

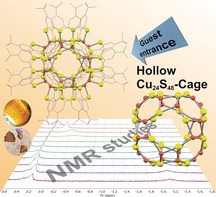
Abstract
Tetramercaptotetrathiacalix[4]arene (LH4) can be used as a coordination platform to bind four CuI ions at the thiolate and thioether S atoms. Donor ligands such as phosphanes can stabilize the resulting [LCu4] units, which then remain monomeric ([(Ph3PCu)4L]). In the absence of donor ligands, they aggregate, providing a hexamer ([LCu4]6) in high yields, with a hollow‐sphere structure formed by an unprecedented Cu24S48 cage that is surrounded by the organic framework of the calixarene chalices. Preliminary NMR experiments with regard to the host‐guest chemistry in solution showed that the compound represents a polytopic host for acetonitrile and methane.
Keywords: calixarene, cavity, cluster, copper, guest molecule
A cluster with a CuI 24 S48 nanocage can be accessed in high yields using mercaptothiacalixarenes as directing ligands. The structure contains 12 Cu2S2 diamond core building blocks featuring exceptionally short Cu⋅⋅⋅Cu distances. Detailed NMR spectroscopic investigations revealed, for instance, that it can host small molecules like acetonitrile and methane.
Calixarenes are a unique class of compounds, which—beside many other applications—are also commonly employed in coordination chemistry as flexible and broadly variable ligands. The parent calix[4]arene has four phenolate functions connected via methylene units and its metal complexes have, for instance, been utilized as mimics of oxide‐supported metal sites.1 Replacement of the methylene groups by sulfur atoms leads to thiacalix[4]arenes featuring a slightly increased ring‐size and their complexes have been extensively studied, too.2 In 1999 a variant has been reported where the hydroxo functions have been replaced by thiol units.3 The resulting tetramercaptotetrathiacalix[4]arene, LH4 (Scheme 1), represents a versatile coordination platform bearing four thiol beside four thioether donors. The thiol groups exhibit weaker hydrogen bonding interactions between each other than the hydroxo groups. In consequence, LH4 can provide a variety of conformations for coordination, but is also less likely to form a single product in complexation studies as compared to calixarenes with hydroxo groups.4 This may be one reason, why the LH4 ligand has hardly been employed in coordination chemistry. Moreover, the redox activity of the thiol groups towards metal ions poses a challenge, too.
Scheme 1.
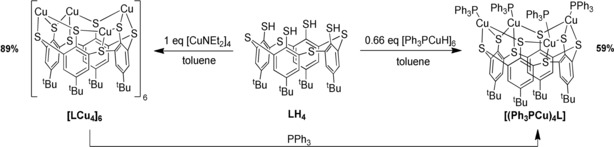
Synthesis of [LCu4]6 and [(Ph3PCu)4L].
Accordingly, examples, where LH4 has been successfully employed as a ligand in transition metal chemistry are so far limited to hexanuclear mercury complexes,5 dinuclear iridium/rhodium complexes with a 1,2‐alternate conformation4 and dinuclear tungsten/molybdenum complexes.6
We were interested in the combination of LH4 with copper(I), which should have a high affinity to the presented soft thiolate and thioether functions. In nature the latter expresses itself in a number of copper proteins, which show cysteine/methionine ligation, such as the family of metallothioneins,7 the blue copper centers or CuA centers.8 Furthermore, a family of bacterial proteins use a Cys‐rich four‐helix bundle to store large quantities of CuI.9 We have therefore explored the binding of copper(I) at LH4, which—having found suitable conditions and reagents—led us reproducibly and in good yields to a unique cluster featuring a hollow‐sphere structure composed of 24 CuI ions connected by six L4− ligands.
Initial experiments employed [Ph3PCuH]6, which represents a readily available source of copper(I) and has the ability to deprotonate the thiol groups on the calixarenes lower rim by the basic hydride ligands. Furthermore, it provides stabilizing triphenylphosphine donors that can occupy vacant coordination sites where needed. After reaction with LH4 in toluene at −80 °C, the 31P NMR spectrum of the reaction solution exhibited a single signal at −0.3 ppm.
The 1H NMR spectrum contained—apart from signals belonging to the PPh3 ligands—a singlet at 7.49 ppm caused by the aromatic protons of the calixarene and a further singlet at 1.08 ppm caused by the tert‐butyl protons, pointing to a high symmetry of the product. Layering of a toluene solution with pentane yielded yellow block‐shaped crystals. Investigation by X‐ray diffraction provided the structure of [(Ph3PCu)4L] (with two co‐crystallized molecules of toluene) as depicted in Figure 1 and Scheme 1. It consists of four copper atoms coordinated by one L4 − ligand. Each of the copper atoms is bound between two thiolate donors and one thioether group on the lower rim of the calixarene, which adopts a so‐called pinched‐cone conformation with C 2v symmetry. Through additional coordination of a triphenylphosphine ligand, each copper atom reaches a tetrahedral coordination sphere. The fact that only singlets are visible in the 1H NMR spectrum suggests that there is either a breathing motion between the pinched‐cone and the cone conformation of the complex or that the more symmetric cone conformer is the more stable one in solution.
Figure 1.
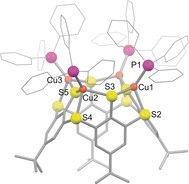
Molecular structure of [(Ph3PCu)4L]. Hydrogen atoms and co‐crystallized solvent molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: Cu1–P1 2.1888(6), Cu1–S2 2.3263(6), Cu1–S3 2.2984(6), Cu2–S5 2.2921(5); S3‐Cu1‐P1 113.24(2), S2‐Cu1‐S3 90.29(2), Cu1‐S3‐Cu2 126.82(2), S4‐Cu2‐S5 88.341(19), Cu2‐S5‐Cu3 148.08(2).19
When for the same reaction only 0.165 equiv of [Ph3PCuH]6 were used (i.e., a LH4:Cu ratio of 1:1), the formation of 0.25 equiv of [(Ph3PCu)4L] was observed by 1H NMR spectroscopy while 0.75 equiv of LH4 remained unreacted. Hence, the coordination of four copper ions by L4− leads to a very stable entity and [(Ph3PCu)4L] represents the thermodynamic sink of the system. Obviously, the strong and bulky triphenylphosphine donors supplied as part of the precursor stabilize the [LCu4] entity as a monomer and we were interested in how it behaves in the absence of such additional donors.
This required a basic copper(I) precursor lacking neutral ligands. In this context copper(I) diethylamide [CuNEt2]4 appeared ideal: it is readily soluble in toluene and capable of deprotonating thiol groups. Hence, 4 equiv of [CuNEt2]4 were reacted with LH4 at −80 °C in toluene, and after warming to r.t. the resulting solution was layered with acetonitrile, yielding large orange octahedral crystals within two days.
A subsequent single crystal X‐ray analysis revealed a structure (Figure 2), which can be considered as a hexamer of the [LCu4] building block found in [(Ph3PCu)4L], that is, [LCu4]6 (as each of the chalices hosts an acetonitrile guest molecule, the product has to be formulated correctly as [(MeCN)@LCu4]6).
Figure 2.

Left: Molecular structure of [(MeCN)@LCu4]6. Although, each of the six calixarene cavities contains one acetonitrile molecule as a guest, only one is displayed and hydrogen atoms as well as co‐crystallized solvent molecules are omitted for clarity, too; right: Structure of the core of [LCu4]6, viewed along the central axis of two opposing [LCu4] units. Selected bond lengths [Å] and angles [°]: Cu1′⋅⋅⋅Cu1# 2.4725(16), Cu1′‐S1′ 2.2524(17), Cu1′‐S2′ 2.2396(18), Cu1′‐S1* 2.4718(17), Cu1′‐S1+ 2.2408(18), S2′‐Cu1′‐S1′ 94.84(6), S1′‐Cu1′‐S1* 98.66(8), S2′‐Cu1′‐S1* 89.86(11), S1*‐Cu1′‐S1+ 114.16(5), Cu1′‐S1*‐Cu1# 63.04(8). Symmetry code: (′) −1+z, −1+x, y; (*) 1−x, −1+z, y; (#) 1−z, 0.5−y, 1.5−x; (+) −1+x, 0.5−y, 1.5−z.19
At the lower rim of each calixarene—as in case of [(Ph3PCu)4L]—an eight‐membered ring has been formed, consisting of the thiolate sulfur atoms and the coordinated copper atoms, which arrange themselves to a square (Scheme 2, left) with a diagonal of 6.5 Å. The four copper‐thiolate moieties inherent to each Cu4S4 square connect to the copper‐thiolate units of four adjacent squares in a formal “dimerization” process forming Cu2S2 diamond core motifs (Scheme 2, middle). The twelve Cu2S2 cores thus created, connect the six Cu4S4 squares to form a hollow sphere in the center of the hexamer. The six chalices of the calixarenes form the outer shell of the cluster, arranged in form of an octahedron. The central Cu24S48 cage (Figure 2) has a diameter of ≈10 Å and the volume of the cavity has been determined as 187 Å3 (see Supporting Information). NMR studies described below show that a molecule of Et2NH is located inside.
Scheme 2.
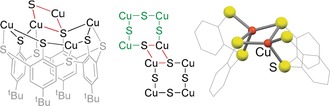
Left/middle: Connectivity of the [Cu4S4] rings that form the cluster core of [LCu4]6. Adjacent [Cu4S4] ring depicted in green; right: Cu2S2 motif, extracted from the crystal structure [(MeCN)@LCu4]6.
It has been observed in the past that thiacalixarenes, containing −OH groups instead of −SH, are capable of stabilizing spherical metal aggregates, but these do not contain purely inorganic cavities (i.e., without ancillary ligands) of a size as observed here.10 Putting the focus on CuI/CuII chalcogenide clusters in combination with smaller ligands11 certainly a variety of representatives is known, partly with interesting electronic properties, caused by Cu2S2 diamond core structural motifs.12 However, again, to the best of our knowledge, none of these clusters can be considered as hollow. The stability of the cavity in [LCu4]6 can be rationalized by a combination of the stiff structural conformation of each of the [LCu4] units and the very stable Cu2S2 entities that can be found in a large variety of copper thiolate complexes. The Cu2S2 motifs found in [LCu4]6 (Scheme 2, right) even display the shortest Cu⋅⋅⋅Cu distances (2.4725(16) Å) among all Cu2S2 motifs found in the CSD13 (where they are mostly longer than 2.6 Å, see Supporting Information); the CuI⋅⋅⋅CuI distance determined for a reduced CuA center (featuring a CuI(μ‐Scys)2CuI core) of 2.51 Å belongs to the shortest representatives.14 DFT calculations on [LCu4]6 reproduced the rather short Cu⋅⋅⋅Cu distances (deviation of 0.083–0.031 Å depending on the functional used, see Supporting Information), and a similar value was predicted for an extract of the structure as depicted in Scheme 2 (right hand side), so that the phenomenon is intrinsic to the Cu2S2 core with this ligation.
To confirm that the structure of [LCu4]6, as determined by X‐ray structure analysis for the solid state, remains intact in solution, DOSY NMR measurements were performed, that is, the diffusion coefficient was used to determine the hydrodynamic radius of the molecules in solution via the stokes Einstein equation (see Supporting Information). A DOSY NMR spectrum of [(Ph3PCu)4L] was also recorded for comparison. The results indicate that no fragmentation of [LCu4]6 occurs upon dissolution in C6D6 or [D8]THF. Addition of Ph3P, however, leads to a break‐up of the structure and conversion to [(Ph3PCu)4L] as shown by 1H NMR spectroscopy (Scheme 1).
The 1H NMR spectrum of [LCu4]6 dissolved in C6D6 exhibits a singlet at 0.99 ppm caused by the tert‐butyl groups. The aromatic protons give rise to two doublets at 7.64 and 7.50 ppm, suggesting—in contrast to the spectrum of [(Ph3PCu)4L]—a rigid conformation in solution. In addition to these signals, the 1H NMR spectrum showed three resonances at 5.01, 4.01, and 3.20 ppm with an integral ratio of 4 to 1 to 6, which did not disappear after drying of the powdered solid under vacuum for 6 d. COSY, HSQC, HMBC and DOSY spectra lead to the conclusion that they have their origin in one molecule of Et2NH that is located in the [LCu4]6 cage (see Supporting Information) and likely has served as a template. Based on this information, an analysis of the voids in the X‐ray structure showed residual electron density in the Cu24S48 cage and thus confirmed the possibility of a heavily disordered molecule of Et2NH present there (see SI and Figure 3). As C4H11N has a van der Waals volume15 of 94 Å3, 93 Å3 of the 187 Å3 cavity remain empty. Considering the suggestion that a packing coefficient of around ≈55 % is favorable for molecular cavities,16 it is unlikely that a stable situation can be reached where other molecules permanently reside in the central cavity beside Et2NH.
Figure 3.
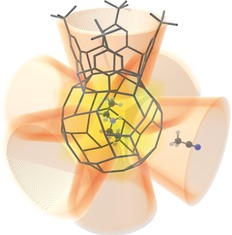
Cavities of [LCu4]6, hosting the guest molecules Et2NH (in the centre of the Cu24S48 cage (yellow)) and MeCN (in the calixarene calices (orange)). For further illustration parts of the concrete structure of [LCu4]6 have been included, namely the frameworks of the central cage and of one of the six calixarene chalices in form of grey sticks. The rest of the molecule is symbolized by truncated cones. Only one of the six MeCN molecules is displayed.
However, also calixarene units can act as hosts for small molecules,17 as already illustrated by the fact that [LCu4]6 crystallized with six acetonitrile molecules located in the chalices (see Figures 2 and 3). Hence, we were interested in its behavior as a host in solution. In this context it was interesting to note that the 1H NMR spectrum of [LCu4]6 (crystals dried under high vacuum for 16 h dissolved in C6D6) exhibits—besides the signals mentioned above—a further resonance at −0.5 ppm that could not be assigned to any proton of [LCu4]6. This signal disappears only after prolonged (60 h) drying of [LCu4]6 under high vacuum. To determine whether it is caused by MeCN, an NMR titration experiment was performed (see Supporting Information). [LCu4]6 was first exposed to a high vacuum for 60 h, so that the region between 0.0 ppm and −2.5 ppm of its 1H NMR spectrum was free of signals. Then the solution was titrated with small quantities of acetonitrile and 1H NMR spectra were recorded after each addition. In the first spectrum, a new signal was detected at −0.8 ppm, shifting with each subsequent addition to higher chemical shifts. This can be rationalized assuming that the signal represents an average of two resonances, which was confirmed by a low temperature NMR experiment (see Supporting Information): When a solution of [LCu4]6 in [D8]toluene/C6D6 with 12 equiv of MeCN was cooled in 10 °C steps to −70 °C, an initial signal at 0.00 ppm disappeared below −10 °C and at −30 °C two new signals evolved at 0.29 ppm and −2.18 ppm with an integral ratio of 2:1. Such an upfield shifted resonance (−2.18 ppm) for methyl protons of acetonitrile suggest their presence in the center of the calix cavity,18 which is a highly shielded region for the acetonitrile molecule, especially when the methyl group is oriented towards the lower rim as revealed for the crystal structure of [(MeCN)@LCu4]6. Accordingly, the signal at −2.18 ppm was assigned to acetonitrile molecules in the guest position within the calixarene ligands of [LCu4]6. The signal at 0.29 ppm can be assigned to free acetonitrile in C6D6/[D8]toluene at −70 °C.
Having found that the central cavity of [LCu4]6 is occupied by a molecule of Et2NH, the question arose whether it can be replaced by other small molecules entering through the calices and whether the latter can incorporate also other molecules than MeCN, rendering [LCu4]6 to a polytopic host (compare Figure 3). Exemplarily, the behavior of methane in contact with [LCu4]6 was tested: CH4 gas was bubbled through a solution of [LCu4]6 (not containing any MeCN) in a mixture of [D8]toluene/C6D6 for 30 s in a Young NMR tube. The 1H NMR spectrum recorded subsequently exhibits a new signal at 0.11 ppm (see Supporting Information). In a low temperature NMR experiment, where the solution was cooled to −70 °C in steps of 10 °C, the signal at 0.11 ppm disappeared below −70 °C. No new signals evolved in the 1H NMR spectrum. A ROESY NMR spectrum of the sample revealed coupling through space between the methane protons and the aromatic protons of [LCu4]6, leading to the conclusion that at least a part of the methane molecules have become guests within the calixarene cavities. The NMR spectra clearly showed that the Et2NH molecule remains in the central Cu24S48 cage.
Since the electron transfer unit CuA contains two cysteinate‐bridged Cu centers and [LCu4]6 contains twelve similar entities, we were finally also interested in the electrochemical properties of [LCu4]6. Cyclovoltametric measurements using THF as the solvent exhibited three reversible oxidation events at 1 E 1/2=−0.475 V, 2 E 1/2=−0.190 V and 3 E 1/2=0.085 V (Figure 4; the events display a quite large peak separation of 0.16 to 0.21 V, but under the same conditions the CoCp2/CoCp2 + couple exhibits a peak separation of 0.17 V, too). This suggests that [LCu4]6 undergoes three reversible one electron processes. Solvents with a larger oxidative window, employed to investigate whether even more electrons can be removed, led to decomposition of the product (DCM) or could not be used due to insufficient solubility of [LCu4]6.
Figure 4.
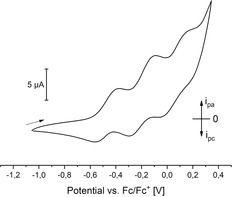
Cyclic voltammogram of [LCu4]6 recorded vs. Fc/Fc+ (100 mm TBAPF6, GC/Pt/Ag electrodes, 100 mV s−1) of [LCu4]6 in THF (1.1 mm) at 293 K.
In conclusion, using the tetramercaptotetrathiacalix[4]arene ligand, we have accessed a novel type of copper(I) thiolate cluster with a unique hollow inorganic ball structure. The central cage cavity hosts an Et2NH molecule and the surrounding calixarene units, which represent windows to the cavity, can host small molecules such as CH4 or MeCN. The results thus stimulate further, more detailed investigations on the host‐guest chemistry of this cluster as well as an extension of the studies to other noble metal ions. Corresponding research is underway.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to the Humboldt‐Universität zu Berlin for financial support and the cluster of excellence UniSysCat for valuable discussions.
N. Frank, A. Dallmann, B. Braun-Cula, C. Herwig, C. Limberg, Angew. Chem. Int. Ed. 2020, 59, 6735.
References
- 1.
- 1a. Floriani C., Chem. Eur. J. 1999, 5, 19–23; [Google Scholar]
- 1b. Floriani C., Floriani-Moro R. in Advances in Organometallic Chemistry, Academic Press, New York, 2001, pp. 167–233; [Google Scholar]
- 1c. Hoppe E., Limberg C., Chem. Eur. J. 2007, 13, 7006–7016. [DOI] [PubMed] [Google Scholar]
- 2. Kumar R., Lee Y. O., Bhalla V., Kumar M., Kim J. S., Chem. Soc. Rev. 2014, 43, 4824–4870. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Rao P., Hosseini M. W., de Cian A., Fischer J., Chem. Commun. 1999, 2169–2170; [Google Scholar]
- 3b. Akdas H., Graf E., Hosseini M. W., Rao P., de Cian A., J. Supramol. Chem. 2002, 2, 21–28. [Google Scholar]
- 4. Hirata K., Suzuki T., Noya A., Takei I., Hidai M., Chem. Commun. 2005, 3718–3720. [DOI] [PubMed] [Google Scholar]
- 5. Akdas H., Graf E., Hosseini M. W., de Cian A., Bilyk A., Skelton B. W., Koutsantonis G. A., Murray I., Harrowfield J. M., White A. H., Chem. Commun. 2002, 1042–1043. [DOI] [PubMed] [Google Scholar]
- 6. Buccella D., Parkin G., Chem. Commun. 2009, 289–291. [DOI] [PubMed] [Google Scholar]
- 7. Henkel G., Krebs B., Chem. Rev. 2004, 104, 801–824. [DOI] [PubMed] [Google Scholar]
- 8. Belle C., Rammal W., Pierre J.-L., J. Inorg. Biochem. 2005, 99, 1929–1936. [DOI] [PubMed] [Google Scholar]
- 9. Straw M. L., Hough M. A., Wilson M. T., Worrall J. A. R., Chem. Eur. J. 2019, 25, 10678–10688. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Bi Y., Du S., Liao W., Coord. Chem. Rev. 2014, 276, 61–72; [Google Scholar]
- 10b. Guan Z.-J., Zeng J.-L., Nan Z.-A., Wan X.-K., Lin Y.-M., Wang Q.-M., Sci. Adv. 2016, 2, e1600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Fuhr O., Dehnen S., Fenske D., Chem. Soc. Rev. 2013, 42, 1871–1906; [DOI] [PubMed] [Google Scholar]
- 11b. Dance I. G., Polyhedron 1986, 5, 1037–1104. [Google Scholar]
- 12. Maji R. C., Das P. P., Bhandari A., Mishra S., Maji M., Ghiassi K. B., Olmstead M. M., Patra A. K., Chem. Commun. 2017, 53, 3334–3337. [DOI] [PubMed] [Google Scholar]
- 13. Groom C. R., Bruno I. J., Lightfoot M. P., Ward S. C., Acta Crystallogr. Sect. B 2016, 72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blackburn N. J., de Vries S., Barr M. E., Houser R. P., Tolman W. B., Sanders D., Fee J. A., J. Am. Chem. Soc. 1997, 119, 6135–6143. [Google Scholar]
- 15. Bondi A., J. Phys. Chem. 1964, 68, 441–451. [Google Scholar]
- 16. Mecozzi S., J. Rebek, Jr. , Chem. Eur. J. 1998, 4, 1016–1022. [Google Scholar]
- 17. Kotzen N., Vigalok A., Supramol. Chem. 2008, 20, 129–139. [Google Scholar]
- 18. Singh N., Kumar M., Hundal G., Tetrahedron 2004, 60, 5393–5405. [Google Scholar]
- 19.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201915882 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary