

Abstract
Objective
Evidence for a role of microglia in the pathogenesis of multiple sclerosis (MS) is growing. We investigated association of microglial markers at time of diagnostic lumbar puncture (LP) with different aspects of disease activity (relapses, disability, magnetic resonance imaging parameters) up to 6 years later in a cohort of 143 patients.
Methods
In cerebrospinal fluid (CSF), we measured 3 macrophage and microglia‐related proteins, chitotriosidase (CHIT1), chitinase‐3–like protein 1 (CHI3L1 or YKL‐40), and soluble triggering receptor expressed on myeloid cells 2 (sTREM2), as well as a marker of neuronal damage, neurofilament light chain (NfL), using enzyme‐linked immunosorbent assay and electrochemiluminescence. We investigated the same microglia‐related markers in publicly available RNA expression data from postmortem brain tissue.
Results
CHIT1 levels at diagnostic LP correlated with 2 aspects of long‐term disease activity after correction for multiple testing. First, CHIT1 increased with reduced tissue integrity in lesions at a median 3 years later (p = 9.6E‐04). Second, CHIT1 reflected disease severity at a median 5 years later (p = 1.2E‐04). Together with known clinical covariates, CHIT1 levels explained 12% and 27% of variance in these 2 measures, respectively, and were able to distinguish slow and fast disability progression (area under the curve = 85%). CHIT1 was the best discriminator of chronic active versus chronic inactive lesions and the only marker correlated with NfL (r = 0.3, p = 0.0019). Associations with disease activity were, however, independent of NfL.
Interpretation
CHIT1 CSF levels measured during the diagnostic LP reflect microglial activation early on in MS and can be considered a valuable prognostic biomarker for future disease activity. ANN NEUROL 2020;87:633–645
Multiple sclerosis (MS) is the most common chronic disease of the central nervous system (CNS) in young adults, causing serious physical disability in adults of working age.1 MS is characterized pathologically by focal areas of inflammation, demyelination, gliosis, and axonal damage. Important patient‐to‐patient heterogeneity is seen for clinical (onset, severity, etc) as well as paraclinical features (laboratory measures, imaging). With more than 11 classes of disease‐modifying treatments currently available, one of the biggest challenges in therapeutic decision making to date is effective stratification of treatment in the face of an uncertain prognosis. The status of “no evidence of disease activity” (NEDA), defined using a composite of absence of relapses, disability progression, and magnetic resonance imaging (MRI) activity, has been suggested as a treatment target and is expanding with the inclusion of additional disease metrics.2 Sensitive, specific, and relatively inexpensive biomarkers in blood and cerebrospinal fluid (CSF) may facilitate the treatment decision‐making, especially if they have prognostic value and predict disease activity, but only a few of them are validated for clinical practice.3
Several biomarkers directly reflect the neurodegenerative and inflammatory processes.3 Neurofilament light chain (NfL), a structural axonal component released upon injury, is one of the most studied biomarkers for follow‐up of disease activity and treatment response in MS.4 Evidence for a role of innate immune cells such as the CNS‐resident macrophages or microglia in MS pathogenesis is growing, and the heterogeneity of microglial subsets is increasingly recognized.5, 6, 7, 8 Hence, we investigate 3 main macrophage and microglia‐related markers in CSF as potential biomarkers: chitotriosidase (also known as chitinase 1 [CHIT1]), chitinase‐3–like protein 1 (CHI3L1, also known as YKL‐40), and soluble triggering receptor expressed on myeloid cells 2 (sTREM2). Both CHIT1 and CHI3L1 are secreted by activated macrophages and are markers of microglial activation.9, 10 However, CHI3L1 is less cell‐type specific than CHIT1 and is produced also by astrocytes, epithelial cells, and neutrophils.9, 11 Finally, TREM2 is selectively expressed by microglia, and a soluble fragment (sTREM2) is released into the CSF upon cleavage of the ectodomain at the cell surface.12
In brief, we measured known CSF microglial markers (CHIT1, CHI3L1, and sTREM2), as well as NfL at time of diagnosis, in the same large cohort of 143 MS patients for whom longitudinal follow‐up by the same expert neurologist was available. We investigated whether protein levels at diagnosis correlate with or predict different aspects of disease activity (relapses, disability, MRI parameters) up to 6 years later, and we shed light on the role of heterogeneous microglia subsets in the MS disease process.
Patients and Methods
Study Population
MS patients were diagnosed based on the 2017 McDonald criteria13 at the Department of Neurology of the University Hospitals Leuven. For 143 patients, leftover CSF samples from the clinical laboratory obtained through lumbar puncture (LP) at the time of diagnosis were available (Table 1). For the genetic analyses, a larger existing cohort of up to 604 patients was available (Supplementary Table 1).14 The study was approved by the Ethics Committee of the University Hospitals Leuven (S9940 and S60222), and patients provided written informed consent.
Table 1.
Study Population Characteristics
| Characteristics | Total Cohort, N = 143 | Subcohorts with Measures of Disease Activity | ||
|---|---|---|---|---|
| Disability, n = 120 | Relapse, n = 104 | MRI, n = 79 | ||
| Gender: female/male (% female) | 88/55 (62) | 75/45 (63) | 68/36 (65) | 50/29 (63) |
| Disease course: BO/PP/unknown (% BO) | 112/25/6 (79) | 97/18/5 (81) | 104/0/0 (100) | 74/2/3 (97) |
| Age at onset, yr, median (IQR) | 34 (25–44) | 34 (24–43) | 30 (23–40) | 32 (24–40) |
| Disease duration at LP, yr, median (IQR) | 0 (0–5) | 1 (0–4) | 1 (0–3) | 0 (0–2) |
| OCB, positive/negative/unknown (% positive) | 124/17/2 (86) | 104/14/2 (87) | 90/12/2 (87) | 69/8/2 (90) |
| IgG index, median (IQR) | 0.87 (0.63–1.33) | 0.87 (0.64–1.33) | 0.90 (0.66–1.36) | 0.89 (0.69–1.41) |
| Time LP to most recent EDSS, yr, median (IQR) | — | 5 (2–7) | — | — |
| EDSS most recent, median (IQR) | — | 2 (1–3.5) | — | — |
| ARMSS most recent, median (IQR) | — | 2.36 (1.01–4.31) | — | — |
| RH before treatment from LP, yr, median (IQR) | — | — | 1 (0–3) | — |
| ARR before treatment, median (IQR) | — | — | 0.90 (0.35–1.85) | — |
| Total RH from LP, yr, median (IQR) | — | — | 6 (3–8) | — |
| ARR total, median (IQR) | — | — | 0.35 (0.21–0.58) | — |
| Time LP to MRI, yr, median (IQR) | — | — | — | 3 (2–5) |
ARMSS = Age‐Related Multiple Sclerosis Severity Score; ARR = annualized relapse rate; BO = bout onset; EDSS = Expanded Disability Status Scale; IgG = immunoglobulin G; IQR = interquartile range; LP = lumbar puncture; MRI = magnetic resonance imaging; OCB = oligoclonal bands; PP = primary progressive; RH = relapse history.
Clinical data (see Table 1) were collected during follow‐up by the same expert treating clinician (B.D.). Relapse was defined according to the 2017 McDonald criteria as the patient‐reported symptoms or objectively observed signs typical of an acute inflammatory demyelinating event in the CNS with duration of at least 24 hours, in the absence of fever or infection. Onset of disease refers to the first symptoms declared. Disease course is distinguished as presenting with relapsing–remitting disease at onset (bout onset, or BO) or presenting with progression from onset (primary progressive). Annualized relapse rate was calculated from onset before start of immunomodulatory treatment over a minimum duration of 90 days, as described previously.14 Total annualized relapse rate is calculated until last follow‐up, independent of treatment. Sensitivity analyses included treatment as covariate. Age‐Related Multiple Sclerosis Severity Score (ARMSS) and Multiple Sclerosis Severity Score (MSSS) were calculated based on the Expanded Disability Status Scale (EDSS) and age or disease duration, respectively.15, 16
Serum and CSF Samples
CSF of MS patients was collected through a standard LP during the diagnostic phase of the disease. Polypropylene tubes were centrifuged for 5 minutes at 4°C to remove any cells and debris, aliquoted in small volumes, and stored at −80°C. Peripheral blood of MS patients was sampled in 5ml serum separation tubes (BD Vacutainer; Becton Dickinson, Franklin Lakes, NJ), and serum was obtained after centrifugation, according to the manufacturer's instructions.
Protein Measurements
We measured protein concentrations in duplicate, blinded to clinical and MRI data. The analysis of CHIT1 and CHI3L1 in CSF and serum samples was performed using commercially available enzyme‐linked immunosorbent assay (ELISA) kits: Circulex Human Chitotriosidase 1 ELISA kit (MBL International, Woburn, MA) and MicroVue YKL40 (Quidel, San Diego, CA), respectively. CSF CHIT1 assay required a 20‐fold dilution and CHI3L1 assay a 2.5‐fold dilution. CSF sTREM2 measurements were performed after 1:4 dilution in sample buffer with a Meso Scale Discovery platform–based assay (MSD, Gaithersburg, MD), as previously reported and validated.17 NfL levels in CSF were measured without any dilution using a previously described electrochemiluminescence protocol that we recently implemented.18
Quality control (QC) for assays and samples was performed as follows. We checked consistency of standard curves across plates, with r = 0.99–1 (p < 2.2E‐16) for all 4 assays. For plate‐to‐plate variability, we calculated the coefficient of variation (CV) based on an internal control sample measured on all plates. Mean values across duplicates were used for analysis, and samples with CV > 25% or biomarker levels below detection limit were excluded. Samples above the detection limit were remeasured using a different dilution factor where possible and were otherwise excluded (Supplementary Table 2). Within‐plate variability was assessed based on CV across duplicates. A sensitivity analysis was performed where levels below or above the detection limit were set to the respective detection limits instead.
MRI and Image Analysis
MRI data were acquired on a 3T MRI scanner (Intera, Ingenia, or Achieva; Philips, Best, the Netherlands) equipped with an 8‐, 15‐, or 32‐channel head coil using parameters summarized in Supplementary Table 3. All sequences were obtained in the context of routine clinical follow‐up at the Neurology Department of University Hospitals Leuven. The magnetization transfer imaging data were obtained by acquiring 2 axial gradient‐echo images with and without an off‐resonance magnetization transfer saturation pulse. From the magnetization transfer imaging data, a magnetization transfer ratio (MTR) image was created by calculating the MTR value as MTR = 100(M0 − Ms)/M0, where Ms and M0 represent the signal intensity with and without application of the saturation pulse, respectively. The MTR and 3‐dimensional (3D) fast fluid‐attenuated inversion recovery (FLAIR) images were coregistered to the 3D T1‐weighted images using statistical parametric mapping (SPM; Welcome Trust Center for Neuroimaging, London, UK; version SPM12). Next, we applied the lesion segmentation toolbox using 20 thresholds for kappa, ranging from 0 to 1 in steps of 0.05.19 This parameter controls the initialization of a lesion growing algorithm, and low values of kappa lead to segmentations of lesions, which can be considered more sensitive but less specific for real lesions, and the reverse is true for high values of kappa. The lesion segmentation toolbox requires the 3D T1 and FLAIR images as input and provides the lesion segmentation as well as a hard segmentation of the brain in gray matter (GM), white matter (WM), and CSF based on the use of the VBM8 toolbox (University of Jena, Thuringia, Germany). Using this hard segmentation, we defined normal‐appearing GM (NAGM) and normal‐appearing WM (NAWM) as voxels belonging to GM and WM, respectively, but not belonging to the lesion segmentation when using kappa = 0. MS lesions were defined based on the lesion segmentation when using kappa = 1. MTR values of the coregistered image were calculated in each tissue class (NAGM, NAWM, and lesions) and plotted in a histogram. Histogram parameters median and peak height (expressed as unit percent) were extracted for further analysis. By assessing the median, we avoided the issue that the MTR value in a class was affected by small coregistration errors or by nearby CSF due to partial volume effects. The peak height reflects tissue heterogeneity in MTR.20 The lesion filling procedure in the lesion segmentation toolbox was used to fill the lesion in the 3D T1 images, and the adapted 3D T1 image was soft segmented using SPM12 with the default settings. Volumes of GM and WM and CSF were calculated as the sum of the soft segmentation classes multiplied with voxel volume. Total intracranial volume was defined as the sum of the volumes of these 3 classes. WM and GM volume were expressed as a percentage of total brain volume.
Genotyping and Quality Checks of CHI3L1 and CHIT1 Variants
DNA was extracted from whole blood using an in‐house protocol based on the ethanol precipitation method. Concentration was measured with Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA), and samples were diluted at 100ng/μl in Tris‐EDTA (TE) 10/1. All participants were genotyped for 700,078 variants using the Infinium HTS assay on Global Screening Array BeadChip (Illumina, San Diego, CA). Genotype calling was done using GenomeStudio V2011.1 software. After performing sample QC using PLINK v1.09b5.4 (April 2018) as part of an ongoing genome‐wide association screen (unpublished data), data were imputed.
First, genotypes were prephased using SHAPEIT2 (v2.12)21 with the European 1000 Genomes October 2014 haplotypes (phase 3 integrated variant set release in NCBI build 37 [hg19] coordinates) as a reference panel (http://www.internationalgenome.org/). For prephasing, default parameters were used. The imputation into the estimated haplotypes was performed using the IMPUTE v2.0 software package22 and all 1000 Genomes October 2014 haplotypes (phase 3 integrated variant set release in NCBI build 37 [hg19] coordinates; http://www.internationalgenome.org/). All samples were prephased and imputed in a single batch to avoid batch effects attributable to the imputation process. To make computation feasible during imputation, each chromosome was split into 5Mb chunks with default parameters.
CHI3L1 expression has previously proven to be a highly heritable, quantitative trait, with 1 functional promoter single nucleotide polymorphism (SNP; rs4950928) explaining 9% of variance in RNA expression and circulating plasma protein levels.23, 24, 25 For CHIT1, a common (minor allele frequency 15%) 24bp duplication located in exon 10 (rs150192398) is known to activate a cryptic 3′ splice site in the same exon. This leads to less stable RNA, an 87 amino acid in‐frame deletion, and lower enzyme levels as well as lower enzyme activity.26, 27, 28, 29 Allele dosages for CHI3L1 variant rs4950928 and the 24bp duplication (rs150192398) in CHIT1 for N = 604 patients were extracted from the post‐QC imputed dataset. Imputation info scores were 0.98 and 0.95, respectively. As QC for correct imputation of the 24bp duplication, we genotyped 101 MS patients by using primers flanking the duplication as previously described27 and obtained 100% of concordance with imputed data.
Brain Tissue Data
Normalized microarray and RNA‐sequencing data of microglial biomarkers in brain tissues5, 6 were obtained from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov) with accession numbers GSE108000 and GSE111972, respectively. For RNA‐seq counts,6 we applied (value +1), as there were values of 0. As is conventional for RNA expression data, logarithmic transformation with base 2 was applied.
Statistical Analysis
All statistical analyses were performed with R version 3.1.1. Normality of the data was assessed with Shapiro–Wilk test. As protein levels were not normally distributed, we applied logarithmic transformation with base 10. For clinical and radiological variables, we consistently applied the inverse rank normal transformation for main analyses, as most variables were not normally distributed. Correlations were calculated, and their significance was tested with the Pearson method.
First, we defined our primary and secondary outcomes. As primary outcome, we tested the association between CSF biomarker levels and the following disease activity parameters: ARMSS, recently developed, as a measure of disability progression16; annualized relapse rate before treatment (minimal untreated follow‐up of 90 days), measuring the relapse activity; volumetric parameters GM, WM, and lesion volume as measures of MRI activity; and MTR parameters NAGM, NAWM, and lesion median and peak height, reflecting tissue integrity, as measures of MRI activity. As secondary outcomes, we analyzed the relationship between our CSF biomarker levels and MSSS, as measure of disease progression15; EDSS, as measure of disability; and annualized relapse rate during entire follow‐up (untreated and treated), as relapse activity parameter.
These associations were assessed using the linear regression model of clinical or neurological variables in function of biomarker levels, including covariates when they were correlated with biomarkers. We used age at MRI, gender (for GM volume only), and MRI protocol as covariate for MRI analysis. Age, gender, and genotype (dosage of the minor allele) were included for CHI3L1 and only genotype for CHIT1. We applied a Bonferroni correction for multiple testing, assuming 4 independent biomarkers correlated with 11 independent primary outcomes, resulting in a threshold of p ≤ 0.0011. Correlations with baseline characteristics were considered descriptive and not included in the multiple testing.
Percentage of variance explained was calculated for associations surviving multiple testing based on a linear regression model as the adjusted r 2 with or without covariates and the r 2 of the full model minus the model only including covariates. Clinical covariates known to be associated with the outcome were included, that is, sex, age at onset, and disease course for inverse ranked disability,30 and age at MRI for median MTR in lesions.31 For the receiver operating characteristics (ROC) curve, patients were divided into fast progressors and slow progressors on the basis of ARMSS ≥5 or ARMSS <5, which reflects the top 50% and lower 50% of disability progression in the global MS study population.16 In our local study population, ARMSS ≥5 reflects about 20% of patients (n = 23). The ROC curve was based on the probabilities of logistic regression analysis (generalized linear model) with CHIT1 and clinical covariates known to be associated with disability (sex, age at onset, initial disease course30) as explanatory variables.
The association between genetic variants and biomarker levels was modeled according to a linear regression model in PLINK v1.90b6.2 (June 2018). Age at MTR and MRI protocol were used as covariates for MRI outcomes.
Nonparametric Kruskal–Wallis test with Dunn post hoc test was used to determine statistical differences between RNA levels of microglial biomarkers in different human brain regions.
Results
We investigated association of microglial markers in CSF at time of diagnostic LP with different aspects of disease activity (relapses, disability, MRI parameters) upon follow‐up in a cohort of 143 newly diagnosed, untreated patients (see Table 1). Median disease duration at diagnostic LP was 0 years. A subset of 55 patients (38%) underwent LP within 1 month of a relapse, in most cases (62%) following a relapse. Steroids were administered to only 4 patients (2.8%) before the LP. Our study cohort was representative of the MS patient population, with 62% of patients being female, 79% of patients presenting with BO disease, a median age at onset of 34 years, and a median ARMSS of 2.
Heterogeneity in CSF Microglia‐Related Biomarkers
We compared 3 microglia‐related markers both at the protein level in CSF (Fig 1A–C) and at the RNA level in brain tissue of MS patients (Fig 2). CHI3L1 correlated strongly (r > 0.5) with CHIT1, both at the protein and RNA level, and more modestly (r = 0.3) with the soluble protein form of TREM2. CHI3L1, and to a lesser extent CHIT1, were associated with CSF hallmarks currently used in the diagnosis of MS, that is, immunoglobulin G (IgG) index and oligoclonal bands (OCBs; Supplementary Table 4). In a multiple linear regression analysis including both markers, associations appeared to be primarily driven by CHI3L1 (data not shown). Only CHIT1 was correlated with the established marker of neuronal damage in MS, NfL (see Fig 1D–F).
Figure 1.
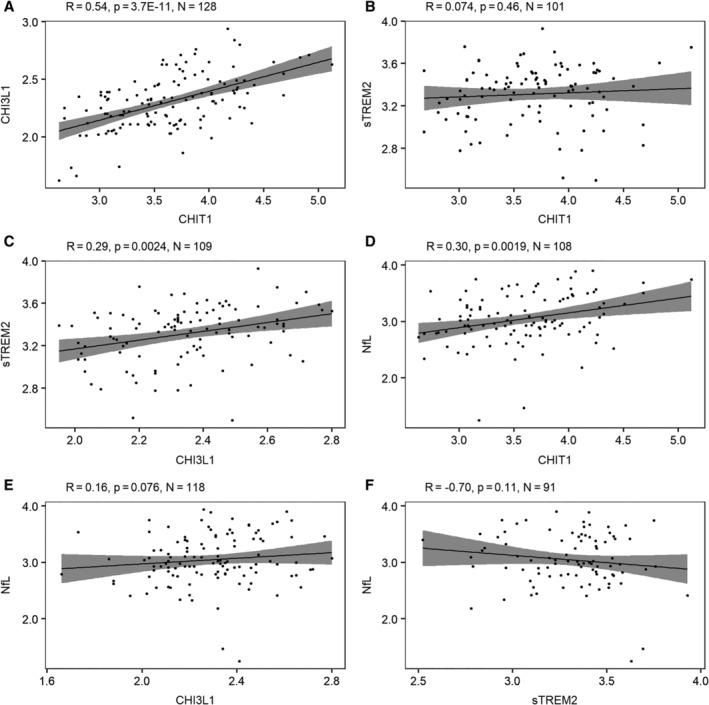
Correlation between cerebrospinal fluid (CSF) microglial and axonal biomarkers. Pairwise comparisons between CSF markers: (A) chitotriosidase (CHIT1; pg/ml) versus chitinase‐3–like protein 1 (CHI3L1; ng/ml), (B) CHIT1 (pg/ml) versus soluble triggering receptor expressed on myeloid cells 2 (sTREM2; pg/ml), (C) CHI3L1 (ng/ml) versus sTREM2 (pg/ml), (D) CHIT1 (pg/ml) versus neurofilament light chain (NfL; pg/ml), (E) CHI3L1 (ng/ml) versus NfL (pg/ml), and (F) sTREM2 (pg/ml) versus NfL (pg/ml). Probability values are derived from Pearson test. Gray shading shows the 95% confidence bands. The logarithmic (base 10) transformation was applied for all biomarkers.
Figure 2.
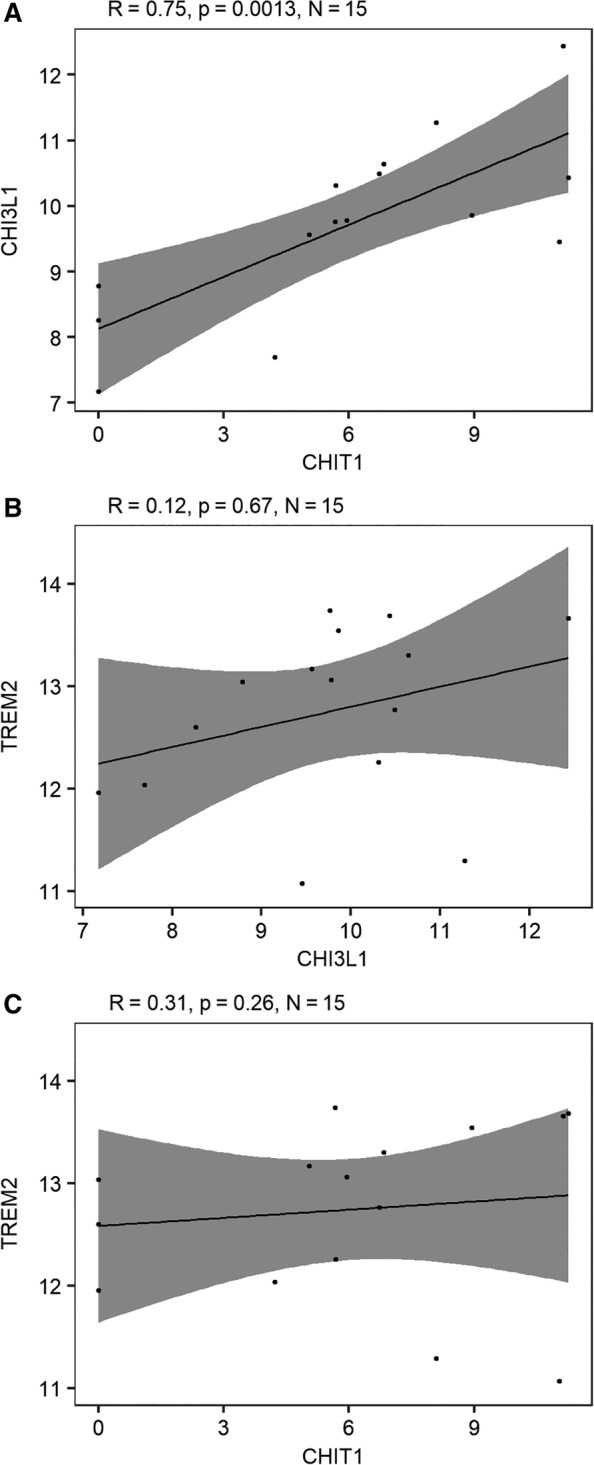
Correlations between microglial marker RNA expression in brain tissue (corpus callosum) of multiple sclerosis patients.6 Pairwise comparisons of gene expression extracted from publicly available data deposited in the Gene Expression Omnibus database originating from Van der Poel et al6: (A) chitotriosidase (CHIT1) versus chitinase‐3–like protein 1 (CHI3L1), (B) CHI3L1 versus triggering receptor expressed on myeloid cells 2 (TREM2), and (C) CHIT1 versus TREM2. Raw values of RNA‐seq counts were replaced with (value +1) because of the presence of zero values before taking the logarithmic transformation with base 2 as conventional in RNA expression data. Probability values are derived from Pearson test. Gray shading shows the 95% confidence bands.
Baseline demographic parameters only influenced CHI3L1, with male sex (p = 0.02) and older age at LP (p = 0.0002) increasing levels. No marker was associated with disease duration at diagnostic LP (p > 0.14) or with the occurrence of a relapse within 1 month of LP (p > 0.63).
CSF CHIT1 Levels at Diagnosis Correlate with Disease Activity Measures at Follow‐up Years Later
Subsequently, we compared how CSF biomarkers measured at time of diagnosis correlated with disease activity parameters at follow‐up many years later, in particular at a median 3 years later for MRI, median 5 years for disability, and median 6 years for relapse activity. In the context of the expanding concept of NEDA, MRI outcomes included volumetric measures for total lesion load (mm3), GM and WM volume (% of total brain volume), as well as MTR in lesions, NAWM, and NAGM. Primary and secondary outcomes were defined, and results are shown in Table 2.
Table 2.
Association of Baseline CSF Biomarkers with Clinical and MRI Parameters at Follow‐up
| NEDA Parameter | Outcome | Variable (Outcome) | NfL | CHIT1 | CHI3L1 | sTREM2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| β | p | β | p | β | p | β | p | |||
| Disability | Primary | ARMSS | 0.18 | 0.39 | 0.70 | 1.2E‐04a | 0.73 | 0.061 | −0.33 | 0.37 |
| Secondary | MSSS | 0.18 | 0.42 | 0.49 | 0.0087b | 0.64 | 0.089 | −0.35 | 0.34 | |
| — | EDSS | 0.22 | 0.68 | 1.62 | 0.00056b | 2.44 | 0.0054b | −1.4 | 0.33 | |
| Relapse | Primary | ARR, untreated | 0.69 | 0.0055b | 0.64 | 0.0015b | 1.12 | 0.0038b | −0.11 | 0.81 |
| Secondary | ARR, all | 0.66 | 0.013b | 0.88 | 1.5E‐05b | 1.19 | 0.0033b | −0.12 | 0.82 | |
| MRI | ||||||||||
| Volumetric | Primary | GM vol | 0.35 | 0.22 | 0.05 | 0.80 | 0.58 | 0.092 | 0.75 | 0.19 |
| — | WM vol | −0.36 | 0.24 | −0.26 | 0.29 | 0.32 | 0.49 | 0.98 | 0.08 | |
| — | Lesion vol (mm3) | 0.39 | 0.18 | 0.70 | 0.0022b | 0.67 | 0.11 | 0.2 | 0.72 | |
| MTR | Primary | Median NAGM | −0.24 | 0.44 | −0.53 | 0.027b | −0.21 | 0.62 | 0.06 | 0.56 |
| — | PH NAGM | −0.37 | 0.26 | −0.44 | 0.046b | −0.59 | 0.17 | 0.21 | 0.73 | |
| — | Median NAWM | −0.27 | 0.40 | −0.61 | 0.015b | −0.17 | 0.70 | −0.92 | 0.11 | |
| — | PH NAWM | −0.04 | 0.89 | −0.18 | 0.46 | −0.16 | 0.71 | 0.38 | 0.53 | |
| — | Median lesion | −0.16 | 0.62 | −0.87 | 9.6E‐04a | −0.61 | 0.21 | −0.76 | 0.23 | |
| — | PH lesion | 0.11 | 0.72 | −0.58 | 0.020b | −0.65 | 0.15 | 0.11 | 0.85 | |
Uncorrected p value from linear regression analysis after applying a logarithmic transformation with base 10 for biomarker measurements, and an inverse rank normal transformation for outcome variables. Primary and secondary outcomes are indicated. For magnetic resonance imaging (MRI) correlations, age at MRI, gender (for gray matter [GM] volume only), and MRI protocol were used as covariates. For chitinase‐3–like protein 1 (CHI3L1), age, gender, and rs4950928 genotype, and for CHIT1 rs150192398 were used as covariates. Volumetric measures are expressed as percentage of total brain volume.
Uncorrected p ≤ 0.0011, passed Bonferroni correction for multiple testing of primary outcomes.
Nominally significant p values (uncorrected p < 0.05).
ARMSS = Age‐Related Multiple Sclerosis Severity Score; ARR = annualized relapse rate; CHIT1 = chitotriosidase; CHI3L1 = chitinase‐3–like protein 1; CSF = cerebrospinal fluid; EDSS = Expanded Disability Status Scale; MSSS = Multiple Sclerosis Severity Score; MTR = magnetization transfer ratio; NAGM = normal appearing gray matter; NAWM = normal appearing white matter; NEDA = no evidence of disease activity; NfL = neurofilament light chain; PH = peak height; sTREM2 = soluble triggering receptor expressed on myeloid cells 2; vol = volume; WM = white matter.
CHIT1 as well as CHI3L1 and NfL levels were associated with relapse rate before treatment (see Table 2). For CHIT1, this association became substantially stronger when considering relapse rate across the entire disease course as evaluated 6 years from LP (see Table 2). A multiple linear regression analysis indicated that CHIT1 correlated with annualized relapse rate independently (Supplementary Table 5). Considering treatment as covariate did not alter these results (data not shown).
The microglia‐related biomarker CHIT1 was remarkably associated at nominal significance (uncorrected p < 0.05) with 7 out of 11 primary outcomes as well as all 3 secondary outcomes. Two associations, with lesion tissue integrity and disability, survived a Bonferroni correction for all primary outcomes, which was even conservative as it assumed all tests were independent.
CHIT1 levels at diagnosis correlated with MRI measures, especially those related to lesions, more than 3 years later. CHIT1 levels increased with increasing MTR abnormalities (decreasing MTR) in lesions (p = 9.6E‐04; see Table 2). CHIT1 levels explained 12% of variance (both with and without covariates) in median lesion MTR across patients (Fig 3).
Figure 3.
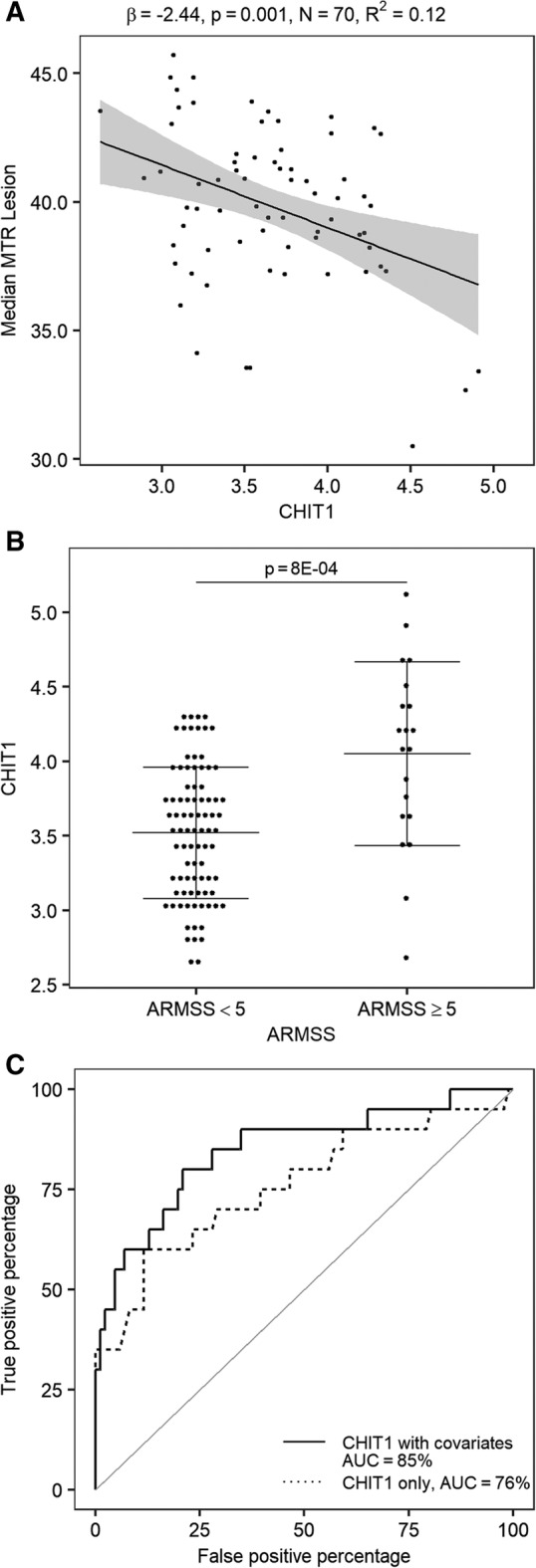
Association of cerebrospinal fluid chitotriosidase (CHIT1) at diagnosis with primary outcome disease activity parameters at follow‐up. (A) Linear regression analysis for CHIT1 (pg/ml) at diagnostic lumbar puncture (LP) versus median lesion magnetization transfer ratio (MTR; untransformed data, P Shapiro–Wilk = 0.03) at a median 3 years (interquartile range [IQR] = 2–5) later including age at magnetic resonance imaging as covariate; gray shading shows the 95% confidence bands. (B) CHIT1 (pg/ml) at diagnostic LP between groups of fast (Age‐Related Multiple Sclerosis Severity Score [ARMSS] ≥ 5) and slow (ARMSS <5) disability progression as measured at a median 5 years (IQR = 2–7) later. Probability value is derived from a logistic regression including known covariates for disability (sex, age at onset, and disease course); horizontal lines indicate mean and error bars. (C) Receiver operating characteristic curves based on CHIT1 alone or CHIT1 including covariates for discrimination of disability progression as defined in (B). The logarithmic (base 10) transformation was applied for CHIT1.
CHIT1 at diagnosis showed a positive association with disability progression as measured by ARMSS 5 years later (p = 1.2E‐04; see Table 2). CHIT1 levels accounted for 8.48% and, when including known clinical covariates, 27.3% of variance in ARMSS rankings across patients. When classifying MS patients into fast and slow progressors on the basis of median ARMSS (ARMSS ≥5 and ARMSS <5), CHIT1 levels were significantly different (p = 8E‐04), and the area under the curve (AUC) in an ROC analysis reached 76% (95% confidence interval [CI] = 62–90%) and 85% (95% CI = 74–96%) without and with known covariates, respectively (see Fig 3). There was no correlation between CSF and serum CHIT1 levels in 78 patients for whom both samples were available (r = 0.03, p = 0.78), emphasizing the need for CSF as a prognostic marker.
Genetics Impacting CSF Microglia Markers
CHI3L1 expression has previously proven to be a highly heritable, quantitative trait, with 1 functional promoter SNP (rs4950928) explaining 9% of variance in RNA expression and circulating plasma protein levels.23 We extended this association to circulating CHI3L1 levels in CSF (β = −0.46, p = 0.014, n = 141).
For CHIT1, a common (minor allele frequency 15%) 24bp duplication located in exon 10 (rs150192398) is known to activate a cryptic 3′ splice site in the same exon. This leads to less stable RNA, an 87 amino acid in‐frame deletion, and lower enzyme levels as well as lower enzyme activity.28, 29 We observed significantly lower CHIT1 levels in duplication carriers (β = −0.28, p = 0.009, n = 121; Fig 4).
Figure 4.
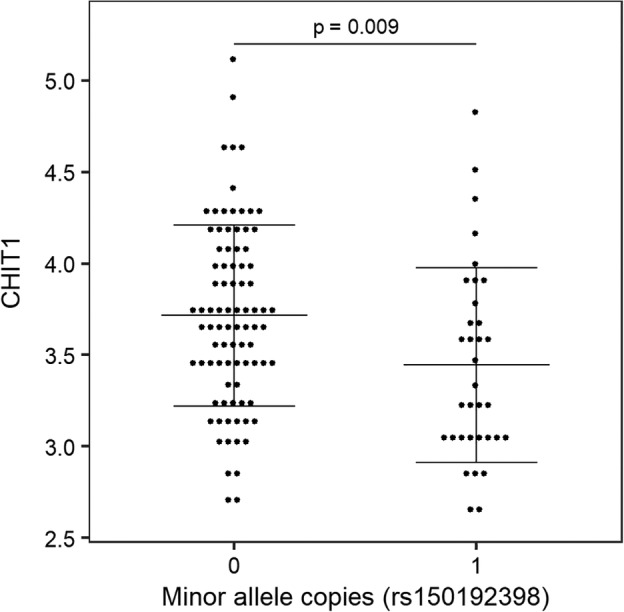
Correlation between chitotriosidase (CHIT1) 24bp duplication (rs150192398) and CHIT1 levels. X‐axis depicts copies of the minor allele (24bp duplication), which was previously demonstrated to affect RNA stability27 and protein levels.29 The logarithmic transformation (base 10) was applied for CHIT1 (pg/ml) levels. Probability value is derived from linear regression model of allele dosage. 0 = homozygous major allele, n = 87; 1 = heterozygous, n = 34. Horizontal lines indicate mean and error bars.
As both variants affect levels, we included them as covariates in main analyses with primary and secondary outcomes (see Table 2). A sensitivity analysis indicated that leaving out these covariates slightly reduced significance levels but overall did not affect conclusions (data not shown). No association was observed between these genetic variants and any of the primary outcomes in a larger genetics study population of up to 604 MS patients (Supplementary Table 6).
CHIT1 Is a Marker of Chronic Active MS Lesions in Postmortem Tissue
Publicly available datasets on postmortem MS brain tissue allowed us to compare RNA levels of microglial biomarkers across brain regions. In the dataset from Van der Poel et al, 6 CHI3L1 was differentially expressed in WM in patients versus WM control tissue. CHIT1 expression was virtually absent in GM and in the majority of WM tissue samples from non‐neurological controls and was therefore excluded from the original analysis.6 Nevertheless, CHIT1 was significantly upregulated in WM of MS patients compared to both GM and WM of controls, whereas no differential expression was seen for TREM2 (Fig 5A).
Figure 5.
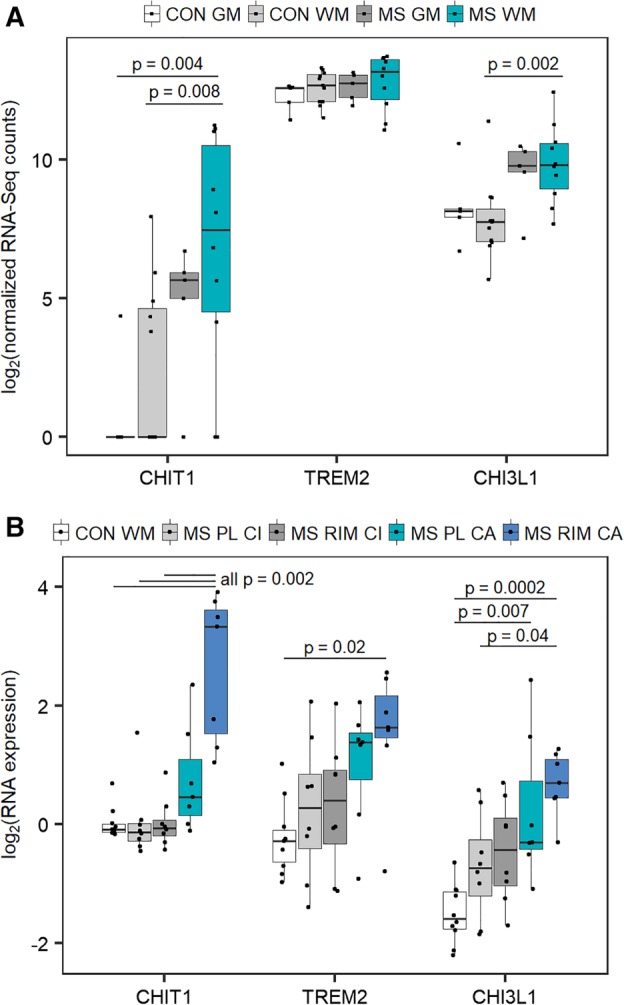
(A) Microglial marker RNA expression in multiple sclerosis (MS) white and gray matter in 15 MS patients and 10 controls. Probability values are derived from nonparametric Kruskal–Wallis test with Dunn post hoc test on publicly available data deposited in the Gene Expression Omnibus database originating from Van der Poel et al.6 Comparisons without p indicated were not significant. Raw values of RNA‐seq counts were replaced with (value +1) because of the presence of zero values before taking the logarithmic transformation with base 2 as conventional in RNA expression data. (B) Microglial marker RNA expression in active and inactive chronic lesions. Probability values are derived from nonparametric Kruskal–Wallis test with Dunn post hoc test on publicly available data deposited in the Gene Expression Omnibus database originating from Hendrickx et al.5 CHIT1 = chitotriosidase; CHI3L1 = chitinase‐3–like protein 1; CON GM = gray matter control; CON WM = white matter control; MS GM = gray matter from MS patients; MS WM = white matter lesions from MS patients; MS PL CA = perilesion of chronic active lesions from MS patients; MS PL CI = perilesion of chronic inactive lesions from MS patients; MS RIM CA = rim of chronic active lesions from MS patients; MS RIM CI = rim of chronic inactive lesions from MS patients; TREM2 = triggering receptor expressed on myeloid cells 2.
The dataset of Hendrickx et al5 subsequently provided more detail on lesions and allowed us to distinguish between the rim and perilesion region around chronic active and inactive lesions. As originally reported, CHIT1 is upregulated 10.2‐fold in the rim of chronic active lesions versus the rim of chronic inactive lesions. Considering here the overview of all markers and tissue types (see Fig 5B), we additionally observed differential expression for CHI3L1 and more modestly for TREM2. Importantly, among the 3 markers, CHIT1 discriminated best between chronic active lesions and chronic inactive lesions, the latter not different from control WM tissue (see Fig 5B).
Discussion
We demonstrated that CHIT1 is a microglial marker upregulated specifically in chronic active MS lesions and that CHIT1 levels measured in the CSF during diagnostic LP correlated with multiple aspects of disease activity 3 to 6 years later. CHIT1 levels explained 12% and 27% of variance in 2 disease activity measures, that is, MTR abnormalities in lesions and disability progression, respectively. Moreover, CHIT1 levels reached an AUC of 85% to distinguish fast and slow progressors. Our data indicate that CHIT1 CSF levels measured only once during the diagnostic LP reflect microglial activation early in the disease and may predict subsequent disease activity. CHIT1 has already been used for 15 years in clinical practice for monitoring disease severity and effectiveness of therapy in Gaucher patients, despite some limitations.32
CHIT1 was additionally associated with NfL, a structural cellular protein in neurons that is released into the extracellular space as a consequence of neuronal damage. Both serum and CSF NfL levels have been consistently associated with long‐term brain atrophy.33, 34 Of note, this effect size is similar to the percentage of variance explained for the associations between CHIT1 and disease activity measures in our work. In contrast, serum and CSF NfL levels measured at 6 to 8 years of disease duration were not associated or were at most weakly associated with future relapse activity, EDSS worsening, or lesions in 2 recent large‐scale studies.33, 34 Similarly, we did not detect an association between NfL levels at diagnosis and any primary outcome, and associations between CHIT1 and disease activity outcomes were importantly independent of NfL. A combination of biomarkers will likely be indicated to capture the different disease processes in MS.
MTR is not currently included as a NEDA parameter, but given its increasing use in clinical trials35 and the extending concept of NEDA, we included it in our study. Demyelination has been proposed to be the major pathologic substrate for decreased MTR within MS lesions, but additional pathologic features may contribute to the MTR signal. Increased numbers of enlarged microglia/macrophages contribute to MTR abnormalities in NAWM and even more prominently in lesions.36 Imaging measures that are more specific for activated microglia and macrophages are being developed, such as position emission tomography imaging using 18kDa translocator protein binding radioligands.7, 36 Correlations between MTR and disease progression have been observed in epidemiological studies. African American MS patients have lower MTR in lesions, NAWM, and NAGM, higher lesion volume, and a higher MSSS compared to white American patients,37 although the direct correlation between these outcomes is not fully understood.
Three microglia‐related markers, CHIT1, CHI3L1, and TREM2, correlated only partially both at the protein level in CSF and at the RNA level in postmortem brain tissue. CHIT1 appeared to be largely undetectable in controls and the best discriminator of chronic active versus chronic inactive lesions. This suggests the 3 markers reflect the increasingly recognized heterogeneity in microglia and macrophages. In postmortem amyotrophic lateral sclerosis (ALS) spinal cord WM, CHIT1 was expressed in cells positive for IBA1, a general marker for microglia and macrophages, and CD68, a marker of phagocytosis in macrophages.10 This is in line with the upregulation of CHIT1 in lipid‐laden macrophages in the lysosomal Gaucher and Fabry diseases,38, 39 its correlation with myelin basic protein in MS,40 and the upregulation of CHIT1 after uptake of myelin from MS patients but not from control individuals.5 In MS active lesions, these phagocytic innate immune cells increase already in initial lesion stages and reach their peak in early/late active lesion areas.41 A major proportion of phagocytes present in early stages of MS lesions are derived from the microglia pool, and with the maturation of active lesions, an increasing proportion of macrophages have entered the lesions from the blood.41 Although CHIT1 function in the CNS is not yet clear, a recent study shows that, like murine CHI3L3, human CHI3L1 and CHIT1 induce oligodendrogenesis in vitro.42 First single‐cell sequencing data emerged from a cancer context, where a branch of intratumoral macrophages was composed of 3 clusters with as top upregulated genes TREM2, APOE, CD68, and CHIT1. Despite this coregulation as part of the same branch, these genes were differentially expressed among the 3 clusters, with CHIT1 correlating the most with increasing intratumoral macrophage activation.43 With the application of single‐cell sequencing on postmortem brain material from patients, it may become possible to distinguish whether CHIT1 in neurological disease is produced by microglia and/or by macrophages that have entered from the periphery but are activated in the CNS. Moreover, it may indicate which subsets of these cells are the main producers and whether other CNS‐specific cell types contribute to the production.
Microglial activation is a hallmark of many neurological diseases, but microglial subsets appear to contribute to a different extent and highlight parallels and differences between diseases. TREM2 variants increase Alzheimer disease (AD) risk and affect disease progression through impairing the proper function of microglia.44 CSF sTREM2 levels are increased in AD and mild cognitive impairment patients compared with controls and correlate strongly with tau pathology.45 Although Piccio et al12 reported elevated sTREM2 levels in MS patients compared to controls, we did not observe any correlation with disease activity. In ALS, CHIT1 is able to discriminate better between patients and healthy controls than CHI3L1 and discriminates ALS from frontotemporal dementia and primary lateral sclerosis, where instead CHI3L1 is more prominently increased. CHIT1 levels and activity in ALS have repeatedly been associated with neurofilament levels and with a faster disease progression rate in large study populations.10, 28, 29, 46, 47, 48 Our results demonstrate the correlation between baseline CSF CHIT1 levels and neuronal damage and prognosis in MS.
We found CHI3L1 levels to increase with age, as also demonstrated for ALS,46, 49 and to be associated with antibody‐related CSF measures such as OCB‐positive status and IgG index but most strongly the number of OCBs. CHI3L1 levels have been reported as a predictor for the conversion from clinically isolated syndrome to MS, although not consistently for longer follow‐up times or for radiologically isolated syndrome.11, 50, 51 This association appears independent of OCB positive/negative status, whereas the number of OCBs was not taken into account.11, 52 Only 1 previous study considered CHIT1 in the context of prognosis in MS but was limited in terms of a smaller study population and of sampling patients with an indication to switch therapies at different time points in disease instead of newly diagnosed patients.53 Baseline (diagnostic stage) CSF CHI3L1 levels have been associated with earlier progression to disability.50, 51 There is a similar trend in our study, but we demonstrated this to be secondary to the substantially stronger association with CHIT1.
Genetic variants are known to explain a substantial fraction of CHIT1 and CHI3L1 levels and/or activity.28, 29 They can be considered as naturally occurring, genetically determined variation in levels or activity preceding disease onset. Taking these genetic variants into account in our analysis did not substantially alter conclusions, and, in contrast to CHIT1 and CHI3L1 levels in a cohort of 143 patients, the functional genetic variants were not associated with any of the primary outcomes in a cohort of up to 604 patients. Although using just 1 genetic variant is a weaker instrument with reduced power compared to the levels itself, this would suggest that increased CHIT1 and CHI3L1 levels are markers for the contribution of microglia to disease rather than that variation in CHIT1 and CHI3L1 levels or activity contributes mechanistically to disease outcomes.
The strengths of our study, which distinguish our work from many related studies, are (1) the investigation of multiple CSF microglial biomarkers as well as NfL in the same large cohort of 143 MS patients, (2) the measurements at time of diagnosis (and thus in untreated patients) instead of at different time points during the disease process, and (3) the ability to correlate potentially prognostic biomarkers at diagnosis with a median 6‐year longitudinal follow‐up of these patients by the same expert neurologist. Our cohort reflects a real‐world clinical setting, but this could also be a drawback as it is therefore less harmonized in terms of follow‐up protocol. This necessarily introduces nonstandardized time points for disability measurements and MRI scans and different MRI scanners and protocols used in clinical practice over time. We attempted to take these limitations into account as much as possible by different measures. Specifically, 1 expert neurologist registered all clinical follow‐up, and we performed sensitivity analyses or included covariates reflecting biological or technical differences. However, validation in a large, prospective study cohort would be required before implementation in clinical practice, and such studies are ongoing.
In summary, our data contribute to the growing recognition of the role of microglia in neurological diseases including MS, highlight the contribution of early microglial activation to subsequent disease progression, and indicate CHIT1 can be considered a good candidate for a prognostic biomarker.
Author Contributions
E.O., B.D., and A.G. contributed to the conception and design of the study; all authors contributed to the acquisition and analysis of data; E.O. and A.G. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Appendix S1. Supporting Information.
Acknowledgment
This study was supported by the research fund of KU Leuven (C24/16/045), the Research Foundation–Flanders (FWO G073415N), the Belgian Charcot Foundation, and MS Liga Vlaanderen. E.O. holds a European Committee for Treatment and Research in Multiple Sclerosis Postdoctoral Fellowship, and L.V.H. holds a PhD Fellowship of the Belgian Charcot Foundation. B.D. is a Clinical Investigator, and M.V. a Research Fellow of the Research Foundation–Flanders.
We thank K. Clysters and C. Thys for their contribution in sample collection and A. Petzold, M. van der Poel, and M. Suarez Calvet for helpful communication on biomarker protocols.
References
- 1. Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502–1517. [DOI] [PubMed] [Google Scholar]
- 2. Giovannoni G, Bermel R, Phillips T, Rudick R. A brief history of NEDA. Mult Scler Relat Disord 2018;20:228–230. [DOI] [PubMed] [Google Scholar]
- 3. Comabella M, Montalban X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol 2014;13:113–126. [DOI] [PubMed] [Google Scholar]
- 4. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018;14:577–589. [DOI] [PubMed] [Google Scholar]
- 5. Hendrickx DAE, van Scheppingen J, van der Poel M, et al. Gene expression profiling of multiple sclerosis pathology identifies early patterns of demyelination surrounding chronic active lesions. Front Immunol 2017;8:1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van der Poel M, Ulas T, Mizee MR, et al. Transcriptional profiling of human microglia reveals grey–white matter heterogeneity and multiple sclerosis‐associated changes. Nat Commun 2019;10:1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ciccarelli O, Barkhof F, Bodini B, et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol 2014;13:807–822. [DOI] [PubMed] [Google Scholar]
- 8. International Multiple Sclerosis Genetics Consortium . Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019;365:eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ziatabar S, Zepf J, Rich S, et al. Chitin, chitinases, and chitin lectins: emerging roles in human pathophysiology. Pathophysiology 2018;25:253–262. [DOI] [PubMed] [Google Scholar]
- 10. Steinacker P, Verde F, Fang L, et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J Neurol Neurosurg Psychiatry 2018;89:239–247. [DOI] [PubMed] [Google Scholar]
- 11. Cantó E, Tintoré M, Villar LM, et al. Chitinase 3‐like 1: prognostic biomarker in clinically isolated syndromes. Brain 2015;138:918–931. [DOI] [PubMed] [Google Scholar]
- 12. Piccio L, Buonsanti C, Cella M, et al. Identification of soluble TREM‐2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008;131:3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17:162–173. [DOI] [PubMed] [Google Scholar]
- 14. Hilven K, Vandebergh M, Smets I, et al. Genetic basis for relapse rate in multiple sclerosis: association with LRP2 genetic variation. Mult Scler J 2018;24:1773–1775. [DOI] [PubMed] [Google Scholar]
- 15. Roxburgh RHSR, Seaman SR, Masterman T, et al. Multiple sclerosis severity score: using disability and disease duration to rate disease severity. Neurology 2005;64:1144–1151. [DOI] [PubMed] [Google Scholar]
- 16. Manouchehrinia A, Westerlind H, Kingwell E, et al. Age Related Multiple Sclerosis Severity Score: disability ranked by age. Mult Scler J 2017;23:1938–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suárez‐Calvet M, Caballero MÁA, Kleinberger G, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med 2016;8:369ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gille B, De Schaepdryver M, Goossens J, et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 2019;45:291–304. [DOI] [PubMed] [Google Scholar]
- 19. Roura E, Oliver A, Cabezas M, et al. A toolbox for multiple sclerosis lesion segmentation. Neuroradiology 2015;57:1031–1043. [DOI] [PubMed] [Google Scholar]
- 20. Fisniku LK, Altmann DR, Cercignani M, et al. Magnetization transfer ratio abnormalities reflect clinically relevant grey matter damage in multiple sclerosis. Mult Scler 2009;15:668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Connell J, Gurdasani D, Delaneau O, et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet 2014;10:e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dixon AL, Liang L, Moffatt MF, et al. A genome‐wide association study of global gene expression. Nat Genet 2007;39:1202–1207. [DOI] [PubMed] [Google Scholar]
- 24. Zhao X, Tang R, Gao B, et al. Functional variants in the promoter region of chitinase 3‐like 1 (CHI3L1) and susceptibility to schizophrenia. Am J Hum Genet 2007;80:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ober C, Tan Z, Sun Y, et al. Effect of variation in CHI3L1 on serum YKL‐40 level, risk of asthma, and lung function. N Engl J Med 2008;358:1682–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Piras I, Melis A, Ghiani ME, et al. Human CHIT1 gene distribution: new data from Mediterranean and European populations. J Hum Genet 2007;52:110–116. [DOI] [PubMed] [Google Scholar]
- 27. Boot RG, Renkema GH, Verhoek M, et al. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem 1998;273:25680–25685. [DOI] [PubMed] [Google Scholar]
- 28. Pagliardini V, Pagliardini S, Corrado L, et al. Chitotriosidase and lysosomal enzymes as potential biomarkers of disease progression in amyotrophic lateral sclerosis: a survey clinic‐based study. J Neurol Sci 2015;348:245–250. [DOI] [PubMed] [Google Scholar]
- 29. Oeckl P, Weydt P, Steinacker P, et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry 2019;90:4–10. [DOI] [PubMed] [Google Scholar]
- 30. Confavreux C, Vukusic S. Age at disability milestones in multiple sclerosis. Brain 2006;129:595–605. [DOI] [PubMed] [Google Scholar]
- 31. Newbould RD, Nicholas R, Thomas CL, et al. Age independently affects myelin integrity as detected by magnetization transfer magnetic resonance imaging in multiple sclerosis. NeuroImage Clin 2014;4:641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weinreb NJ, Aggio MC, Andersson HC, et al. Gaucher disease type 1: revised recommendations on evaluations and monitoring for adult patients. Semin Hematol 2004;41:15–22. [DOI] [PubMed] [Google Scholar]
- 33. Kuhle J, Plavina T, Barro C, et al. Neurofilament light levels are associated with long‐term outcomes in multiple sclerosis. Mult Scler J (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cantó E, Barro C, Zhao C, et al. Association between serum neurofilament light chain levels and long‐term disease course among patients with multiple sclerosis followed up for 12 years. JAMA Neurol 2019;76:1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y, Salter A, Wallström E, et al. Evolution of clinical trials in multiple sclerosis. Ther Adv Neurol Disord 2019;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moll NM, Rietsch AM, Thomas S, et al. Multiple sclerosis normal‐appearing white matter: pathology‐imaging correlations. Ann Neurol 2011;70:764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weinstock‐Guttman B, Ramanathan M, Hashmi K, et al. Increased tissue damage and lesion volumes in African Americans with multiple sclerosis. Neurology 2010;74:538–544. [DOI] [PubMed] [Google Scholar]
- 38. Boven LA, van Meurs M, Boot RG, et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol 2004;122:359–369. [DOI] [PubMed] [Google Scholar]
- 39. Vedder AC, Cox‐Brinkman J, Hollak CEM, et al. Plasma chitotriosidase in male Fabry patients: a marker for monitoring lipid‐laden macrophages and their correction by enzyme replacement therapy. Mol Genet Metab 2006;89:239–244. [DOI] [PubMed] [Google Scholar]
- 40. Møllgaard M, Degn M, Sellebjerg F, et al. Cerebrospinal fluid chitinase‐3‐like 2 and chitotriosidase are potential prognostic biomarkers in early multiple sclerosis. Eur J Neurol 2016;23:898–905. [DOI] [PubMed] [Google Scholar]
- 41. Zrzavy T, Hametner S, Wimmer I, et al. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017;140:1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Starossom SC, Campo Garcia J, Woelfle T, et al. Chi3l3 induces oligodendrogenesis in an experimental model of autoimmune neuroinflammation. Nat Commun 2019;10:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Azizi E, Carr AJ, Plitas G, et al. Single‐cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018;174:1293–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McQuade A, Blurton‐Jones M. Microglia in Alzheimer's disease: exploring how genetics and phenotype influence risk. J Mol Biol 2019;431:1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brosseron F, Traschütz A, Widmann CN, et al. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer's disease. Alzheimers Res Ther 2018;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thompson AG, Gray E, Thézénas M‐L, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol 2018;83:258–268. [DOI] [PubMed] [Google Scholar]
- 47. Thompson AG, Gray E, Bampton A, et al. CSF chitinase proteins in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2019;90:1215–1220. [DOI] [PubMed] [Google Scholar]
- 48. Gille B, De Schaepdryver M, Dedeene L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry 2019;90:1338–1346. [DOI] [PubMed] [Google Scholar]
- 49. Andrés‐Benito P, Domínguez R, Colomina MJ, et al. YKL40 in sporadic amyotrophic lateral sclerosis: cerebrospinal fluid levels as a prognosis marker of disease progression. Aging 2018;10:2367–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Comabella M, Fernández M, Martin R, et al. Cerebrospinal fluid chitinase 3‐like 1 levels are associated with conversion to multiple sclerosis. Brain 2010;133:1082–1093. [DOI] [PubMed] [Google Scholar]
- 51. Martínez MAM, Olsson B, Bau L, et al. Glial and neuronal markers in cerebrospinal fluid predict progression in multiple sclerosis. Mult Scler 2015;21:550–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dalla Costa G, Passerini G, Messina MJ, et al. Clinical significance of the number of oligoclonal bands in patients with clinically isolated syndromes. J Neuroimmunol 2015;289:62–67. [DOI] [PubMed] [Google Scholar]
- 53. Novakova L, Axelsson M, Khademi M, et al. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing‐remitting multiple sclerosis. J Neurochem 2017;141:296–304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.