely preserved compared to cystobactamid 861‐2 (5) and even considerably higher for strain E. coli BW25113.
Table 2.
Biological activity of urea derivatives 16, 23‐25 and 27 compared to cystobactamid 861–2 (5) (MIC values in μg mL−1; nd=not determined).[a]
|
Strain |
16 |
23 |
24 |
25 |
27 |
5 |
|---|---|---|---|---|---|---|
|
S. aureus Newman |
16 |
>64 |
>64 |
>64 |
>64 |
1 |
|
E. coli BW25113 |
≤0.03 |
nd |
1 |
nd |
>64 |
0.25 |
|
E. coli ΔacrB |
≤0.03 |
nd |
≤0.03 |
nd |
>64 |
≤0.003 |
|
E. coli ΔtolC |
nd |
>64 |
nd |
>64 |
nd |
≤0.003 |
|
E. coli DSM‐1116 |
nd |
>64 |
nd |
>64 |
nd |
0.2 |
|
P. aeruginosa PA14 |
1 |
>64 |
>64 |
>64 |
>64 |
1 |
|
PA14ΔmexAB |
1 |
>64 |
>64 |
>64 |
>64 |
0.25 |
[a] Values for ciprofloxacin (CP) are listed in Table 3.
Likewise, antibacterial properties of triazole‐modified cystobactamid derivatives 45–52 were evaluated (Table 3). Principally, exchange of the amide group by a triazole ring does not lead to loss of antibacterial activity, thus this modification can be regarded as a successful strategy to generate cystobactamid derivatives with enhanced chemical and biological stabilities between rings C and D.9 Furthermore, this set of new analogues reveals fine‐tuning of antibacterial activity by structural changes in ring A. Interestingly, when ring A represents a pyridine ring with N positioned in the para and meta positions relative to ring B as in compounds 50 and 51 substantial reduction of antibacterial activity against S. aureus and P. aeruginosa is observed. This is not the case for pyridine derivative 48, in which the nitrogen atom is located in the ortho position. When extending the length between rings A and B by insertion of an ethylene unit as in derivative 52 reduced antibiotic activity is also observed. On the other hand, triazole derivatives 45–47 as well as fluoro derivative 49 bearing a cyanide group in the para position are still highly active in a comparable range to cystobactamid 861‐2 (5). Importantly, the activity spans across all four strains tested. The important finding of this study is that the triazole group is a good substitute for the amide linkage between rings C and D.
Table 3.
Biological activity of triazole derivatives 45–52 compared to cystobactamid 861‐2 (5) and CP (MIC values in μg mL−1).
|
Strain |
45 |
46 |
47 |
48 |
49 |
50 |
51 |
52 |
5 |
CP |
|---|---|---|---|---|---|---|---|---|---|---|
|
E. coli BW25113 |
≤0.03 |
≤0.03 |
≤0.03 |
0.125 |
≤0.03 |
1 |
≤0.03 |
0.25 |
≤0.03 |
0.03 |
|
E. coli ΔacrB |
≤0.03 |
≤0.03 |
≤0.03 |
≤0.03 |
≤0.03 |
0.25 |
0.25 |
0.125 |
0.06 |
≤0.003 |
|
S. aureus Newman |
1 |
1 |
8 |
4 |
1 |
>64 |
64 |
>64 |
0.125 |
0.2 |
|
P. aeruginosa PA14 |
4 |
4 |
8 |
8 |
4 |
>64 |
32 |
>64 |
1 |
0.2 |
|
PA14ΔmexAB |
4 |
4 |
8 |
4 |
4 |
>64 |
32 |
>64 |
0.125 |
0.01 |
Conclusion
In summary, we report on the chemical synthesis of thirteen new analogues derived from cystobactamid 861‐2 (5). These derivatives differ from the most potent natural member of the cystobactamids by exchange of the amide groups by either the urea group or by a triazole ring. These first structure activity studies (SAR) for natural methoxyaspartate bearing cystobactamids reveal that the urea function can be introduced between rings A and B but not between rings D and E for preserving antibacterial activity. We also show that the triazole ring can serve as a substitute for the amide group in cystobactamids. Finally, ring A can be modified in various manners without substantial loss of activity. This work provides a first information on future directions of medicinal chemistry programmes on the cystobactamids and matches recent findings on the albicidins.10
Experimental Section
The experimental section below covers general procedures for the synthesis and analytic data of all new cystobactamid derivatives 16, 23–25, 27 and 45–52. All other synthetic procedures, general information and analytical data including copies of NMR spectra are found in the supporting information.
General procedure V for tert‐Bu ester hydrolysis (according to Scheme 1)
Precooled TFA (0.02 m) was added slowly to ester (1.0 equiv) at 0 °C. The mixture was warmed up to rt over a period 30 min, then it was stirred between 30 min and 6 h. The reaction was terminated by addition of Et2O. The precipitate was filtered, washed with an excess of Et2O and dried under high vacuum.
4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(3‐(4‐cyanophenyl)ureido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)benzamido)‐2‐hydroxy‐3‐isopropoxybenz‐amido)benzoic acid (16): Following to the general procedure mentioned above phenol S4 (see SI; 29.8 mg, 32.6 μmol, 1.0 equiv) was stirred for 5.5 h. Acid 16 was obtained as a colourless amorphous solid (22.5 mg, 26.3 μmol, 81 %). [α]D 22=+23.1° (c 1.1, MeOH); 1H NMR (400 MHz, [D6]DMSO): δ=12.3 (1 H, br. s, OH), 10.6 (1 H, br. s, NH), 10.6 (1 H, s, NH), 9.40 (1 H, s, NH), 9.38 (1 H, s, NH Urea), 9.26 (1 H, s, NH Urea), 8.37 (1 H, d, J=8.1 Hz, NHCH), 7.98–7.95 (4 H, m, ArH), 7.88–7.82 (7 H, m, ArH), 7.76–7.70 (3 H, m, ArH), 7.66 (2 H, m, ArH), 7.58 (2 H, m, ArH), 7.51 (2 H, d, J=26.7 Hz, CONH 2), 4.90 (1 H, t, J=8.1 Hz, CHNH), 4.55 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.31 (3 H, s, OCH 3), 1.27 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (100 MHz, [D6]DMSO): δ=170.9 (q, CONH2), 168.8 (q, CONH), 168.5 (q, CONH), 166.9 (q, CO2H), 165.5 (q, CONH), 164.2 (q, CONH), 154.1 (q, C‐Ar), 151.9 (q, NHCONH), 144.0 (q, C‐Ar), 142.4 (q, C‐Ar), 142.3 (q, C‐Ar), 142.0 (q, C‐Ar), 137.0 (q, C‐Ar), 136.3 (q, C‐Ar), 133.3 (2C, t, C‐Ar), 130.2 (2C, t, C‐Ar), 128.5 (2C, t, C‐Ar), 128.5 (q, C‐Ar), 128.3 (2C, t, C‐Ar), 127.1 (q, C‐Ar), 126.3 (q, C‐Ar), 122.8 (t, C‐Ar), 120.7 (2C, t, C‐Ar), 119.3 (q, CN), 119.0 (2C, t, C‐Ar), 118.2 (2C, t, C‐Ar), 117.5 (2C, t, C‐Ar), 112.4 (t, C‐Ar), 112.2 (q, C‐Ar), 103.5 (q, C‐Ar), 80.0 (t, CHOMe), 74.9 (t, CH(CH3)2), 57.7 (p, OCH3), 55.7 (t, CHNH), 22.3 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H41N8O11 [M+H]+: 857.2895; found 857.2896.
4‐(4‐(3‐(4‐((2S,3R)‐4‐Amino‐3‐methoxy‐2‐(4‐(4‐nitrobenzamido)benzamido)‐4‐oxobutan‐amido)phenyl)ureido)‐2‐hydroxy‐3‐isopropoxybenzamido)‐benzoic acid (23): Following the general procedure mentioned above phenol S6 (see SI; 9.0 mg, 9.64 μmol, 1.0 equiv) was stirred for 6 h. Acid 23 was obtained as a colourless amorphous solid (5.8 mg, 6.62 μmol, 69 %). [α]D 24=+38.7° (c 0.6, DMSO); 1H NMR (500 MHz, [D6]DMSO +2 μL DCO2D): δ=10.8 (1 H, s, NH), 10.6 (1 H, br. s, NH), 10.2 (1 H, s, NH), 9.61 (1 H, s, NH Urea), 8.40–8.37 (3 H, m, ArH, NHCH), 8.33 (1 H, s, NH Urea), 8.20 (2 H, m, ArH), 7.96 (2 H, m, ArH), 7.92–7.84 (5 H, m, ArH), 7.84 (2 H, m, ArH), 7.80 (1 H, d, J=8.5 Hz, ArH), 7.60 (2 H, m, ArH), 7.49 (2 H, d, J=46.1 Hz, CONH 2), 7.42 (2 H, m, ArH), 4.89 (1 H, t, J=8.5 Hz, CHNH), 4.63 (1 H, hept, J=6.0 Hz, CHMe2), 4.07 (1 H, d, J=7.3 Hz, CHOMe), 3.30 (3 H, s, OCH 3), 1.30 ppm (6 H, d, J=6.0 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO +2 μL DCO2D): δ=171.1 (q, CONH2), 169.0 (q, CONH), 167.6 (q, CONH), 167.0 (q, CO2H), 165.4 (q, CONH), 164.3 (q, CONH), 154.5 (q, C‐Ar), 152.0 (q, NHCONH), 149.3 (q, C‐Ar), 142.1 (q, C‐Ar), 141.7 (q, C‐Ar), 140.4 (q, C‐Ar), 139.1 (q, C‐Ar), 135.0 (q, C‐Ar), 133.7 (q, C‐Ar), 132.9 (q, C‐Ar), 130.2 (2C, t, C‐Ar), 129.4 (2C, t, C‐Ar), 129.2 (q, C‐Ar), 128.3 (2C, t, C‐Ar), 126.2 (q, C‐Ar), 123.6 (2C, t, C‐Ar), 123.1 (t, C‐Ar), 120.8 (2C, t, C‐Ar), 120.2 (2C, t, C‐Ar), 119.7 (2C, t, C‐Ar), 118.7 (2C, t, C‐Ar), 109.3 (q, C‐Ar), 108.2 (t, C‐Ar), 80.2 (t, CHOMe), 74.2 (t, CH(CH3)2), 57.7 (p, OCH3), 55.6 (t, CHNH), 22.0 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C43H40N8O13Na [M+Na]+: 899.2613; found 899.2613.
4‐(4‐(3‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(4‐cyanobenzamido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)phenyl)ureido)‐2‐hydroxy‐3‐isopropoxybenz‐amido)benzoic acid (24): Following the general procedure mentioned above phenol S7 (see SI; 14.0 mg, 15.3 μmol, 1.0 equiv) was stirred for 30 min. Acid 24 was obtained as a yellow amorphous solid (13.0 mg, 15.2 μmol, 99 %). [α]D 23=+37.2° (c 0.3, DMSO); 1H NMR (500 MHz, [D6]DMSO +2 μL DCO2D): δ=12.5 (1 H, s, OH), 10.7 (1 H, s, NH), 10.5 (1 H, s, NH), 10.1 (1 H, s, NH), 9.61 (1 H, s, NH Urea), 8.38 (1 H, d, J=8.1 Hz, NHCH), 8.34 (1 H, s, NH Urea), 8.13 (2 H, m, ArH), 8.04 (2 H, m, ArH), 7.96 (2 H, m, ArH), 7.91–7.83 (7 H, m, ArH), 7.81 (1 H, d, J=9.2 Hz, ArH), 7.60 (2 H, m, ArH), 7.52–7.41 (4 H, m, ArH, CONH 2), 4.88 (1 H, t, J=8.1 Hz, CHNH), 4.62 (1 H, hept, J=6.1 Hz, CHMe2), 4.06 (1 H, d, J=8.1 Hz, CHOMe), 3.30 (3 H, s, OCH 3), 1.30 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO +2 μL DCO2D): δ=171.1 (q, CONH2), 169.0 (q, CONH), 167.6 (q, CONH), 166.9 (q, CO2H), 165.3 (q, CONH), 164.5 (q, CONH), 154.3 (q, C‐Ar), 151.9 (q, NHCONH), 142.0 (q, C‐Ar), 141.7 (q, C‐Ar), 139.1 (q, C‐Ar), 138.7 (q, C‐Ar), 134.9 (q, C‐Ar), 133.7 (q, C‐Ar), 132.5 (2C, t, C‐Ar), 130.2 (2C, t, C‐Ar), 129.0 (q, C‐Ar), 128.6 (2C, t, C‐Ar), 128.3 (2C, t, C‐Ar), 126.1 (q, C‐Ar), 123.0 (t, C‐Ar), 120.8 (2C, t, C‐Ar), 120.6 (q, C‐Ar), 120.1 (2C, t, C‐Ar), 119.6 (2C, t, C‐Ar), 118.7 (2C, t, C‐Ar), 118.3 (q, CN), 114.1 (q, C‐Ar), 109.2 (q, C‐Ar), 108.2 (t, C‐Ar), 80.1 (t, CHOMe), 74.3 (t, CH(CH3)2), 57.7 (p, OCH3), 55.5 (t, CHNH), 21.9 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H40N8O11Na [M+Na]+: 879.2714; found 879.2714.
4‐(4‐(3‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(3‐(4‐cyanophenyl)ureido)benzamido)‐3‐methoxy‐4‐oxobutanamido)phenyl)ureido)‐2‐hydroxy‐3‐isopropoxybenz‐amido)benzoic acid (27): Following the general procedure mentioned above phenol S8 (see SI; 9.0 mg, 9.70 μmol, 1.0 equiv) was stirred for 30 min. Acid 27 was obtained as a colourless amorphous solid (8.4 mg, 9.70 μmol, 99 %). [α]D 23=+12.7° (c 1.1, MeOH); 1H NMR (600 MHz, [D6]DMSO +2 μL DCO2D): δ=12.5 (1 H, br. s, OH), 10.5 (1 H, br. s, NH), 10.1 (1 H, s, NH), 9.61 (1 H, s, NH Urea), 9.37 (1 H, s, NH Urea), 9.25 (1 H, s, NH Urea), 8.34 (1 H, s, NH Urea),8.30 (1 H, d, J=8.1 Hz, NHCH), 7.96 (2 H, m, ArH), 7.89 (1 H, d, J=9.0 Hz, ArH), 7.85–7.80 (5 H, m, ArH), 7.74 (2 H, m, ArH), 7.65 (2 H, m, ArH), 7.60–7.55 (4 H, m, ArH), 7.52–7.41 (4 H, m, ArH, CONH 2), 4.86 (1 H, t, J=8.1 Hz, CHNH), 4.62 (1 H, hept, J=6.2 Hz, CHMe2), 4.06 (1 H, d, J=8.1 Hz, CHOMe), 3.30 (3 H, s, OCH 3), 1.30 ppm (6 H, d, J=6.2 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO +2 μL DCO2D): δ=171.1 (q, CONH2), 169.0 (q, CONH), 167.7 (q, CONH), 166.9 (q, CO2H), 165.4 (q, CONH), 154.3 (q, C‐Ar), 152.0 (q, NHCONH), 151.9 (q, NHCONH), 144.0 (q, C‐Ar), 142.3 (q, C‐Ar), 142.0 (q, C‐Ar), 139.1 (q, C‐Ar), 134.9 (q, C‐Ar), 133.7 (q, C‐Ar), 133.3 (2C, t, C‐Ar), 132.8 (q, C‐Ar), 130.2 (2C, t, C‐Ar), 128.5 (2C, t, C‐Ar), 127.3 (q, C‐Ar), 126.2 (q, C‐Ar), 123.0 (t, C‐Ar), 120.8 (2C, t, C‐Ar), 120.1 (2C, t, C‐Ar), 119.3 (q, CN), 118.7 (2C, t, C‐Ar), 118.2 (2C, t, C‐Ar), 117.5 (2C, t, C‐Ar), 109.2 (q, C‐Ar), 108.2 (t, C‐Ar), 103.5 (q, C‐Ar), 80.1 (t, CHOMe), 74.3 (t, CH(CH3)2), 57.7 (p, OCH3), 55.5 (t, CHNH), 21.9 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H42N9O11 [M+H]+: 872.3004; found 872.3004.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐3‐methoxy‐2‐(4‐(4‐nitrobenzamido)benzamido)‐4‐oxo‐butanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐isopropoxy‐benzamido)‐benzoic acid (45): Following to the general procedure mentioned above phenol S18 (see SI; 8.5 mg, 9.03 μmol, 1.0 equiv) was stirred for 6 h. Acid 45 was obtained as a colourless amorphous solid (6.2 mg, 7.00 μmol, 78 %). [α]D 24=+37.1° (c 0.1, DMSO); 1H NMR (500 MHz, [D6]DMSO): δ=12.9 (1 H, br. s, CO2 H), 12.2 (1 H, br. s, OH), 10.8 (1 H, br. s, NH), 10.8 (1 H, s, NH), 10.4 (1 H, s, NH), 8.93 (1 H, s, CH Triazol), 8.44 (1 H, d, J=8.1 Hz, NHCH), 8.39 (2 H, m, ArH), 8.21 (2 H, m, ArH), 8.00–7.87 (11 H, m, ArH), 7.79 (2 H, m, ArH), 7.50 (2 H, d, J=39.2 Hz, CONH 2), 7.37 (1 H, d, J=8.6 Hz, ArH), 4.92 (1 H, t, J=8.1 Hz, CHNH), 4.24 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.02 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.5 (q, CONH), 166.9 (q, CO2H), 165.4 (q, CONH), 164.2 (q, CONH), 154.5 (q, C‐Ar), 149.3 (q, C‐Ar), 146.3 (q, C‐ArTriazol), 141.9 (q, C‐Ar), 141.7 (q, C‐Ar), 140.3 (q, C‐Ar), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 134.4 (q, C‐Ar), 130.7 (2C, t, C‐Ar), 130.3 (q, C‐Ar), 129.4 (2C, t, C‐Ar), 128.7 (2C, t, C‐Ar), 128.3 (q, C‐Ar), 126.5 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (t, C‐Ar), 123.6 (2C, t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.7 (2C, t, C‐Ar), 118.4 (q, C‐Ar), 114.8 (t, C‐Ar), 80.1 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.7 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H40N9O12 [M+H]+: 886.2796; found 886.2798.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(4‐cyanobenzamido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐iso‐propoxybenzamido)‐benzoic acid (46): Following the general procedure mentioned above phenol S19 (see SI; 10.1 mg, 11.0 μmol, 1.0 equiv) was stirred for 3 h. Acid 46 was obtained as a beige amorphous solid (7.4 mg, 8.55 μmol, 78 %). [α]D 25=+55.9° (c 0.2, DMSO); 1H NMR (600 MHz, [D6]DMSO): δ=12.8 (1 H, br. s, CO2 H), 12.2 (1 H, br. s, OH), 10.8 (1 H, br. s, NH), 10.7 (1 H, s, NH), 10.4 (1 H, s, NH), 8.93 (1 H, s, CH Triazol), 8.43 (1 H, d, J=8.1 Hz, NHCH), 8.13 (2 H, m, ArH), 8.05 (2 H, m, ArH), 7.99–7.87 (11 H, m, ArH), 7.79 (2 H, m, ArH), 7.50 (2 H, d, J=45.0 Hz, CONH 2), 7.34 (1 H, d, J=7.0 Hz, ArH), 4.92 (1 H, t, J=8.1 Hz, CHNH), 4.26 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.02 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.4 (q, CONH), 166.8 (q, CO2H), 165.4 (q, CONH), 164.5 (q, CONH), 154.7 (q, C‐Ar), 146.2 (q, C‐ArTriazol), 141.9 (q, C‐Ar), 141.7 (q, C‐Ar), 138.9 (q, C‐Ar), 138.8 (q, C‐Ar), 138.7 (q, C‐Ar), 134.3 (q, C‐Ar), 132.5 (2C, t, C‐Ar), 130.3 (2C, t, C‐Ar), 129.0 (q, C‐Ar), 128.6 (2C, t, C‐Ar), 128.3 (2C, t, C‐Ar), 126.4 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 123.5 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.6 (2C, t, C‐Ar), 118.4 (q, C‐Ar), 118.3 (q, CN), 114.6 (q, C‐Ar), 114.0 (t, C‐Ar), 80.1 (t, CHOMe), 75.6 (t, CH(CH3)2), 57.7 (p, OCH3), 55.6 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C45H40N9O10 [M+H]+: 866.2898; found 866.2897.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(4‐fluorobenzamido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐iso‐propoxybenzamido)‐benzoic acid (47): Following the general procedure mentioned above phenol S20 (see SI; 20.1 mg, 22.0 μmol, 1.0 equiv) was stirred for 3.5 h. Acid 47 was obtained as an orange amorphous solid (19.1 mg, 19.6 μmol, 89 %). [α]D 24=+27.6° (c 0.9, DMSO); 1H NMR (600 MHz, [D6]DMSO): δ=12.2 (1 H, br. s, OH), 10.8 (1 H, s, NH), 10.5 (1 H, s, NH), 10.4 (1 H, s, NH), 8.93 (1 H, s, CH Triazol), 8.41 (1 H, d, J=8.1 Hz, NHCH), 8.07–8.05 (2 H, m, ArH), 8.00–7.87 (11 H, m, ArH), 7.79 (2 H, m, ArH), 7.50 (2 H, d, J=47.2 Hz, CONH 2), 7.40–7.36 (3 H, m, ArH), 4.92 (1 H, t, J=8.1 Hz, CHNH), 4.25 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.03 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.5 (q, CONH), 166.9 (q, CO2H), 165.0 (q, CONH), 164.7 (q, CONH), 164.4 (d, J=311.0 Hz, C‐F), 158.2 (TFA), 154.5 (q, C‐Ar), 146.3 (q, C‐ArTriazol), 142.1 (q, C‐Ar), 141.9 (q, C‐Ar), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 134.4 (q, C‐Ar), 131.1 (d, J=2.8 Hz, C‐Ar), 130.5 (2C, d, J=9.1 Hz, C‐Ar), 130.3 (2C, t, C‐Ar), 129.8 (TFA), 128.6 (q, C‐Ar), 128.2 (2C, t, C‐Ar), 126.5 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 123.5 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.5 (2C, t, C‐Ar), 118.4 (q, C‐Ar), 115.4 (2C, d, J=21.8 Hz, C‐Ar), 114.8 (t, C‐Ar), 80.1 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.6 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H40N8O10F [M+H]+: 859.2851; found 859.2851.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(5‐cyanopicolinamido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐iso‐propoxybenzamido)benzoic acid (48): Following the general procedure mentioned above phenol S21 (see SI; 21.8 mg, 23.6 μmol, 1.0 equiv) was stirred for 6 h. Acid 48 was obtained as a yellow amorphous solid (8.2 mg, 9.47 μmol, 40 %). [α]D 24=+27.2° (c 0.8, DMSO); 1H NMR (600 MHz, [D6]DMSO): δ=12.9 (1 H, br. s, CO2 H), 12.2 (1 H, br. s, OH), 11.0 (1 H, s, NH), 10.8 (1 H, s, NH), 10.4 (1 H, s, NH), 9.22 (1 H, dd, J=0.7, 2.0 Hz, ArH), 8.93 (1 H, s, CH Triazol), 8.61 (1 H, dd, J=2.0, 8.2 Hz, ArH), 8.43 (1 H, d, J=8.1 Hz, NHCH), 8.31 (1 H, dd, J=0.7, 8.2 Hz, ArH), 8.06 (2 H, m, ArH), 7.99–7.87 (9 H, m, ArH), 7.79 (2 H, m, ArH), 7.50 (2 H, d, J=43.0 Hz, CONH 2), 7.37 (1 H, d, J=8.6 Hz, ArH), 4.91 (1 H, t, J=8.1 Hz, CHNH), 4.24 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.02 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.5 (q, CONH), 166.8 (q, CO2H), 165.4 (q, CONH), 161.7 (q, CONH), 154.5 (q, C‐Ar), 152.3 (q, C‐Ar), 151.5 (t, C‐Ar), 146.3 (q, C‐ArTriazol), 142.3 (t, C‐Ar), 141.9 (q, C‐Ar), 141.0 (q, C‐Ar), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 134.4 (q, C‐Ar), 130.3 (2C, t, C‐Ar), 129.3 (q, C‐Ar), 128.2 (2C, t, C‐Ar), 126.5 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 123.5 (t, C‐Ar), 122.6 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.9 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 118.4 (q, C‐Ar), 116.7 (q, CN), 114.8 (t, C‐Ar), 111.8 (q, C‐Ar), 80.1 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.6 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H39N10O10 [M+H]+: 867.2851; found 867.2857.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(4‐cyano‐3‐fluorobenzamido)benzamido)‐3‐methoxy‐4‐oxobutanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐isopropoxybenzamido)‐benzoic acid (49): Following the general procedure mentioned above phenol S22 (see SI; 22.4 mg, 23.8 μmol, 1.0 equiv) was stirred for 2 h. Acid 49 was obtained as a beige amorphous solid (21.0 mg, 23.8 μmol, quant.). [α]D 24=+39.5° (c 1.3, DMSO); 1H NMR (600 MHz, [D6]DMSO): δ=12.8 (1 H, br. s, CO2 H), 12.2 (1 H, br. s, OH), 10.8 (1 H, s, NH), 10.8 (1 H, s, NH), 10.4 (1 H, s, NH), 8.93 (1 H, s, CH Triazol), 8.45 (1 H, d, J=8.1 Hz, NHCH), 8.16–8.13 (1 H, m, ArH), 8.09–8.07 (1 H, m, ArH), 8.00–7.96 (4 H, m, ArH), 7.93–7.87 (8 H, m, ArH), 7.79 (2 H, m, ArH), 7.51 (2 H, d, J=47.9 Hz, CONH 2), 7.37 (1 H, d, J=8.6 Hz, ArH), 4.92 (1 H, t, J=8.1 Hz, CHNH), 4.25 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.03 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.5 (q, CONH), 166.9 (q, CO2H), 165.4 (q, CONH), 163.2 (q, CONH), 162.2 (d, J=256.3 Hz, C‐F), 154.5 (q, C‐Ar), 146.3 (q, C‐ArTriazol), 141.9 (q, C‐Ar), 141.5 (d, J=6.1 Hz, CCONH), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 134.4 (d, J=7.0 Hz, CHCCN), 130.7 (q, C‐Ar), 130.3 (2C, t, C‐Ar), 129.2 (q, C‐Ar), 128.8 (q, C‐Ar), 128.3 (2C, t, C‐Ar), 126.5 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 124.7 (d, J=3.4 Hz, CHCHCCONH), 123.5 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.7 (2C, t, C‐Ar), 118.4 (q, CN), 115.8 (d, J=21.3 Hz, CHCF), 114.8 (t, C‐Ar), 113.6 (q, C‐Ar), 102.9 (d, J=15.3 Hz, CCN), 80.1 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.7 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C45H39N9O10F [M+H]+: 884.2804; found 884.2808.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(isonicotinamido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐isopropoxy‐benzamido)‐benzoic acid (50): Following the general procedure mentioned above phenol S23 (see SI; 16.5 mg, 18.4 μmol, 1.0 equiv) was stirred for 4 h. Acid 50 was obtained as a yellow amorphous solid (13.7 mg, 16.3 μmol, 89 %). [α]D 24=+22.6° (c 1.4, DMSO); 1H NMR (500 MHz, [D6]DMSO): δ=12.2 (1 H, br. s, OH), 10.8 (1 H, s, NH), 10.8 (1 H, s, NH), 10.4 (1 H, s, NH), 8.93 (1 H, s, CH Triazol), 8.85 (2 H, m, ArH), 8.44 (1 H, d, J=8.0 Hz, NHCH), 8.13–7.87 (13 H, m, ArH), 7.79 (2 H, m, ArH), 7.50 (2 H, d, J=31.9 Hz, CONH 2), 7.37 (1 H, d, J=8.6 Hz, ArH), 4.92 (1 H, t, J=8.0 Hz, CHNH), 4.25 (1 H, hept, J=6.1 Hz, CHMe2), 4.09 (1 H, d, J=8.0 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.03 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.5 (q, CONH), 166.9 (q, CO2H), 165.4 (q, CONH), 164.1 (q, CONH), 154.5 (q, C‐Ar), 149.8 (2C, t, C‐Ar), 146.3 (q, C‐ArTriazol), 141.9 (q, C‐Ar), 141.5 (q, C‐Ar), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 134.4 (q, C‐Ar), 130.3 (2C, t, C‐Ar), 129.2 (q, C‐Ar), 128.3 (2C, t, C‐Ar), 126.5 (q, C‐Ar), 125.9 (q, C‐Ar),125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 123.5 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 122.0 (2C, t, C‐Ar), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.7 (2C, t, C‐Ar), 118.4 (q, C‐Ar), 114.8 (t, C‐Ar), 80.1 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.7 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C43H40N9O10 [M+H]+: 842.2898; found 842.2903.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(6‐cyanonicotinamido)benzamido)‐3‐methoxy‐4‐oxo‐butanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐iso‐propoxybenzamido)benzoic acid (51): Following the general procedure mentioned above phenol S24 (see SI; 24.4 mg, 26.5 μmol, 1.0 equiv) was stirred for 5 h. Acid 51 was obtained as a beige amorphous solid (15.6 mg, 18.0 μmol, 68 %). [α]D 22=+25.1° (c 1.5, DMSO); 1H NMR (600 MHz, [D6]DMSO): δ=12.8 (1 H, br. s, CO2 H), 12.2 (1 H, br. s, OH), 10.9 (1 H, br. s, NH), 10.8 (1 H, s, NH), 10.4 (1 H, s, NH), 9.24–9.23 (1 H, m, ArH), 8.93 (1 H, s, CH Triazol), 8.55–8.53 (1 H, m, ArH), 8.45 (1 H, d, J=8.1 Hz, NHCH), 8.26–8.25 (1 H, m, ArH), 8.00–7.87 (11 H, m, ArH), 7.79 (2 H, m, ArH), 7.50 (2 H, d, J=46.3 Hz, CONH 2), 7.37 (1 H, d, J=8.6 Hz, ArH), 4.92 (1 H, t, J=8.1 Hz, CHNH), 4.25 (1 H, hept, J=6.0 Hz, CHMe2), 4.09 (1 H, d, J=8.1 Hz, CHOMe), 3.32 (3 H, s, OCH 3), 1.03 ppm (6 H, d, J=6.0 Hz, CH(CH 3)2); 13C NMR (125 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 168.2 (q, CONH), 167.5 (q, CONH), 166.8 (q, CO2H), 165.3 (q, CONH), 163.0 (q, CONH), 154.5 (q, C‐Ar), 150.2 (t, C‐Ar), 146.3 (q, C‐ArTriazol), 141.9 (q, C‐Ar), 141.5 (q, C‐Ar), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 137.3 (t, C‐Ar), 134.5 (q, C‐Ar), 134.4 (q, C‐Ar), 133.4 (q, C‐Ar), 130.3 (2C, t, C‐Ar), 129.2 (q, C‐Ar), 128.8 (t, C‐Ar), 128.3 (2C, t, C‐Ar), 126.5 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 123.5 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.6 (2C, t, C‐Ar), 118.4 (q, C‐Ar), 117.1 (q, CN), 114.8 (t, C‐Ar), 80.0 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.6 (t, CHNH), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C44H39N10O10 [M+H]+: 867.2851; found 867.2858.
4‐(4‐(4‐(4‐((2S,3R)‐4‐Amino‐2‐(4‐(3‐(4‐cyanophenyl)‐propanamido)‐benz‐amido)‐3‐methoxy‐4‐oxobutanamido)phenyl)‐1H‐1,2,3‐triazol‐1‐yl)‐2‐hydroxy‐3‐isopropoxy‐benzamido)benzoic acid (52): Following the general procedure mentioned above phenol S25 (see SI; 19.8 mg, 20.9 μmol, 1.0 equiv) was stirred for 4.5 h. Acid 52 was obtained as a grey amorphous solid (18.6 mg, 20.9 μmol, quant.). [α]D 24=+20.8° (c 1.9, DMSO); 1H NMR (400 MHz, [D6]DMSO): δ=12.8 (1 H, br. s, CO2 H), 12.2 (1 H, br. s, OH), 10.8 (1 H, s, NH), 10.3 (1 H, s, NH), 10.2 (1 H, s, NH), 8.92 (1 H, s, CH Triazol), 8.35 (1 H, d, J=8.1 Hz, NHCH), 8.00–7.95 (3 H, m, ArH), 7.93–7.87 (4 H, m, ArH), 7.82–7.75 (6 H, m, ArH), 7.67 (2 H, m, ArH), 7.53–7.45 (4 H, m, ArH, CONH 2), 7.37 (1 H, d, J=8.6 Hz, ArH), 4.90 (1 H, t, J=8.1 Hz, CHNH), 4.25 (1 H, hept, J=6.1 Hz, CHMe2), 4.07 (1 H, d, J=8.1 Hz, CHOMe), 3.31 (3 H, s, OCH 3), 3.01 (2 H, t, J=7.5 Hz, ArCH 2CH2), 2.71 (2 H, t, J=7.5 Hz, ArCH2CH 2), 1.02 ppm (6 H, d, J=6.1 Hz, CH(CH 3)2); 13C NMR (100 MHz, [D6]DMSO): δ=171.0 (q, CONH2), 170.4 (q, CONH), 168.2 (q, CONH), 167.5 (q, CONH), 166.8 (q, CO2H), 165.4 (q, CONH), 154.5 (q, C‐Ar), 147.3 (q, C‐Ar), 146.3 (q, C‐ArTriazol), 142.1 (q, C‐Ar), 141.9 (q, C‐Ar), 138.9 (q, C‐Ar), 138.7 (q, C‐Ar), 134.4 (q, C‐Ar), 132.2 (2C, t, C‐Ar), 130.3 (2C, t, C‐Ar), 129.5 (2C, t, C‐Ar), 128.3 (2C, t, C‐Ar), 128.0 (q, C‐Ar), 126.5 (q, C‐Ar), 125.8 (2C, t, C‐Ar), 125.4 (q, C‐Ar), 123.5 (t, C‐Ar), 122.6 (t, C‐ArTriazol), 120.6 (2C, t, C‐Ar), 119.8 (2C, t, C‐Ar), 119.0 (q, C‐Ar), 118.4 (q, CN), 118.2 (2C, t, C‐Ar), 114.8 (t, C‐Ar), 108.9 (q, C‐Ar), 80.1 (t, CHOMe), 75.7 (t, CH(CH3)2), 57.7 (p, OCH3), 55.6 (t, CHNH), 37.1 (s, ArCH2 CH2), 30.6 (s, ArCH2CH2), 21.8 ppm (2C, p, CH(CH3)2); HRMS (ESI): m/z calc. for C47H43N9O10 [M+H]+: 916.3031; found 916.3029.
Biological activity
Minimal inhibitory concentrations (MIC). MIC values were determined in standard microbroth dilution assays as described elsewhere.1, 2
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the German Center for Infection Research (DZIF) and the Bundesministerium für Bildung und Forschung (BMBF; project OpCyBac).
T. Planke, K. Cirnski, J. Herrmann, R. Müller, A. Kirschning, Chem. Eur. J. 2020, 26, 4289.
References
- 1. Baumann S., Herrmann J., Raju R., Steinmetz H., Mohr K. I., Hüttel S., Harmrolfs K., Stadler M., Müller R., Angew. Chem. Int. Ed. 2014, 53, 14605–14609; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14835–14839. [Google Scholar]
- 2.
- 2a. Hüttel S., Testolin G., Herrmann J., Planke T., Gille F., Moreno M., Stadler M., Brönstrup M., Kirschning A., Müller R., Angew. Chem. Int. Ed. 2017, 56, 12760–12764; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12934–12938. This paper also provides an extended list of new cyctobactamids; [Google Scholar]
- 2b. Moreno M., Elgaher W. A. M., Herrmann J., Schläger N., Hamed M. M., Baumann S., Müller R., Hartmann R. W., Kirschning A., Synlett 2015, 26, 1175–1178. [Google Scholar]
- 3.
- 3a. von Eckardstein L., Petras D., Dang T., Cociancich S., Sabri S., Grätz S., Kerwat D., Seidel M., Pesic A., Dorrestein P. C., Royer M., Weston J. B., Süssmuth R. D., Chem. Eur. J. 2017, 23, 15316–15321; [DOI] [PubMed] [Google Scholar]
- 3b. Kerwat D., Grätz S., Kretz J., von Eckardstein L., Seidel M., Kunert M., Weston J. B., Süssmuth R. D., ChemMedChem 2016, 11, 1899; [DOI] [PubMed] [Google Scholar]
- 3c. Grätz S., Kerwat D., Kretz J., von Eckardstein L., Semsary S., Seidel M., Kunert M., Weston J. B., Süssmuth R. D., ChemMedChem 2016, 11, 1499; [DOI] [PubMed] [Google Scholar]
- 3d. Petras D., Kerwat D., Pesic A., Hempel B., von Eckardstein L., Semsary S., Arasté J., Marguerettaz M., Royer M., Cociancich S., Süssmuth R. D., ACS Chem. Biol. 2016, 11, 1198; [DOI] [PubMed] [Google Scholar]
- 3e. Vieweg L., Kretz J., Pesic A., Kerwat D., Graetz S., Royer M., Cociancich S., Mainz A., Süssmuth R. D., J. Am. Chem. Soc. 2015, 137, 7608; [DOI] [PubMed] [Google Scholar]
- 3f. Kretz J., Kerwat D., Schubert V., Grätz S., Pesic A., Semsary S., Cociancich S., Royer M., Süssmuth R. D., Angew. Chem. Int. Ed. 2015, 54, 1969–1973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1992. [Google Scholar]
- 4. Cheng B., Müller R., Trauner D., Angew. Chem. Int. Ed. 2017, 56, 12755–12759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12929–12933. [Google Scholar]
- 5. Planke T., Moreno M., Fohrer J., Gille F., Kanakis A., Norris M. D., Siebke M., Wang L. L., Hüttel S., Müller R., Kirschning A., Org. Lett. 2019, 21, 1359–1363. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Hitotsuyanagi Y., Motegi S., Fukaya H., Takeya K., J. Org. Chem. 2002, 67, 3266–3271; [DOI] [PubMed] [Google Scholar]
- 6b. Hitotsuyanagi Y., Motegi S., Hasuda T., Takeya K., Org. Lett. 2004, 6, 1111–1114. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Bonandi E., Christodoulou M. S., Fumagalli G., Perdicchia D., Rastelli G., Passarella D., Drug Discov. Today 2017, 22, 1572–1581; [DOI] [PubMed] [Google Scholar]
- 7b. Mohammed I., Kummetha I. R., Singh G., Sharova N., Lichinchi G., Dang J., Stevenson M., Rana T. M., J. Med. Chem. 2016, 59, 7677–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Testolin G., Cirnski K., Rox K., Prochnow H., Fetz V., Grandclaudon C., Mollner T., Baiyoumy A., Ritter A., Leitner C., Krull J., van den Heuvel J., Vassort A., Sordello S., Hamed M. M., Elgaher W. A. M., Herrmann J., Hartmann R. W., Müller R., Brönstrup M., Chem. Sci. DOI: 10.1039/c9sc04769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bock V. D., Hiemstra H., van Maarseveen J. H., Eur. J. Org. Chem. 2006, 51–68. [Google Scholar]
- 10. Behroz I., Durkin P., Grätz S., Seidel M., Rostock L., Spinczyk M., Weston J. B., Süssmuth R. D., Chem. Eur. J. 2019, 25, 16538–16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary