

Abstract
Redox control is lost when the antioxidant defense system cannot remove abnormally high concentrations of signaling molecules, such as reactive oxygen species (ROS). Chronically elevated levels of ROS cause oxidative stress that may eventually lead to cancer and cardiovascular and neurodegenerative diseases. In this review, we focus on redox effects in the vascular system. We pay close attention to the subcompartments of the vascular system (endothelium, smooth muscle cell layer) and give an overview of how redox changes influence those different compartments. We also review the core aspects of redox biology, cardiovascular physiology, and pathophysiology. Moreover, the topic‐specific knowledgebase DES‐RedoxVasc was used to develop two case studies, one focused on endothelial cells and the other on the vascular smooth muscle cells, as a starting point to possibly extend our knowledge of redox control in vascular biology.
Keywords: cardiovascular diseases, cardiovascular system, reactive oxygen species, redox
Abbreviations
- Ang II
angiotensin II
- AP‐1
activator protein
- CVD
cardiovascular disease
- EC
endothelial cells
- EGFR
epidermal growth factor receptor
- ET‐1
endothelin‐1
- H2O2
hydrogen peroxide
- MMP
matrix metalloproteinase
- NF‐κB
nuclear factor‐kappaB
- NO
nitric oxide
- ONOO−
peroxynitrite
- OxS
oxidative stress
- PDGF
platelet‐derived growth factor
- PDGFR‐β
platelet‐derived growth factor receptor beta
- PTK
protein tyrosine kinases
- PTP
protein tyrosine phosphatase
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rats
- SM‐MHC
smooth muscle‐myosin heavy chain
- TF
tissue factor
- TF‐FVIIa
tissue factor‐FVIIa
- TFPI
tissue factor pathway inhibitor
- t‐PA
tissue‐type plasminogen activator
- u‐PA
urokinase‐type plasminogen activator
- VSMC
vascular smooth muscle cells
- XO
xanthine oxidase
1. INTRODUCTION
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) stimulate redox homeostasis mechanisms, and thus, they are considered critical signaling molecules for maintaining cellular homeostasis. However, these signaling molecules can be harmful, as well. That is, when ROS/RNS formation outpaces the ROS/RNS removal processes, oxidative stress (OxS) occurs. Chronic OxS leads to several pathologies.1, 2 In the vascular system, ROS is produced by the vascular smooth muscle cells (VSMCs), endothelial cells (EC), and adventitial cells, among others.3 The endothelium regulates the vascular homeostatic system, as it is the barrier that separates blood and tissue. However, when the antioxidant defense system does not remove the excess ROS, it can cause endothelial dysfunction.4 This perturbation of the homeostatic functioning of the vascular EC leads to pathologies such as tissue ischemia and atherothrombosis.5 ROS is also involved in other pathophysiological processes such as hypertension, inflammation, and vascular remodeling,6, 7, 8, 9 which leads to vascular pathologies, including atherosclerosis, arterial hypertension, and cardiovascular diseases (CVDs). Moreover, ROS‐generating systems also facilitate the development of diabetes mellitus, obesity, and hypercholesterolemia, which increases the risk of developing vascular pathologies.2
1.1. ROS in general
Reactive species are molecules interacting with various biomolecules, including nucleic acids, lipids, and proteins, and generating electron‐deficient species, leading to cell and tissue damages. Different endogenous metabolic and biochemical reactions and exogenous sources, such as ionizing radiation, generate these reactive species. The term “reactive oxygen species” refers to oxygen metabolites, such as superoxide anion radical (O2 •−), singlet oxygen (1O2), hydroxyl radical (OH• ─), and perhydroxyl radical (HO2 •), which are yielded by the reduction of the oxygen molecule (O2) with two uncoupled electrons.10, 11, 12 Furthermore, ROS with an unpaired electron are free radicals; hence, they are also named oxygen radicals or oxygen‐free radicals,12 and free radicals are chemical species that are capable of independent existence with one or more unpaired electron. The unpaired electron transforms the chemical reactivity of the molecule, leading to its increased reactivity compared with the nonradical form of the molecule.10, 13 Thus, free‐radical forms of ROS can remove electrons from other molecules to gain stability and by that causes new free radical formation, initiating a chain reaction cascade, which consequently impairs the cell/tissue functioning.14 In addition, the term ROS cover all oxygen‐containing reactive species, namely, alkoxyl radical (LO•), peroxyl radical (LOO⁃), lipid hydroperoxide (LOOH), peroxynitrite anion (ONOO─), hypochlorous acid (HOCl), and ozone (O3),11, 12, 13 from which some are without unpaired electrons (hydrogen peroxide (H2O2), ONOO─, HOCl, and O3), and as such, they are not free radicals.13
In biological systems, ROS produced by cellular metabolism exert both deleterious, cytotoxic effects, as well as beneficial effects13 such as antimicrobial activity,15 metabolic pathways regulation,16 and cell signaling.17 Redox signaling represents pathways in which ROS or some other reactive species serve as messengers to induce cellular responses via redox reactions, implicated in many different physiological and pathological processes in the organism. Thus, ROS can provoke redox modulation of protein kinase cascades and transcription factors leading to different cellular responses, such as cell proliferation and differentiation, altered expression, or synthesis of cytokine and/or adhesion molecules.12 Owing to the mostly deleterious nature of ROS, mammals, including humans, have developed a robust antioxidant protection system through the action of enzymes such as Cu─Zn superoxide dismutase (Cu─Zn SOD), catalase, glutathione peroxidase, glutathione reductase, and glutathione‐S‐transferase. Moreover, several diet compounds, particularly from fruits and vegetables, also exhibit antioxidant activities.12 An imbalance between ROS generation and removal rate leads to OxS,12 and chronic OxS leads to different pathologies, including the vascular pathophysiology.1, 2
1.2. Vascular physiology in general
The vascular system is finely synchronized to adjust the blood flow to the metabolic requirements of the body and is made up of a heart and a closed network of vessels (arteries, veins, and capillaries) that supply oxygen and nutrients to every tissue to maintain cellular homeostasis. Endothelium lines the interior surface of blood vessels—a single layer of homogenous EC layer being in connection with blood and lymph from the circulation and represents a surface (blood/endothelium interface) with roughly 300–1,000 m2, whereas the mass is approximately 110 g.18, 19 VSMC surround the endothelium, establishing the outer layer of arteries. The fibroelastic connective layer and internal elastic lamina located below endothelium provide flexibility and stability for EC. Perivascular cells (pericytes) also surround the EC—multipotential stem cells with the possibility to differentiate into different types of cells among other VSMC, fibroblasts, osteoblasts, connective tissue cells, and adipocytes, providing the EC and VSMC with a stable microenvironment and contractile ability.20
EC have a vital function in tissue homeostasis by the regulation of solute and macromolecule transport through the vessels. A glycocalyx layer on the EC surface provides a locally charged barrier to cell and protein movement from the blood through endothelium under physiological conditions.21 The endothelium also regulates and maintains vascular tone through interaction with the peripheral nervous system and by synthesizing and releasing vasodilatory factors such as endothelin‐1 (ET‐1), thromboxane, endothelium‐derived hyperpolarizing factor, nitric oxide (NO), and prostacyclin.22 Another endothelium function is hemostasis regulation. That is, the endothelium synthesizes compounds, which maintain blood fluidity and are involved in the formation of nonthrombogenic surface and coagulation processes. EC are exposed to lipids (present in the circulation and accumulating in the subendothelial regions) and are also involved in immunological and inflammatory processes, which are associated with atherosclerosis and occlusive vascular disorders.23
Furthermore, the endothelium is responsible for the reaction to pathophysiological conditions such as infection or trauma of the neighboring tissues, vessel remodeling, and growth,24, 25, 26 therefore having significant diagnostic and therapeutic potential. EC is capable of synthesizing most of the proteins that constitute the basal lamina and enzymes involved in extracellular matrix remodeling such as matrix metalloproteinase (MMP), which is vital for the blood vessel plasticity and angiogenesis.27 Thus, endothelium represents an essential and dynamic endocrine organ due to its different functions as alluded to above.
The fibrinolytic system is a system that restores a blood vessel when the blood clot is not needed, and it includes an inactive proenzyme, the plasminogen, and an active form, plasmin that is responsible for fibrin degradation. The activity of plasminogen activators (PA) such as tissue‐type PA (t‐PA) and urokinase‐type PA (u‐PA) regulates the fibrinolytic potential of the vasculature.28 Activation of the EC and the proinflammatory and procoagulant response lead to the synthesis and release of u‐PA.29, 30 The constitutively produced protein of EC, the PA inhibitor type I (PAI‐1), suppresses t‐PA as well as u‐PA. The activity of PAI‐1 is an independent risk factor for CVD.31 The EC synthesized and released 13‐hydroxyoctadecadienoic acid, and the vasodilator factors prostacyclin and NO, which prevents adhesion, aggregation, and activation of platelets.32 EC also acts as a natural anticoagulant by expressing the receptor for thrombin, the thrombomodulin, responsible for thrombin conversion into anticoagulant protein from a procoagulant protease. The thrombin bound to thrombomodulin lids to protein C activation, which then binds to the endothelial protein C receptor. Protein C (blood coagulation factor XIV or autoprothrombin IIA) is an anticoagulant serine protease, which participates in coagulation of the blood. Specifically, activated protein C associates with its cofactor, protein S, and inhibit the coagulation process through the inactivation of coagulation factors FVa and FVIIIa.33 EC surface has heparin‐like sulfated molecules of glycosaminoglycan, which bind/activate the antithrombin that is the FXa and thrombin leading inhibitor.
Furthermore, the inhibition of the tissue factor‐FVIIa (TF‐FVIIa) complex occurs by its interaction with a specific polypeptide, the tissue factor pathway inhibitor (TFPI), and formation of a stable quaternary complex TFPI‐TF‐FVIIa‐FXa.34 Thus, EC prevent blood clotting via a mechanism that involves the heparin‐like molecule thrombomodulin, together with NO and prostacyclin. When an endothelial injury occurs, EC stop the secretion of coagulation and aggregation factors, and synthesize and secrete a large multimeric protein called the von Willebrand factor, which initiates the hemostasis maintenance after injury.35 In addition, EC produces the lipid‐mediator platelet‐activating factor, which activates platelets and their attachment to EC.36
VSMC represent one of the most frequent types of cells in arteries. VSMC are also essential for the homeostasis of the vasculature, as well as for the vasculature contractions and relaxations, which are responsible for the blood vessel luminal diameter alterations to maintain blood pressure within the normal range. VSMC contractility is controlled regularly by exchanging two different phenotypes.37 Different states of VSMC are noted among both the VSMC of the same and various blood vessels. The mature, differentiated VSMC are contractile, and their phenotype is elongated and spindle‐shaped, and these VSMC display low rates of migration and proliferation and express increased levels of proteins that are important for contractility. VSMC shuttle to a dedifferentiated synthetic phenotype with rhomboid shape in certain physiological conditions, including pregnancy or exercise, and after injury of the vasculature, which is vital during vessel remodeling.38 Dedifferentiated VSMC are exceptionally proliferative and migratory, and synthesizes extracellular matrix proteins, like elastin and collagen.39 Based on all these properties, VSMC are capable of regulating the vessel diameter in short terms and in long terms VSMC are responsible for adaptation by structural remodeling through changing the number of cells and the constitution of connective tissue. The VSMC marker proteins that are most significant in defining VSMC phenotypes include cellular retinol binding protein‐1, smooth muscle myosin heavy chain (SM‐MHC), α‐smooth muscle actin, SMemb/nonmuscle MHC‐B, and smoothelin A and B.38 A few of these proteins are structural components of the contractile apparatus or act as the contraction regulators. Generally, the loss of proteins essential to the contractile phenotype indicates the synthetic phenotype. The expression of the two marker proteins, SM‐MHC, and smoothelin, characterizes a mature contractile VSMC phenotype.40
2. EFFECTS OF ROS
The human body uses a cellular antioxidant defense system to neutralize free radicals and, consequently, OxS. In OxS, ROS are involved directly or indirectly in the macromolecules deterioration, along with oxidative deterioration of nucleic acids, proteins, and lipids.41 However, ample evidence suggests that ROS directly activate OxS‐responsive pathways regulating different cellular processes, additionally leading to the progression of diseases. OxS is associated with carcinogenesis,42 neurodegeneration,43 atherosclerosis, diabetes,3 and aging.44 ROS are also critical physiological modulators of the redox state signaling molecules.45 ROS changes the function of the protein by the mechanism that includes the modifications of cysteine (Cys) residues, which are redox‐reactive. Cys residue oxidation promotes the formation of reactive sulfenic acid (─SOH) that can interact with nearby Cys‐forming disulfide (─S─S─) bonds or endure additional oxidation to sulfinic (─SO2H) or sulfonic (─SO3H) acid.46 These modifications (except sulfonic acid and to a lower extent sulfinic acid) reverse specific reducing systems including peroxiredoxin and thioredoxin, suggesting that these modifications are involved in redox sensing and signaling and regulation of protein structure/function.47 Numerous studies show that ROS influence many different signaling pathways involving molecules, such as tyrosine and Rho kinases, mitogen‐activated protein (MAP) kinases, transcription factors (HIF‐1, nuclear factor‐kappaB [NF‐κB], and activator protein‐1 [AP‐1]), as well as protein tyrosine phosphatases (PTPs) involved in cardiovascular, neural, and renal cell function.48, 49 ROS elevates the concentration of intracellular free Ca2+ ([Ca2+]i) by the activation of ion channels, and ROS upregulates the expression of proinflammatory and proto‐oncogene genes (Figure 1).50
Figure 1.
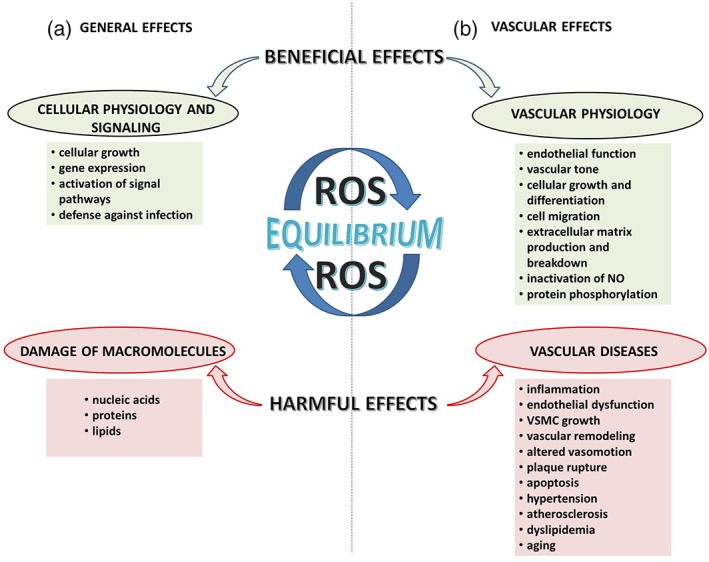
Effects of ROS in physiological and pathophysiological conditions
3. REDOX SIGNALING IN THE VASCULAR PATHOPHYSIOLOGY
In vascular tissue, diverse systems generating ROS interrelate to establish a combined redox modulation.51 Nonetheless, the majority of tissue damage and pathology is a result of excessive ROS production, which leads to inflammation in the vascular tissues, while excessively low levels of ROS interrupt the oxidant physiological role in vasodilation and cellular growth.52 Furthermore, it has been suggested that “intermediate” ROS levels could facilitate the physiological response in the vasculature, while chronically elevated ROS level characteristic for OxS could be associated with CVD.53 Hence, ROS participate in the pathophysiology of the vascular disease, but also regulate vascular function in healthy vessels54 (Table 1). In addition, ROS is part of an adaptive response in vascular diseases.87 In Table 2, we summarized some of the effects of ROS on vascular physiology in animal and human studies. These distinctive roles of ROS may be a consequence of the different physicochemical properties of ROS. For example, H2O2 biological half‐life is expanded compared with the half‐life of O2 − and OH− and can diffuse through lipid membranes, and the charge of the O2 − molecule prevents it from crossing the cell membranes except for the possibility to cross the membrane through ion channels.3 Besides, reductive stress (RS) is another deleterious factor that disturbs the redox state and promotes vascular pathophysiology.103, 104, 105 Furthermore, RS increases s‐glutathionylation of proteins, and that may be involved in uncoupling of eNOS that elevates O2 − production and induces OxS.103, 104, 105, 106
Table 1.
Some ROS species important for vascular physiology and pathophysiology
| ROS | Mechanism of generation/enzymatic source | Physiological concentrations | Reference | Elevated concentrations | Reference |
|---|---|---|---|---|---|
| •O2 |
|
|
55 |
|
|
|
|
||||
|
|
56 | |||
|
58 | ||||
| H2O2 |
|
|
55, 66 |
|
|
|
55 | ||||
|
55 | ||||
|
55 | ||||
|
55 | ||||
| 55, 72, 73, 74, 75, 76 | |||||
| 77 | |||||
| •OH |
|
|
|
||
| ONOO− |
|
|
|
Note: Under physiological conditions, ROS in the vasculature are produced in a controlled manner at low concentrations and function as signaling molecules. Increased ROS production leads to pathological conditions of vascular system.
Abbreviations: •OH, hydroxyl radical; BH4, tetrahydrobiopterin; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; H2O2, hydrogen peroxide; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; O2, superoxide anion; ONOO−, peroxynitrite; sGC, soluble guanylate cyclase; SOD, superoxide dismutase; VSMC, vascular smooth muscle cells.
Table 2.
The effects of ROS on vascular physiology‐ animal and human studies
| Effects of ROS | Condition and/or treatment | Species | Ref. | ||
|---|---|---|---|---|---|
|
↑ OxS ↑ XO activity |
|
dTGR SDR |
88 | |
|
↑ XO activity | SHR | 89 | ||
|
↑ XO activity |
|
SHR DSS rats |
90, 91 | |
|
↑ O2 − ↑ p22phox mRNA |
|
SHR WKY |
92 | |
|
↑ ROS |
|
Rat VSMC | 93 | |
|
↑ O2 − |
|
94 | ||
|
↑H2O2 ↑O2 − |
|
95 | ||
|
↑ O2 − |
|
Hypertensive DSS rat |
58 | |
|
↑ superoxide production ↑ ET‐1 |
|
DOCA‐salt hypertensive mice and rats | 96, 97 | |
|
↑ O2 − |
|
Mice/ Aortic rings |
46 | |
|
↑ ROS |
|
Nox1‐deficient (−/Y) mice |
98 | |
|
↑ superoxide production | VSMC TgSMCnox1 | 99 | ||
|
↑ O2 − | p47phox−/−deficient mice | 63 | ||
|
↑ ROS |
|
iNOS−/− mice | 100 | |
|
‐ |
|
C57BL/6 TG and WT mice |
101 | |
|
↑ O2 − production |
|
Hypertensive and normotensive patients | 102 | |
|
↑ ROS | ‐ | Hypertensive and normotensive patients | 53 | |
Abbreviations: ↑, increased; ↓ decreased; Ang II, angiotensin II; DOCA, salt‐deoxycorticosterone acetate‐salt; DSS, Dahl salt‐sensitive; dTGR, double‐transgenic rats; ET‐1, endothelin‐1; iNOS−/−, iNOS‐deficient; ONOO−, peroxynitrite; OxS, oxidative stress; PMA, phorbol myristate acetate; SDR, Sprague Dawley rats; SHR, spontaneously hypertensive rats; TG, transgenic; TgSMCnox1, transgenic mice overexpressing Nox1 in smooth muscle cells; VSMC, vascular smooth muscle cell; WKY, Wistar‐Kyoto rats; WT, wild type (nontransgenic); XO, xanthine oxidase.
3.1. Xanthine and NAD(P)H oxidase as the culprit of ROS in vascular pathophysiology
The most prominent origins of ROS in the vascular tissue are xanthine oxidase (XO) and NAD(P)H oxidase. NAD(P)H oxidase regulates the conversion of xanthine dehydrogenase (XDH) to XO. The modulation of XDH to XO reduces oxygen to O2 − and H2O2. XO regulates the oxidation process of hypoxanthine and xanthine and generates O2 − in the vascular endothelium.3, 107
The increase in XO activity has been associated with endothelial dysfunction in rats overexpressing renin and angiotensinogen from implanted human genes.88 On the other hand, in spontaneously hypertensive rats (SHR), increased activity of XO was found in the microcirculation of the mesentery.89 The increased XO activity has also been found in the renal tissue of SHR and Dahl salt‐sensitive (DSS) rats, while the discovery that allopurinol can correct the hypertrophy of renal and cardiac tissue in SHR without lowering the blood pressure implies the involvement of XO in organ damage induced by elevated blood pressure.90, 91 Models for ischemia–reperfusion injury and heart failure also demonstrated the involvement of XO.3
NAD(P)H oxidase is an enzyme complex that uses NADH/NADPH as the donor of an electron in the reduction of O2 and consequent formation of superoxide. The membrane‐bound p22phox subunit is crucial for the NAD(P)H oxidase complex activation and is accompanied by an elevated production of O2 − in humans with coronary artery disease.108 The increased levels of OxS byproducts, together with elevated oxidative DNA damage, were found in samples obtained from patients with arterial hypertension.53
The mononuclear cells isolated from the blood of patients with essential hypertension exhibited increased O2 − production after treatment with angiotensin II (Ang II) or ET‐1 when compared with controls.102 The majority of studies investigating NAD(P)H oxidase activation in vascular cells focused on Ang II signaling, which implicates receptor tyrosine kinases, protein kinase C, c‐Src, and phospholipase D.109 The expression/activity of NAD(P)H oxidase was found to be increased in rats with hypertension caused by Ang II treatment.94 A NAD(P)H oxidase inhibitor, decreasing both vascular O2− synthesis and hypertension in mice, supports this finding.110 The NAD(P)H oxidase Nox1 isoform participates in the Ang II‐induced hypertensive response. The reduced production of O2 − and the blunted effect of Ang II on the blood pressure were both found in Nox1‐deficient mice.98
On the other hand, the overexpression of Nox1 enhances Ang II‐induced O2 − formation, VSMC hypertrophy, and hypertension in transgenic mice.99 Furthermore, in cytosolic NAD(P)H p47phox subunit‐deficient mice, the endothelial dysfunction, O2 − production, and Ang II‐induced hypertensive response were blunted.63 Moreover, NAD(P)H oxidase was associated with raised O2 − production in the arterial tissue of SHR.92
The produced ROS from NAD(P)H can react with products of nitric oxide synthase (NOS), which may further amplify vascular function. For example, O2 − generated from NAD(P)H oxidase reacts with NO, forming peroxynitrite (ONOO−), which consequently induces changes in the NOS enzyme subunits resulting in further O2 − formation and the declination of NO bioavailability, which impairs endothelium‐dependent relaxation in a model of salt‐induced hypertensive rats.111 ROS synthesized by NAD(P)H oxidase is required for iNOS expression in microvascular EC.112
The membrane‐bound gp91phox‐containing NAD(P)H oxidase is identified as a source of excessive O2 − linked with elevated activity of the renin–angiotensin system as shown in renovascular113 and salt‐induced hypertension114 models. Treatments with the angiotensin receptor blocker and with the gp91phox NAD(P)H oxidase inhibitor restrained the elevated O2 − aortic generation and the expression of the proinflammatory molecules in a model of hypertensive DSS rats. Importantly, ROS‐induced injury was shown to lead to further ROS formation while proinflammatory cytokines activated the system of NAD(P)H oxidase.58 The JAK/STAT cascade, which regulates the transcription of proinflammatory genes, is also upregulated by ROS synthesized by the Ang II and platelet‐derived growth factor (PDGF) activity. Inhibition of the NAD(P)H oxidase subunit p47phox inhibits Ang II‐induced activation of JAK/STAT signaling and interleukin‐6, which suggests that the NAD(P)H oxidase activity generates the ROS involved in this pathway.115
Furthermore, ROS produced at one subcellular locus can initiate ROS synthesis in another locus by signal transmission. For example, mitochondria‐derived ROS production activates ROS formation by NAD(P)H oxidase.116 The NAD(P)H oxidase increased activity in pulmonary microvascular EC produces extracellular O2 −. The extracellular O2 − moves through chloride channel 3 into cells, where it triggers Ca2+ mobilization and mitochondrial O2 − production. Thus, ROS generated from endothelial NAD(P)H oxidase mediates intracellular signaling.117, 118
The ET‐1 also induces an increase in ROS generation by the activation of NAD(P)H oxidase pathway. ET‐1 via ETA receptor activation increases O2 − production in the DOCA‐salt hypertensive rat model, while the ETA receptor inhibition decreases the production of vascular superoxide.96, 97 Moreover, endothelium‐restricted human ET‐1 overexpression was shown to cause dysfunction of the endothelium and vascular remodeling in mice through the NAD(P)H oxidase pathway.101 Besides, NAD(P)H oxidase pathway, dysfunction of endothelium, and vascular remodeling can be the result of an activated extracellular signal–regulated kinases (ERK) signaling pathway. Numerous studies have shown that increased level of oxLDL induces accumulation of ROS, which stimulates ERK phosphorylation, upregulates the expression of endothelial transcriptional factor AP‐1 and ET‐1, and, consequently, promotes the progression of atherosclerosis.119, 120, 121
Activation of NAD(P)H oxidase is also involved in the pathogenesis of abdominal aortic aneurysm. That is, samples obtained from subjects suffering from aneurysms of the abdominal aorta exhibit overexpressed NADPH oxidase and raised level of O2 − in the endothelium and within the aortic wall. ROS synthesized by the NAD(P)H oxidase activity facilitate the inflammatory process in the aortic endothelium and stimulate the extracellular matrix impairment through the MMP‐2 activation and apoptosis of VSMC. A murine aneurysm model was used to show that inhibition of ROS mitigates the formation of the aneurysm.100 Also, the cyclic stretch of VSMC stimulates the expression of MMP‐2. This response is blunted in murine cells deficient in the p47phox subunit of NAD(P)H oxidase, thus implying the function of ROS generated by the NAD(P)H oxidase in MMP‐2 signaling in VSMC.122 Moreover, Ang II induces MMP‐2 via the p47phox subunit.123 Also, ROS generated by macrophage‐derived foam cells modifies the activity of MMP‐2 and MMP‐9 in vitro.124
Various growth factors (such as PDGF and transforming growth factor β) and hormones are involved in the modulation of NAD(P)H oxidase activity/expression, while ROS regulates the activity of many signaling enzymes (tyrosine and MAP kinases, PTPs) in the vasculature.3 Redox signaling also mediates the activity of transcription factors in the vascular cells. In addition, the laminar flow and the shear stress influence the ROS activity in both physiological and pathophysiological settings.
3.2. The effects of ROS on tyrosine and MAP kinases, and PTPs in vascular pathophysiology
ROS regulates the tyrosine kinases (receptor and nonreceptor forms) in the vasculature. Receptor tyrosine kinases, PDGF receptor beta (PDGFR‐β), and the epidermal growth factor receptor (EGFR) require ligand‐stimulated signal transduction. It appears that H2O2 generation is mandatory for signal transmission through PDGFR‐β.125, 126 Furthermore, Ang II stimulates EGFR and PDGFR‐β through transactivation. The nonreceptor tyrosine kinase Src also contributes to the H2O2‐dependent Ang II‐stimulated EGFR transactivation.127 Activation of tyrosine kinases regulates NAD(P)H oxidase, which additionally amplifies ROS production in the vascular tissue. The transactivation of EGFR and PDGFR‐β stimulates MAP kinases in VSMC.128
PDGF and Ang II activate MAP kinases in the vascular tissue.129 H2O2 and O2 − activate MAP kinases in VSMC.95 H2O2 stimulates p38 MAP kinase, required for redox‐sensitive signal transduction initiated by Ang II in VSMC.130 ROS implicated in MAP kinase activation induced by Ang II has also been derived from NAD(P)H oxidase in VSMC.93
Protein tyrosine phosphorylation is also affected by ROS. Protein tyrosine kinases (PTK) and PTPs regulate tyrosine phosphorylation. The complex of PTP enzymes dephosphorylates PTK substrate proteins and counteracts PTK activity. Both receptor and nonreceptor PTPs are susceptible to O2 − and H2O2. Exposure of cells to ROS inactivates PTP through oxidation of Cys residue in its structure and increases protein phosphorylation.131 However, this process is reversible, and PTPs is present in two states, either with a reduced or with oxidized Cys. Ang II may influence the oxidation of PTP‐activated NAD(P)H oxidase and O2 − generation.132, 133 Also, Ang II stimulates the expression of the vascular cell adhesion molecule‐1 via NF‐κB activation through ROS signaling.134
3.3. ROS‐induced activation of transcription factors in vascular pathophysiology
The activation of ROS by NF‐κB occurs through the activation of inhibitory κB kinases. The NF‐κB signaling modulates the expression of genes involved in inflammation and stimulates the ROS generation. In diabetic mice, the activation of NF‐κB increased the generation of the proinflammatory cytokine, the tumor necrosis factor‐alpha, which in turn elevated O2 − production by NAD(P)H oxidase.135 Ang‐II stimulated endothelial ROS generation through the stimulation of NF‐κB signaling pathway in DSS rats.136 One of the sources of ROS (O2 − and H2O2) generated through NF‐κB signaling is NAD(P)H oxidase. The NF‐κB cis‐acting elements regulate the promoter of p22phoxNAD(P)H oxidase gene in human aortic smooth muscle cells.137 The findings that revealed that NF‐κB enters the mitochondria of the obese mouse with diabetes and increases mitochondrial O2 − suggest the nontranscriptional role of NF‐κB in ROS production.138 Since NF‐κB activity leads to elevated ROS production, while ROS activates NF‐κB, the ROS generated by NAD(P)H oxidase will upregulate NF‐κB, initiating a positive feedback mechanism for ROS generation.108
4. HEMODYNAMICS AND REDOX SIGNALING
Redox signaling likely also influences the mechanical forces acting on the vascular wall. That is, elevated intraluminal flow stimulates the generation of O2 − and H2O2 in intact blood vessels. In VSMCs, shear stress was demonstrated to induce ROS generation.139 The ROS activity in cultured EC during oscillatory shear stress is derived from the activation of XO. The attenuation of O2 − production in the vasculature affected by oscillatory shear was found in cells lacking NAD(P)H oxidase activity, or by application of oxypurinol, a specific blocker of XO.107 However, the exact mechanism is still undetermined and may implicate NAD(P)H, and XO and/or mitochondrial enzymes.132 Laminar shear upregulates the expression of the extracellular SOD and cytosolic copper and zinc SOD in human aortic EC, and may regulate protective vascular response.140 Finally, ROS, particularly O2 − and H2O2, can diminish the efficacy of the antioxidant system, leading to the promotion of OxS. These signaling molecules play a significant role in the pathophysiology of the vasculature.
5. DEVELOPING LEADS TO EXTEND OUR UNDERSTANDING OF REDOX CONTROL OF VASCULAR BIOLOGY
Data‐mining and text‐mining techniques have been used to explore the information contained in published biomedical literature. Advancements in these techniques have led to the development of several topic‐specific knowledgebases (KB),141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159 including the first topic‐specific KB for redox control of vascular systems, named DES‐RedoxVasc.160 DES‐RedoxVasc was constructed using the search query: (human OR mouse OR rat OR mammal*) AND (radical* OR peroxide* OR “reductive stress” OR ROS OR “reactive oxygen species” OR RNS OR “reactive nitrogen species” OR redox OR “reduction–oxidation reaction” OR oxidative OR nitrosative OR peroxide* OR superoxide* OR detoxifi* OR antioxid* OR “polyunsaturated fatty acids” OR “arachidonic acid” OR “linoleic acid” OR hydroperoxide* OR “hypochlorous acid” OR peroxynitrit* flavoprot* OR xanthine oxidase* OR “cytochromes P450” OR catalase* OR sulfiredoxin* OR peroxiredoxin*) AND (“angina pectoris” OR anemia OR aneurysm* OR angio* OR arter* OR atrial OR atrioventricular OR aort* OR bradycardia OR blood OR brain OR circulati* OR clogging OR cardio* OR coronary OR edema OR heart OR ischemic OR hemo* OR hypertension OR leukemia OR leuko* OR macroangiopathy OR microangiopathy OR neovascularization OR occlusion OR pericardi* OR sepsis OR “sickle cell” OR tachycardia OR tachyarrhythmia OR thromb* OR vaso OR vein* OR ventricular OR vascular* OR vessel*) to retrieve all literature specifically focused on research related to redox effects on the cardiovascular system in mammalian organisms. This allowed for the retrieval and analyses of published information from 233′399 PubMed (based on article abstracts) and PubMed Central documents (based on the complete text in the article) linked to redox processes in the cardiovascular system. Users can easily explore the analyzed documents in DES‐RedoxVasc through links between various concepts from 28 topic‐specific dictionaries such as pathways, diseases, genes/proteins, miRNAs, toxins, drugs, biological processes, molecular functions, and so on. DES‐RedoxVasc can be used to search for hypotheses and potentially new knowledge in vascular biology. A published example in 160 shows that the semantic similarity tool in this KB linked ZFAS1 (long non‐coding RNA) and miR‐27b, even though there is no literature linking the functioning of miR‐27b and ZFAS1. Nonetheless, miR‐27b and ZFAS1 are linked to different vascular pathologies, and the DIANA tool, LncBase Predicted v.2,161 predicts that ZFAS1 binds hsa‐miR‐27b‐3p, which supports the possibility that the link suggested by DES‐RedoxVasc may be correct. The possible role of ZFAS1 in the fine tuning of levels of miR‐27B, a microRNA that is known to be responsive to OxS, has not been explored.160 As a consequence, we used DES‐RedoxVasc to develop additional case studies.
Case Study 1: Here, we characterize the biological functions of VSMC relative to chemical substances in the literature in connection with both VSMC and gene ontology biological functions that were associated with VSMC. As such, Figure 2 gives an overview of substances that are vital for homeostasis or can disturb the proper functioning of VMSC
Figure 2.
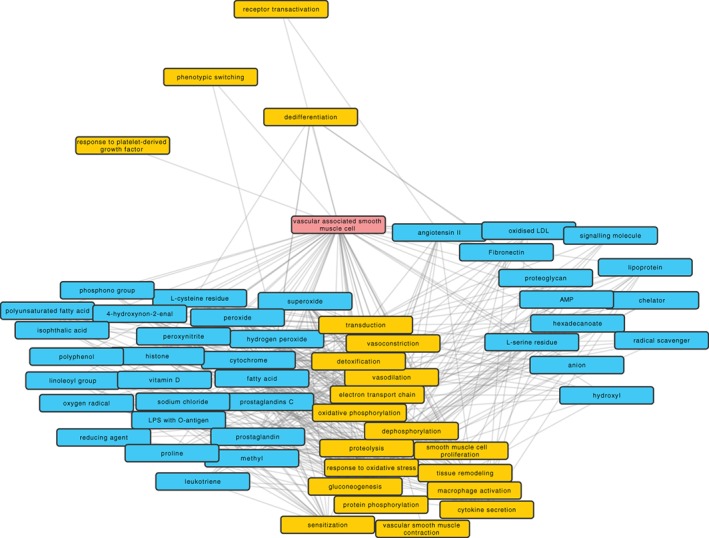
Chemical substances connecting vascular smooth muscle cells (VSMC) and gene ontology (GO) biological functions. Chemical substances from ChEBI are shown with a blue background color and GO biological functions with yellow color. The transparency of the edge indicates the frequency of co‐occurrence of the connected terms. The graph was formed by extracting all ChEBI or GO term co‐occurrences with “vascular associated smooth muscle cell” from the DES‐RedoxVasc KB. The connection between CheBI and GO terms already extracted in the first step was also added. A general filter was applied to the edges to have at least two articles reporting co‐occurrence. The layout of the graph was first force‐directed by the amount of co‐occurrence between two terms, but then manually adjusted for readability of the nodes
The following redox‐related molecules discussed in this manuscript were retrieved: “oxygen radical,” “superoxide,” and “hydrogen peroxide.” However, DES‐RedoxVasc points out additional molecules that should be explored (via https://www.cbrc.kaust.edu.sa/des-rv/) in this context. For instance, it would be interesting to look at the relationship between “H2O2” and “Ang II,” as Ang II and other chemotactic factors are dependent on H2O2 for their release.162 It is therefore of interest knowing which GO terms are linked to both H2O2 and Ang II. In the DES‐RedoxVasc KB, they are linked to vasodilation, vasoconstriction, macrophage activation, as well as VSMC that were the starting point of this exploration.
Case Study 2: Studies have demonstrated that both dietary PUFA and fish oils exhibit a protective role in CVD.163, 164, 165, 166 PUFAs provide several benefits including modulating lipid metabolism, reducing the production of inflammatory cytokine, and facilitating improvements in vascular EC function.167 PUFAs also transiently increase the levels of ROS that activate the OxS‐response transcription factor NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2) in pigment epithelial cells from the human retina,168 which transcribe several antioxidant genes. PUFAs were also shown to modulate noncoding RNAs169, 170 that may be the mechanism used to mediate the chemoprotective and antioncogenic properties of PUFAs. Thus, we here also used DES‐RedoxVasc to explore the microRNAs linked to vascular ECs, OxS response, and n‐3 polyunsaturated fatty acids (PUFA) (see Figure 3). We start this exploration by clicking the “Enriched Concepts” link in DES‐RedoxVasc. On this page, we used the search bar to filter the concept “vascular endothelial cell,” and then used this concept right‐click menu to generate a network (see Figure 3). On the “Network” page, we selected four dictionaries including “HFO Ontology (Bioportal) Heart Failure Ontology,” “Pathways (KEGG, Reactome, UniPathway, Panter),” and “Human microRNAs” dictionaries. Then, the “vascular endothelial cell” node was highlighted to expand the associated concepts with links from the selected dictionaries. Then, we only selected the “Human microRNAs” dictionary (as microRNAs linked to vascular ECs, OxS response, and PUFAs are the focus of this case study), and highlighted the “Oxidative stress response” node to expand/link the associated concepts from the “Human microRNAs” dictionary. We repeated this step for other nodes including the “Polyunsaturated Fatty Acids,” “MIR3178,” and “MIR937.” All additional nodes with two or more edges (“MIR34A,” “MIR20B,” and “MIR7A2”), generated in step 3, were also expanded/linked with their associated concepts in the “Human microRNAs” dictionary. We then pruned the nodes using the connectivity threshold of 2 (see Figure 3).
Figure 3.
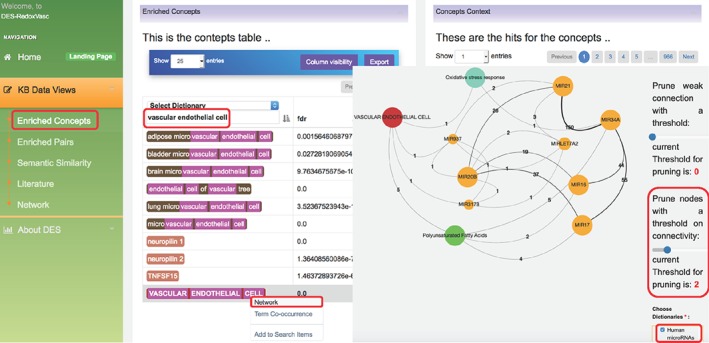
DES‐RedoxVasc network illustrating microRNAs that may affect the relationship among “VASCULAR ENDOTHELIAL CELL”, “Oxidative stress response”, and “Polyunsaturated Fatty Acids.” The orange circles denote concepts from the “human microRNAs” dictionary; the green circles denote concepts from the “HFO Ontology (Bioportal) Heart Failure Ontology” dictionary; the blue circles denote concepts from the “Pathways (KEGG, Reactome, UniPathway, Panter)” dictionary; and the red circles denote concepts from the “Human Anatomy” dictionary
Several of the noncoding RNAs (“MIR15B” [miR‐15b], “MIR16” [miR‐16], “MIR17” [miR‐17], and “MIR20B” [miR‐20b]) retrieved by DES‐RedoxVasc are modulated by PUFAs. That is, three different PUFA‐treated glioblastoma cell lines consistently exhibited an increase in the levels of miR‐20b, and decreased levels of microRNA (miR‐16 and miR‐17) that induces apoptosis‐specific expression changes.169 In addition, the level of miR‐15b increases in rats injected with the colon carcinogen and azoxymethane, and fed by fish oil/n‐3 PUFA rich diets.171
This report by 171 is interesting as the generation of mitochondrial ROS is promoted by miR‐15b, as well as mitochondrial dysfunction, through the inhibition of SIRT4 (exclusively localized in mitochondria).172 SIRT4 is also associated with photoaged skin and stress‐induced cellular senescence, which linked senescence‐associated mitochondrial dysfunction to both miR‐15b and SIRT4.173 Moreover, miR‐20b upregulated by PUFAs directly targets AKT3, and AKT3 silencing decreases the levels of VEGF.174 Specifically, in primary ECs, VEGF stimulation and the downstream mitochondrial biogenesis process required AKT3, and the blockade of AKT3 also reduces PGC‐1α‐dependent gene expression.175 This suggests that even though mitochondrial biogenesis is tightly interlinked to antioxidant systems, both miR‐15b and miR‐20b prevent mitochondrial functioning under unregulated OxS conditions. This decrease in mitochondrial biogenesis substantially decreases dysfunctional mitochondria produced by excessive ROS, which can be viewed as a quality‐control process.
Figure 3 further shows that both the “MIR20B” and “VASCULAR ENDOTHELIAL CELL” nodes are further linked to two additional microRNAs, “MIR3178” (miR‐3,178) and “MIR937” (miR‐937), which suggest that they may function in the same or a closely linked mechanism. For now, we know that the levels of miR‐20b, miR‐937, and miR‐3,178 are upregulated in a human vascular EC line (EAhy926) infected with the DENV‐2 (TR1751 strain),176 which may be a consequence of a mechanism counteracting the DENV infection‐induced OxS.177
6. CONCLUDING REMARKS
Endothelial ROS production has a prominent role in regulating the vascular redox homeostatic mechanisms in the vascular system. By understanding the ROS/RNS removal processes, we may influence the overall perturbing homeostatic functioning of the vascular endothelial system and the underlining consequence of a disbalance portraying as OxS in which it elevates inflammation and vascular remolding, activating the events for CVD.
The most prevailing players in vascular ROS pathology are XO and NAD(P)H oxidase, with links to various models of heart failure, ischemia injury, and DNA damage in patients with arterial hypertension, and so on. Interestingly, NAD(P)H was found to be in an ROS MMP‐2 signaling pathway in VSMC, suggesting some interwoven pathways of redox signaling, PTK and phosphatase activation, growth factors, vasoactive hormones, and transcription factors. To broaden our understanding of the redox control of the vascular endothelial system, we used text mining and data mining techniques to explore new intricacies of information embedded in the biomedical literature that we may not be aware of at first glance. By retrieving redox relating molecules from the DES‐RedoxVasc, we established new links and relationships between different components, such as with hydrogen peroxide and with Ang II. The results also suggest an overlapping interplay of processes that connect mitochondrial biogenesis and antioxidant systems with miR‐3178 and miR‐937. This shows that a cell expresses a quality control mechanism in the Redox homeostatic mechanisms of the vascular endothelial system. By using data mining in a review process, we have also deepened our understanding to our view the intricacies of how ROS is utilized in the vascular endothelial system redox system and also easier to see new pathways for improved treatment and more adequate prevention of cardiovascular‐related diseases.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
ACKNOWLEDGMENTS
This work is part of the collaboration between the Laboratory of Radiobiology and Molecular Genetics, Vinca Institute of Nuclear Sciences, University of Belgrade, Belgrade, Serbia and King Abdullah University of Science and Technology (KAUST), Computational Bioscience Research Center (CBRC), Thuwal, Saudi Arabia. This work has been supported by grants No. 173033 (E.R.I.) from the Ministry of Education, Science and Technological Development, Republic of Serbia and by the KAUST grant OSR#4129 (to E.R.I. and V.B.B.), which also supported M.O., S.Z., E.S.M., and V.P.B. V.B.B. has been supported by the King Abdullah University of Science and Technology Base Research Fund (BAS/1/1606‐01‐01), and V.B.B. and M.E. by KAUST Office of Sponsored Research (OSR) Awards No. FCC/1/1976‐24‐01 and FCC/1/1976‐17‐01. M.O., S.Z., E.S.M, A.T., J.S., and V.P.B. have been supported by the grants No. 173033 (E.R.I.) and No. 173034 from the Ministry of Education, Science and Technological Development, Republic of Serbia.
Obradovic M, Essack M, Zafirovic S, et al. Redox control of vascular biology. BioFactors. 2020;46:246–262. 10.1002/biof.1559
Milan Obradovic and Magbubah Essack contributed equally to this study.
Funding information KAUST Office of Sponsored Research, Grant/Award Numbers: FCC/1/1976‐17‐01, FCC/1/1976‐24‐01; King Abdullah University of Science and Technology Base Research Fund, Grant/Award Number: BAS/1/1606‐01‐01; KAUST, Grant/Award Number: OSR#4129; Ministry of Education, Science and Technological Development, Republic of Serbia, Grant/Award Numbers: 173034, 173033
Contributor Information
Milan Obradovic, Email: obradovicmilan@hotmail.com.
Magbubah Essack, Email: magbubah.essack@kaust.edu.sa.
REFERENCES
- 1. Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev. 2016;2016:1245049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zinkevich NS, Gutterman DD. ROS‐induced ROS release in vascular biology: Redox‐redox signaling. Am J Physiol Heart Circ Physiol. 2011;301:H647–H653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–258. [DOI] [PubMed] [Google Scholar]
- 4. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, et al. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. [DOI] [PubMed] [Google Scholar]
- 5. Khazaei M, Moien‐Afshari F, Laher I. Vascular endothelial function in health and diseases. Pathophysiology. 2008;15:49–67. [DOI] [PubMed] [Google Scholar]
- 6. Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;51:1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bir SC, Kolluru GK, Fang K, Kevil CG. Redox balance dynamically regulates vascular growth and remodeling. Semin Cell Dev Biol. 2012;23:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tabet F, Schiffrin EL, Callera GE, et al. Redox‐sensitive signaling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP‐2 in vascular smooth muscle cells from SHR. Circ Res. 2008;103:149–158. [DOI] [PubMed] [Google Scholar]
- 9. Togliatto G, Lombardo G, Brizzi MF. The future challenge of reactive oxygen species (ROS) in hypertension: From bench to bed side. Int J Mol Sci. 2017;18:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahsan H, Ali A, Ali R. Oxygen free radicals and systemic autoimmunity. Clin Exp Immunol. 2003;131:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Halliwell B. Antioxidants in human health and disease. Annu Rev Nutr. 1996;16:33–50. [DOI] [PubMed] [Google Scholar]
- 12. Li R, Jia Z, Trush MA. Defining ROS in biology and medicine. React Oxyg Species (Apex). 2016;1:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(2012) Free Radical Biomedicine: Principles, Clinical Correlations, and Methodologies. UAE, Bentham Science Publishers. [Google Scholar]
- 14. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97:8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. D'Autreaux B, Toledano MB. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. [DOI] [PubMed] [Google Scholar]
- 18. Pries AR, Kuebler WM. Normal endothelium. Handb Exp Pharmacol. 2006;176 Pt1:1–40. [DOI] [PubMed] [Google Scholar]
- 19. Jaffe EA. Cell biology of endothelial cells. Hum Pathol. 1987;18:234–239. [DOI] [PubMed] [Google Scholar]
- 20. Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Hinsbergh VW. Endothelium‐‐role in regulation of coagulation and inflammation. Semin Immunopathol. 2012;34:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feletou M. Integrated systems physiology: From molecule to function to disease. San Rafael (CA): Morgan and Claypool Life Sciences, 2011. [Google Scholar]
- 23. Chistiakov DA, Revin VV, Sobenin IA, Orekhov AN, Bobryshev YV. Vascular endothelium: Functioning in norm, changes in atherosclerosis and current dietary approaches to improve endothelial function. Mini Rev Med Chem. 2015;15:338–350. [DOI] [PubMed] [Google Scholar]
- 24. Rafieian‐Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int J Prev Med. 2014;5:927–946. [PMC free article] [PubMed] [Google Scholar]
- 25. Rajendran P, Rengarajan T, Thangavel J, et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9:1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Ierssel SH, Jorens PG, Van Craenenbroeck EM, Conraads VM. The endothelium, a protagonist in the pathophysiology of critical illness: Focus on cellular markers. Biomed Res Int. 2014;2014:985813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fischer C, Schneider M, Carmeliet P. Principles and therapeutic implications of angiogenesis, vasculogenesis and arteriogenesis. Handb Exp Pharmacol. 2006;176 Pt2:157–212. [DOI] [PubMed] [Google Scholar]
- 28. Fay WP, Garg N, Sunkar M. Vascular functions of the plasminogen activation system. Arterioscler Thromb Vasc Biol. 2007;27:1231–1237. [DOI] [PubMed] [Google Scholar]
- 29. Brown NJ. Blood pressure reduction and tissue‐type plasminogen activator release. Hypertension. 2006;47:648–649. [DOI] [PubMed] [Google Scholar]
- 30. Prager GW, Breuss JM, Steurer S, Mihaly J, Binder BR. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro‐uPA) activation on the surface of endothelial cells. Blood. 2004;103:955–962. [DOI] [PubMed] [Google Scholar]
- 31. Nordt TK, Bode C. Impaired endogenous fibrinolysis in diabetes mellitus: Mechanisms and therapeutic approaches. Semin Thromb Hemost. 2000;26:495–501. [DOI] [PubMed] [Google Scholar]
- 32. Zhang G, Xiang B, Dong A, et al. Biphasic roles for soluble guanylyl cyclase (sGC) in platelet activation. Blood. 2011;118:3670–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gale AJ, Cramer TJ, Rozenshteyn D, Cruz JR. Detailed mechanisms of the inactivation of factor VIIIa by activated protein C in the presence of its cofactors, protein S and factor V. J Biol Chem. 2008;283:16355–16362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor‐alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci U S A. 2013;110:17838–17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lenting PJ, Casari C, Christophe OD, Denis CV. von Willebrand factor: The old, the new and the unknown. J Thromb Haemost. 2012;10:2428–2437. [DOI] [PubMed] [Google Scholar]
- 36. Pearson JD. Endothelial cell function and thrombosis. Baillieres Best Pract Res Clin Haematol. 1999;12:329–341. [DOI] [PubMed] [Google Scholar]
- 37. Hao H, Gabbiani G, Bochaton‐Piallat ML. Arterial smooth muscle cell heterogeneity: Implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol. 2003;23:1510–1520. [DOI] [PubMed] [Google Scholar]
- 38. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 39. Zhu SB, Zhu J, Zhou ZZ, Xi EP, Wang RP, Zhang Y. TGF‐beta1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am J Transl Res. 2015;7:2764–2774. [PMC free article] [PubMed] [Google Scholar]
- 40. Chamley‐Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev. 1979;59:1–61. [DOI] [PubMed] [Google Scholar]
- 41. Young IS, Woodside JV. Antioxidants in health and disease. J Clin Pathol. 2001;54:176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS‐mediated mechanisms: A radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. [DOI] [PubMed] [Google Scholar]
- 43. Shukla V, Mishra SK, Pant HC. Oxidative stress in neurodegeneration. Adv Pharmacol Sci. 2011;2011:572634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010;40:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sakamoto K, Iwasaki K, Sugiyama H, Tsuji Y. Role of the tumor suppressor PTEN in antioxidant responsive element‐mediated transcription and associated histone modifications. Mol Biol Cell. 2009;20:1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roos G, Messens J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–326. [DOI] [PubMed] [Google Scholar]
- 48. Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. [DOI] [PubMed] [Google Scholar]
- 49. Xu S, Touyz RM. Reactive oxygen species and vascular remodelling in hypertension: Still alive. Can J Cardiol. 2006;22:947–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)‐induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vincent A, Crozatier M. Neither too much nor too little: Reactive oxygen species levels regulate drosophila hematopoiesis. J Mol Cell Biol. 2010;2:74–75. [DOI] [PubMed] [Google Scholar]
- 53. Redon J, Oliva MR, Tormos C, Giner V, Chaves J, et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41:1096–1101. [DOI] [PubMed] [Google Scholar]
- 54. Matoba T, Shimokawa H, Nakashima M, et al. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Breton‐Romero R, Lamas S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014;2:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Price DT, Vita JA, Keaney JF Jr. Redox control of vascular nitric oxide bioavailability. Antioxid Redox Signal. 2000;2:919–935. [DOI] [PubMed] [Google Scholar]
- 57. Fukai T, Ushio‐Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Touyz RM, Briones AM. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens Res. 2011;34:5–14. [DOI] [PubMed] [Google Scholar]
- 59. Griendling KK, Sorescu D, Lassegue B, Ushio‐Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. [DOI] [PubMed] [Google Scholar]
- 60. Touyz RM. Oxidative stress and vascular damage in hypertension. Curr Hypertens Rep. 2000;2:98–105. [DOI] [PubMed] [Google Scholar]
- 61. Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 62. Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. [DOI] [PubMed] [Google Scholar]
- 63. Landmesser U, Cai H, Dikalov S, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Landmesser U, Harrison DG. Oxidative stress and vascular damage in hypertension. Coron Artery Dis. 2001;12:455–461. [DOI] [PubMed] [Google Scholar]
- 65. Diez J, Laviades C, Orbe J, Zalba G, Lopez B, et al. The A1166C polymorphism of the AT1 receptor gene is associated with collagen type I synthesis and myocardial stiffness in hypertensives. J Hypertens. 2003;21:2085–2092. [DOI] [PubMed] [Google Scholar]
- 66. Ushio‐Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. [DOI] [PubMed] [Google Scholar]
- 67. Stone JR, Collins T. The role of hydrogen peroxide in endothelial proliferative responses. Endothelium. 2002;9:231–238. [DOI] [PubMed] [Google Scholar]
- 68. Antunes F, Cadenas E. Cellular titration of apoptosis with steady state concentrations of H(2)O(2): Submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic Biol Med. 2001;30:1008–1018. [DOI] [PubMed] [Google Scholar]
- 69. Zafari AM, Ushio‐Fukai M, Akers M, et al. Role of NADH/NADPH oxidase‐derived H2O2 in angiotensin II‐induced vascular hypertrophy. Hypertension. 1998;32:488–495. [DOI] [PubMed] [Google Scholar]
- 70. Shimokawa H, Matoba T. Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pharmacol Res. 2004;49:543–549. [DOI] [PubMed] [Google Scholar]
- 71. Fujiki T, Shimokawa H, Morikawa K, et al. Endothelium‐derived hydrogen peroxide accounts for the enhancing effect of an angiotensin‐converting enzyme inhibitor on endothelium‐derived hyperpolarizing factor‐mediated responses in mice. Arterioscler Thromb Vasc Biol. 2005;25:766–771. [DOI] [PubMed] [Google Scholar]
- 72. Fujimoto S, Asano T, Sakai M, et al. Mechanisms of hydrogen peroxide‐induced relaxation in rabbit mesenteric small artery. Eur J Pharmacol. 2001;412:291–300. [DOI] [PubMed] [Google Scholar]
- 73. Thengchaisri N, Kuo L. Hydrogen peroxide induces endothelium‐dependent and ‐independent coronary arteriolar dilation: Role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol. 2003;285:H2255–H2263. [DOI] [PubMed] [Google Scholar]
- 74. Burke TM, Wolin MS. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am J Physiol. 1987;252:H721–H732. [DOI] [PubMed] [Google Scholar]
- 75. Ray R, Murdoch CE, Wang M, et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–1376. [DOI] [PubMed] [Google Scholar]
- 76. Cai H, Li Z, Davis ME, Kanner W, Harrison DG, Dudley SC Jr. Akt‐dependent phosphorylation of serine 1179 and mitogen‐activated protein kinase kinase/extracellular signal‐regulated kinase 1/2 cooperatively mediate activation of the endothelial nitric‐oxide synthase by hydrogen peroxide. Mol Pharmacol. 2003;63:325–331. [DOI] [PubMed] [Google Scholar]
- 77. Rubanyi GM. Vascular effects of oxygen‐derived free radicals. Free Radic Biol Med. 1988;4:107–120. [DOI] [PubMed] [Google Scholar]
- 78. Lipinski B. Hydroxyl radical and its scavengers in health and disease. Oxid Med Cell Longev. 2011;2011:809696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4‐hydroxy‐2‐nonenal. Oxid Med Cell Longev. 2014;2014:360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ferdinandy P, Schulz R. Peroxynitrite: Toxic or protective in the heart? Circ Res. 2001;88:E12–E13. [DOI] [PubMed] [Google Scholar]
- 81. Ferdinandy P. Peroxynitrite: Just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol. 2006;148:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ronson RS, Nakamura M, Vinten‐Johansen J. The cardiovascular effects and implications of peroxynitrite. Cardiovasc Res. 1999;44:47–59. [DOI] [PubMed] [Google Scholar]
- 83. Graves JE, Bates JN, Kooy NW, Lewis SJ. Vasodilator actions of the endothelium‐derived relaxing factor L‐S‐nitrosocysteine in anaesthetized rats are markedly diminished by peroxynitrite. Clin Exp Pharmacol Physiol. 2005;32:1137–1141. [DOI] [PubMed] [Google Scholar]
- 84. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li J, Su J, Li W, Liu W, Altura BT, Altura BM. Peroxynitrite induces apoptosis in canine cerebral vascular muscle cells: Possible relation to neurodegenerative diseases and strokes. Neurosci Lett. 2003;350:173–177. [DOI] [PubMed] [Google Scholar]
- 86. Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, et al. Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: Implications in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:2623–2629. [DOI] [PubMed] [Google Scholar]
- 87. Ungvari Z, Bailey‐Downs L, Gautam T, et al. Adaptive induction of NF‐E2‐related factor‐2‐driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol. 2011;300:H1133–H1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mervaala EM, Cheng ZJ, Tikkanen I, Lapatto R, Nurminen K, et al. Endothelial dysfunction and xanthine oxidoreductase activity in rats with human renin and angiotensinogen genes. Hypertension. 2001;37:414–418. [DOI] [PubMed] [Google Scholar]
- 89. Suzuki H, DeLano FA, Parks DA, et al. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc Natl Acad Sci U S A. 1998;95:4754–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Laakso J, Mervaala E, Himberg JJ, Teravainen TL, Karppanen H, et al. Increased kidney xanthine oxidoreductase activity in salt‐induced experimental hypertension. Hypertension. 1998;32:902–906. [DOI] [PubMed] [Google Scholar]
- 91. Laakso JT, Teravainen TL, Martelin E, Vaskonen T, Lapatto R. Renal xanthine oxidoreductase activity during development of hypertension in spontaneously hypertensive rats. J Hypertens. 2004;22:1333–1340. [DOI] [PubMed] [Google Scholar]
- 92. Zalba G, Beaumont FJ, San Jose G, Fortuno A, Fortuno MA, et al. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension. 2000;35:1055–1061. [DOI] [PubMed] [Google Scholar]
- 93. Viedt C, Soto U, Krieger‐Brauer HI, et al. Differential activation of mitogen‐activated protein kinases in smooth muscle cells by angiotensin II: Involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol. 2000;20:940–948. [DOI] [PubMed] [Google Scholar]
- 94. Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, et al. Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Baas AS, Berk BC. Differential activation of mitogen‐activated protein kinases by H2O2 and O2‐ in vascular smooth muscle cells. Circ Res. 1995;77:29–36. [DOI] [PubMed] [Google Scholar]
- 96. Heimlich JB, Speed JS, Bloom CJ, O'Connor PM, Pollock JS, et al. ET‐1 increases reactive oxygen species following hypoxia and high‐salt diet in the mouse glomerulus. Acta Physiol (Oxf). 2015;213:722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li L, Fink GD, Watts SW, et al. Endothelin‐1 increases vascular superoxide via endothelin(A)‐NADPH oxidase pathway in low‐renin hypertension. Circulation. 2003;107:1053–1058. [DOI] [PubMed] [Google Scholar]
- 98. Matsuno K, Yamada H, Iwata K, et al. Nox1 is involved in angiotensin II‐mediated hypertension: A study in Nox1‐deficient mice. Circulation. 2005;112:2677–2685. [DOI] [PubMed] [Google Scholar]
- 99. Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, et al. Nox1 overexpression potentiates angiotensin II‐induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. [DOI] [PubMed] [Google Scholar]
- 100. Xiong W, Mactaggart J, Knispel R, et al. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis. 2009;202:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Amiri F, Virdis A, Neves MF, et al. Endothelium‐restricted overexpression of human endothelin‐1 causes vascular remodeling and endothelial dysfunction. Circulation. 2004;110:2233–2240. [DOI] [PubMed] [Google Scholar]
- 102. Fortuno A, Olivan S, Beloqui O, San Jose G, Moreno MU, et al. Association of increased phagocytic NADPH oxidase‐dependent superoxide production with diminished nitric oxide generation in essential hypertension. J Hypertens. 2004;22:2169–2175. [DOI] [PubMed] [Google Scholar]
- 103. de Haan JB. Limiting reductive stress for treating in‐stent stenosis: The heart of the matter? J Clin Invest. 2014;124:5092–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Perez‐Torres I, Guarner‐Lans V, Rubio‐Ruiz ME. Reductive stress in inflammation‐associated diseases and the pro‐oxidant effect of antioxidant agents. Int J Mol Sci. 2017;18:pii:E2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bubb KJ, Birgisdottir AB, Tang O, Hansen T, Figtree GA. Redox modification of caveolar proteins in the cardiovascular system‐ role in cellular signalling and disease. Free Radic Biol Med. 2017;109:61–74. [DOI] [PubMed] [Google Scholar]
- 106. Galougahi KK, Liu CC, Gentile C, Kok C, Nunez A, et al. Glutathionylation mediates angiotensin II‐induced eNOS uncoupling, amplifying NADPH oxidase‐dependent endothelial dysfunction. J Am Heart Assoc. 2014;3:e000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. McNally JS, Davis ME, Giddens DP, et al. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285:H2290–H2297. [DOI] [PubMed] [Google Scholar]
- 108. Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. [DOI] [PubMed] [Google Scholar]
- 109. Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. [DOI] [PubMed] [Google Scholar]
- 110. Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. [DOI] [PubMed] [Google Scholar]
- 111. Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu F, Tyml K, Wilson JX. iNOS expression requires NADPH oxidase‐dependent redox signaling in microvascular endothelial cells. J Cell Physiol. 2008;217:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox‐containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109:1795–1801. [DOI] [PubMed] [Google Scholar]
- 114. Zhou MS, Adam AG, Jaimes EA, Raij L. In salt‐sensitive hypertension, increased superoxide production is linked to functional upregulation of angiotensin II. Hypertension. 2003;42:945–951. [DOI] [PubMed] [Google Scholar]
- 115. Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK‐STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–C1652. [DOI] [PubMed] [Google Scholar]
- 116. Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006;281:36228–36235. [DOI] [PubMed] [Google Scholar]
- 117. Hawkins BJ, Madesh M, Kirkpatrick CJ, Fisher AB. Superoxide flux in endothelial cells via the chloride channel‐3 mediates intracellular signaling. Mol Biol Cell. 2007;18:2002–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II‐mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. [DOI] [PubMed] [Google Scholar]
- 119. Duan J, Xu H, Dai S, et al. Phytoestrogen alpha‐zearalanol inhibits homocysteine‐induced endothelin‐1 expression and oxidative stress in human umbilical vein endothelial cells. Atherosclerosis. 2008;197:549–555. [DOI] [PubMed] [Google Scholar]
- 120. Xu H, Duan J, Dai S, Wu Y, Sun R, Ren J. Alpha‐Zearalanol attenuates oxLDL‐induced ET‐1 gene expression, ET‐1 secretion and redox‐sensitive intracellular signaling activation in human umbilical vein endothelial cells. Toxicol Lett. 2008;179:163–168. [DOI] [PubMed] [Google Scholar]
- 121. Xu H, Duan J, Wang W, et al. Reactive oxygen species mediate oxidized low‐density lipoprotein‐induced endothelin‐1 gene expression via extracellular signal‐regulated kinase in vascular endothelial cells. J Hypertens. 2008;26:956–963. [DOI] [PubMed] [Google Scholar]
- 122. Grote K, Flach I, Luchtefeld M, et al. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase‐2 (MMP‐2) via NAD(P)H oxidase‐derived reactive oxygen species. Circ Res. 2003;92:e80–e86. [DOI] [PubMed] [Google Scholar]
- 123. Luchtefeld M, Grote K, Grothusen C, et al. Angiotensin II induces MMP‐2 in a p47phox‐dependent manner. Biochem Biophys Res Commun. 2005;328:183–188. [DOI] [PubMed] [Google Scholar]
- 124. Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage‐derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mehdi MZ, Azar ZM, Srivastava AK. Role of receptor and nonreceptor protein tyrosine kinases in H2O2‐induced PKB and ERK1/2 signaling. Cell Biochem Biophys. 2007;47:1–10. [DOI] [PubMed] [Google Scholar]
- 126. Mendelson K, Swendeman S, Saftig P, Blobel CP. Stimulation of platelet‐derived growth factor receptor beta (PDGFRbeta) activates ADAM17 and promotes metalloproteinase‐dependent cross‐talk between the PDGFRbeta and epidermal growth factor receptor (EGFR) signaling pathways. J Biol Chem. 2010;285:25024–25032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chen K, Vita JA, Berk BC, Keaney JF Jr. C‐Jun N‐terminal kinase activation by hydrogen peroxide in endothelial cells involves SRC‐dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:16045–16050. [DOI] [PubMed] [Google Scholar]
- 128. Saito Y, Berk BC. Transactivation: A novel signaling pathway from angiotensin II to tyrosine kinase receptors. J Mol Cell Cardiol. 2001;33:3–7. [DOI] [PubMed] [Google Scholar]
- 129. Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease‐dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem. 2001;276:7957–7962. [DOI] [PubMed] [Google Scholar]
- 130. Ushio‐Fukai M, Alexander RW, Akers M, Griendling KK. p38 mitogen‐activated protein kinase is a critical component of the redox‐sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. [DOI] [PubMed] [Google Scholar]
- 131. Stoker AW. Protein tyrosine phosphatases and signalling. J Endocrinol. 2005;185:19–33. [DOI] [PubMed] [Google Scholar]
- 132. De Keulenaer GW, Chappell DC, Ishizaka N, Nerem RM, Alexander RW, et al. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: Role of a superoxide‐producing NADH oxidase. Circ Res. 1998;82:1094–1101. [DOI] [PubMed] [Google Scholar]
- 133. Murphy TV, Spurrell BE, Hill MA. Tyrosine phosphorylation following alterations in arteriolar intraluminal pressure and wall tension. Am J Physiol Heart Circ Physiol. 2001;281:H1047–H1056. [DOI] [PubMed] [Google Scholar]
- 134. Tummala PE, Chen XL, Sundell CL, et al. Angiotensin II induces vascular cell adhesion molecule‐1 expression in rat vasculature: A potential link between the renin‐angiotensin system and atherosclerosis. Circulation. 1999;100:1223–1229. [DOI] [PubMed] [Google Scholar]
- 135. Gao X, Belmadani S, Picchi A, et al. Tumor necrosis factor‐alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007;115:245–254. [DOI] [PubMed] [Google Scholar]
- 136. Zhou MS, Schulman IH, Raij L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt‐sensitive hypertension: Role of nuclear factor kappa B activation. J Hypertens. 2010;28:527–535. [DOI] [PubMed] [Google Scholar]
- 137. Manea A, Manea SA, Gafencu AV, Raicu M. Regulation of NADPH oxidase subunit p22(phox) by NF‐kB in human aortic smooth muscle cells. Arch Physiol Biochem. 2007;113:163–172. [DOI] [PubMed] [Google Scholar]
- 138. Pantano C, Reynaert NL, van der Vliet A, Janssen‐Heininger YM. Redox‐sensitive kinases of the nuclear factor‐kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. [DOI] [PubMed] [Google Scholar]
- 139. Laurindo FR, Pedro Mde A, Barbeiro HV, et al. Vascular free radical release. ex vivo and in vivo evidence for a flow‐dependent endothelial mechanism. Circ Res. 1994;74:700–709. [DOI] [PubMed] [Google Scholar]
- 140. Inoue N, Ramasamy S, Fukai T, Nerem RM, Harrison DG. Shear stress modulates expression of Cu/Zn superoxide dismutase in human aortic endothelial cells. Circ Res. 1996;79:32–37. [DOI] [PubMed] [Google Scholar]
- 141. Kordopati V, Salhi A, Razali R, et al. DES‐mutation: System for exploring links of mutations and diseases. Sci Rep. 2018;8:13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Salhi A, Negrao S, Essack M, Morton MJL, Bougouffa S, et al. DES‐TOMATO: A knowledge exploration system focused on Tomato species. Sci Rep. 2017;7:5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Salhi A, Essack M, Alam T, et al. DES‐ncRNA: A knowledgebase for exploring information about human micro and long noncoding RNAs based on literature‐mining. RNA Biol. 2017;14:963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bin Raies A, Mansour H, Incitti R, Bajic VB. Combining position weight matrices and document‐term matrix for efficient extraction of associations of methylated genes and diseases from free text. PLoS One. 2013;8:e77848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bin Raies A, Mansour H, Incitti R, Bajic VB. DDMGD: The database of text‐mined associations between genes methylated in diseases from different species. Nucleic Acids Res. 2015;43:D879–D886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Dawe AS, Radovanovic A, Kaur M, et al. DESTAF: A database of text‐mined associations for reproductive toxins potentially affecting human fertility. Reprod Toxicol. 2012;33:99–105. [DOI] [PubMed] [Google Scholar]
- 147. Essack M, Radovanovic A, Bajic VB. Information exploration system for sickle cell disease and repurposing of hydroxyfasudil. PLoS One. 2013;8:e65190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Essack M, Radovanovic A, Schaefer U, et al. DDEC: Dragon database of genes implicated in esophageal cancer. BMC Cancer. 2009;9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kwofie SK, Radovanovic A, Sundararajan VS, Maqungo M, Christoffels A, Bajic VB. Dragon exploratory system on hepatitis C virus (DESHCV). Infect Genet Evol. 2011;11:734–739. [DOI] [PubMed] [Google Scholar]
- 150. Kwofie SK, Schaefer U, Sundararajan VS, Bajic VB, Christoffels A. HCVpro: Hepatitis C virus protein interaction database. Infect Genet Evol. 2011;11:1971–1977. [DOI] [PubMed] [Google Scholar]
- 151. Maqungo M, Kaur M, Kwofie SK, et al. DDPC: Dragon database of genes associated with prostate cancer. Nucleic Acids Res. 2011;39:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sagar S, Kaur M, Dawe A, Seshadri SV, Christoffels A, et al. DDESC: Dragon database for exploration of sodium channels in human. BMC Genomics. 2008;9:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sagar S, Kaur M, Radovanovic A, Bajic VB. Dragon exploration system on marine sponge compounds interactions. J Chem. 2013;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kaur M, Radovanovic A, Essack M, et al. Database for exploration of functional context of genes implicated in ovarian cancer. Nucleic Acids Res. 2009;37:D820–D823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Salhi A, Essack M, Radovanovic A, et al. DESM: Portal for microbial knowledge exploration systems. Nucleic Acids Res. 2016;44:D624–D633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Bajic VB, Veronika M, Veladandi PS, et al. Dragon plant biology explorer. A text‐mining tool for integrating associations between genetic and biochemical entities with genome annotation and biochemical terms lists. Plant Physiol. 2005;138:1914–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chowdhary R, Zhang J, Tan SL, Osborne DE, Bajic VB, Liu JS. PIMiner: A web tool for extraction of protein interactions from biomedical literature. Int J Data Min Bioinform. 2013;7:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chowdhary R, Tan SL, Zhang J, Karnik S, Bajic VB, Liu JS. Context‐specific protein network miner‐‐an online system for exploring context‐specific protein interaction networks from the literature. PLoS One. 2012;7:e34480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Pan H, Zuo L, Choudhary V, et al. Dragon TF association miner: A system for exploring transcription factor associations through text‐mining. Nucleic Acids Res. 2004;32:W230–W234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Essack M, Salhi A, Stanimirovic J, Tifratene F, Raies AB, et al. Literature‐based enrichment insights into redox control of vascular biology. Oxid Med Cell Longev. 2019;2019:1769437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA‐LncBase v2: Indexing microRNA targets on non‐coding transcripts. Nucleic Acids Res. 2016;44:D231–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Taverne YJ, Bogers AJ, Duncker DJ, Merkus D. Reactive oxygen species and the cardiovascular system. Oxid Med Cell Longev. 2013;2013:862423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Yang BC, Saldeen TG, Bryant JL, Nichols WW, Mehta JL. Long‐term dietary fish oil supplementation protects against ischemia‐reperfusion‐induced myocardial dysfunction in isolated rat hearts. Am Heart J. 1993;126:1287–1292. [DOI] [PubMed] [Google Scholar]
- 164. Sergiel JP, Martine L, Raederstorff D, Grynberg A, Demaison L. Individual effects of dietary EPA and DHA on the functioning of the isolated working rat heart. Can J Physiol Pharmacol. 1998;76:728–736. [DOI] [PubMed] [Google Scholar]
- 165. Rodrigo R, Korantzopoulos P, Cereceda M, et al. A randomized controlled trial to prevent post‐operative atrial fibrillation by antioxidant reinforcement. J Am Coll Cardiol. 2013;62:1457–1465. [DOI] [PubMed] [Google Scholar]
- 166. Abeywardena MY, Patten GS. Role of omega3 long‐chain polyunsaturated fatty acids in reducing cardio‐metabolic risk factors. Endocr Metab Immune Disord Drug Targets. 2011;11:232–246. [DOI] [PubMed] [Google Scholar]
- 167. Weber P, Raederstorff D. Triglyceride‐lowering effect of omega‐3 LC‐polyunsaturated fatty acids‐‐a review. Nutr Metab Cardiovasc Dis. 2000;10:28–37. [PubMed] [Google Scholar]
- 168. Johansson I, Monsen VT, Pettersen K, et al. The marine n‐3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy. 2015;11:1636–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Farago N, Feher LZ, Kitajka K, Das UN, Puskas LG. MicroRNA profile of polyunsaturated fatty acid treated glioma cells reveal apoptosis‐specific expression changes. Lipids Health Dis. 2011;10:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Mandal CC, Ghosh‐Choudhury T, Dey N, Choudhury GG, Ghosh‐Choudhury N. miR‐21 is targeted by omega‐3 polyunsaturated fatty acid to regulate breast tumor CSF‐1 expression. Carcinogenesis. 2012;33:1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Davidson LA, Wang N, Shah MS, Lupton JR, Ivanov I, Chapkin RS. n‐3 polyunsaturated fatty acids modulate carcinogen‐directed non‐coding microRNA signatures in rat colon. Carcinogenesis. 2009;30:2077–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhu Y, Yan Y, Principe DR, Zou X, Vassilopoulos A, Gius D. SIRT3 and SIRT4 are mitochondrial tumor suppressor proteins that connect mitochondrial metabolism and carcinogenesis. Cancer Metab. 2014;2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lang A, Grether‐Beck S, Singh M, et al. MicroRNA‐15b regulates mitochondrial ROS production and the senescence‐associated secretory phenotype through sirtuin 4/SIRT4. Aging (Albany NY). 2016;8:484–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Qin B, Liu J, Liu S, Li B, Ren J. MiR‐20b targets AKT3 and modulates vascular endothelial growth factor‐mediated changes in diabetic retinopathy. Acta Biochim Biophys Sin (Shanghai). 2016;48:732–740. [DOI] [PubMed] [Google Scholar]
- 175. Wright GL, Maroulakou IG, Eldridge J, et al. VEGF stimulation of mitochondrial biogenesis: Requirement of AKT3 kinase. FASEB J. 2008;22:3264–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Bavia L, Mosimann AL, Aoki MN, Duarte Dos Santos CN. A glance at subgenomic flavivirus RNAs and microRNAs in flavivirus infections. Virol J. 2016;13:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Soundravally R, Hoti SL, Patil SA, et al. Association between pro‐inflammatory cytokines and lipid peroxidation in patients with severe dengue disease around defervescence. Int J Infect Dis. 2014;18:68–72. [DOI] [PubMed] [Google Scholar]