

Abstract
The demand for allosteric targeting of nuclear receptors is high, but examples are limited, and structural information is scarce. The retinoic acid‐related orphan receptor gamma t (RORγt) is an important transcriptional regulator for the differentiation of T helper 17 cells for which the first, and some of the most promising, examples of allosteric nuclear receptor modulation have been reported and structurally proven. In a 2015 patent, filed by the pharmaceutical company Glenmark, a new class of small molecules was reported that act as potent inverse agonists for RORγt. A compound library around the central thienopyrazole scaffold captured a clear structure‐activity relationship, but the binding mechanism of this new class of RORγt modulators has not been elucidated. Using a combination of biochemical and X‐ray crystallography studies, here the allosteric mechanism for the inverse agonism for the most potent compound, classified in the patent as “example 13”, is reported, providing a strongly desired additional example of allosteric nuclear receptor targeting.
Keywords: Nuclear Receptors, RORγt, Allosteric Modulators, Structure Elucidation, Drug Discovery
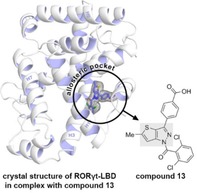
Example 13: Inhibition of the RAR‐related orphan receptor gamma t (RORγt) nuclear receptor has proven to be a novel approach for targeting autoimmune diseases. Recently, RORγt was shown to feature an allosteric pocket. Using X‐ray crystallography, we now show that compound 13, a promising thienopyrazole analogue of previously reported RORγt modulators, exerts its inverse agonism by binding to this allosteric pocket.
Introduction
Nuclear receptors (NRs) are a family of ligand‐dependent transcription factors that constitute an important class of drug target.1, 2 Within this family, the retinoic acid‐related orphan receptor gamma t (RORγt) has garnered much attention as an intervention point to treat autoimmune diseases such as multiple sclerosis, psoriasis and inflammatory bowel disease. A significant characteristic of these diseases is the excessive production of the pro‐inflammatory cytokine interleukin 17 (IL‐17) by Th17 cells. RORγt plays a key role in Th17 cell differentiation. Disruption of the IL‐17 signaling pathway using recently FDA‐approved monoclonal antibodies already demonstrated to be an effective strategy for the treatment of psoriasis.3, 4, 5 Therefore, inhibition of RORγt would be a highly promising alternative therapeutic strategy.6, 7, 8 Numerous small molecules have been reported that effectively inhibit RORγt, which is transcriptionally active even in the absence of any endogenous agonist.9, 10, 11 Typically, such inverse agonists bind to the orthosteric NR binding pocket; a pocket conserved across many NRs, but with concomitant challenges in achieving NR subtype selectivity and competition with endogenous ligands. Allosteric modulation of NR activity offers a promising novel concept for addressing such challenges and for novel modes of NR drug action in general.12, 13, 14 However, examples of structurally characterized and sufficiently potent NR allosteric modulators are scarce. The discovery and development of such compounds is therefore urgently needed in order to steer their molecular design process and further our conceptual understanding of allosteric NR modulation.13
Recently, allosteric modulation of RORγt was shown to be a promising approach for drug development,15, 16, 17 featuring examples of allosteric ligands with high NR subtype selectivity and absence of competition with the endogenous ligands.18 The first example to emerge was the indazole MRL‐871 (Figure 1), a potent inverse agonist. Originally disclosed by Merck Sharp and Dohme in 2012,19 its allosteric mode of action was characterized three years later.15 Glenmark Pharmaceuticals used the MRL‐871 core as the basis for an in silico scaffold hopping screen in search of a novel but similarly potent compound class.20 This led to the development of a family of thienopyrazole inverse agonists with nanomolar activity which were disclosed in a 2015 patent, where ‘compound 13’ was the most potent example (Figure 1).21 Although a comprehensive structure–activity relationship (SAR) study was conducted around the thienopyrazole scaffold, the binding mode to RORγt was not reported.
Figure 1.
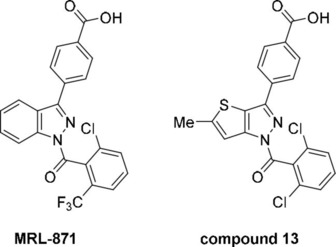
The chemical structures of allosteric RORγt inverse agonists MRL‐871 (MSD) and compound 13 (Glenmark).
MRL‐871 and 13 only differ with any significance in their central heteroaromatic core (Figure 1). Due to the similarity between the two compounds, we postulated that the thienopyrazole series shared the same allosteric binding mode as indazole MRL‐871. Here, we report X‐ray crystallography and match this with biochemical binding data, confirming an allosteric mode of action for the exemplar thienopyrazole 13. The structural characterization of new classes of allosteric RORγt inverse agonist is of great value in terms of better understanding SAR with respect to this allosteric pocket. Furthermore, it contributes to our growing conceptual understanding of NR allosteric modulation in general.
Results and Discussion
In recent work to develop novel allosteric RORγt inverse agonists we reported the synthesis and inverse agonistic behavior of compound 13, which was used as a reference molecule.16 In a time‐resolved FRET (TR‐FRET) coactivator recruitment assay we reported that compound 13 effectively inhibited coactivator peptide binding, and thus the background constitutive activity of RORγt, in a dose‐dependent manner with an IC50 value of 425±61 nM (Figure 2A).16 This was less potent than indicated in the original patent disclosure where an IC50 value of <50 nM was reported using a similar assay format.21 In agreement with previously reported values, MRL‐871 was found to be ∼50 times more potent (7.8±0.5 nM). To determine if compound 13 has an allosteric mode of action analogous to MRL‐871, we performed a competition TR‐FRET assay. Compound 13 titrations were performed in the presence of increasing concentrations of the well‐characterized orthosteric agonist cholesterol (Figure 2B). Consequently, the addition of cholesterol stabilizes the active conformation, thereby increasing the FRET signal in the absence of compound 13. The binding curves revealed that the presence of the agonist does not lower the inhibitory potency of 13, but instead even slightly enhances it (IC50=269±19 nM in the presence of 1.0 μM cholesterol).16 The absence of competition between cholesterol and 13 suggests that the binding mode of 13 is independent to that of cholesterol and therefore that 13 likely binds to an allosteric pocket on RORγt. Next, we sought evidence to prove that compound 13 was indeed binding to the same allosteric site as MRL‐871. For this, we performed a ligand displacement assay using a previously reported MRL‐871‐derived fluorescent probe that is known to bind the previously published allosteric site. Compound 13 effectively displaced the probe in a dose‐dependent manner (Figure 2C).16 These results are thus a strong indication that compound 13 binds to this specific allosteric pocket. Nevertheless, probe displacement could also be caused by changes in protein conformation induced by binding of compound 13 binding to another site on RORγt.
Figure 2.
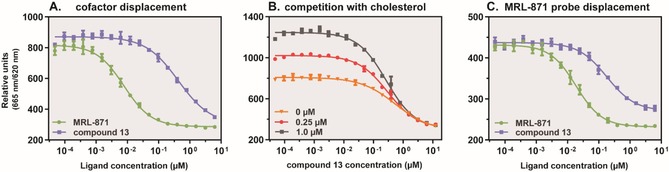
Bio‐chemical assay data for MRL‐871 and compound 13. (A) Dose‐response curves from the TR‐FRET coactivator recruitment assay for MRL‐871 and compound 13; (B) Dose‐response curves from the competitive TR‐FRET coactivator recruitment assay for compound 13, with fixed concentrations of cholesterol (0 μM, 0.25 μM and 1.0 μM); (C) Dose‐response curves from the ligand displacement TR‐FRET assay for MRL‐871 and compound 13, using a fluorescently labelled MRL probe. Data adapted from Meijer et al. 2020.16
To now unambiguously confirm the binding mode of compound 13, we determined the structure of the 13‐RORγt binary complex using protein X‐ray crystallography. A C455H mutant of the ligand‐binding domain (LBD) of human RORγt was used for this study; the region surrounding this native cysteine is important for crystal packing of RORγt in complex with allosteric inverse agonists.15, 16 RORγt and compound 13 were co‐crystallized and crystals grew as hexagonal bipyramids overnight using sitting‐drop vapor diffusion. The data collection and refinement statistics of the crystal are provided in Table S1. The crystal structure (PDB: 6TLM) shows the complete LBD of RORγt and reveals that compound 13 indeed binds in the anticipated allosteric pocket and that the orthosteric pocket is devoid of ligand (Figure 3). Of specific and notable interest is the folding of helix 12, in a conformation precluding coactivator binding. The carboxylic acid of 13 interacts with Q329 and two backbone amides of A497 and F498 located in the loop between helix 11 and 12 of RORγt. The binding mode of 13 to RORγt is comparable to that of MRL‐871. A first notable difference in the binding to RORγt of both ligands lies in the increased bulk of compound 13 toward helix 4 of RORγt, resulting from the methyl substituent on the thienopyrazole core. This induces a shift of helix 4, correlated with a displacement of helix 9 (Figure 3C and Figure 4). A second, and most pronounced, difference can be observed in the loop between helix 11 and 12. Compared to MRL‐871, the benzoic acid moiety of compound 13 is orientated differently in the pocket, which is connected to a change in the overall fold of this loop. In particular, F498 changes conformation and thereby flips away from helix 4. A comparable behavior is observed for the recently documented RORγt allosteric inverse agonist FM26, where a pyrrole introduces bulk toward helix 4 (Figure S1).16 Both 13 and FM26 induce a fold of the loop between helix 11 and 12 that has F498 in the “flipped‐out” conformation. In contrast to compound 13, the pyrrole of FM26 makes an additional interaction with the backbone carbonyl groups of L353 and K354, which can explain the higher affinity of FM26 for RORγt (IC50=264±23 nM).16
Figure 3.
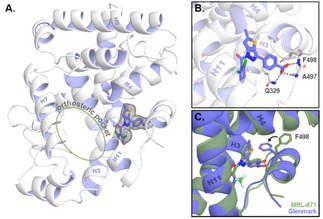
Co‐crystal structure of RORγt with compound 13 (PDB code: 6TLM). (A) The tertiary structure. The final 2Fo−Fc electron density map around compound 13 is shown as an isomesh contoured at 1σ; (B) Close‐up of the allosteric pocket. The polar interactions between RORγt and compound 13 are shown as a grey dotted line. (C) Overlay of the crystal structure of RORγt bound to compound 13 (blue) and RORγt bound to MRL‐871 (green; PDB: 5C4O).
Figure 4.
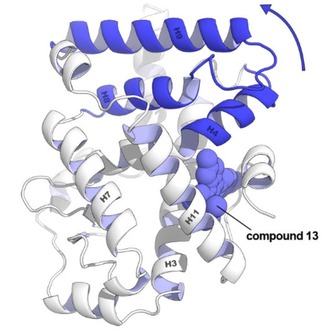
Conformational change in the tertiary structure of RORγt comparing the complex with compound 13 and MRL‐871. The methyl substituent on the thienopyrazole increases the bulk toward helix 4. The helices that change position compared to the MRL‐871 structure are coloured dark blue and are moving in the arrow's direction.
Comparison of the IC50 values of compound 13, FM26 and MRL‐871 would suggest that steric bulk toward helix 4 does not favor the affinity of the allosteric modulator for RORγt (Figure S1). MSD's patent application showed that bulky substitutions on the 4 and 5 position of the indazole moiety also resulted in a reduced affinity for the receptor.19 Glenmark did disclose the less bulky hydrogen‐substituted thienopyrazole, but the biochemical activity of this analog was not evaluated. However, various compounds were evaluated incorporating bulkier amide substituents replacing the methyl group.21 Such compounds generally showed a lower or no binding affinity for RORγt.
Conclusions
In summary, using X‐ray crystallography and supported by biochemical studies, Glenmark's compound 13 was shown to bind to an allosteric pocket of RORγt. The binding mode of compound 13 is similar to MRL‐871 and the recently reported FM26, with small but notable differences. The structural data imply that the lower affinity of 13 for RORγt relates to additional bulk on the thienopyrazole core pointing to RORγt helix 4, which leads to changes in the overall protein fold. Such changes are likely to affect the dynamics of the protein and stability of the specific fold. The new structural data expand the collection of crystallized ligands binding to allosteric binding pockets on NRs, and specifically on RORγt. The resulting new insights will aid in the understanding of reported compounds classes, potentially also addressing the same allosteric RORγt pocket and in the development of new compound classes with more diverse chemotypes or optimized pharmacodynamics profile.
Experimental Section
General
The synthesis of compound 13 and the biochemical assays have been described earlier.16
RORγtC455H‐LBD expression and purification for crystallography
The expression and purification of RORγtC455H‐LBD were performed as described earlier.16 In short, a pET15b vector encoding for RORγt LBD (AA 265–507) incorporating a C455H mutation was transformed into E. Coli BL21 (DE3) cells. These cells were cultured in 2x YT medium supplied with 0.05 % antifoam SE‐15 (Sigma Aldrich) with 100 μg/mL ampicillin. After an OD600 of 0.6 is reached, protein expression was induced by adding 0.25 mM IPTG. The protein expression continued overnight at 15 °C. Centrifugation was used to collect the cells which were lysed using an Emulsiflex‐C3 homogenizer (Avestin). The resulting solution was purified using Ni‐NTA affinity chromatography. The elution fraction was dialyzed overnight in buffer A without imidazole and thrombin was added to remove the purification tag. The purified sample was subsequently purified using gel filtration. Fractions containing RORγtC455H were collected and concentrated before being flash‐cooled and stored at −80 °C.
X‐ray crystallography
Compound 13 was dissolved in 50 % DMSO and 50 % EtOH to a final concentration of 20 mM. One equivalent of compound 13 was added to the RORγt protein solution and the mixture was placed on ice. After 1 hour incubation, centrifugation at 20.000 RCF for 20 minutes at 4 °C was used to remove protein and ligand precipitate. Sitting‐drop vapor diffusion was used to generate crystals using 800 nL of the protein‐ligand solution and 400 nL crystallization solution (1.6 M (NH4)2SO4 and 0.1 M Tris (at pH 8.5)). Crystals grew as hexagonal bipyramids to their final size overnight. Because the crystallization solution was not cryogenic, the crystal was briefly transferred to a cryo‐solution (1.6 M (NH4)2SO4, 0.1 M Tris, 25 % glycerol and 200 μM compound 13 (at pH 8.5)) before being flash‐cooled. The crystal was measured at the i24 microfocus beamline of the Diamond Light Source (Oxford, United Kingdom). Initial data processing was performed using the CCP4i2 suite (version 7.0.078).22 DIALS (2.0.2) was used to integrate the data.23 Because the diffraction was anisotropic, STARANISO was used to correct the data.24 Aimless was used to scale the corrected data.25 Using the RORγt crystal structure in complex with allosteric ligand FM26 (PDB: 6SAL) as a search model for molecular replacement, PHASER was used to phase the data and ligand restraints were generated using AceDRG.16, 26, 27 REFMAC and COOT were used for sequential refinement and model building.28, 29 Final refinement was performed using phenix.refine from the Phenix software suite (version 1.16_3459).30, 31 Figures were made with PyMOL (version 2.2.3, Schrödinger).32
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Alex A. A. Vos and Dr. Lech‐Gustav Milroy for the synthesis of compound 13, Dr. Marcel Scheepstra and Rowin J.P.M. de Visser for the synthesis of the AlexaFluor647‐labeled MRL‐871 probe, and Dr. Christian Ottmann for valuable discussions. We also want to thank the tutors of the DLS‐CCP4 Data Collection and Structure Solution Workshop 2017 at the Diamond Light Source (Oxfordshire, UK). This work was funded by The Netherlands Organization for Scientific Research (NWO) via Gravity program 024.001.035 and VICI grant 016.150.366 by the European Union through H2020‐MSCA IEF, grant number 705188.
R. M. J. M. de Vries, R. G. Doveston, F. A. Meijer, L. Brunsveld, ChemMedChem 2020, 15, 561.
References
- 1. Huang P., Chandra V., Rastinejad F., Annu. Rev. Physiol. 2010, 72, 247–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Santos R., Ursu O., Gaulton A., Bento A. P., Donadi R. S., Bologa C. G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T. I., Overington J. P., Nat. Rev. Drug Discov. 2016, 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mease P. J., Van Der Heijde D., Ritchlin C. T., Okada M., Cuchacovich R. S., Shuler C. L., Lin C. Y., Braun D. K., Lee C. H., Gladman D. D., Ann. Rheum. Dis. 2017, 76, 79–87.27553214 [Google Scholar]
- 4. Attia A., Abushouk A. I., Ahmed H., Gadelkarim M., Elgebaly A., Hassan Z., Abdel-Daim M. M., Negida A., Clin. Drug Invest. 2017, 37, 439–451. [DOI] [PubMed] [Google Scholar]
- 5. Hueber W., Patel D. D., Dryja T., Wright A. M., Koroleva I., Bruin G., Antoni C., Draelos Z., Gold M. H., Durez P., Tak P. P., Gomez-Reino J. J., Foster C. S., Kim R. Y., Samson C. M., Falk N. S., Chu D. S., Callanan D., Nguyen Q. D., Rose K., Haider A., Di Padova F., Sci. Transl. Med. 2010, 2, 52ra72. [DOI] [PubMed] [Google Scholar]
- 6. Lock C., Hermans G., Pedotti R., Brendolan A., Schadt E., Garren H., Langer-Gould A., Strober S., Cannella B., Allard J., Klonowski P., Austin A., Lad N., Kaminski N., Galli S. J., Oksenberg J. R., Raine C. S., Heller R., Steinman L., Nat. Med. 2002, 8, 500–508. [DOI] [PubMed] [Google Scholar]
- 7. Burkett P. R., Kuchroo V. K., Cell 2016, 167, 1669. [DOI] [PubMed] [Google Scholar]
- 8. Duerr R. H., Taylor K. D., Brant S. R., Rioux J. D., Silverberg M. S., Daly M. J., Steinhart A. H., Abraham C., Regueiro M., Griffiths A., Dassopoulos T., Bitton A., Yang H., Targan S., Datta L. W., Kistner E. O., Schumm L. P., Lee A. T., Gregersen P. K., Barmada M. M., Rotter J. I., Nicolae D. L., Cho J. H., Science. 2006, 314, 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fauber B. P., Magnuson S., J. Med. Chem. 2014, 57, 5871–5892. [DOI] [PubMed] [Google Scholar]
- 10. Qiu R., Wang Y., J. Med. Chem. 2018, 61, 5794–5804. [DOI] [PubMed] [Google Scholar]
- 11. Sun N., Guo H., Wang Y., Expert Opin. Ther. Pat. 2019, 29, 663–674. [DOI] [PubMed] [Google Scholar]
- 12. Moore T. W., Mayne C. G., Katzenellenbogen J. A., Mol. Endocrinol. 2010, 24, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tice C. M., Zheng Y. J., Bioorg. Med. Chem. Lett. 2016, 26, 4157–4164. [DOI] [PubMed] [Google Scholar]
- 14. Fernandez E. J., Pharmacol. Ther. 2018, 183, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scheepstra M., Leysen S., van Almen G. C., Miller J. R., Piesvaux J., Kutilek V., van Eenennaam H., Zhang H., Barr K., Nagpal S., Soisson S. M., Kornienko M., Wiley K., Elsen N., Sharma S., Correll C. C., Trotter B. W., van der Stelt M., Oubrie A., Ottmann C., Parthasarathy G., Brunsveld L., Nat. Commun. 2015, 6, 8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meijer F. A., Doveston R. G., de Vries R. M. J. M., Vos G. M., Vos A. A. A., Leysen S., Scheepstra M., Ottmann C., Milroy L.-G., Brunsveld L., J. Med. Chem. 2020, 63, 241–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang X., Dulubova I., Reisman S. A., Hotema M., Lee C.-Y. I., Liu L., McCauley L., Trevino I., Ferguson D. A., Eken Y., Wilson A. K., Wigley W. C., Visnick M., Bioorg. Med. Chem. Lett. 2020, 30, 126967. [DOI] [PubMed] [Google Scholar]
- 18. Meijer F. A., Leijten-van de Gevel I. A., De Vries R. M. J. M., Brunsveld L., Mol. Cell. Endocrinol. 2019, 485, 20–34. [DOI] [PubMed] [Google Scholar]
- 19.W. F. J. Karstens, M. van der Stelt, J. Cals, R. C. R. G. Azevedo, K. J. Barr, H. Zhang, R. T. Beresis, D. Zhang, X. Duan, (Merck Sharp & Dohme Corp. and N. V. Organon), WO2012106995A1, 2012.
- 20. Gege C., Expert Opin. Ther. Pat. 2015, 25, 1215–1221. [DOI] [PubMed] [Google Scholar]
- 21.S. S. Chaudari, A. Thomas, S. V. Dhone, N. Khairatkar-Joshi, M. Bajpai, (Glenmark Pharmceuticals), WO2015008234A1, 2015.
- 22. Potterton L., Agirre J., Ballard C., Cowtan K., Dodson E., Evans P. R., Jenkins H. T., Keegan R., Krissinel E., Stevenson K., Lebedev A., McNicholas S. J., Nicholls R. A., Noble M., Pannu N. S., Roth C., Sheldrick G., Skubak P., Turkenburg J., Uski V., von Delft F., Waterman D., Wilson K., Winn M., Wojdyr M., Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clabbers M. T. B., Gruene T., Parkhurst J. M., Abrahams J. P., Waterman D. G., Acta Crystallogr. Sect. D 2018, 74, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.I. J. Tickle, C. Flensburg, P. Keller, W. Paciorek, A. Sharff, C. Vonrhein, G. Bricogne, STARANISO, Global Phasing Ltd., Cambridge (United Kingdom), 2018.
- 25. Evans P. R., Murshudov G. N., Acta Crystallogr. Sect. D 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy A. J., Acta Crystallogr. Sect. D 2007, 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Long F., Nicholls R. A., Emsley P., Gražulis S., Merkys A., Vaitkus A., Murshudov G. N., Acta Crystallogr. Sect. D 2017, 73, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., Acta Crystallogr. Sect. D 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Acta Crystallogr. Sect. D 2012, 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liebschner D., Afonine P. V., Baker M. L., Bunkoczi G., Chen V. B., Croll T. I., Hintze B., Hung L. W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., Richardson J. S., Richardson D. C., Sammito M. D., Sobolev O. V., Stockwell D. H., Terwilliger T. C., Urzhumtsev A. G., Videau L. L., Williams C. J., Adamsa P. D., Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. The PyMOL Molecular Graphics System, Version 2.2.3, Schrödinger LLC, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary