

Abstract
The decomposition of state‐of‐the‐art lithium ion battery (LIB) electrolytes leads to a highly complex mixture during battery cell operation. Furthermore, thermal strain by e.g., fast charging can initiate the degradation and generate various compounds. The correlation of electrolyte decomposition products and LIB performance fading over life‐time is mainly unknown. The thermal and electrochemical degradation in electrolytes comprising 1 m LiPF6 dissolved in 13C3‐labeled ethylene carbonate (EC) and unlabeled diethyl carbonate is investigated and the corresponding reaction pathways are postulated. Furthermore, a fragmentation mechanism assumption for oligomeric compounds is depicted. Soluble decomposition products classes are examined and evaluated with liquid chromatography‐high resolution mass spectrometry. This study proposes a formation scheme for oligo phosphates as well as contradictory findings regarding phosphate‐carbonates, disproving monoglycolate methyl/ethyl carbonate as the central reactive species.
Keywords: 13C-labeling, electrolyte decomposition pathways, LC-MS, lithium ion battery
Thermal and electrochemical degradation reactions of a common lithium ion battery electrolyte (ethylene carbonate/diethyl carbonate + LiPF6) were investigated by using isotope labeling studies. Reaction pathways are postulated as well as a fragmentation mechanism assumption for oligomeric compounds depicted.

Introduction
Lithium ion batteries (LIBs) are the energy storage technology of choice for portable electronics and the E‐mobility sector.1, 2, 3 Challenging demands on LIBs like fast charging, long‐term cycling stability and safety features can be approached by specifically tailored electrolyte formulations.4, 5 The state‐of‐the‐art electrolyte typically consists of lithium hexafluorophosphate dissolved in a binary mixture of cyclic and linear organic carbonates.6 In this regard, the advantages of electrolytes should not be reduced to the physical properties of high ionic conductivity and Li+ solvation by the initial electrolyte composition, but also specific interphase formation capabilities via degradation products can be addressed, enabling high performance LIBs. Particularly, the formation process and the properties of the solid electrolyte interphase (SEI) and cathode electrolyte interphase (CEI) on the surfaces of the negative electrode, usually graphite7, 8, 9 and positive electrode, commonly a lithium transition metal oxide,10, 11, 12 respectively, are fundamental for the performance of LIBs.5, 13, 14 CEI and SEI are formed during the first (formation) cycles. The SEI consists of organic and inorganic electrolyte decomposition products.6, 15, 16 The correlation of electrolyte decomposition products and LIB performance beyond the interphase formation is basically unknown. Consequently, the identification of electrolyte‐soluble decomposition products and reaction pathways can be beneficial to improve the LIB system. Laruelle and co‐workers identified species, proposed decomposition pathways and concluded that the electrolyte degradation is a diverse and progressive process.17, 18, 19, 20, 21 The proposed reactive species monoglycolate methyl/ethyl carbonate18 was claimed the key compound for the initiation of the decomposition (“intermediary A”).19 The decomposition of LiPF6‐based electrolytes to organo(fluoro)phosphates (O(F)Ps) was investigated intensively.22, 23, 24, 25 Subsequent studies addressed reductively and oxidatively generated products and evolving gases separately26, 27 as well as the identification of further decomposition compounds.28, 29
Besides electrochemical decomposition, thermal stress at elevated temperatures (60–80 °C) is another reason for a ring opening polymerization of ethylene carbonate (EC) and a comparable variety of species.25, 30, 31, 32, 33 Overall, the identified soluble compound classes in electrolytes covered (i) oligo carbonates, (ii) carbonate ether co‐oligomers, (iii) glycols, (iv) O(F)Ps, (v) phosphate‐carbonate‐intermixtures and (vi) oligo phosphates, resulting in various different species.25, 28, 29, 34, 35
Particularly, the decomposition route to oligo phosphates via “intermediary A” was difficult to define. Recently, Wang, Xu, Eichhorn and co‐workers corrected the previous assumption of lithium ethyl di‐carbonate (LEDC) as a main SEI component to the analogous monocarbonate species.36 Lithium ethyl monocarbonate (LEMC) presumably provides high reactivity and is an integral component of electrochemical degradation assumptions in this study.37
In this work, the two major degradation processes of LIB electrolytes are investigated. Therefore, thermally‐ and electrochemically‐induced degradation products are addressed, particularly by isotopically labeled electrolyte components, such as 13C3‐EC and unlabeled diethyl carbonate (DEC), analogously to the NMR analysis approaches of Grey and co‐workers.38, 39 The focus was to clarify the origin of soluble oligomeric compounds in LIB electrolytes and to evaluate previous decompositions pathways by a 13C‐isotope labeling study. The structure elucidation was conducted with liquid chromatography‐mass spectrometry (LC‐MS) as it provides high resolution MS and fragmentation capabilities. This study confirms postulated reaction pathways, reveals contradictories with “intermediary A” and harmonizes findings to new decomposition routes considering LEMC as reactive species. The obtained insights contribute to the general understanding of the electrolyte decomposition process in LIBs, potentially resulting in the redesign or tailoring of electrolyte formulations and additives in order to suppress/enhance certain reactions for specific LIB applications.
Results and Discussion
In previous studies, a large variety of similar as well as different decomposition product species was identified in thermally/electrochemically aged samples.27, 28, 29, 40
One representative LIB electrolyte formulation ((13C3‐)EC/DEC, 30/70 wt. %; 1 mol L−1 LiPF6) was investigated after thermal degradation in a sealed container or electrochemical aging consisting of three formation cycles and >500 cycles. To decipher the molecular origin of electrolyte decomposition products, the exclusion of reaction products of labeled EC and unlabeled DEC solvent reagents during the respective decomposition was imperative. Therefore, gas chromatography‐mass spectrometry (GC‐MS) measurements were conducted and yielded unaltered 13C3‐EC and DEC species after both aging processes (Figure S1 in the Supporting Information). Hence, decomposition products that contain isotope‐labeled carbons can be solely attributed to EC degradation. Based on this conclusion, LC‐IT‐TOF‐MS investigations were conducted to decipher the carbon origin either 13C3‐EC or DEC of formed species. The possibility to perform multiple fragmentation reactions (MSn) combined with high resolution MS of the respective decomposition product (precursor ion) allowed the determination of the 13C positions. Decomposition pathways for thermal and electrochemical aging were postulated using reactive species described in literature and obtained LC‐IT‐TOF‐MS results.5, 19, 36 Accordingly, 13C‐EC‐originating entities are labeled in red, whereas unlabeled DEC‐originating entities are shown in black. Furthermore, the formed adduct regions in the mass spectra are indicated with different background colors ([M+H]+ in blue, [M+NH4]+ in red, [M+Na]+ in green). Structural findings and 13C position allocations were confirmed via GC‐atmospheric pressure chemical ionization‐MS (for experimental data and one representative mass spectrum we refer to the Supporting Information). In the following sections, thermal and electrochemical aging pathways of the major decomposition classes are addressed separately and findings are correlated with pathways described in literature. For clarity reasons, only one representative of each decomposition class is discussed in detail.
Thermal aging
In contrast to electrochemical aging, thermal aging of electrolytes led to the formation of carbonate ether co‐oligomers with decreased carbonate content and ethylene glycol oligomers to a large extent. The spectra of a co‐oligomer containing one carbonate subunit and the corresponding fragmentation pattern are depicted in Figure 1.
Figure 1.

LC‐IT‐TOF‐MS spectra of one carbonate ether co‐oligomer (top left) and one ethylene glycol oligomer (bottom left) as well as the respective MS2 information (right). Regions of varying adducts are indicated with different colors. The selected precursor ions are colored according to the formed adduct, fragment structures are shown and characteristic fragments are labeled in yellow. The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
The formed adduct patterns revealed two major signals with a total mass shift of +6/7 Da compared to the unlabeled species. Signals with low intensities at ±1 Da to these two signals relate to the purity of 13C3‐EC (99 % isotope enrichment) and the additional occurrence of the natural 13C/12C ratio (1.1 %). Consequently, only major signals were considered for the discussion of the decomposition product formation.
The MS2 spectra of the most intensive proton adduct signal showed two characteristic fragments with 13C‐labeled carbonyl carbons and a molecular termination with a DEC‐based ethyl group (Figure 1 top; m/z 92.0321, m/z 120.0652). Regarding this finding, the two major signals of the co‐oligomer originated from a 13C‐labeled and an unlabeled carbonyl carbon. The higher intensity of the labeled species indicated a prevalent EC‐based decomposition pathway. The ethylene glycol oligomer (n=3) showed only one signal for the respective adducts with a mass shift of +6 Da compared to the unlabeled species (Figure 1, top and bottom). The fragment pattern revealed 13C‐labeled vinylene groups (m/z 93.0739) and a maximum of two unlabeled carbon atoms in all fragments, indicating high intramolecular distance of the four unlabeled carbons. The combined information regarding the co‐oligomer and the ethylene glycol indicated an EC‐based elongation of the ethylene glycol subunits and a DEC‐based termination in thermal aging experiments. These assumptions were conveyed to carbonate oligomers for the confirmation and postulation of a thermal decomposition pathway. Exemplarily, the MS1/2 spectra of a carbonate oligomer with four carbonate subunits are depicted in Figure 2. The formed sodium adducts showed mass shifts of +7,8,9 and 10 Da (m/z 412.1234, 413.1277, 414.1312 and 415.1346) compared to the unlabeled carbonate oligomer.
Figure 2.
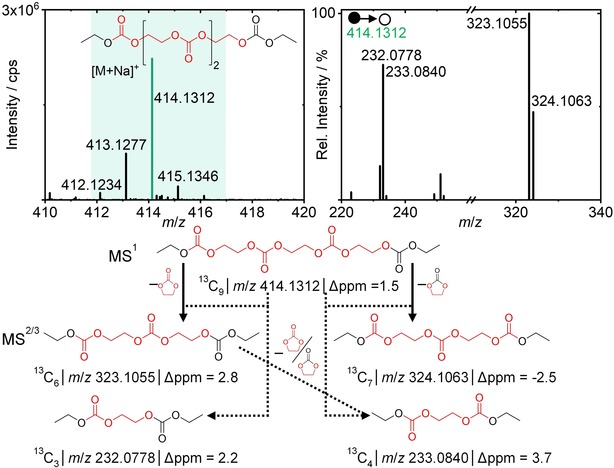
LC‐IT‐TOF‐MS and MS2 spectra of a carbonate oligomer (top). The selected precursor ion is colored according to the formed adduct and the allocation of obtained m/z to fragment structures via an intramolecular rearrangement depicted (bottom). The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
In comparison to Figure 1, the labeling variation in Figure 2 increased for higher carbonyl carbon content. Threefold 13C2 labeled ethylene conjunctions between four carbonate subunits and a different labeling degree of the carbonyl carbons (0–4) provided the explanation for the +7, 8, 9 and 10 Da observation. Remarkably, the threefold labeled carbonyl species showed the highest intensity, whereas the fourfold labeled carbonyl carbon compound was less intense compared to twofold labeled. Signals of singly labeled and unlabeled species were low or below detection limits. Hence, the polymerization in thermal aging is based on an EC elongation mechanism and a predominant termination via DEC.
Considering a single bond cleavage starting from the terminal groups, the obtained fragment information was contradictory. The threefold labeled species cannot be explained with this fragmentation assumption since all obtained fragments of the precursor ion (13C9 12C5 carbon content) required 12C4‐5 remaining in the fragment structure. Varied positions of the unlabeled carbons within the molecule entities or possible isomers did not allow the clarification of main fragments. Furthermore, the assumption of arbitrary 13C positions and possible isomeric structures would result in a more complex MS2 spectrum. Consequently, an alternative fragmentation mechanism was postulated (Figure 2). An intramolecular rearrangement and simultaneous loss of a neutral leaving group (a stoichiometric equivalent to 13C3‐EC or 13C2‐EC) served as a possible explanation for the observed spectra. Minor signals in the MS2 spectra (not shown) within a distance of 45 Da (intramolecular loss of 13CO2) to the main fragments strengthened the postulated fragmentation mechanism of prior intramolecular rearrangements. Carbonate oligomers with three carbonate subunits revealed an analogous fragment pattern.
On condition that intramolecular rearrangements occurred, the formation mechanism of carbonate oligomers was in good agreement with findings for co‐oligomers and ethylene glycols. Nuyken and Pask showed multiple polymerization mechanisms for cyclic monomers analogous to EC.41 A polymer scheme was postulated based on a cationic ring opening polymerization of EC according to Ariga et al. (Figure 3),42 which could also be initiated via polarization with the lewis acid PF5,19, 33 or follow an anionic ring opening polymerization initiated via lithium alkoxides.43 Two polymerization routes after the initial nucleophilic attack lead to carbonate oligomers (green arrows), ethylene glycols and mixed products (light blue arrows). Since only the terminated products (blue background) were accessible for LC‐MS measurements, the explicit initiation reaction and elongation cascade were postulated to match the obtained results. To generate further insights into the presence of reactive species, the application of in situ techniques (e.g., IR, Raman and NMR) are possible approaches but will face challenges regarding sensitivity as well as the reliable assignment of signals without chromatographic separation in the highly complex mixture of decomposition products in LIB electrolytes with >300 possible compounds.25, 34, 35 Possible unlabeled termination molecules were DEC and lithium alkoxides (originating from DEC). Resulting leaving groups were postulated stoichiometrically.
Figure 3.

Proposed thermal decomposition route forming carbonates, carbonate ethers and ethylene glycol oligomers via an EC‐based cationic ring opening polymerization reaction and Li+ coordination.41, 42 Postulated leaving groups were assigned stoichiometrically. The reaction for the reaction initiation (yellow), elongation (green) and termination (blue) are labeled. The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
In general, the following conclusions regarding thermal electrolyte solvent decomposition can be drawn:
The conjunction of all‐solvent decomposition products is EC‐based.
Ethylene glycols are formed via polymerization of EC and subsequent decarboxylation.
The origin of carbonyl carbons can be EC and DEC, while DEC carbonyl carbons are mainly integrated during the termination step.
The termination of the reaction cascade is always DEC‐originated.
Lithium alkoxide species originate from the linear carbonate solvent.
The hydrolysis of the conducting salt LiPF6 and the subsequent reaction with lithium alkoxides to organo(fluoro)phosphates is addressed in the next section, comparing the thermal and electrochemical formation of triethyl phosphate (TEP) as representative species of this compound class.
Electrochemical aging
Electrolytes were extracted after cell formation and long‐term cycling in order to investigate electrochemical degradation products, since some species are more pronounced after cell formation compared to >500 cycles and vice versa. Despite many similar carbonate oligomers and carbonate ether co‐oligomers were formed after thermal and electrochemical aging (the latter is often also called: cyclic aging), further classes of decomposition products indicated a varied decomposition pathway with another reactive species. In particular, the main differences to thermally generated decomposition products were the formation of oligo phosphates and the incorporation of carbonates and phosphates.
The organophosphate TEP formed after thermal aging (top) and electrochemical aging (bottom) is shown in Figure 4. For both aging procedures, the unlabeled species was the most pronounced one, while the spectrum of TEP after battery cell formation also contained one or two labeled ethoxy side chains. Consequently, the resulting formation pathway needed EC‐based lithium alkoxides, although their formation was excluded indirectly via GC‐MS measurements. EC‐based lithium alkoxides would result in 13C2/4‐labeled DEC via exchange reactions (transesterification) during battery cell operation.44 Hence, the formation pathway of unlabeled TEP is based on DEC‐derived alkoxides and the pathway of labeled TEP remains unresolved. Nevertheless, this TEP formation pathway is constituted only to a minor extent in LIBs. One possible origin might be a side reaction product of the oligo phosphate formation or reactions based on radical chemistry which could result in a manifold of (new and minor) decomposition products.45, 46
Figure 4.

LC‐IT‐TOF‐MS and MS2 spectra of TEP after thermal (top) and electrochemical (bottom) aging after battery cell formation. The selected precursor ions are colored according to the formed adduct, fragment structures are shown as well as characteristic fragments (yellow). The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
Oligo phosphates and phosphate carbonates are depicted in Figure 5. The respective oligo phosphate species, only observed after >500 cycles in low intensity, revealed a mass shift of +2 Da for the main signal and +4 Da for a minor signal compared to the unlabeled species. The MS2 information showed a 13C‐labeled vinylene phosphate fragment (m/z 127.0070) which can be distinctly differentiated from unlabeled ethyl phosphate (Figure 4; m/z 127.0134) via high resolution MS. Hence, m/z 127.0070 was a characteristic fragment and indicated the presence of a 13C ethylene bridge between phosphates. The +4 Da signal (m/z 339.1111) can be ascribed to unresolved side reaction parallel to the observation for TEP, since the intensities exceed the values of natural 13C abundance.
Figure 5.
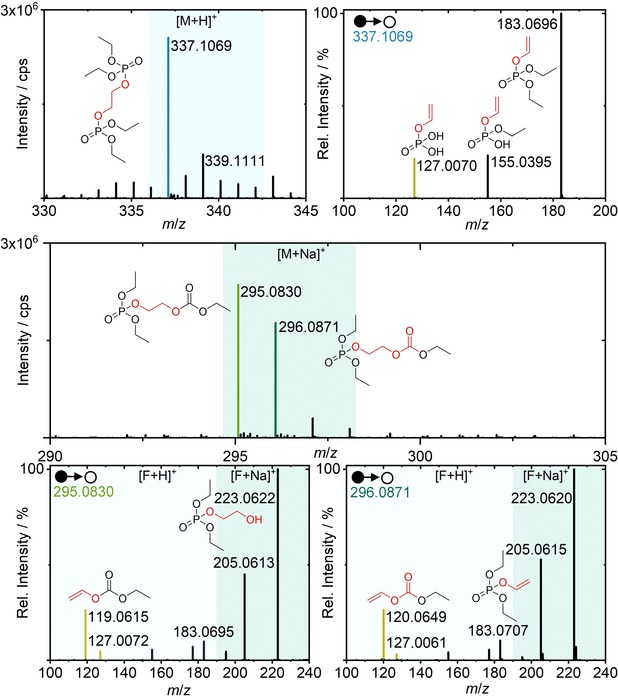
LC‐IT‐TOF‐MS and MS2 spectra of an oligo phosphate after >500 cycles (top) and a phosphate‐carbonate after cell formation (center, bottom). The selected precursor ions are colored according to the formed adduct, fragment structures are shown as well as characteristic fragments labeled in yellow. The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
The phosphate‐carbonate decomposition product showed two major signals (m/z 295.0830 and 296.9871), with a mass shift of +2 Da and +3 Da compared to the unlabeled species. The fragmentation of the [M+Na]+ adducts revealed also fragment ions ([F+H]+) which can be explained via sodium adduct formation at the P=O as well as at the carbonyl entity as discussed in Figure S2 of the Supporting Information. Characteristic fragments for both signals (Figure 5; yellow) allowed an exact allocation of the 13C positions in both species. Remarkably, the compound with +2 Da (m/z 295.0830) was predominantly formed. Hence, the formation pathway proposed by Gachot et al. via an alkoxide‐EC intermediary revealed to be contradictory to findings of the study.19 Wang, Xu, Eichhorn and colleagues recently reported the presence of LEMC as main SEI component.36 The proposed electrochemical formation pathways for phosphate‐carbonates, oligo phosphates and solvent decomposition products were postulated based on LEMC as reactive initiator of a nucleophilic attack (Figures 6 and 8). The possibility of a decarboxylation of LEMC supports the obtained signals of the MS1 spectra of the phosphate‐carbonate with uniform formation of 13C‐labeled and unlabeled carbonyl species in contrast to the postulated “intermediary A” which would result in completely 13C‐labeled species. Analogously to Figure 3, the decomposition route is based on literature‐known initiation reactions and the observed termination products of this study. Consequently, the process of elongation and termination was postulated.
Figure 6.
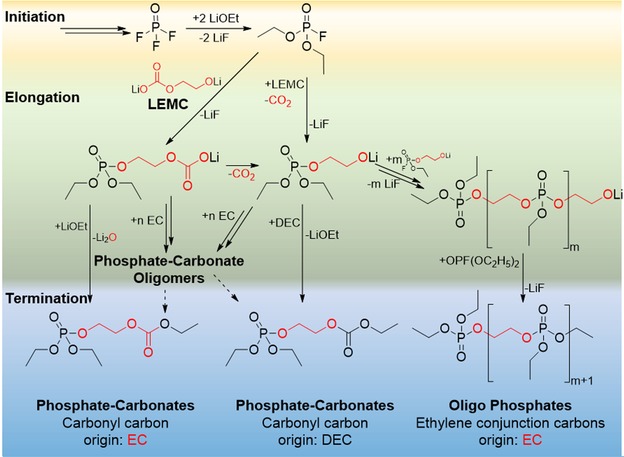
Proposed electrochemical decomposition route forming phosphate‐carbonates with odd and even numbers of carbonyl carbons as well as oligo phosphates. Postulated leaving groups were assigned stoichiometrically. The termination of phosphate‐carbonates is only presented for n=0. The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
Figure 8.
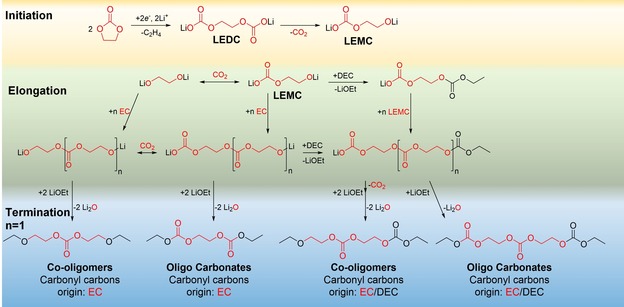
Proposed electrochemical decomposition route via a single‐electron reduction pathway of EC, forming carbonate oligomers and carbonate ether oligomers with odd and even numbers of carbonyl carbon.17 Postulated leaving groups were assigned stoichiometrically. The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
Electrochemical aging did not result in significant intensities of ethylene glycols and therefore only carbonate oligomers and co‐oligomers were assessed in this study. Exemplarily, the MS1/2 spectra of a carbonate oligomer with three carbonate subunits after three formation cycles are depicted in Figure 7. Analogous to results in section 3.1, a total mass shift of +5, 6 and 7 Da (m/z 322.1009, 323.1045 and 324.1068) compared to the unlabeled species was detected, indicating a different content of 13C carbonyl carbons. Furthermore, the intramolecular rearrangement and cleavage of an EC equivalent during fragmentation was also observed for m/z 323.1045. The formation process of oligo carbonates and co‐oligomers is depicted in Figure 8.
Figure 7.

LC‐IT‐TOF‐MS and MS2 spectra of an oligo carbonate (top) and carbonate ether co‐oligomer after cell formation (bottom). The selected precursor ions are colored according to the formed adduct and fragment structures are shown, indicating multiple formation pathways via 13C‐labeling. The origin of molecular entities is marked (red=13C3‐EC‐origin, black=unlabeled DEC‐origin).
The exemplarily shown carbonate ether co‐oligomer in Figure 7 (bottom) contained total mass shifts of +4, 5 and 6 Da (m/z 277.1087, 278.1120 and 279.1135) compared to the unlabeled species. Both, unlabeled and single labeled carbonyl carbon containing compounds comprised the main species of co‐oligomers. Furthermore, the fragmentation pattern points to a varying 13C position and/or a coeluting structural isomer with an ethylene glycol subunit between the carbonate entities. The first possibility is discussed in Figure 7. The formation of intramolecular unlabeled carbonyl carbons can occur via a twofold LEMC attack on DEC. Hence, the process leading to carbonate ether co‐oligomers in LIBs revealed to be highly complex and does not follow only one main decomposition route. This conclusion was in agreement with the overall low intensities detected for this product class in electrolytes.
In order to summarize the findings regarding electrochemical electrolyte decomposition in LIB cells after formation and after >500 cycles, the following conclusions can be drawn:
The conjunction of solvent decomposition products and oligo phosphates is EC‐based.
Ethylene glycol subunits form via EC polymerization with subsequent decarboxylation.
The origin of carbonyl carbons can be EC and DEC.
The termination of the reaction cascade is predominantly lithium alkoxide‐based.
Alkoxide species originate from the linear carbonate solvent.
The formation of O(F)Ps is predominantly DEC‐originated parallel to thermal aging.
Overall, the generation of decomposition products in LIBs can be explained by a reaction cascade based on LEMC, while thermal aging is based on a ring opening polymerization, without incorporating the formation of conducting salt‐solvent mixed decomposition products and oligo phosphates.
The comparison of short‐term and long‐term cycling revealed that the 13C distribution within the respective decomposition product class did not change. Thus, the possibility of progressive substitution reactions of products and reactive intermediates (resulting in an increased 13C degree after >500 cycles) was not observed.
Conclusion
In this study, electrolyte decomposition reactions were investigated regarding thermal and electrochemical degradation pathways with 13C3‐labeled EC. Although similar carbonate‐based compounds were identified, a different nature of formation could was identified, and possible pathways postulated that are in accordance with literature. For both aging processes, an EC‐derived elongation of decomposition products was observed, although different degrees of 13C content were observed. Thus, multiple formation pathways for a specific product could be identified. While the chain growth in case of electrochemical aging was predominantly terminated via DEC‐derived lithium alkoxides, in thermal aging DEC termination prevailed. Moreover, the assumption of LEMC as reactive species in LIBs enabled the explanation of oligo phosphate formation and the counterintuitive 13C carbonyl distribution of phosphate‐carbonates. This study shed light on occurring decomposition reactions during thermal stress and battery operation. Furthermore, the obtained insights can be implemented in the design of tailored electrolyte additives to suppress the formation of potentially toxic O(F)Ps and performance impairing oligomeric solvent decomposition products. Improvement strategies focusing on an increased thermal as well as electrochemical stability can be detrimental, since the substitution of established electrolyte components (e.g., EC, LiPF6) could possibly result in new side reactions or an aggravated electrochemical performance. Moreover, the correlation of formed decomposition products and battery cell performance requires further investigation. Overall, the diminution of protic impurities (in particular H2O) using scavenging materials (e.g., electrolyte additives, separator/active material coatings) could decrease the LiPF6 hydrolysis and consequently the electrolyte decomposition.47
Experimental Section
Identification of thermally induced LIB electrolyte decomposition products was performed on a Nexera X2 UHPLC system (Shimadzu, Kyoto, Japan) hyphenated to a LCMS‐IT‐TOF™ (Shimadzu). Reversed‐phase chromatography was conducted on a ZORBAX SB‐C18 column (100×2.1 mm, 1.8 μm; Agilent Technologies, Inc., Santa Clara, CA, USA) at 40 °C and a flow rate of 0.5 mL min−1. The mobile phase gradient consisted of water (A) and acetonitrile (B). The optimized gradient started with 5 % B from 0 to 1.8 min and increased to 60 % within 12.2 min. Subsequently, the mobile phase was kept constant at 60 % for 2 min. Finally, the column equilibrated at 5 % B for 4 min. The injection volume was 5 μL. To protect the IT‐TOF™ mass spectrometer from high concentrations of conducting salt LiPF6 and thermally induced acidic decomposition products, the flow line was switched to the MS after 1.8 min. Ionization was performed in ESI(+) mode at 4.5 kV. Curved desolvation line and heat block temperature were 230 °C, the drying gas pressure at 100 kPa and the nebulizer gas flow was 1.5 L min−1. The ion trap operated in automatic MS2 mode with an ion accumulation time of 10 ms in MS1 and 40 ms in MS2, leading to a loop time of 260 ms. The mass range was set to m/z 100–400 in MS1 and m/z 50–400 in MS2 for the low mass window, m/z 200–1000 in MS1 and m/z 150–1000 in MS2 for the high mass window. The collision induced dissociation energy and gas flow were set to 25 %. LCMSsolution 3.80 (Shimadzu) was used for LC‐MS instrument control, data acquisition and data evaluation.
Chemicals and materials: The investigated electrolytes contained EC and DEC in a ratio of 30/70 wt % as well as 1 mol L−1 LiPF6. Electrolytes with unlabeled EC for reference (ELref) and with 13C3‐labeled EC (EL ) were used. EC (Targray Technology International Inc., Kirkland, Canada, battery grade, 99.99 %), DEC (BASF SE, battery grade, 99.98 %), 13C3 labeled EC (Sigma Aldrich, Chemie GmbH, Steinheim, Germany, 99 at % 13C, 97 %) and LiPF6 (BASF SE, battery grade, ≥99.99 %) were used for the preparation of LIB electrolytes. Acetonitrile (VWR International GmbH, Darmstadt, Germany; LC‐MS grade) and deionized water (Millipore, Molsheim, France; 18.2 MΩ cm−1) were used for eluents and sample dilution. All chemicals were used as received.
Thermal degradation: Gas‐tight 10 mL aluminum vials were cleaned with isopropanol/MilliQ and dried in reduced pressure at 60 °C for 2 hours. 100 μL battery grade SelectiLyte™ electrolyte (BASF, Ludwigshafen am Rhein, Germany) was stored at 80 °C for 3 weeks. The containers were opened in a dry room (dew point of −65 °C, H2O <10 mL m−3). Samples were diluted 1:20 with acetonitrile prior to measurements.
Cell assembly and electrochemical operation: LIB pouch cells (Li‐Fun NCM622||graphite) were used for the investigation after cell formation and >500 cycles (Li‐Fun Technology Co. Ltd, Zhuzhou City, China). Cells were filled with 700 μL ELref/EL and sealed at 165 °C, 90 kPa for 5 s. Three constant current constant voltage (CCCV) charge and constant current (CC) discharge cycles at 0.2 C in a voltage window of 3.0–4.2 V were performed for cell formation. For long‐term cycling CCCV charge and CC discharge at 1C were applied after three formation cycles. After opening of cells in a glove box (O2, H2O<0.1 ppm) and electrolyte extraction via centrifugation of the separator stack, samples were diluted 1:20 with acetonitrile prior to measurements.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the German Federal Ministry of Education and Research for funding the project “Cell‐Fi” (03XP0069B) and Shimadzu Deutschland GmbH for IT‐TOF‐MS fragmentation discussions.
J. Henschel, C. Peschel, S. Klein, F. Horsthemke, M. Winter, S. Nowak, Angew. Chem. Int. Ed. 2020, 59, 6128.
References
- 1. Schmuch R., Wagner R., Hörpel G., Placke T., Winter M., Nat. Energy 2018, 3, 267–278. [Google Scholar]
- 2. Kwade A., Haselrieder W., Leithoff R., Modlinger A., Dietrich F., Droeder K., Nat. Energy 2018, 3, 290–300. [Google Scholar]
- 3. Winter M., Barnett B., Xu K., Chem. Rev. 2018, 118, 11433–11456. [DOI] [PubMed] [Google Scholar]
- 4. Cekic-Laskovic I., von Aspern N., Imholt L., Kaymaksiz S., Oldiges K., Rad B. R., Winter M., Top. Curr. Chem. 2017, 375, 1–64. [DOI] [PubMed] [Google Scholar]
- 5. Xu K., Chem. Rev. 2014, 114, 11503–11618. [DOI] [PubMed] [Google Scholar]
- 6. Xu K., Chem. Rev. 2004, 104, 4303–4417. [DOI] [PubMed] [Google Scholar]
- 7. Li H., Zhou H., Chem. Commun. 2012, 48, 1201–1217. [DOI] [PubMed] [Google Scholar]
- 8. Obrovac M. N., Chevrier V. L., Chem. Rev. 2014, 114, 11444–11502. [DOI] [PubMed] [Google Scholar]
- 9. Placke T., Kloepsch R., Dühnen S., Winter M., J. Solid State Electrochem. 2017, 21, 1939–1964. [Google Scholar]
- 10. Minato T., Abe T., Prog. Surf. Sci. 2017, 92, 240–280. [Google Scholar]
- 11. Schipper F., Erickson E. M., Erk C., Shin J.-Y., Chesneau F. F., Aurbach D., J. Electrochem. Soc. 2017, 164, A6220–A6228. [Google Scholar]
- 12. Erickson E. M., Schipper F., Penki T. R., Shin J.-Y., Erk C., Chesneau F.-F. F., Markovsky B., Aurbach D., Erickson E. M., Erk C., Shin J.-Y., Chesneau F.-F. F., Aurbach D., J. Electrochem. Soc. 2017, 164, A6341–A6348. [Google Scholar]
- 13. Winter M., Z. Phys. Chem. 2009, 223, 1395–1406. [Google Scholar]
- 14. Qian Y., Niehoff P., Börner M., Grützke M., Mönnighoff X., Behrends P., Nowak S., Winter M., Schappacher F. M., J. Power Sources 2016, 329, 31–40. [Google Scholar]
- 15. Peled E., Menkin S., J. Electrochem. Soc. 2017, 164, A1703–A1719. [Google Scholar]
- 16. Gauthier M., Carney T. J., Grimaud A., Giordano L., Pour N., Chang H. H., Fenning D. P., Lux S. F., Paschos O., Bauer C., Maglia F., Lupart S., Lamp P., Shao-Horn Y., J. Phys. Chem. Lett. 2015, 6, 4653–4672. [DOI] [PubMed] [Google Scholar]
- 17. Laruelle S., Pilard S., Guenot P., Grugeon S., Tarascon J.-M., J. Electrochem. Soc. 2004, 151, A1202–A1209. [Google Scholar]
- 18. Gireaud L., Grugeon S., Pilard S., Guenot P., Tarascon J. M., Laruelle S., Anal. Chem. 2006, 78, 3688–3698. [DOI] [PubMed] [Google Scholar]
- 19. Gachot G., Grugeon S., Armand M., Pilard S., Guenot P., Tarascon J. M., Laruelle S., J. Power Sources 2008, 178, 409–421. [Google Scholar]
- 20. Sasaki T., Abe T., Iriyama Y., Inaba M., Ogumi Z., J. Electrochem. Soc. 2005, 152, A2046–A2050. [Google Scholar]
- 21. Gireaud L., Grugeon S., Laruelle S., Pilard S., Tarascon J.-M., J. Electrochem. Soc. 2005, 152, A850–A857. [Google Scholar]
- 22. Plakhotnyk A. V., Ernst L., Schmutzler R., J. Fluorine Chem. 2005, 126, 27–31. [Google Scholar]
- 23. Campion C. L., Li W., Euler W. B., Lucht B. L., Ravdel B., DiCarlo J. F., Gitzendanner R., Abraham K. M., Electrochem. Solid-State Lett. 2004, 7, A194–A197. [Google Scholar]
- 24. Campion C. L., Li W., Lucht B. L., J. Electrochem. Soc. 2005, 152, A2327–A2334. [Google Scholar]
- 25. Henschel J., Schwarz J. L., Glorius F., Winter M., Nowak S., Anal. Chem. 2019, 91, 3980–3988. [DOI] [PubMed] [Google Scholar]
- 26. Metzger M., Strehle B., Solchenbach S., Gasteiger H. A., J. Electrochem. Soc. 2016, 163, A798–A809. [Google Scholar]
- 27. Sahore R., Dogan F., Bloom I. D., Chem. Mater. 2019, 31, 2884–2891. [Google Scholar]
- 28. Tochihara M., Nara H., Mukoyama D., Yokoshima T., Momma T., Osaka T., J. Electrochem. Soc. 2015, 162, A2008–A2015. [Google Scholar]
- 29. Takeda S., Morimura W., Liu Y. H., Sakai T., Saito Y., Rapid Commun. Mass Spectrom. 2016, 30, 1754–1762. [DOI] [PubMed] [Google Scholar]
- 30. Kraft V., Weber W., Grützke M., Winter M., Nowak S., RSC Adv. 2015, 5, 80150–80157. [Google Scholar]
- 31. Stenzel Y. P., Horsthemke F., Winter M., Nowak S., Separations 2019, 6, 26. [Google Scholar]
- 32. Nowak S., Winter M., J. Electrochem. Soc. 2015, 162, A2500–A2508. [Google Scholar]
- 33. Sloop S. E., Kerr J. B., Kinoshita K., J. Power Sources 2003, 119–121, 330–337. [Google Scholar]
- 34. Henschel J., Dressler J. M., Winter M., Nowak S., Chem. Mater. 2019, 31, 9970–9976. [Google Scholar]
- 35. Henschel J., Peschel C., Günter F., Reinhart G., Winter M., Nowak S., Chem. Mater. 2019, 31, 9977–9983. [Google Scholar]
- 36. Wang L., Menakath A., Han F., Wang Y., Zavalij P. Y., Gaskell K. J., Borodin O., Iuga D., Brown S. P., Wang C., Xu K., Eichhorn B. W., Nat. Chem. 2019, 11, 789–796. [DOI] [PubMed] [Google Scholar]
- 37. Parimalam B. S., MacIntosh A. D., Kadam R., Lucht B. L., J. Phys. Chem. C 2017, 121, 22733–22738. [Google Scholar]
- 38. Jin Y., Kneusels N. H., Grey C. P., J. Phys. Chem. Lett. 2019, 10, 6345–6350. [DOI] [PubMed] [Google Scholar]
- 39. Michan A. L., Leskes M., Grey C. P., Chem. Mater. 2016, 28, 385–398. [Google Scholar]
- 40. Schultz C., Vedder S., Winter M., Nowak S., Anal. Chem. 2016, 88, 11160–11168. [DOI] [PubMed] [Google Scholar]
- 41. Nuyken O., Pask S. D., Polymers 2013, 5, 361–403. [Google Scholar]
- 42. Ariga T., Takata T., Endo T., Macromolecules 1997, 30, 737–744. [Google Scholar]
- 43. Lee J. C., Litt M. H., Macromolecules 2000, 33, 1618–1627. [Google Scholar]
- 44. Petibon R., Rotermund L. M., Dahn J. R., J. Power Sources 2015, 287, 184–195. [Google Scholar]
- 45. Shkrob I. A., Zhu Y., Marin T. W., Abraham D., J. Phys. Chem. C 2013, 117, 19255–19269. [Google Scholar]
- 46. Horsthemke F., Friesen A., Ibing L., Klein S., Winter M., Nowak S., Electrochim. Acta 2019, 295, 401–409. [Google Scholar]
- 47. Wiemers-Meyer S., Jeremias S., Winter M., Nowak S., Electrochim. Acta 2016, 222, 1267–1271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary