

Summary
It has been hypothesised that vegetative desiccation tolerance in resurrection plants evolved via reactivation of the canonical LAFL (i.e. LEC1, ABI3, FUS3 and LEC2) transcription factor (TF) network that activates the expression of genes during the maturation of orthodox seeds leading to desiccation tolerance of the plant embryo in most angiosperms. There is little direct evidence to support this, however, and the transcriptional changes that occur during seed maturation in resurrection plants have not previously been studied. Here we performed de novo transcriptome assembly for Xerophyta humilis, and analysed gene expression during seed maturation and vegetative desiccation. Our results indicate that differential expression of a set of 4205 genes is common to maturing seeds and desiccating leaves. This shared set of genes is enriched for gene ontology terms related to abiotic stress, including water stress and abscisic acid signalling, and includes many genes that are seed‐specific in Arabidopsis thaliana and targets of ABI3. However, while we observed upregulation of orthologues of the canonical LAFL TFs and ABI5 during seed maturation, similar to what is seen in A. thaliana, this did not occur during desiccation of leaf tissue. Thus, reactivation of components of the seed desiccation program in X. humilis vegetative tissues likely involves alternative transcriptional regulators.
Keywords: desiccation tolerance, resurrection plant, seed maturation, LAFL, ABI3, Xerophyta humilis, Xerophyta viscosa
Significance Statement
A key question is whether vegetative desiccation tolerance in resurrection plants evolved through the ectopic expression of seed maturation transcription factors (TFs) in leaves. While the full‐length ABI3 and ABI5 master seed TFs are expressed at high levels in the maturing seed of Xerophyta humilis, they are silent in desiccating leaves. Desiccation tolerance in Xerophyta leaves must instead have evolved by rewiring of the vegetative stress response pathway to activate seed genes.
Introduction
Plant adaptations to water stress form a spectrum that includes drought avoidance, drought tolerance and, at the extreme end of the scale, vegetative desiccation tolerance (VDT; Alpert, 2005). VDT is defined as survival after loss of > 95% of cellular water (Gaff and Oliver, 2013). It is very common in early diverging plants, such as mosses, liverworts and hornworts, where it is believed to have been the ancestral state, and vital for the colonisation of land by the ancestors of terrestrial plants (Oliver et al., 2000). With the evolution and improvement of the plant vascular system, VDT was gradually lost from tracheophyte lineages. However, VDT re‐evolved independently in monocot and eudicot plant families, and is known to occur in over 135 taxonomically diverse species across 44 genera. These plant species are commonly referred to as ‘resurrection plants’, and are found across central and southern Africa, Europe, Asia, South America and Australia (Behnke et al., 2013; Rakić et al., 2014; Xiao et al., 2015). A key question is how VDT evolved multiple times in these diverse plant lineages and whether it can be engineered into crop plants.
Although desiccation tolerance (DT) was lost in the vegetative tissues of vascular plants, the processes and networks involved in primitive VDT were not abandoned completely. Instead, these mechanisms were co‐opted by early vascular plants for abiotic stress responses and the protection of their reproductive propagules (Oliver et al., 2000). Over 95% of angiosperms and gymnosperms produce orthodox seeds that contain DT embryos (Gaff and Oliver, 2013). Although the leaves and roots of most angiosperms do not exhibit VDT, there is nonetheless a narrow developmental window post‐germination in which seedlings are able to re‐establish DT in response to stress (Buitink et al., 2006; Maia et al., 2011; Lyall et al., 2014). This phenomenon has been observed in the seedlings of several species, including Cucumis sativus, Impatiens walleriana and Triticum aestivum (Bruggink and van der Toorn, 1995, 1997; Farrant et al., 2004), and has been investigated in both Arabidopsis thaliana and Medicago truncatula (Terrasson et al., 2013; Maia, 2014; Dekkers et al., 2015).
In orthodox seeds, the onset of embryonic DT occurs during the late maturation stage of embryogenesis as a pre‐programmed developmental transition (Angelovici et al., 2010). Maturation and maturation drying are characterised by the accumulation of storage compounds (sugars, triglycerides and seed storage proteins), late embryogenesis abundant proteins (LEAs) and anti‐oxidants, which act as both osmoprotectants during desiccation and nutrient reserves during germination (Bewley and Black, 1994; Vicente‐Carbajosa and Carbonero, 2005; Dekkers et al., 2015; Leprince et al., 2017). Notably, many of the genes involved in seed maturation are also upregulated during the re‐acquisition of DT in germinating seedlings and in desiccating leaves of resurrection plants (Illing et al., 2005; Buitink et al., 2006; Terrasson et al., 2013; Maia, 2014), but not in the leaves of desiccation‐sensitive plant species. It has been proposed that the similarity in protection mechanisms of seed maturation, seedling DT and VDT is evidence for a common regulatory mechanism between these processes, possibly via co‐option of the seed maturation gene networks during vegetative stress in resurrection plants (Oliver et al., 2000; Illing et al., 2005; VanBuren, 2017; Costa, Cooper, et al., 2017).
In A. thaliana, the canonical LAFL network of seed master transcriptional regulators LEAFY COTYLEDON 1 (LEC1), ABSCISIC ACID INSENSITIVE 3 (ABI3), FUSCA 3 (FUS3) and LEAFY COTYLEDON 2 (LEC2) (Santos‐Mendoza et al., 2008; North et al., 2010) activates the expression of seed maturation genes in response developmental cues and abscisic acid (ABA) signalling. The B3 DNA binding domain transcription factor (TF) ABI3 plays a central role in the maturation response and has functional orthologues in many angiosperm species (Hattori et al., 1994; Shiota et al., 1998; Suzuki et al., 2001; Footitt et al., 2003; Carbonero et al., 2016). Seeds of abi3 mutants are desiccation‐sensitive in A. thaliana and M. truncatula (Nambara et al., 1995; Terrasson et al., 2013). ABI3 orthologues have also been identified in basal land plants, including Physcomitrella patens (Khandelwal et al., 2010), and abi3 deletion mutants of P. patens are unable to recover from desiccation, suggesting that this TF is a conserved and ancestral regulator of DT (Marella et al., 2006; Khandelwal et al., 2010; Komatsu et al., 2013).
The ABI3 protein is comprised of several conserved structural domains: the acidic regions A1 and A2, separated by a Pro‐, Ser‐ and Thr‐rich (PST) region, and the basic domains B1, B2 and B3 (Giraudat et al., 1992). The A1 and A2 domains are transcriptional activators (McCarty et al., 1991; Gagete et al., 2009), while the B3 DNA‐binding domain binds the RY motif (CATGCA) commonly found in the promoters of seed maturation genes (Suzuki et al., 1997; Sasnauskas et al., 2018). The B2 domain contains a nuclear localisation signal and appears to have transactivation capability (Marella et al., 2006), but can also mediate gene expression indirectly via the ABA response element (ABRE), most likely through protein−protein interactions (Ezcurra et al., 2000). The B1 domain can interact with other proteins, especially those of the bZIP TF family and in particular ABI5 (Nakamura et al., 2001), which is known to bind to ABREs. ABI5, a member of the ABA‐responsive (group A) bZIP family of TFs, acts downstream of the canonical LAFL network and, like ABI3, is essential for seed desiccation (Santos‐Mendoza et al., 2008). Furthermore, both ABI3 and ABI5 are important for the re‐acquisition of DT in germinating seedlings in response to ABA signalling (Lopez‐Molina et al., 2001; Terrasson et al., 2013).
The genomes of several resurrection plant species have been sequenced in recent years, including the monocots Oropetium thomaeum and Xerophyta viscosa, and the eudicots Boea hygrometrica and Lindernia brevidens (VanBuren et al., 2015, 2018; Wen et al., 2015; Costa, Ligterink, et al., 2017). RNA‐Seq analysis of the transcriptional changes occurring during desiccation of leaf tissue has also been performed for a number of additional species, including Craterostigma plantagineum (Rodriguez et al., 2010), Sporobolus stapfianus (Yobi et al., 2017), B. hygrometrica (Xiao et al., 2015) and Haberlea rhodopensis (Gechev et al., 2013). While these studies have demonstrated that many genes induced during seed maturation are also upregulated in desiccating leaves of resurrection plants, the question as to how these seed maturation genes are reactivated in desiccating leaves remains unanswered. It has been speculated that members of the canonical LAFL network perform this role (Costa, Cooper, et al., 2017; VanBuren, 2017), but there is little direct evidence to support this hypothesis. For example, although X. viscosa homologues of many of the A. thaliana genes directly regulated by ABI3 (the ABI3 regulon) were upregulated in both leaves and seedlings during desiccation or ABA treatment, transcript levels of two X. viscosa ABI3 orthologues were not (Costa, Ligterink, et al., 2017). Furthermore, no transcriptomic analysis of seed maturation in resurrection plants has been undertaken. Thus, it is not possible to know whether the transcriptional reprogramming that occurs during this developmental process is analogous to that occurring in A. thaliana, or whether the same TFs regulate genes important for both seed and vegetative DT in resurrection plants.
In this study we present an RNA‐Seq transcriptome assembly and analysis of gene expression during seed maturation and vegetative desiccation in the monocot resurrection plant Xerophya humilis. Our results suggest that the expression of seed‐specific genes during VDT is highly unlikely to result from simple reactivation of the canonical LAFL network or known seed regulatory TFs (such as ABI5). Instead, we hypothesise that in vegetative tissues the expression of the ABI3 regulon is activated by a desiccation‐responsive pathway, which may include members of the ABRE‐binding factor (ABF) family of TFs.
Results
Stages of development of Xerophyta humilis seeds
Seed development in X. humilis takes 2–3 weeks from fertilisation to dry seed. The early embryogenesis stages, characterised by a slow increase in seed size, occurred between 1 and 4 days after fertilisation (DAF1−DAF4; Figure 1). Seed size increased rapidly thereafter (DAF5−DAF8), which likely represents the switch from embryogenesis to seed filling and early maturation, as observed in other angiosperms (Dam et al., 2009; Garg et al., 2017). Over the next few days, during the mid‐maturation stage (DAF9−DAF12), the size of the seeds did not change substantially, and they lost their green colouration. Late maturation occurred from DAF13 to DAF17 when seeds shrunk and turned brown. Dry seeds (DAF14+) were wrinkled and spilled easily from the seed pod (Figure 1). For this study, seed developmental stages representing early seed maturation (DAF6; small, green seeds), mature green (DAF11; large, green seeds) and mature dry seeds (DAF17; shrunken, brown seeds with little moisture) were selected for RNA‐Seq.
Figure 1.
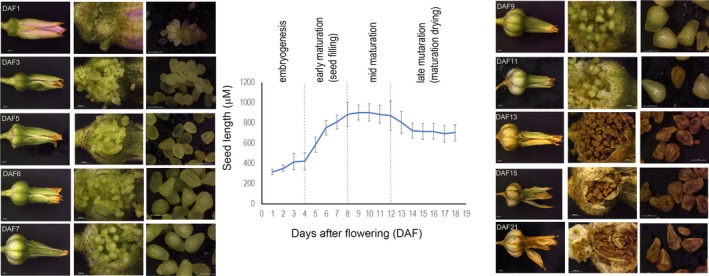
Overview of Xerophyta humilis seed development.
Left and right: Images showing X. humilis flowers, seed pods and seeds at different days after fertilisation (DAF).
Centre: X. humilis seed length as development progresses. Seeds show a rapid increase in size between DAF4 and DAF8, consistent with early maturation and seed filling. Seed length is stable during mid maturation (DAF8−DAF12) and drops slightly as seeds lose water during late maturation (DAF12+). For this study, seeds were sampled at DAF6 (early maturation), DAF11 (mid maturation) and DAF17 (late maturation/dry). A range of 34–161 seeds were measured from across three seed pods for each DAF; error bars are standard deviation.
Assembly and annotation of the Xerophyta humilis seed and leaf transcriptome
Independent RNA‐Seq experiments were performed on RNA extracted from X. humilis seeds during three stages of seed maturation (DAF6, DAF11 and DAF17, totalling 9 libraries and 193 million read pairs) and from leaves harvested at five relative water contents (RWCs) during a course of vegetative desiccation (100%, 80%, 60%, 40% and 5% RWC, totalling 15 libraries and 350 million read pairs).
After trimming, filtering and error correction, the reads from both experiments were combined for de novo transcriptome assembly. Over 500 000 contigs (350 000 Trinity genes) were assembled in the raw assembly, with an N50 of 1198 bp and an average read mapping rate of 81% (seed libraries) and 77% (leaf libraries). RapClust was used to remove low‐evidence contigs and cluster transcripts based on fragment equivalence classes computed by Salmon (Patro et al., 2017). We leveraged a draft X. humilis genome (Schlebusch, 2019) to collapse these groups by combining transcript clusters that mapped to the same genomic locus. The resultant RapClust assembly consisted of 120 158 genes (where each ‘gene’ consists of one or more alternatively spliced transcripts). Putative open reading frames (ORFs) for each gene were determined using TransDecoder, which identified 25 391 genes containing a complete ORF and a further 35 868 that contained a partial ORF. Representative transcripts from each gene were defined based on length of the longest predicted complete ORF or, if lacking an ORF, the longest contig sequence.
TransRate and the Benchmarking Universal Single‐Copy Orthologs (BUSCO) programs were used to assess the quality and completeness of the de novo transcriptome assemblies, respectively. The TransRate scores of the raw assembly were low due to the very large number of low‐evidence contigs (assembled from background reads, and reads containing SNPs and errors). Both scores improved substantially when using the RapClust assembly, and the optimised score fell within the 25–50th percentile of the optimised scores calculated for 155 published assemblies by Smith‐Unna et al. (2016; Figure S1a). In order to assess the completeness and integrity of the assembly, the transcriptome was scanned for the presence of 1440 plant BUSCOs. Over 70% of BUSCOs were present as full‐length sequences in the RapClust assembly, with 90% of all BUSCOs present as complete or partial sequences (Figure S1b).
To predict potential gene identity and function, a representative transcript from each gene (encoding the longest ORF) was compared with protein sequences in the Swissprot and Uniref90 databases. In total, 40 567 (34%) transcripts had a significant blast match at an e‐value of < 1e‐6. Of these, the vast majority (80%) were against plant proteins (Figure S1c). The remaining top blast hits (20%) were against proteins of fungal origin, but < 0.5% of reads in each library mapped against these sequences suggesting that they likely originated from low‐level fungal contamination of the field‐collected plants. Annotations were assigned using Blast2GO, resulting in the successful annotation of 34 783 genes using the default evidence weightings, matching to 19 956 individual protein entries in the Swissprot and Uniref90 public databases.
Comparison of gene expression during seed maturation and vegetative desiccation tolerance
Principal component analysis (PCA) of regularised log‐normalised read count data showed that the RNA‐Seq libraries separated primarily by experiment (tissue of origin) and secondarily by water content (Figure 2). In leaf tissues, there was a clear separation of samples at higher water contents (100%, 80% and 60% RWC) and samples during the later stages of desiccation (40% and 5% RWC), suggestive of a large shift in gene expression. Recently, it has been argued that a RWC of 40% represents the boundary between dehydration and desiccation, as defined by physiological and molecular changes observed across several resurrection plants (Zhang and Bartels, 2018).
Figure 2.
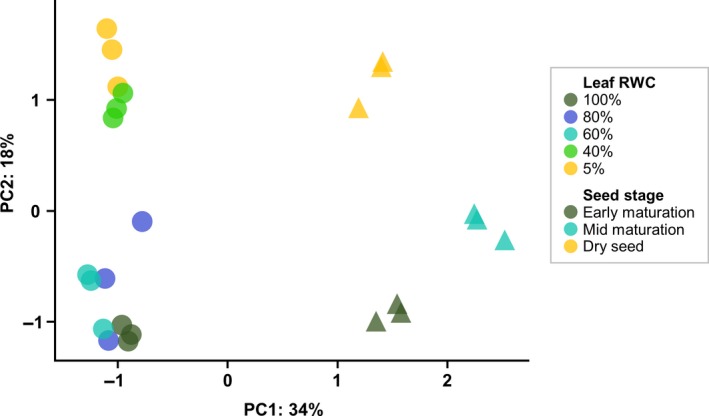
Principal component analysis (PCA) plot of seed and leaf RNA‐Seq libraries.
Plots were generated using regularised log‐normalised read counts. PC1 separates samples on experiment (tissue type) and PC2 by water content.
The degree to which the transcriptional reprogramming occurring during seed maturation and VDT overlaps in resurrection plants is unknown. To address this, we identified genes that were differentially expressed (DE) during VDT and/or seed maturation, and then compared their expression profiles during the two processes. DE genes were grouped into simple expression clusters based on the point of maximal expression. In leaves this resulted in three groups: downregulated (maximal expression at 100% RWC); early dehydration‐induced (maximal expression at 80% or 60% RWC); and desiccation‐induced (maximal expression at 40% or 5% RWC) as defined by Zhang and Bartels (2018). For seed maturation, we used only two groups: downregulated genes (maximal expression during early maturation) and upregulated genes (genes with maximal expression at either mid or late maturation). The average expression profile for the genes in each group can be seen in Figure 3. To validate the RNA‐Seq data, we used quantitative polymerase chain reaction (qPCR) to independently analyse the expression of three genes that are upregulated during desiccation in leaf tissue according to our DeSEQ2 analysis, and observed good agreement between the expression values derived from the two methods (Figure S2).
Figure 3.
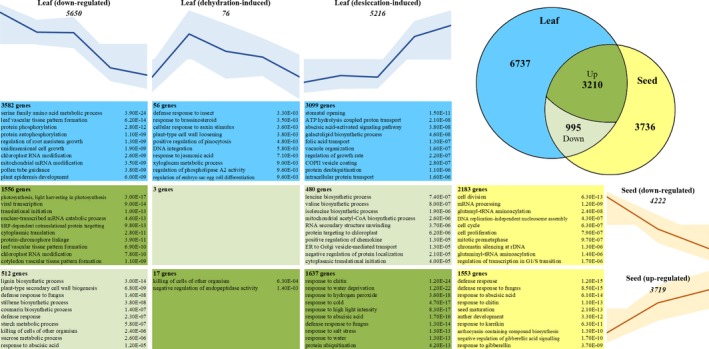
Number, overlap and gene ontology (GO) enrichment of genes differentially expressed (DE) during vegetative desiccation tolerance (VDT) and seed maturation.
Genes only DE in leaf tissue (blue) are grouped based on expression pattern as downregulated (most highly expressed at 100% relative water content (RWC)), dehydration‐induced (most highly expressed at 80–60% RWC) or desiccation‐induced (most highly expressed at 40–5% RWC). Seed‐specific DE genes (yellow) are classified as downregulated (expressed at early maturation) or upregulated (most highly expressed mid−late maturation). Gene sets that are DE in both tissues are shown in green, with the darker shade indicating those that display the same direction of regulation during VDT and seed maturation. Expression profiles show the average scaled expression with standard deviation (shaded) for each gene in that cluster. The number of genes in each group is indicated, as are the 10 most significantly over‐represented GO terms in that set, in descending order of significance (P < 0.01).
A total of 14 678 X. humilis genes were DE, of which 6737 (46%) were DE in drying leaves only, 3736 (25%) in maturing seeds only, and 4205 (28%) during both processes (Figure 3). The number of genes that were specifically induced during the early dehydration phase in leaves (80% and 60% RWC) was very low (76 genes). Of the 4205 genes DE in both seed and leaf, 76% showed the same direction of DE (i.e. either upregulated in both tissues or downregulated in both; Figure 3). These results suggest that there is indeed a degree of overlap in the transcriptional reprogramming that occurs during VDT and seed maturation in X. humilis.
To identify the potential roles that might be played by these gene sets, we looked for enrichment of GO terms within them. Interestingly, GO terms associated with abiotic stress, including water stress, are not enriched in the leaf‐ or seed‐specific gene sets. Instead, most of the GO terms that might be expected to be associated with VDT were only associated with genes that were DE during both vegetative desiccation and seed maturation (Figure 3). For example, as a poikilochlorophyllous resurrection plant, X. humilis ceases photosynthesis and dismantles its thylakoid membranes during desiccation (Ingle et al., 2008). GO terms related to photosynthesis and response to light were enriched only in the genes downregulated during both seed maturation and VDT. This suggests that many of the genes involved in photosynthetic and chloroplast structural changes during VDT may be shared with processes in the seed. Similarly, the 1637 genes that are upregulated during vegetative desiccation and seed maturation were enriched for a broad range of terms associated with response to stress, including response to water, water deprivation, cold, H2O2, high light, chitin and salt − terms that are absent from other gene sets (Figure 3).
Homologues of Arabidopsis thaliana seed genes are upregulated during both seed maturation and vegetative desiccation tolerance in Xerophyta humilis
While both seed maturation and VDT in X. humilis involve the upregulation of a shared subset of genes, it is still unclear to what extent VDT‐associated genes in X. humilis are canonically seed‐specific in desiccation‐sensitive plants. We thus analysed the expression of the X. humilis homologues of a set of genes previously identified as seed‐specific in A. thaliana. Our X. humilis transcriptome contains 204 potential homologues to 152 of the 289 seed‐specific genes identified by Le et al. (2010), of which 136 (67%) were DE (Figure 4a; Data S1).
Figure 4.
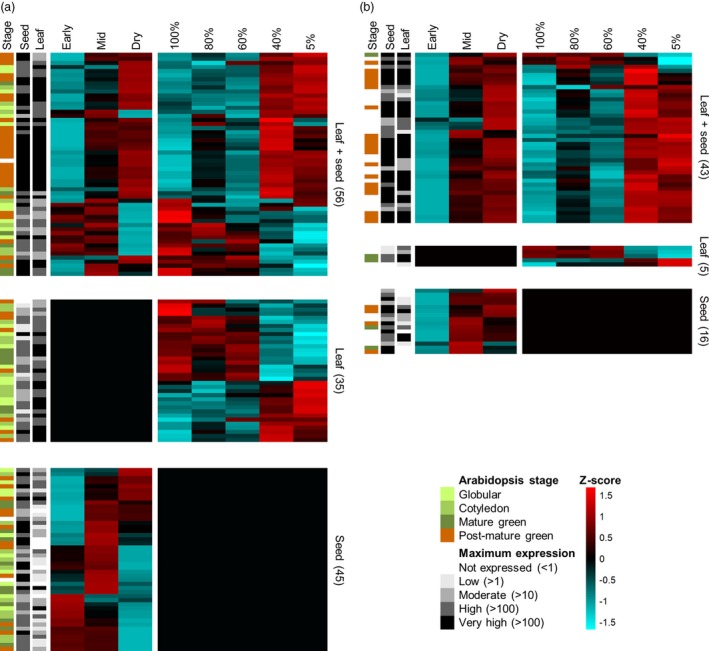
Expression patterns of Xerophyta humilis homologues of Arabidopsis thaliana seed‐specific and ABI3 regulon genes.
Heatmaps profiling expression of A. thaliana seed‐specific genes (a) and 64 putative homologues of the ABI3 regulon in X. humilis (b) during seed maturation and vegetative desiccation tolerance (VDT). Genes that are not differentially expressed (DE) during either one of the processes have been given a scaled expression value (z‐score) of 0 in that tissue. Annotations on the left of both graphs represent whether the homologue in A. thaliana reached maximum expression during the globular (GLOB), cotyledon (COT), mature green (MG) or post‐mature green (PMG) stage of seed development in that species, based on data from Le et al. (2010). The read count at the maximum expression level during seed maturation (‘Seed’) and VDT (‘Leaf’) for each given gene is shown in a grey scale.
In general, genes that were DE in both tissues (56 in total) displayed the same expression profile (up in seed and leaf, or vice versa; Figure 4a). Additionally, in the leaf, there is again a clear switch in gene expression that occurs for these genes between 60% and 40% RWC. Thirty‐five A. thaliana seed‐specific genes were not identified as being DE during seed maturation in X. humilis despite showing a change in expression during VDT (Figure 4a). However, the majority of these genes reached peak expression early during A. thaliana seed development (globular, cotyledon or mature green stages) and, as our RNA‐Seq analysis was restricted to maturing seed tissues only, they may have been activated earlier in X. humilis seed development and not identified as DE in this study. Homologues of genes identified as being seed‐specific in Arabidopsis, but which were not DE in either X. humilis seeds or leaves, are listed in Data S1, together with their average expression. Absent from this list were genes linked to constitutive desiccation protection in some resurrection plants; for example, LEAs, dehydrins, antioxidant enzymes or transketolases (Oliver et al., 2005; Dinakar and Bartels, 2013).
The 37 genes upregulated during both seed maturation and VDT (Figure 4a, upper segment) contained a significantly higher proportion of genes that were specific to the post‐mature green (PMG) stage of A. thaliana seed development (24/37 genes) compared with genes with other expression patterns (25/99 genes; χ 2 P = 0.0004). All these PMG‐associated genes were homologues of genes from the A. thaliana ABI3 regulon (genes directly regulated by the seed‐specific TF ABI3). In A. thaliana, RNA‐Seq and ChIP‐chip data have been used to identify a set of 98 target genes that constitute the ABI3 regulon (Mönke et al., 2012). We expanded our search of seed‐specific genes to include all X. humilis homologues of the 98 A. thaliana ABI3 regulon genes, several of which are not included in the Le et al. (2010) dataset. We identified 70 potential homologues in X. humilis to 62 of these genes, of which 64 (91%) were DE in one or both tissues (16 in the seed, five in the leaf and 43 in both tissues). The majority were upregulated during late seed maturation and at RWC below 60% in the leaf, and generally had very high expression levels (Figure 4b), supporting the idea that the ABI3 regulon is activated during both processes.
LAFL regulators and ABI5 are active during seed maturation but not induced during vegetative desiccation tolerance
The induction of homologues of canonically seed‐specific genes during VDT in X. humilis is consistent with the hypothesis that VDT evolved through de‐repression or reactivation of seed maturation transcriptional regulators in vegetative tissue. To test this hypothesis, we queried our transcriptome for orthologues of members of the canonical LAFL regulatory network; specifically, the genes LEC1, ABI3, FUS3 and LEC2, and the downstream group‐A bZIP ABI5. We identified one or more co‐orthologues to each of these, and the evolutionary relationships between these sequences in X. humilis and other plant species are shown in Figure S3. Two X. humilis LEC1 co‐orthologues were expressed during early seed maturation and upregulated during mid‐maturation, consistent with the known role of this TF as a regulator of seed development. In contrast, these genes were barely expressed in leaf tissue irrespective of RWC, with mean normalised read counts < 10 across all RWC (Figure 5). Two putative orthologues of the monocot LEC2‐like genes and a putative orthologue of monocot FUS3 were also identified in X. humilis. Both XhLEC2‐like genes were downregulated during seed maturation, while XhFUS3 was upregulated specifically during mid‐maturation. As with the XhLEC1 genes, the normalised read counts of XhFUS3 and XhLEC2 transcripts in leaves were extremely low (mean count < 6) across all RWCs (Figure 5).
Figure 5.
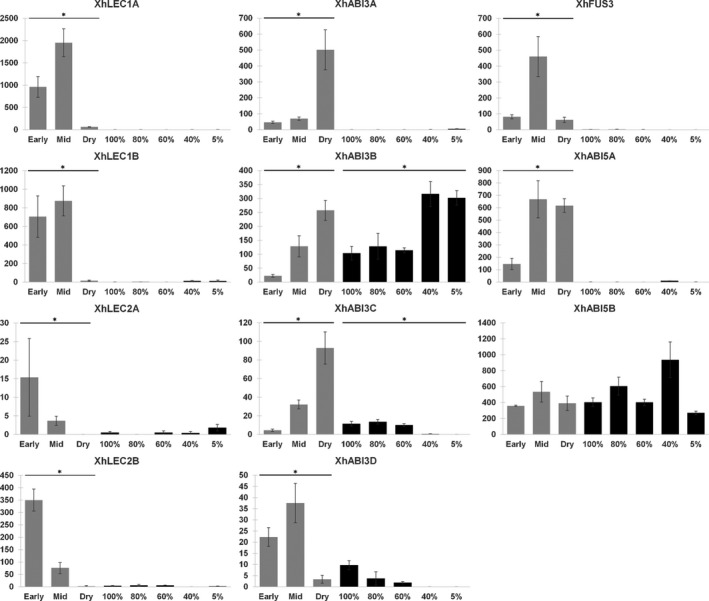
Expression of Xerophyta humilis LAFL and ABI5 genes.
DESeq2 normalised counts of X. humilis seed regulators during seed maturation (grey) and vegetative desiccation tolerance (VDT; black), marked with an asterisk if the change was significant in that tissue. Counts from both tissues are displayed on the same axis but are derived from different experiments. Error bars are standard error. (*) indicates significant at FDR < 0.01.
Interestingly, we found that X. humilis possesses at least four genes with homology to ABI3 (Figure S3), three of which (XhABI3A‐C) were upregulated during late seed maturation, while one (XhABI3D) was upregulated during earlier stages of seed development (Figure 5). XhABI3B was also expressed in leaves and upregulated during desiccation, but levels of the other ABI3 transcripts were barely detectable in leaves (Figure 5). Lastly, two transcripts with similarity to the A. thaliana and rice ABI5 genes were identified in X. humilis (Figure S3). XhABI5A was significantly upregulated during mid and late seed maturation but expressed at very low levels in the leaf desiccation series, while XhABI5B was not DE in either tissue but was consistently expressed throughout both seed maturation and VDT (Figure 5). The seed‐associated patterns of gene expression observed for the canonical LAFL TFs are consistent with a role in the regulation of seed maturation in X. humilis (as in other plant species) but not as regulators of the VDT response.
Duplication of ABI3 and loss of the B3 domain in Xerophyta resurrection plants
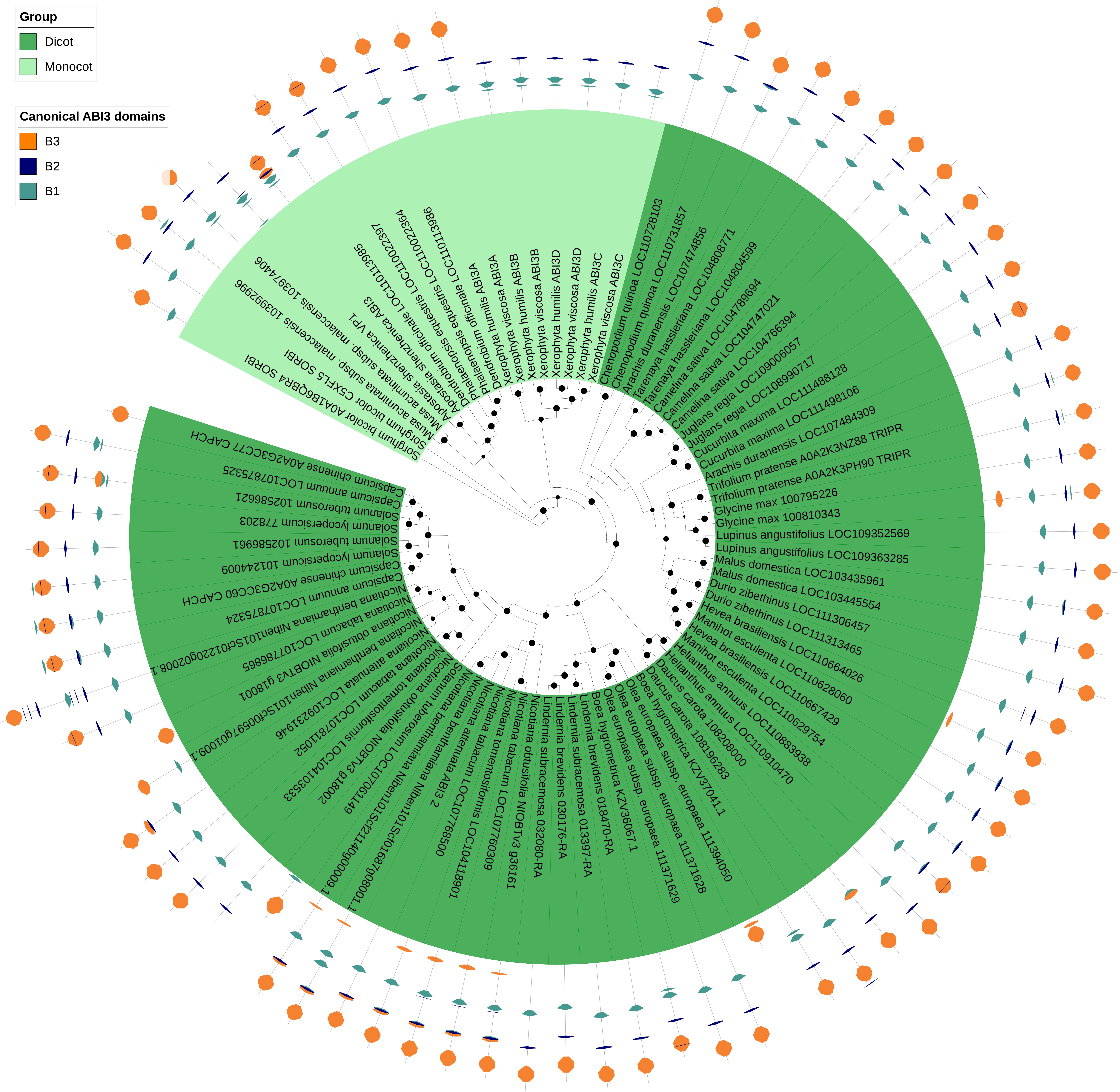
We identified four paralogues of ABI3 in X. humilis. A comparison of the protein‐coding regions of the representative transcripts for these four paralogues showed that only one, XhABI3A, possessed a B3 DNA‐binding domain (Figure 6). The remaining three encode a truncated ABI3 protein due to the presence of a stop codon directly after the B2 domain in their transcripts (Figure 6), including XhABI3B which was upregulated during both seed maturation and VDT (Figure 5). To verify that these transcripts were not artefacts from the de novo assembly process, 3′‐RACE was performed using primers designed to sites between the B1 and B2 domains for each ABI3 paralogue on pooled cDNA generated from leaves and seeds. We were successful in cloning the 3′ downstream regions of the two most highly expressed ABI3 transcripts: the B3‐containing transcript XhABI3A and the truncated transcript XhABI3B, confirming their presence and transcription termination positions (Data S2). To determine whether the increased copy number of ABI3 is unique to X. humilis, we searched for similar sequences in the X. viscosa genome assembly using BLAST (Costa, Ligterink, et al., 2017). Four ABI3 paralogues were also detected, each on a separate scaffold of the X. viscosa genome, of which three also encode proteins lacking the B3 domain due to the presence of a stop codon directly after the B2 domain (Figure 6). The number and position of exons for each X. viscosa ABI3 paralogue was determined using the complete X. viscosa genome assembly (Costa, Ligterink, et al., 2017). The XvABI3A transcript has a similar structure to Arabidopsis ABI3, with the B1, B2 and B3 domains spread across multiple exons. In contrast, the duplicated paralogues possess only a single exon and lack the exons containing the B3 domain (Figure S4a). Expression analysis showed that of these four paralogues, only XvABI3B was expressed at appreciable levels in leaves and expression increased during desiccation (Figure S4b). Partial fragments to a putative XhABI3B orthologue in Xerophyta elegans were identified in a transcriptome dataset obtained from the 1000 Plants project (http://www.onekp.com/). This transcript also contained a stop codon directly after the B2 domain (Figure 6).
Figure 6.
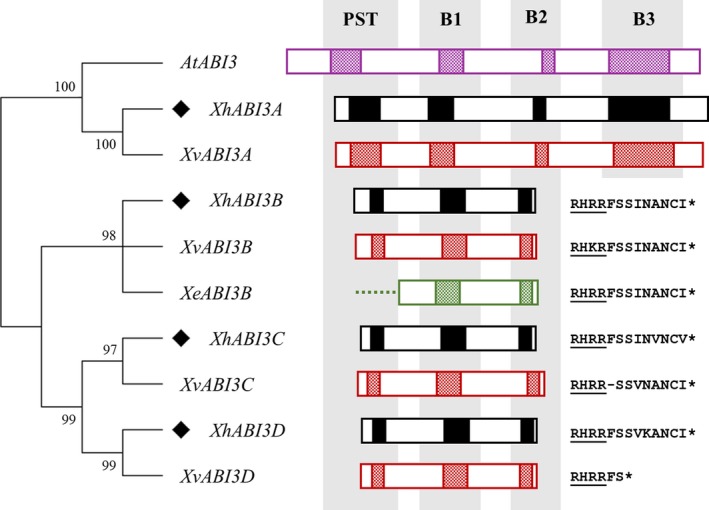
Duplication of ABI3 in Xerophyta and loss of the B3 domain.
Maximum‐likelihood phylogenetic tree showing the relationship between AtABI3 and the predicted ABI3 paralogues in Xerophyta humilis, X. viscosa and X. elegans, using full‐length amino acid sequences and 1000 bootstrap replicates. Positions of the conserved PST, B1, B2 and B3 domains are given by shaded segments. The X. elegans ABI3B sequence corresponds to a partial open reading frame (ORF) that is missing the N‐terminal region (dashed line). The conserved stop codon sequences for the Xerophyta‐specific ABI3B, ABI3C and ABI3D genes are shown on the right, with the terminal region of the B2 domain underlined. Protein alignments and phylogenetic analysis performed using Clustal Omega and RAxML.
Xerophyta are a paleohexaploid genus, likely having undergone an ancient whole genome duplication event 55−40 Mya (Goldblatt and Poston, 1988; Alcantara et al., 2018). Nonetheless, the presence of multiple ABI3 paralogues is unusual. An analysis of B3 domain‐containing TF ABI3 orthologues (Group 71729at3193) from OrthoDB (Kriventseva et al., 2018) shows that most angiosperms and many polyploid species encode only a single copy of ABI3. Species with duplicated ABI3 tended to have only two copies, though a handful of species contain more (Figure S5). ABI3 duplication is also less prominent in monocot species compared with dicot species, being observed so far in only Xerophyta, Sorghum bicolor, Musa acuminata and the orchid species Apostasia shenzhenica, Dendrobium officianale and Phalaenopsis equestris (Figure S5). Only two putative ABI3s, from A. shenzhenica and Nicotiana obtusifolio, were predicted to lack a B3 domain due to a premature stop codon – though there is no additional evidence that these transcripts are expressed (Figure S5). We could not find any other similarly truncated ABI3 protein sequences in the NCBI protein database.
Promoters of Xerophyta ABI3 regulon homologues are enriched for the ABRE but not RY motif
The majority of canonical ABI3 regulon genes were upregulated during seed maturation and VDT in X. humilis (Figure 4b), and also during VDT in X. viscosa (Costa, Ligterink, et al., 2017). In stark contrast, during VDT only one LAFL paralogue – an unusual ABI3‐like gene lacking the B3 domain – was induced in X. humilis and X. viscosa, while the transcript encoding a full‐length ABI3 protein was barely detectable (Figures 5 and S4). The B3 domain of ABI3 recognises the RY motif (CATGCA), which is enriched in the promoters of its target genes in a range of plant species (Reidt et al., 2000). To determine whether control of the ABI3 regulon in Xerophyta resurrection plants may differ from that in A. thaliana, we looked for evidence of altered enrichment of regulatory motifs in the promoters of ABI3 regulon genes between A. thaliana and X. viscosa, for which a complete genome is available (Costa, Ligterink, et al., 2017).
We found 105 putative homologues in X. viscosa to 62 of the 98 A. thaliana ABI3 regulon genes. Enriched motif identification was performed with DREME, using the regions 500 bp upstream of the TSS of the 62 A. thaliana genes and the 105 putative homologues in X. viscosa as input. As expected, the RY motif was enriched within the A. thaliana promoters, as was the ABRE (ACGTG[G/T][C/A]) (Figure 7a). A third enriched motif, TTT[G/T]GTT, shows similarity to the Pollen Box (PB) motif identified in a few pollen‐specific genes (Okada et al., 2000), consistent with the fact that several ABI3 regulon genes have been shown to be expressed in stamen/maturing pollen tissue in A. thaliana (Mönke et al., 2012). In contrast, while there was enrichment of the ABRE motif in the X. viscosa promoters, the RY motif was not enriched (Figure 7a). Another motif enriched in the X. viscosa gene promoters, similar to the heptamer sequence AGATATT, is associated with gene expression during abiotic stress (drought, heat, salt, oxidative and wounding stress) and with the expression of several stress‐related TFs, including the ABF bZIP AtABF1 (atted.jp/data/cis/AGATATT.html; Obayashi et al., 2006).
Figure 7.
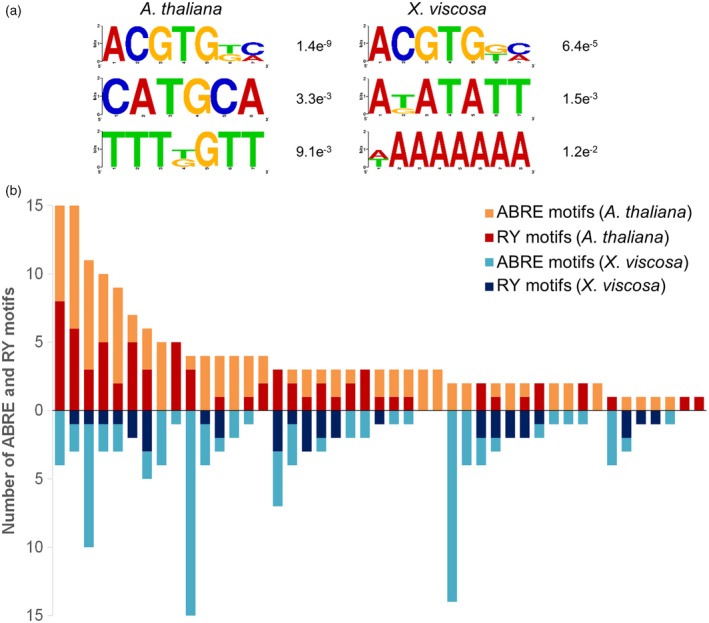
The RY and ABRE motifs in the promoters of Arabidopisis thaliana and Xerophyta viscosa ABI3 regulon genes.
(a) Over‐represented motifs in the upstream 500‐bp promoter regions of the ABI3 regulon genes in both species, as determined by DREME. The ABRE (ACGTG) is enriched in both species, whereas the RY element (CATGCA) is enriched in only A. thaliana.
(b) Total number of ABRE and RY motifs present in the 500‐bp upstream regions of A. thaliana ABI3 regulon genes, and all equivalent orthologues found in X. viscosa.
To account for the presence of duplicated co‐orthologues that may have diverged in their promoter cis‐element composition, we also analysed the total number of RY and ABRE motifs present within each set of genes when combined into orthogroups (defined here as a collection of A. thaliana and X. viscosa genes that have the same top blast matches to each other). The 62 A. thaliana genes could be divided into 45 orthogroups, and the total number of RY and ABRE motifs within the orthogroup gene promoters within 500 bp of the TSS on either strand was determined (Figure 7b). The RY motif was far more prominent in the A. thaliana gene promoters than those of X. viscosa, despite the smaller number of sequences (72 RY motifs across 62 A. thaliana promoters versus 37 RY motifs across 105 X. viscosa promoters; Figure 7b). In contrast, the ABRE featured prominently across orthogroups from both species. To test whether the 500 bp limit was too restrictive, we also investigated the distribution of RY and ABRE motifs up to 3000 bp upstream of the TSS in genes in both species. The RY element was enriched within the first 200 bp upstream of the tested A. thaliana genes but showed no such strong peak in X. viscosa (Figure S6a), while the ABRE is found within the first 200–300 bp of the promoters of both species (Figure S6b). This suggests that seed maturation genes in Xerophyta may be under the control of a different transcriptional regulatory network than in A. thaliana, one where the RY motif is less central.
ABI3 classically activates its target genes by binding directly to the RY element. However, there is evidence that ABI3 may also regulate gene expression through actions independent of the B3 DNA‐binding domain, for example via interaction with other TFs, including bZIPs, mediated by the B1/B2 domains (Ezcurra et al., 2000; Nakamura et al., 2001). Suzuki et al. (2014) investigated the B3‐dependent and B3‐independent activation of a small number of seed‐specific genes in transgenic A. thaliana by overexpression of VP1 (the maize ABI3 orthologue) with or without a functional B3 domain. B3‐independent genes included several oleosins, LEAs and Responsive to Desiccation 29B. In contrast, activation of the 2S‐albumins and CRUCIFERIN C (AtCRUC) required the presence of the B3 domain. While no 2S‐albumins were detected in the X. humilis transcriptome, both XhCRUC orthologues were only induced during seed maturation and not expressed at all in leaves (Figure S8), correlating with the lack of full‐length ABI3 (XhABI3A) in these tissues. In contrast, orthologues of the B3‐independent genes (two oleosins and a gene similar to AtEM1) were induced during late seed maturation and during VDT in leaves when only the truncated ABI3 (XhABI3B) is expressed (Figure S8).
A bZIP transcription factor can drive expression from seed gene promoters in Xerophyta humilis
As a first step in dissecting the transcriptional networks involved in seed maturation and VDT in X. humilis, we investigated the changes in expression of TF genes during these two processes. TFs present in the X. humilis transcriptome were identified using the program iTAK, resulting in the classification of 695 TF‐encoding genes across 61 families. Over 60% of these (446) were DE during seed maturation or VDT (Figure 8), with 130 DE during both processes. In general, TF families were not unique to either tissue type or biological process (Figure 8).
Figure 8.
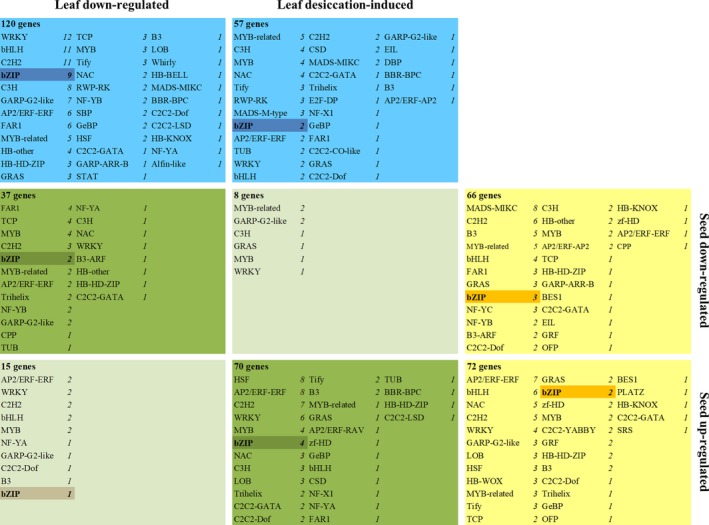
Xerophyta humilis transcription factors (TFs) differentially expressed (DE) during seed maturation and vegetative desiccation tolerance (VDT).
Overlap between TFs expressed in seeds and leaves, using the same criteria as Figure 4, but omitting the dehydration‐induced leaf cluster that contained only a single TF. The number of genes in each group is indicated, as well as the numbers of each TF family present and the presence or absence of bZIP TFs (shaded).
The ABRE was the most highly enriched motif in the promoters of X. viscosa seed maturation genes (Figure 7a), and ABA signalling is known to be important for the regulation of VDT (Zhang and Bartels, 2018). Gene regulation via the ABRE is controlled by bZIP TFs in A. thaliana, particularly those of the group A sub‐family, which includes ABI5 (Dröge‐Laser et al., 2018). Of the four group A bZIPs DE in our X. humilis experiments, a single TF (XhABFA) was highly upregulated in leaves during desiccation (< 60% RWC), and to a lesser extent in seeds after mid‐maturation (Figures 9a and S7). To determine whether XhABFA might be able to activate expression of X. humilis homologues of the ABI3 regulon, we used protoplast transactivation assays to test whether it was able to drive transcription from the first 350 bp of the X. humilis PER1, ECP63 and DSI‐1VOC promoters. These genes are expressed exclusively during seed development in Arabidopsis (Yang et al., 1997; Aalen, 1999; Mulako et al., 2008), but are activated during seed development as well as in leaves in response to desiccation in X. humilis (Figure S7). PER1 and ECP63 are known ABI3 targets in A. thaliana (Haslekås et al., 2003; Mönke et al., 2012), and both PER1 and DSI‐1VOC are expressed ectopically in homozygous fie (a subunit of PRC2) Arabidopsis mutants (Bouyer et al., 2011). The cloned promoters of these genes have multiple ABRE elements (Figure S9). Arabidopsis protoplasts transfected with the XhPER1‐LUC, XhECP63‐LUC and XhDSI‐1VOC‐LUC reporter plasmids displayed significantly higher firefly luciferase (LUC) reporter activity when co‐transfected with the effector plasmid containing XhABFA than did those co‐transfected with the empty effector plasmid (Figure 9b). To demonstrate that transactivation by XhABFA is sequence selective, we also transfected protoplasts with a XhAHL23‐LUC (AT HOOK PROTEIN 23) reporter plasmid. This gene was not DE in X. humilis during seed maturation or VDT (Figure S7), nor does its promoter contain any ABRE elements (Figure S9). LUC activity was uniformly low in these protoplasts when co‐transfected with either the ABFA effector vector or the empty vector control, indicating that ABFA was not able to drive transcription from this promoter (Figure 9b).
Figure 9.
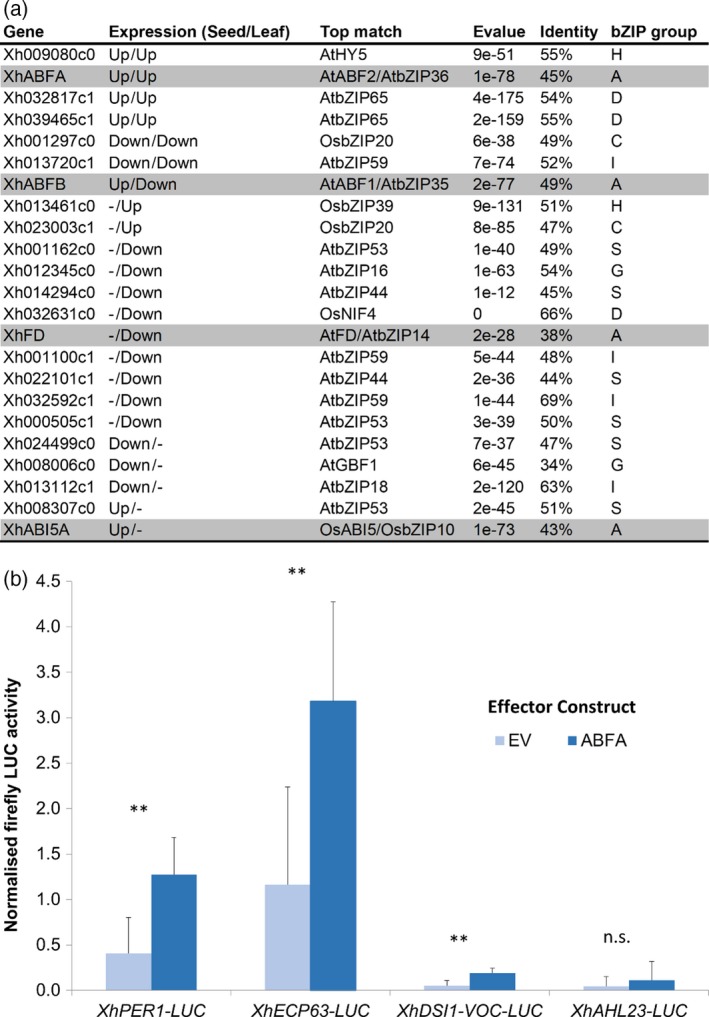
XhABFA activates transcription from ‘seed gene’ promoters.
(a) Differentially regulated bZIP transcription factors (TFs) in Xerophyta humilis leaves and/or seeds, together with most likely bZIP group classification based on top BLASTP hit against the Swissprot database. Group A bZIPs have been shaded.
(b) Protoplasts from 4‐week‐old Arabidopsis thaliana plants were co‐transfected with three vectors: 35S‐GFP, Firefly LUC and 35S‐Renilla LUC. The GFP vector either contained 35S‐XhABFA or it was an empty vector ‘EV’ control. The Firefly LUC reporter contained one of four promoter elements: XhPER1, XhECP63, XhDSI‐1VOC or XhAHL23. Firefly LUC activity was determined 22 h post‐transfection and normalised to the geometric mean value of the Renilla LUC and GFP signals for each transfection. Values shown are mean Firefly LUC activities ± SD from nine biological repeats (except XhECP63‐LUC where n = 8 and XhAHL23‐LUC where n = 5). The P‐values shown are from a two‐tailed t‐test with Bonferroni correction (** P < 0.01).
Discussion
The favoured hypothesis for the origin of VDT in angiosperms is that the gene regulatory networks that activate the DT response in orthodox seeds have been co‐opted by the vegetative stress response in resurrection plants, allowing them to mimic the protective responses used by drying embryos in their adult tissues (Costa, Cooper, et al., 2017; VanBuren, 2017). Although there is ample evidence demonstrating the expression of seed maturation‐specific genes in the drying vegetative tissues of resurrection plants (Illing et al., 2005; Rodriguez et al., 2005; VanBuren et al., 2015; Costa, Ligterink, et al., 2017), it is unknown whether these genes are indeed activated as a consequence of the reactivation of conserved seed‐specific TFs. Arguments have been made for the involvement of the important seed regulators ABI3 and ABI5 during VDT, as these genes have been shown to be important for re‐acquisition of DT in germinating seedlings; however, neither is upregulated during VDT in resurrection plants (Dekkers et al., 2015; Costa, Ligterink, et al., 2015). Furthermore, no previous study has analysed the transcriptional changes during seed maturation in a resurrection plant to see if they are indeed similar to those occurring in model plants, making it impossible to determine whether the hypothesis of co‐option of seed TFs is correct.
To test this hypothesis, we assembled and quantified the dehydration transcriptomes of X. humilis seeds during maturation and leaves during desiccation. While there are clearly patterns of differential gene expression and biological processes that are unique to each, approximately 28% of DE genes are common to both seed maturation and VDT − with 75% of these displaying the same direction of regulation (Figure 3). Within these overlapping genes, a subset of 1637 that were upregulated during both seed maturation and late desiccation contained the majority of enriched GO terms related to abiotic stress responses, which were absent from all other gene sets (Figure 3). Our expression results also highlight a large shift in gene expression that occurs in desiccating leaves between 60% and 40% RWC, which is clearly noticeable in both the PCA plot of the RNA‐Seq data (Figure 2) and when analysing groups of VDT‐responsive genes (Figure 4). Similar observations have been made in other resurrection species, and it has been recently proposed that the physiological and gene expression changes that occur in resurrection plants around and after 40% RWC define the desiccation response (Zhang and Bartels, 2018). The clear activation of a single core set of abiotic stress response genes in both seed maturation and VDT is consistent with the argument that seed maturation and VDT share a common protection response, activated between 60% and 40% RWC in leaves and during mid/late seed maturation.
Our results provide further support for the observation that numerous genes that are seed‐specific in A. thaliana are upregulated during VDT in the leaves of a resurrection plant. Of the 289 seed‐specific genes identified in A. thaliana (Le et al., 2010), we found 204 potential homologues expressed in X. humilis, of which 45% are DE during VDT and 50% during seed maturation, with 27% common to both processes (Figure 4a). Furthermore, the majority of homologues of the A. thaliana ABI3 regulon genes (59 of 70) were upregulated during late seed maturation in X. humilis, consistent with their role as important constituents of maturing seeds in other plants, and nearly 60% of these genes were also upregulated in leaves during VDT (Figure 4b). However, while we identified orthologues of the canonical LAFL TFs LEC1, ABI3, FUS3 and LEC2 and the bZIP ABI5 in X. humilis, which were upregulated during seed maturation, the transcripts of these genes were barely detectable in leaves; instead, only transcripts for an ABI3 paralogue lacking the B3 DNA‐binding domain (Figure 5) showed a significant increase in abundance in leaves during desiccation. These data strongly suggest that the ABI3 regulon and other seed maturation genes are activated by a different regulatory network in seeds versus leaves in X. humilis.
Interestingly, we found that both X. humilis and X. viscosa possess four paralogues of ABI3 in their genomes. Xh/XvABI3A has all the functional domains identified in AtABI3, and is highly expressed during seed development, but not in leaves. However, the other three ABI3 genes lack the B3 domain entirely due to the presence of a common stop codon, though they maintain N‐terminal acidic regions separated by a PST domain, as well as the complete B1 and B2 domains (Figure 6). We could not find similarly truncated ABI3 genes in the NCBI protein database, but did find a partial transcript matching the truncated XhABI3B gene in X. elegans (Figure 6). The X. elegans RNA was sampled from young shoots, and the failure to detect either full‐length ABI3A or the other two truncated ABI3 genes is consistent with the absence of these transcripts in leaves of X. humilis.
The ABI3 transcript is known to be post‐transcriptionally modified in multiple species − including the formation of splice variants lacking the B3 and/or other domains (McKibbin et al., 2002; Fan et al., 2007; Gagete et al., 2009; Sugliani et al., 2010). However, our analysis of the three truncated X. viscosa ABI3 transcripts indicated that they were derived from three genomic scaffolds that lack any sequences with homology to the B3 domain, indicating that these are not splicing variants but rather paralogues in which the B3 domain has been lost. Additionally, unlike full‐length XvABI3A, transcripts of the other three genes in X. viscosa contain only a single exon. Phylogenetic analysis of these genes implies that the duplication events from which they arose predates the speciation of X. humilis and X. viscosa and occurred after the mutation resulting in the truncation (Figure 6). The expression, maintenance and further duplication in Xerophyta of the ABI3 genes bearing this mutation implies some functional activity of the encoded truncated protein and associated benefit to the ancestral lineages, possibly through neofunctionalisation of the duplicated copies. The truncated ABI3 genes are expressed in planta; two of the three truncated XhABI3 transcripts were induced during seed maturation, and one of these was also expressed in leaves and upregulated during late desiccation (Figure 5).
The B3 domain of ABI3 is known to bind to the seed‐specific RY motif, which is found in many of the promoters of seed maturation genes directly regulated by ABI3 in A. thaliana. A comparison of the promoter regions of the ABI3 regulon genes in both A. thaliana and X. viscosa revealed that the RY motif was diminished in the promoters of X. viscosa seed genes, even when accounting for gene duplications, whereas the ABRE was enriched in the promoters from both species (Figure 7). The question arises as to what role these truncated XhABI3 paralogues might play during VDT. ABI3 proteins lacking the B3 domain can still regulate gene expression, most likely via interaction with other TFs, including bZIPs, mediated by the B1/B2 domains. Maize ABI3 orthologues lacking a functional B3 domain can complement the desiccation‐intolerant phenotype of abi3 Arabidopsis mutant seeds, and constitutive overexpression of this protein results in the upregulation of a subset of ABI3‐regulated genes (Suzuki et al., 2014). Orthologues of these B3‐independent genes (two oleosins and a gene similar to AtEM1) were induced during VDT in X. humilis, when only the truncated XhABI3B was expressed, whereas X. humilis CRUC genes (orthologues of the B3‐dependent target AtCRUC) were induced only during seed maturation and not during VDT (Figure S8). It is thus possible that XhABI3B could be involved in regulation of gene expression during VDT despite lacking the B3 domain, although further work is required to determine if this is the case. The B1 and B2 domain of ABI3 are known to be important for interactions with ABI5 during seed maturation. However, ABI5 is not DE during desiccation in X. humilis leaves. Instead, ABI3B might interact with one or more other bZIP TFs that bind to the ABRE. ABA signalling is an important element of both seed maturation and VDT (Santos‐Mendoza et al., 2008; Zhang and Bartels, 2018), and X. viscosa ABI3 regulon promoters, while depleted for the RY motif, are still enriched for the ABRE (Figure S6). GO terms related to ABA signalling were enriched not only in the set of genes upregulated specifically during seed maturation, but also in the set upregulated during both seed maturation and vegetative desiccation (Figure 3). This is consistent with a central role for ABA signalling during these responses, and implies an important role for ABRE‐binding TFs during the acquisition of DT seeds and leaves of X. humilis. The ABI5 gene and its paralogues, such as EEL, are known to regulate seed genes during seed maturation and, consistent with this, an XhABI5 orthologue was upregulated at this time point (Figure 5). However, no ABI5 orthologue was found to be upregulated during VDT. Instead a single group‐A bZIP (XhABFA), with similarity to the A. thaliana ABF TFs, was strongly induced during VDT (and to a lesser extent during seed maturation) making it a promising candidate for ABA‐dependent regulation of seed‐specific genes in the leaf tissues of X. humilis. Using protoplast transactivation assays we have demonstrated that XhABFA can drive expression from the X. humilis PER1, ECP63 and DSI‐1VOC promoters (Figure 9b); the orthologues of these genes are only expressed during seed maturation in desiccation‐sensitive plants. Furthermore, experimental evidence supports PER1 and ECP63 as direct ABI3 target genes (Mönke et al., 2012). Further experiments are required to validate the binding of XhABFA to these promoters in a chromatin‐dependent context, and to determine whether this occurs only during vegetative desiccation or also during seed maturation in X. humilis.
The ABF genes in A. thaliana constitute a semi‐redundant family of abiotic stress response factors specifically regulating the ABA response in vegetative tissues, in contrast to ABI5‐related genes that are expressed in embryonic tissues (Kim, 2006; Yoshida et al., 2010; Yoshida et al., 2015). If an X. humilis ABF TF is responsible for activating seed gene expression during VDT, it would suggest that at least part of the seed maturation network in Xerophyta has been captured by the vegetative stress response pathway rather than by ectopic expression of seed maturation regulators.
Experimental Procedures
Plant material
Xerophyta humilis plants were collected from Borakalalo Nature Reserve (Northwest Province, South Africa) and transported in a desiccated state to the University of Cape Town (North West Provincial Government Permit 062 NW‐12; Cape Nature Permit AAA007‐01733). The dried plant mats were transferred to a total of three growth trays (60 × 450 × 220 mm) containing soil from the collection site and maintained in a glasshouse under ambient light conditions after rehydration.
For leaf RNA‐Seq, the plant trays were transferred to a climate‐controlled plant growth chamber (Conviron Adaptis A350) 3 weeks prior to sampling to acclimatise to the experimental conditions: 16‐h‐long day, temperature setting of 22°C, an average luminosity of 250 µmol m−2 sec−1 and watering three times a week. Undamaged leaves were sampled from clusters of plantlets across all trays at the same time mid‐morning over a period of approximately 2 weeks after the cessation of watering. Sampled leaves were split down the mid‐vein, with half used for RNA isolation and half for RWC determination. The RWC of each leaf was calculated using the following equation: RWC = (Wwet – Wdry)/(Wdry × TWavg), where Wwet is the wet weight of the sampled leaf tissue, Wdry is the dry weight of the same tissue after 3 days at 60°C, and TWavg is the average turgor weight of a X. humilis leaf. A TWavg value was calculated from the average of 15 control leaves across all three trays, using the equation: TWavg = (Wturgid – Wdry)/Wdry, where Wturgid is the turgid weight of a leaf soaked in distilled water overnight at 4°C, and Wdry is the dry weight of that leaf.
In order to collect X. humilis seed material, pollen was transferred between flowering plants with gentle brushing, and fertilised flowers were marked with cotton thread and allowed to ripen on the parent plant. Seeds were dissected from seed pods and photographed on a Nikon Stereoscope Zoom Microscope (SMZ1500). Seeds were measured using NIS‐Elements (Nikon) digital 3D imaging software. Seeds from at least three seed pods per DAF stage were imaged, with a range of 34−161 seeds measured per pod. For RNA‐Seq, seed pods were harvested at 6, 11 or 17 DAF, flash frozen in liquid nitrogen and stored at −80°C.
RNA extraction and sequencing
RNA extraction from individual leaves was performed using the Qiagen RNeasy Plant Mini Kit following the manufacturer’s instructions. Leaf RNA extracts at each RWC determined to be of sufficient quality by gel electrophoresis and RIN analysis (BioAnalyser) were pooled to form three independent biological samples, with each pool consisting of equimolar extracts from one−five individual leaves from plants across any of the three plant trays. The leaf RWCs for the RNA‐Seq experiments were 100% (fully hydrated), 80%, 60%, 40% and 5% (fully desiccated). RNA extraction from seed tissue was performed using a modified SDS‐Trizol extraction protocol, using the entire contents of a seed pod (approximately 200 seeds, following removal of any parental tissue) as starting material (Wang et al., 2012). Three biological replicates at each developmental stage (DAF6, DAF11 and DAF17) were created by combining extractions from two or three independent seed pod RNA extractions from different plants.
Purified RNA was treated with RNAstable LD (Biomatrica) and shipped to the Beijing Genomic Institute (BGI) for sequencing. Sequencing libraries were prepared using the Illumina TruSeq Stranded Total RNA with Ribo‐Zero Plant kit (Illumina), and 150 bp fragment size. Sequencing was performed over eight lanes of an Illumina flow cell using a HiSeq2000 sequencing instrument and a 90‐bp paired‐end amplification protocol (leaf RNA) or a HiSeq4000 with 150‐bp paired‐end amplification protocol (seed RNA).
Transcriptome assembly and annotation
Assembly and assembly post‐processing were performed on a high‐performance computing cluster (Dakon) at the UCT Computational Biology Division, Cape Town, South Africa. Read quality was assessed using the FastQC software package (v0.10.1; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). For each library, Illumina adaptors and low‐quality nucleotides were removed with Trimmomatic (Bolger et al., 2014), using a sliding window size of 4, a PHRED quality score cut‐off of 20 and a minimum read length of 36. Orphaned reads lacking a read pair were discarded from the dataset. Error correction of the remaining paired reads was performed using SEECER (v1.0.3; Le et al., 2013) using a kmer size of 25. Although multiple assemblers were tested, de novo transcriptome assembly was ultimately performed using Trinity (v2.0.6; Grabherr et al., 2011) in stranded mode. For transcriptome assembly, the corrected reads for both the leaf and seed libraries were combined and normalised using the Trinity in silico normalisation pipeline in paired‐end mode, with a kmer size of 25, a maximum coverage of 50 and a PCTS of 200.
Reads were mapped to the Trinity de novo transcriptome using Salmon (v0.8.1; Patro et al., 2017). Contigs were initially clustered using RapClust (Srivastava et al., 2016), and the clustered genes were further refined using genomic information derived from a draft X. humilis genome (Schlebusch, 2019). The quality of the raw and clustered transcriptome assembly was calculated according to TransRate (Smith‐Unna et al., 2016), while completeness was assessed using the BUSCO pipeline (Simão et al., 2015).
Transcriptome annotation
The full set of assembled transcripts was compared with known plant protein sequences with NCBI blastx (Camacho et al., 2009) against local installations of the Swissprot and Uniref90 databases. The blastx parameters were tailored to suit those required for downstream Blast2GO analysis, with an e‐value cut‐off of 1e‐6 (https://www.blast2go.com/support/blog/22-blast2goblog/111-format-fasta-file-blast). ORF prediction was done using the Trinity suite program TransDecoder (Haas et al., 2013). Representative transcripts from each gene were selected based on length of the longest predicted complete ORF or, if lacking an ORF, the longest contig sequence. Protein sequences derived from representative transcripts were used as input to a local installation of InterproScan (Zdobnov and Apweiler, 2001), and the blast results for each representative transcript as well as the identified InterPro terms were imported into Blast2GO Pro (Conesa et al., 2005). Gene ontology terms for each gene were assigned from the representative transcripts using the recommended mapping/annotation settings, followed by Annex annotation augmentation.
Differential expression and gene ontology analysis
Gene‐level expression estimation and DE analysis were performed using DESeq2 (v1.8.1; Love et al., 2014). The tximport R package (Soneson et al., 2015) was used to import Salmon transcript level abundance estimates and convert them to RapClust gene‐level counts (Love et al., 2017). DE testing was performed independently on the seed and leaf read data using a log‐likelihood ratio test with a false discovery rate cut‐off of 0.01. Only genes with an adjusted q‐value < 0.01 and a log2 fold‐change of at least 1 were considered as DE in either dataset. GO enrichment was performed using the topGO R package (Alexa and Rahnenfuhrer, 2010), using the ‘Fisher’ statistical test and ‘weight01’ algorithm. Only GO terms with an unadjusted P‐value of < 0.01 were considered significant.
Quantitative polymerase chain reaction validation of RNA‐Seq data
Reverse transcriptase (RT)‐qPCR was used to validate the RNA‐Seq data for XhABFA and two known ABI3 regulon genes XhPER1 and XhCal1 (Figure S2), in leaves at five RWCs (100, 80, 60, 40 and 5%). qPCR was performed using a RotorGene RG3000A instrument (Corbett Research, Australia). Reactions consisted of 1 μL template cDNA, 5 μL Kapa SYBR FAST Universal 2 × qPCR Master Mix (Kapa Biosystems, South Africa), and 200 nm of each primer in a final volume of 10 μL. Amplification conditions included an initial step at 95°C for 3 min, followed by 40 cycles of 95°C for 5 sec, primer annealing at 60°C for 30 sec, and elongation at 72°C for 1 sec. Melt curve analysis confirmed that the individual amplified products corresponded to a single DNA fragment. The relative expression level of each gene of interest was calculated with the RotorGene 6000 series software v1.7 using the standard curve method, with normalisation to the reference gene XhMSRB5 (peptide methionine sulfoxide reductase B5), which was not DE during leaf desiccation. Details of the primers used are given in Table S1.
Transcriptome analysis
ABI3 regulon genes in A. thaliana were obtained based on the analysis performed by Mönke et al. (2012), and the list of seed‐specific A. thaliana genes as well as seed‐stage expression levels was obtained from Le et al. (2010). Protein, nucleotide and promoter (500 bp and 3000 bp) sequences for both lists of genes were retrieved from the TAIR online database. Homologues between these genes and proteins in X. humilis and X. viscosa were identified using blastp as described in Costa, Ligterink, et al. (2017) in X. viscosa: all hits with an e‐value within 10‐10 of the gene with the lowest e‐value, with an e‐value threshold cut‐off of 10‐20. Closely related genes that shared the same homologues were grouped into putative orthogroups for analysis (Costa et al., 2017). Protein multi‐alignment was performed using Clustal Omega (Sievers and Higgins, 2014), and phylogenetic trees of specific gene families were created with RaxML (Stamatakis, 2014). Xerophyta viscosa promoter sequences for homologous ABI3 regulon genes were obtained from the published X. viscosa genome (version 3, 2017; Costa, Ligterink, et al., 2017). Prediction of enriched motif sequences in A. thaliana and X. viscosa promoter sets was done using a local installation of the DREME package from the MEME Suite (Bailey, 2011), using as input the 500 bp and 3000 bp sequence upstream of the TSS of each gene.
Putative X. humilis TF genes were identified using a local installation of iTAK and the iTAK TF database (Zheng et al., 2016). The full set of predicted best ORFs in the X. humilis transcriptome was used as input.
ABI3 orthologues were identified using blast in X. elegans transcriptome assembly data for this species available from the 1000 Plants project (http://www.onekp.com/public_data.html; Leebens‐Mack et al., 2019), and from other resurrection species using the relevant published genomes (VanBuren et al., 2015, 2018; Xiao et al., 2018).
3′‐RACE PCR
3′‐RACE was used to confirm the unusual transcript structure of the identified XhABI3 genes. Four primers were designed, corresponding to the region between the B1 and B2 domains of each gene: (XhABI3A) 5′‐CGTGACCACCCAGCCGTTTT‐3′, (XhABI3B) 5′‐CCGTCATCCCCTGCTCCAAG‐3′, (XhABI3C) 5′‐TCCCTTTCCAACAACCCCGTTTC‐3′ and (XhABI3D) 5′‐AGCCACCGCATTTCCCAGAC‐3′. RACE PCR was run on cDNA pooled from both seed and leaf tissue, using the SMARTer 3′‐RACE Kit (Clontech) with standard controls and procedures except using only half reaction volumes. Only transcripts corresponding to XhABI3A and XhABI3B were amplified in these reactions, which were verified by Sanger sequencing.
Analysis of Xerophyta viscosa RNA‐Seq data
ABI3 orthologues in the X. viscosa genome were identified using BLAST and the XhABI3 sequences as queries. XvABI3 transcript architecture was plotted using the AnnotationSketch module in GenomeTools (Gremme et al., 2013). Gene expression was calculated by aligning the reads obtained from a leaf desiccation experiment (PRJNA295811; Costa, Ligterink et al., 2017) to the genome using STAR and the X. viscosa annotation file updated to include the XvABI3 gene coordinates. Read counts were determined using featureCounts (Liao et al., 2013), and DE was analysed using DESeq2 and the same parameters as for X. humilis.
Protoplast transactivation assays
The X. humilis RNA‐Seq assembly transcript for XhABFA was used to design PCR primers (Table S2) to amplify the full‐length XhABFA coding region from cDNA prepared from desiccating X. humilis leaves. The amplified PCR product was cloned into pENTR1A and recombined into the GATEWAY‐compatible expression vector pUC19‐35S‐Rfa‐35S‐sGFP (Li et al., 2012) generating pUC19‐35S‐ABFA‐35S‐sGFP.
The promoter regions for XhPER1, XhECP63, XhDSI‐1VOC and XhAHL23 were identified by aligning the transcripts for these genes, assembled from the RNA‐Seq data, to the partial genome assembly (Schlebusch, 2019; Figure S9). PCR primers (Table S2) were designed to amplify 297–347‐bp promoter regions from genomic DNA. The PCR products were cloned into the pENTR1A vector, before being recombined into the GATEWAY‐compatible Firefly luciferase vector pGWL7. All cloned promoters were sequenced in both directions to confirm their identity. PlantCare (Lescot et al., 2002) was used to identify ABRE elements in these promoters, while a search for the RY element (CATGCA; Guerriero et al., 2009) was done manually. Promoter images were drawn using AnnotationSketch, part of the GenomeTools package (Gremme et al., 2013).
Protoplasts were isolated from 4‐week‐old A. thaliana grown under short‐day conditions and transfected with 10 µg of effector (pUC19‐35S‐ABFA‐35S‐sGFP), 8 µg reporter plasmid (XhPER1‐LUC, XhECP63‐LUC, XhDSI1VOC‐LUC or XhAHL23‐LUC) DNA and 2 μg of the control plasmid (pBS‐35S‐Ala‐Rluc) according to the protocol developed by the Sheen Lab (Yoo et al., 2007). Firefly LUC activity 22 h post‐transfection was determined using the dual‐luciferase reporter assay (Promega) in a SpectraMax Paradigm microplate reader and normalised to the geometric mean of the GFP and Renilla luciferases signal measured in the same device. The resulting values were then scaled by dividing each value by the average of the normalised values across the whole experiment.
Validation of promoter regions
Splinkerette PCR (Devon et al., 1995) was used to independently check the integrity of the XhPER1 and XhDSI‐1VOC promoter regions relative to the assembled mRNA transcripts. Xerophyta humilis genomic DNA was digested with EcoRI and XbaI, and ligated to Splinkerette adaptors (Devon et al., 1995). Splk0 (5′‐TCGTACGAGAATCGCTGTCC‐3′) and Splk1 (5'‐ACTATCGTCGTCTCACCGCTCTC‐3') forward PCR primers were used in combination with reverse primers PER1‐gsp0 (5′‐ACCAATGATGTGCAGAGCTCGC‐3′) and PER1‐gsp1 (5'‐ACACAGCGCATTTGCTGGCC‐3') or DSI1‐gsp0 (5′‐ ACGACTCCGTCACGTCCTTCACATA G‐3′) and DSI1‐gsp1 (5′‐ ACGACTCCGTCACGTCCTTCACATA G‐3′) in nested PCR reactions, to amplify regions upstream of the coding regions for XhPER1 and XhDSI‐1VOC, respectively. Splinkerette PCR products were cloned into pGEM®‐T Easy Vector (Promega) and sequenced by Sanger sequencing and aligned to the X. humilis assembly (Figure S9).
Conflict of interests
The authors declare no conflict of interest.
Author contributions
NI, RAI and RL conceptualised the study and designed the experiments with input from SAS and SGH. RL performed the majority of the experimental work. JP and SGH performed the protoplast transfection experiments. JP performed the 3′‐RACE and MP the RT‐qPCR validation experiments. NI characterised the seed development series, and the promoters used in the protoplast experiments. SAS worked closely with RL on bioinformatics analysis. RL curated the data and made the datasets available to the public. RL, RAI and NI wrote the manuscript with input from all authors.
Data statement
All data referred to are included in the manuscript or supplementary materials of this manuscript.
Supporting information
Figure S1. Xerophyta humilis transcriptome assembly scores and taxonomic annotation.
Figure S2. Quantitative PCR analysis of mRNA levels of XhABFA, XhCAL and XhPER1 during desiccation of X. humilis.
Figure S3. Evolutionary relationship of LAFL genes across X. humilis and other species.
Figure S4. Conservation of ABI3 paralogues in X. viscosa.
Figure S5. Domain structure of ABI3 paralogues in angiosperms with duplicated ABI3.
{kind=link}
Figure S6. Average motif distance from the TSS in A. thaliana and X. viscosa ABI3 regulon genes.
Figure S7. Expression of XhABFA and putative target genes.
Figure S8. Expression of several X. humilis seed genes.
Figure S9. Summary of promoter regions used in protoplast experiments.
Table S1. Primers for quantitative PCR validation of RNA‐Seq data.
Table S2. PCR primers to amplify the XhABFA coding region, or promoter regions for XhPER1, XhECP63, XhDSI‐1VOC or XhAHL23 for cloning into vectors for plant protoplast experiments.
Data S1. Expression data for genes analysed in Figure 4.
Data S2. ABI3 3’‐RACE sequences.
Acknowledgements
The authors thank Prof. Nicola Mulder and Gerrit Botha (UCT) for giving them access to their computational resources and for technical assistance, Evan D. Milborrow (UCT) for collecting the X. humilis samples used for preparation of the leaf RNA‐Seq libraries, and Prof. Vincent Chiang (North Carolina State University) for kindly providing pUC19‐35S‐Rfa‐35S‐sGFP. Wendy Stone, Faezah Davids, Zanele Lwana, Tatenda Dube, Emang Molojwane assisted with the Splinkerette experiments. This study was funded by the University of Cape Town, and the National Research Foundation via Incentive Funding (UID 103423) to NI, and a National Bioinformatics Functional Genomics grant (UID 93697) to RI.
Data Availability Statement
Raw sequence reads have been submitted to the SRA at NCBI BioProject ID PRJNA505754. Supplementary data including transcriptome sequences, representative transcript and protein sequences, gene functional annotations and gene expression values can be found at https://doi.org/10.6084/m9.figshare.c.4691234
References
- Aalen, R.B. (1999) Peroxiredoxin antioxidants in seed physiology. Seed Sci. Res. 9, 285–295. [Google Scholar]
- Alcantara, S. , Ree, R.H. and Mello‐Silva, R. (2018) Accelerated diversification and functional trait evolution in Velloziaceae reveal new insights into the origins of the campos rupestres’ exceptional floristic richness. Ann. Bot. 122, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa, A. and Rahnenfuhrer, J. (2010) topGO: enrichment analysis for gene ontology. R Packag. version, 2. [Google Scholar]
- Alpert, P. (2005) The limits and frontiers of desiccation‐tolerant life. Integr. Comp. Biol. 45, 685–695. [DOI] [PubMed] [Google Scholar]
- Angelovici, R. , Galili, G. , Fernie, A.R. and Fait, A. (2010) Seed desiccation: a bridge between maturation and germination. Trends Plant Sci. 15, 211–218. [DOI] [PubMed] [Google Scholar]
- Bailey, T.L. (2011) DREME: motif discovery in transcription factor ChIP‐seq data. Bioinformatics, 27, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke, H.D. , Hummel, E. , Hillmer, S. , Sauer‐Gürth, H. , Gonzalez, J. and Wink, M. (2013) A revision of African Velloziaceae based on leaf anatomy characters and rbcL nucleotide sequences. Bot. J. Linn. Soc. 172, 22–94. [Google Scholar]
- Bewley, J.D. and Black, M.J. (1994) Seeds: Physiology of Development and Germination. New York, NY: Plenum Press. [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer, D. , Roudier, F. , Heese, M. et al (2011) Polycomb repressive complex 2 controls the embryo‐to‐seedling phase transition. PLoS Genet. 7, e1002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggink, T. and van der Toorn, P. (1995) Induction of desiccation tolerance in germinated seeds. Seed Sci. Res. 5, 1–4. [Google Scholar]
- Bruggink, T. and van der Toorn, P. (1997) Induction of desiccation tolerance in germinated impatiens seeds enables their practical use In Basic and Applied Aspects of Seed Biology (Ellis R.H., Black M., Murdoch A.J. and Hong T.D., eds.). Dordrecht: Springer, 30, pp. 461–467. [Google Scholar]
- Buitink, J. , Leger, J.J. , Guisle, I. et al (2006) Transcriptome profiling uncovers metabolic and regulatory processes occurring during the transition from desiccation‐sensitive to desiccation‐tolerant stages in Medicago truncatula seeds. Plant J. 47, 735–750. [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. and Madden, T.L. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonero, P. , Iglesias‐Fernandez, R. and Vicente‐Carbajosa, J. (2016) The AFL subfamily of B3 transcription factors: evolution and function in angiosperm seeds. J. Exp. Bot. 68, 871–880. [DOI] [PubMed] [Google Scholar]
- Conesa, A. , Götz, S. , García‐Gómez, J.M. , Terol, J. , Talón, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Costa, M.‐C.D. , Cooper, K. , Hilhorst, H.W.M. and Farrant, J.M. (2017) Orthodox seeds and resurrection plants: two of a kind? Plant Physiol. 175, 00760.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, M.‐C.D. , Ligterink, W. , Derks, M.F.L. et al (2017) A footprint of desiccation tolerance in the genome of Xerophyta viscosa . Nat. Plants, 3, 17 038. [DOI] [PubMed] [Google Scholar]
- Dam, S. , Laursen, B.S. , Ørnfelt, J.H. et al (2009) The proteome of seed development in the model legume Lotus japonicus . Plant Physiol. 149, 1325–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers, B.J.W. , Costa, M.C.D. , Maia, J. , Bentsink, L. , Ligterink, W. and Hilhorst, H.W.M. (2015) Acquisition and loss of desiccation tolerance in seeds: from experimental model to biological relevance. Planta, 241, 563–77. [DOI] [PubMed] [Google Scholar]
- Devon, R.S. , Porteous, D.J. and Brookes, A.J. (1995) Splinkerettes–improved vectorettes for greater efficiency in PCR walking. Nucleic Acids Res. 23, 1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinakar, C. and Bartels, D. (2013) Desiccation tolerance in resurrection plants: new insights from transcriptome, proteome and metabolome analysis. Front. Plant Sci. 4, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge‐Laser, W. , Snoek, B.L. , Snel, B. and Weiste, C. (2018) The Arabidopsis bZIP transcription factor family—an update. Curr. Opin. Plant Biol. 45, 36–49. [DOI] [PubMed] [Google Scholar]
- Ezcurra, I. , Wycliffe, P. , Nehlin, L. , Ellerstrom, M. and Rask, L. (2000) Transactivation of the Brassica napus napin promoter by ABI3 requires interaction of the conserved B2 and B3 domains of ABI3 with different cis‐elements: B2 mediates activation through an ABRE, whereas B3 interacts with an RY/G‐box. Plant J. 24, 57–66. [DOI] [PubMed] [Google Scholar]
- Fan, J. , Niu, X. , Wang, Y. , Ren, G. , Zhuo, T. , Yang, Y. , Lu, B.‐R. and Liu, Y. (2007) Short, direct repeats (SDRs)‐mediated post‐transcriptional processing of a transcription factor gene OsVP1 in rice (Oryza sativa). J. Exp. Bot. 58, 3811–3817. [DOI] [PubMed] [Google Scholar]
- Farrant, J.M. , Bailly, C. , Leymarie, J. , Hamman, B. , Côme, D. and Corbineau, F. (2004) Wheat seedlings as a model to understand desiccation tolerance and sensitivity. Physiol. Plant. 120, 563–574. [DOI] [PubMed] [Google Scholar]
- Footitt, S. , Ingouff, M. , Clapham, D. and von Arnold, S. (2003) Expression of the viviparous 1 (Pavp1) and p34cdc2 protein kinase (cdc2Pa) genes during somatic embryogenesis in Norway spruce (Picea abies [L.] Karst). J. Exp. Bot. 54, 1711–1719. [DOI] [PubMed] [Google Scholar]
- Gaff, D.F. and Oliver, M. (2013) The evolution of desiccation tolerance in angiosperm plants: a rare yet common phenomenon. Funct. Plant Biol. 40, 315–328. [DOI] [PubMed] [Google Scholar]
- Gagete, A.P. , Riera, M. , Franco, L. and Rodrigo, M.I. (2009) Functional analysis of the isoforms of an ABI3‐like factor of Pisum sativum generated by alternative splicing. J. Exp. Bot. 60, 1703–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, R. , Singh, V.K. , Rajkumar, M.S. , Kumar, V. and Jain, M. (2017) Global transcriptome and coexpression network analyses reveal cultivar‐specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 91, 1088–1107. [DOI] [PubMed] [Google Scholar]
- Gechev, T.S. , Benina, M. , Obata, T. et al (2013) Molecular mechanisms of desiccation tolerance in the resurrection glacial relic Haberlea rhodopensis . Cell. Mol. Life Sci. 70, 689–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudat, J. , Hauge, B.M. , Valon, C. , Smalle, J. , Parcy, F. and Goodman, H.M. (1992) Isolation of the Arabidopsis ABI3 gene by positional cloning. Plant Cell, 4, 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldblatt, P. and Poston, M.E. (1988) Observations on the chromosome cytology of Velloziaceae. Ann. Missouri Bot. Gard. 192–195. [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. et al (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremme, G. , Steinbiss, S. and Kurtz, S. (2013) GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinforma. 10, 645–656. [DOI] [PubMed] [Google Scholar]
- Guerriero, G. , Martin, N. , Golovko, A. , Sundström, J.F. , Rask, L. and Ezcurra, I. (2009) The RY/Sph element mediates transcriptional repression of maturation genes from late maturation to early seedling growth. New Phytol. 184, 552–565. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Papanicolaou, A. , Yassour, M. et al. (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslekås, C. , Grini, P.E. , Nordgard, S.H. , Thorstensen, T. , Viken, M.K. , Nygaard, V. and Aalen, R.B. (2003) ABI3 mediates expression of the peroxiredoxin antioxidant AtPER1 gene and induction by oxidative stress. Plant Mol. Biol. 53, 313–326. [DOI] [PubMed] [Google Scholar]
- Hattori, T. , Terada, T. and Hamasuna, S.T. (1994) Sequence and functional analyses of the rice gene homologous to the maize Vp1. Plant Mol. Biol. 24, 805–810. [DOI] [PubMed] [Google Scholar]
- Illing, N. , Denby, K.J. , Collett, H. , Shen, A. and Farrant, J.M. (2005) The signature of seeds in resurrection plants: a molecular and physiological comparison of desiccation tolerance in seeds and vegetative tissues. Integr. Comp. Biol. 45, 771–787. [DOI] [PubMed] [Google Scholar]
- Ingle, R.A. , Collett, H. , Cooper, K. , Takahashi, Y. , Farrant, J.M. and Illing, N. (2008) Chloroplast biogenesis during rehydration of the resurrection plant Xerophyta humilis: parallels to the etioplast‐chloroplast transition. Plant. Cell Environ. 31, 1813–1824. [DOI] [PubMed] [Google Scholar]
- Khandelwal, A. , Cho, S.H. , Marella, H. , Sakata, Y. , Perroud, P.‐F. , Pan, A. and Quatrano, R.S. (2010) Role of ABA and ABI3 in desiccation tolerance. Science, 327, 546. [DOI] [PubMed] [Google Scholar]
- Kim, S.Y. (2006) The role of ABF family bZIP class transcription factors in stress response. Physiol. Plant. 126, 519–527. [Google Scholar]
- Komatsu, K. , Suzuki, N. , Kuwamura, M. et al (2013) Group A PP2Cs evolved in land plants as key regulators of intrinsic desiccation tolerance. Nat. Commun. 4, 2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva, E.V. , Kuznetsov, D. , Tegenfeldt, F. , Manni, M. , Dias, R. , Simão, F.A. and Zdobnov, E.M. (2018) OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, B.H. , Cheng, C. , Bui, A.Q. et al (2010) Global analysis of gene activity during Arabidopsis seed development and identification of seed‐specific transcription factors. Proc. Natl Acad. Sci. USA, 107, 8063–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, H.‐S. , Schulz, M.H. , McCauley, B.M. , Hinman, V.F. and Bar‐Joseph, Z. (2013) Probabilistic error correction for RNA sequencing. Nucleic Acids Res. 41, e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens-Mack, J.H. , Barker, M.S. , Carpenter, E.J. et al (2019) One thousand plant transcriptomes and the phylogenomics of green plants. Nature, 574, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leprince, O. , Pellizzaro, A. , Berriri, S. and Buitink, J. (2017) Late seed maturation: drying without dying. J. Exp. Bot. 68, 827–841. [DOI] [PubMed] [Google Scholar]
- Lescot, M. , Déhais, P. , Thijs, G. , Marchal, K. , Moreau, Y. , de Peer, Y. , Rouzé, P. and Rombauts, S. (2002) PlantCARE, a database of plant cis‐acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Lin, Y.‐C. , Sun, Y.‐H. , Song, J. , Chen, H. , Zhang, X.‐H. , Sederoff, R.R. and Chiang, V.L. (2012) Splice variant of the SND1 transcription factor is a dominant negative of SND1 members and their regulation in Populus trichocarpa . Proc. Natl Acad. Sci. USA, 109, 14 699–14 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G.K. and Shi, W. (2013) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lopez‐Molina, L. , Mongrand, S. and Chua, N.H. (2001) A postgermination developmental arrest checkpoint is mediated by abscisic acid and requires the ABI5 transcription factor in Arabidopsis. Proc. Natl Acad. Sci. USA, 98, 4782–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Anders, S. and Huber, W. (2017) Analyzing RNA‐seq data with DESeq2. R Packag. Ref. Man.
- Lyall, R. , Ingle, R.A. and Illing, N. (2014) The window of desiccation tolerance shown by early‐stage germinating seedlings remains open in the resurrection plant, Xerophyta viscosa. PLoS One, 9, e93093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia, J. (2014) Unravelling desiccation tolerance in germinated Arabidopsis seeds. Wageningen University; https://edepot.wur.nl/302759 [Google Scholar]
- Maia, J. , Dekkers, B.J.W. , Provart, N.J. , Ligterink, W. and Hihorst, H.W.M. (2011) The re‐establishment of desiccation tolerance in germinated Arabidopsis thaliana seeds and its associated transcriptome. PLoS ONE, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella, H.H. , Sakata, Y. and Quatrano, R.S. (2006) Characterization and functional analysis of ABSCISIC ACID INSENSITIVE3‐like genes from Physcomitrella patens . Plant J. 46, 1032–1044. [DOI] [PubMed] [Google Scholar]
- McCarty, D.R. , Hattori, T. , Carson, C.B. , Vasil, V. , Lazar, M. and Vasil, I.K. (1991) The Viviparous‐1 developmental gene of maize encodes a novel transcriptional activator. Cell, 66, 895–905. [DOI] [PubMed] [Google Scholar]
- McKibbin, R.S. , Wilkinson, M.D. , Bailey, P.C. , Flintham, J.E. , Andrew, L.M. , Lazzeri, P.A. , Gale, M.D. , Lenton, J.R. and Holdsworth, M.J. (2002) Transcripts of Vp‐1 homeologues are misspliced in modern wheat and ancestral species. Proc. Natl. Acad. Sci. USA, 99, 10 203–10 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mönke, G. , Seifert, M. , Keilwagen, J. et al (2012) Toward the identification and regulation of the Arabidopsis thaliana ABI3 regulon. Nucleic Acids Res. 40, 8240–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulako, I. , Farrant, J.M. , Collett, H. and Illing, N. (2008) Expression of Xhdsi‐1VOC, a novel member of the vicinal oxygen chelate (VOC) metalloenzyme superfamily, is up‐regulated in leaves and roots during desiccation in the resurrection plant Xerophyta humilis (Bak) Dur and Schinz. J. Exp. Bot. 59, 3885–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, S. , Lynch, T.J. and Finkelstein, R.R. (2001) Physical interactions between ABA response loci of Arabidopsis. Plant J. 26, 627–635. [DOI] [PubMed] [Google Scholar]
- Nambara, E. , McCourt, P. and Naito, S. (1995) A regulatory role for the ABI3 gene in the establishment of embryo maturation in Arabidopsis thaliana . Development, 121, 629–636. [Google Scholar]
- North, H. , Baud, S. , Debeaujon, I. et al (2010) Arabidopsis seed secrets unravelled after a decade of genetic and omics‐driven research. Plant J. 61, 971–981. [DOI] [PubMed] [Google Scholar]
- Obayashi, T. , Kinoshita, K. , Nakai, K. , Shibaoka, M. , Hayashi, S. , Saeki, M. , Shibata, D. , Saito, K. and Ohta, H. (2006) ATTED‐II: a database of co‐expressed genes and cis elements for identifying co‐regulated gene groups in Arabidopsis. Nucleic Acids Res. 35, D863–D869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, T. , Sasaki, Y. , Ohta, R. , Onozuka, N. and Toriyama, K. (2000) Expression of Bra r 1 gene in transgenic tobacco and Bra r 1 promoter activity in pollen of various plant species. Plant Cell Physiol. 41, 757–766. [DOI] [PubMed] [Google Scholar]
- Oliver, M. , Tuba, Z. and Mishler, B. (2000) The evolution of vegetative desiccation tolerance in land plants. Plant Ecol. 85–100. [Google Scholar]
- Oliver, M.J. , Velten, J. and Mishler, B.D. (2005) Desiccation tolerance in bryophytes: a reflection of the primitive strategy for plant survival in dehydrating habitats? Integr. Comp. Biol. 45, 788–799. [DOI] [PubMed] [Google Scholar]
- Patro, R. , Duggal, G. , Love, M.I. , Irizarry, R.A. and Kingsford, C. (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nat. Methods, 14, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakić, T. , Lazarević, M. , Jovanović, Z.S. , Radović, S. , Siljak‐Yakovlev, S. , Stevanović, B. and Stevanović, V. (2014) Resurrection plants of the genus Ramonda: prospective survival strategies ‐ unlock further capacity of adaptation, or embark on the path of evolution? Front. Plant Sci. 4, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidt, W. , Wohlfarth, T. , Ellerstrom, M. , Czihal, A. , Tewes, A. , Ezcurra, I. , Rask, L. and Baumlein, H. (2000) Gene regulation during late embryogenesis: the RY motif of maturation‐specific gene promoters is a direct target of the FUS3 gene product. Plant J. 21, 401–408. [DOI] [PubMed] [Google Scholar]
- Rodriguez, M.C.S. , Edsgärd, D. , Hussain, S.S. , Alquezar, D. , Rasmussen, M. , Gilbert, T. , Nielsen, B.H. , Bartels, D. and Mundy, J. (2010) Transcriptomes of the desiccation‐tolerant resurrection plant Craterostigma plantagineum . Plant J. 63, 212–228. [DOI] [PubMed] [Google Scholar]
- Santos‐Mendoza, M. , Dubreucq, B. , Baud, S. , Parcy, F. , Caboche, M. and Lepiniec, L. (2008) Deciphering gene regulatory networks that control seed development and maturation in Arabidopsis. Plant J. 54, 608–620. [DOI] [PubMed] [Google Scholar]
- Sasnauskas, G. , Manakova, E. , Lapenas, K. , Kauneckaite, K. and Siksnys, V. (2018) DNA recognition by Arabidopsis transcription factors ABI 3 and NGA 1. FEBS J. 285, 4041–4059. [DOI] [PubMed] [Google Scholar]
- Schlebusch, S.A. (2019) Events that shape genomes. University of Cape Town; Available at: https://open.uct.ac.za/handle/11427/29115. [Google Scholar]
- Shiota, H. , Satoh, R. , Watabe, K. , Harada, H. and Kamada, H. (1998) C‐ABI3, the carrot homologue of the Arabidopsis ABI3, is expressed during both zygotic and somatic embryogenesis and functions in the regulation of embryo‐specific ABA‐inducible genes. Plant Cell Physiol. 39, 1184–1193. [DOI] [PubMed] [Google Scholar]
- Sievers, F. and Higgins, D.G. (2014) Clustal Omega, accurate alignment of very large numbers of sequences In Multiple Sequence Alignment Methods. Methods in Molecular Biology (Methods and Protocols) (Russell D., ed.). Totowa, NJ: Humana Press, 1079, pp. 105–116. [DOI] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smith‐Unna, R. , Boursnell, C. , Patro, R. , Hibberd, J.M. and Kelly, S. (2016) TransRate: reference‐free quality assessment of de novo transcriptome assemblies. Genome Res. 26, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson, C. , Love, M.I. and Robinson, M.D. (2015) Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Research, 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, A. , Sarkar, H. , Malik, L. and Patro, R. (2016) Accurate, fast and lightweight clustering of de novo transcriptomes using fragment equivalence classes. arXiv Prepr. arXiv1604.03250.
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugliani, M. , Brambilla, V. , Clerkx, E.J.M. , Koornneef, M. and Soppe, W.J.J. (2010) The conserved splicing factor SUA controls alternative splicing of the developmental regulator ABI3 in Arabidopsis. Plant Cell, 221, 1936–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M. , Kao, C.Y. and McCarty, D.R. (1997) The conserved B3 domain of VIVIPAROUS1 has a cooperative DNA binding activity. Plant Cell, 9, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M. , Kao, C.‐Y. , Cocciolone, S. and McCarty, D.R. (2001) Maize VP1 complements Arabidopsis abi3 and confers a novel ABA/auxin interaction in roots. Plant J. 28, 409–418. [DOI] [PubMed] [Google Scholar]
- Suzuki, M. , Wu, S. , Li, Q. and McCarty, D.R. (2014) Distinct functions of COAR and B3 domains of maize VP1 in induction of ectopic gene expression and plant developmental phenotypes in Arabidopsis. Plant Mol. Biol. 85, 179–191. [DOI] [PubMed] [Google Scholar]
- Terrasson, E. , Buitink, J. , Righetti, K. et al (2013) An emerging picture of the seed desiccome: confirmed regulators and newcomers identified using transcriptome comparison. Front. Plant Sci. 4, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buren, R. (2017) Desiccation tolerance: seedy origins of resurrection. Nat. Plants, 3, 17 046. [DOI] [PubMed] [Google Scholar]
- Van Buren, R. , Bryant, D. , Edger, P.P. et al (2015) Single‐molecule sequencing of the desiccation‐tolerant grass Oropetium thomaeum . Nature, 527, 508–511. [DOI] [PubMed] [Google Scholar]
- Van Buren, R. , Wai, C.M. , Pardo, J. , Giarola, V. , Ambrosini, S. , Song, X. and Bartels, D. (2018) Desiccation tolerance evolved through gene duplication and network rewiring in Lindernia. Plant Cell, 30, 2943–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente‐Carbajosa, J. and Carbonero, P. (2005) Seed maturation: developing an intrusive phase to accomplish a quiescent state. Int. J. Dev. Biol. 49, 645–651. [DOI] [PubMed] [Google Scholar]
- Wang, Guifeng , Wang, Gang , Zhang, X. , Wang, F. and Song, R. (2012) Isolation of high quality RNA from cereal seeds containing high levels of starch. Phytochem. Anal. 23, 159–163. [DOI] [PubMed] [Google Scholar]
- Wen, W. , Li, K. , Alseekh, S. et al (2015) Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. Plant Cell, 27, 1839–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, L. , Yang, G. , Zhang, L. et al (2015) The resurrection genome of Boea hygrometrica: a blueprint for survival of dehydration. Proc. Natl. Acad. Sci. USA, 112, 5833–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Saitou, T. , Komeda, Y. , Harada, H. and Kamada, H. (1997) Arabidopsis thaliana ECP63 encoding a LEA protein is located in chromosome 4. Gene, 184, 82–88. [PubMed] [Google Scholar]
- Yobi, A. , Schlauch, K.A. , Tillett, R.L. , Yim, W.C. , Espinoza, C. , Wone, B.W.M. , Cushman, J.C. and Oliver, M.J. (2017) Sporobolus stapfianus: insights into desiccation tolerance in the resurrection grasses from linking transcriptomics to metabolomics. BMC Plant Biol. 17, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, S.‐D. , Cho, Y.‐H. and Sheen, J. (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565. [DOI] [PubMed] [Google Scholar]
- Yoshida, T. , Fujita, Y. , Sayama, H. , Kidokoro, S. , Maruyama, K. , Mizoi, J. , Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2010) AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE‐dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J. 61, 672–85. [DOI] [PubMed] [Google Scholar]
- Yoshida, T. , Fujita, Y. , Maruyama, K. , Mogami, J. , Todaka, D. , Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2015) Four Arabidopsis AREB/ABF transcription factors function predominantly in gene expression downstream of SnRK2 kinases in abscisic acid signalling in response to osmotic stress. Plant. Cell Environ. 38, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov, E.M. and Apweiler, R. (2001) InterProScan–an integration platform for the signature‐recognition methods in InterPro. Bioinformatics, 17, 847–848. [DOI] [PubMed] [Google Scholar]
- Zhang, Q. and Bartels, D. (2018) Molecular responses to dehydration and desiccation in desiccation‐tolerant angiosperm plants. J. Exp. Bot. 69, 3211–3222. [DOI] [PubMed] [Google Scholar]
- Zheng, Y. , Jiao, C. , Sun, H. et al (2016) iTAK: a program for genome‐wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant, 9, 1667–1670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Xerophyta humilis transcriptome assembly scores and taxonomic annotation.
Figure S2. Quantitative PCR analysis of mRNA levels of XhABFA, XhCAL and XhPER1 during desiccation of X. humilis.
Figure S3. Evolutionary relationship of LAFL genes across X. humilis and other species.
Figure S4. Conservation of ABI3 paralogues in X. viscosa.
Figure S5. Domain structure of ABI3 paralogues in angiosperms with duplicated ABI3.
Figure S6. Average motif distance from the TSS in A. thaliana and X. viscosa ABI3 regulon genes.
Figure S7. Expression of XhABFA and putative target genes.
Figure S8. Expression of several X. humilis seed genes.
Figure S9. Summary of promoter regions used in protoplast experiments.
Table S1. Primers for quantitative PCR validation of RNA‐Seq data.
Table S2. PCR primers to amplify the XhABFA coding region, or promoter regions for XhPER1, XhECP63, XhDSI‐1VOC or XhAHL23 for cloning into vectors for plant protoplast experiments.
Data S1. Expression data for genes analysed in Figure 4.
Data S2. ABI3 3’‐RACE sequences.
Data Availability Statement
Raw sequence reads have been submitted to the SRA at NCBI BioProject ID PRJNA505754. Supplementary data including transcriptome sequences, representative transcript and protein sequences, gene functional annotations and gene expression values can be found at https://doi.org/10.6084/m9.figshare.c.4691234