

Abstract
Aim
Patients with primary Sjögren's syndrome (pSS) have an increased risk of developing diffuse large B‐cell lymphoma (DLBCL), which is an aggressive and heterogeneous non‐Hodgkin lymphoma. This study aimed to characterize DLBCLs in patients with pSS.
Method
We identified 18 patients with DLBCL and pSS over a 22‐year period. Based on the 2016 WHO guidelines, we characterized DLBCL based on immunohistochemical tests using a broad panel of antibodies, and an Epstein‐Barr virus (EBV) test using in situ hybridization.
Results
The median time from initial pSS symptom onset to the DLBCL diagnosis was 20.5 years and the median time from the pSS diagnosis until the DLBCL diagnosis was 14 years. After the lymphoma diagnosis, the median overall survival was 3 months (range: 0‐212 months) and the 5‐year overall survival rate was 37.5%. Thirteen DLBCLs were re‐classified as DLBCL, not otherwise specified (NOS) in nine cases; EBV‐positive DLBCL, NOS in two cases; and T‐cell/histiocyte‐rich large B‐cell lymphoma in two cases. Five cases of DLBCLs were not re‐classified because their EBV status was unknown. The Hans algorithm, which uses a combination of staining for CD10, BCL6, and MUM1, was used to classify the DLBCLs into the germinal center B‐cell (GCB) subtype for three cases and the non‐GCB subtype for nine cases.
Conclusion
These results indicate that DLBCL tends to occur late in pSS cases and is mainly related to the non‐GCB subtype of DLBCL.
Keywords: clinicopathological findings, diffuse large B‐cell lymphoma, primary Sjögren's syndrome, prognostication, subtyping
1. INTRODUCTION
Sjögren's syndrome (SS) is a chronic autoimmune disease which damages the exocrine glands, notably the salivary and lacrimal glands. It is histologically characterized by lymphocytic infiltration and destruction of the exocrine glands and can occur alone (primary SS; pSS) or in association with other autoimmune rheumatic diseases. Recent research has suggested that SS may be associated with the development of diffuse large B‐cell lymphoma (DLBCL), with the relative risk of DLBCL development in patients with pSS ranging from 2.0 to 6.57.1, 2 The broad variability in this risk can be attributed to differences in the SS diagnosis criteria and follow‐up duration. Nevertheless, DLBCL is an aggressive and heterogeneous lymphoid tumor which affects the quality of life and prognosis of patients with pSS. Morphological, immunophenotypic, molecular, and clinical studies have subdivided DLBCL into morphological variants, molecular subtypes, and distinct disease entities that have different prognoses and treatment choices.3 Cases involving DLBCL that do not fulfill the criteria for the specific disease entities are referred to as DLBCL, not otherwise specified (NOS). However, there are few studies that have focused on characterizing DLBCL which occurs in patients with underlying pSS, and the present study aimed to describe the characteristics of DLBCL in a group of consecutive patients with confirmed diagnoses of pSS.
2. PATIENTS AND METHODS
Between June 1997 and June 2019, this retrospective study identified 2021 adult patients (>18 years old) who were diagnosed with SS at the VA Nasonova Research Institute of Rheumatology (Moscow, Russia). These patients included 25 with DLBCL. We excluded seven patients because they had SS with concomitant rheumatoid arthritis (RA), systemic lupus erythematosus, or systemic scleroderma. The medical records for the remaining 18 cases were reviewed to obtain information regarding age, gender, clinical presentation, laboratory findings, the time from the manifestation of SS to the diagnosis of DLBCL, relevant imaging studies, lymphoma treatment, follow‐up interval, and outcome.
All patients underwent diagnostic testing for xerostomia (unstimulated whole saliva flow), xerophthalmia (Schirmer's test, tear break‐up time, and Rose Bengal staining), and lip minor salivary gland biopsy. Immunologic tests that included antinuclear antibodies were performed using indirect immunofluorescence and tissue cryostat sections of the liver. Rheumatoid factor was detected via nephelometry. Precipitating antibodies against the extractable nuclear antigens Ro/SS‐A and La/SS‐B were detected via enzyme‐linked immunosorbent assay. All subjects included in this study fulfilled the consensus criteria for pSS.4
The DLBCLs were identified via surgical biopsy of lymph nodes or extranodal tissues. The tissue specimens were fixed in 10% formalin, routinely processed, and embedded in paraffin. The original hematoxylin‐eosin‐stained slides were used in all cases. Immunohistochemical (IHC) tests were performed using the formalin‐fixed paraffin‐embedded tissues. The broad antibody panel involved the following antibodies used at the manufacturer‐recommended dilutions: anti‐CD2 (clone MRQ‐11, Cell Marque), CD3 (clone F7.2.38, Dako), CD5 (clone 4C7, Dako), CD7 (clone MRQ‐56, Cell Marque), CD15 (clone C3D1, Dako), CD20 (clone L26, Dako), CD30 (clone Ber‐H2, Dako), CD43 (clone MT1, Cell Marque), cyclin D1 (clone SP4, Cell Marque), BCL2 (clone 124, Cell Marque), BCL6 (clone EP278, Cell Marque), MUM1 (clone MRO‐8, Cell Marque), PAX5 (clone DAK‐Pax5, Dako), HGAL (clone MRQ‐49, Cell Marque), c‐MYC (clone EP121, Cell Marque), and Ki‐67 (clone SP6, Cell Marque). After dewaxing and heat‐induced antigen retrieval, the sections were stained by using an Autostainer Link 48 (Dako) according to the manufacturer's instructions. The expressions of HGAL, CD10, BCL6, and MUM1 were considered positive if ≥30% of the tumor cells exhibited positive staining.5 The expression of BCL2 was considered positive if ≥50% of the tumor cells exhibited positive staining.3 The expression of MYC was considered positive if ≥40% of the tumor cell nuclei exhibited positive staining.3
In situ hybridization was performed on formalin‐fixed paraffin sections to detect Epstein‐Barr virus (EBV) in lymphoma tissue in 13 cases; five cases were not evaluated due to the lack of or poor quality of samples. To detect EBV‐encoded small nuclear ribonucleic acid (EBER), a mixture of fluorescein‐conjugated oligodeoxyribonucleotides complementary to the two EBERs (EBER; Dako) was used.
In cases 1, 13, and 15, a c‐MYC/8q24 gene locus translocation test was performed on paraffin sections using fluorescence in situ hybridization (FISH) with the LSI c‐MYC Break Apart Probe (Abbott) and the LSI c‐MYC/IgH Dual Fusion Translocation Probe.
All patients were staged according to the Ann Arbor staging system and stratified according to the original International Prognostic Index (IPI).6 The DLBCLs were also re‐classified according to the 2016 WHO classification of tumors of hematopoietic and lymphoid tissues.3 According to the revised response criteria for malignant lymphomas, overall survival (OS) was defined as the time from the diagnosis until death because of any cause.7
2.1. Statistical analysis
Categorical variables are reported as number (%) and continuous variables are reported as median (range). OS was estimated using the Kaplan‐Meier method (time from the lymphoma diagnosis until the date of death or the last follow‐up) and was compared using the log‐rank test. All research was performed in accordance with the relevant guidelines.
3. RESULTS
Between June 1997 and June 2019, we identified 18 eligible patients with pSS‐related DLBCL. The patients’ clinical characteristics, immunological data, and lymphoma risk factors are shown in Table 1. All 18 patients were women, and the median age at the onset of the initial pSS symptoms was 34 years (range: 20‐61 years). The median time from the onset of the initial pSS symptoms to the DLBCL diagnosis was 20.5 years (range: 6‐38 years), and the median time from the pSS diagnosis to the DLBCL diagnosis was 14 years (range: 1‐27 years). The median age at the DLBCL diagnosis was 55 years (range: 39‐83 years). A monoclonal component in serum and/or urine was detected in 47% of the tested patients (8/17), based on trace secretion. Serum specimens from three cases contained paraprotein immunoglobulin M (IgM) kappa. Urine specimens from four patients contained Bens‐Jones protein, kappa type. One patient had a combination of paraprotein IgM kappa in the serum specimen and Bens‐Jones protein, kappa type in the urine specimen. Cryoglobulinemia was observed in 82% of the tested cases (14/17), and a decreased level of complement C4 was observed in 43% of the tested patients (3/7).
Table 1.
Baseline characteristics, immunological data, and lymphoma risk factors of the 18 patients with primary Sjögren's syndrome and diffuse large B‐cell lymphoma
| Case no. | Gender | Age (y) at initial SS symptoms | Anti‐SSA/Ro | Anti‐SSB/La | RF | ANA | Cryoglobulinemia | Lymphopeniaa | Low C4 complement | Monoclonal component |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 20 | − | − | + | + | − | − | − | − |
| 2 | F | 30 | NA | NA | + | + | + | + | NA | − |
| 3 | F | 35 | NA | NA | + | + | − | − | NA | − |
| 4 | F | 20 | + | − | + | + | + | − | NA | − |
| 5 | F | 61 | NA | NA | + | + | + | + | NA | − |
| 6 | F | 46 | + | + | + | + | + | − | − | − |
| 7 | F | 21 | NA | NA | + | + | + | – | NA | + (BJκ) |
| 8 | F | 29 | NA | NA | + | + | + | + | NA | − |
| 9 | F | 33 | + | + | − | + | NA | NA | NA | NA |
| 10 | F | 47 | NA | NA | + | + | + | NA | NA | + (IgMκ) |
| 11 | F | 23 | NA | NA | + | + | + | NA | NA | + (BJκ) |
| 12 | F | 52 | NA | NA | + | + | + | − | NA | + (BJκ) |
| 13 | F | 55 | + | − | + | + | + | – | − | − |
| 14 | F | 49 | − | − | + | + | − | + | − | + (BJκ) |
| 15 | F | 40 | + | − | + | + | + | + | + | + (IgMκ + BJκ) |
| 16 | F | 21 | + | − | + | + | + | + | + | + (IgMκ) |
| 17 | F | 21 | + | + | + | + | + | NA | NA | − |
| 18 | F | 43 | − | − | + | + | + | NA | + | + (IgMκ) |
| Total | 100% | Median age: 34 y (range: 20‐61) | 70% (7/10) | 30% (3/10) | 94% (17/18) | 100% (18/18) | 82% (14/17) | 46% (6/13) | 43% (3/7) | 47% (8/17) |
Abbreviations: −, negative; +, positive; ANA, antinuclear antibodies; BJ, Bence Jones protein; Ig, immunoglobulin; NA, not available; RF, rheumatoid factor.
Lymphocyte count < 1000/μL at pSS diagnosis.
The pathological findings of DLBCLs are shown in Table 2. Morphological evaluations revealed a centroblastic variant of DLBCL in cases 2, 4‐12, and 15, while an anaplastic variant of DLBCL was observed in case 3. Cases 16‐18 had a limited number of scattered large B cells (CD20+, CD15−, CD30−) embedded in a background of abundant histiocytes and CD3 + and CD5 + T cells. Case 16 was initially misinterpreted as having T‐cell/histiocyte‐rich large B‐cell lymphoma (THRLBCL), but detection of EBV in large cells allowed us to regard this case as a polymorphic variant of EBV‐positive DLBCL. The histology and immunophenotype in cases 17 and 18 were consistent with THRLBCL. Cases 1, 13, and 14 had predominantly medium‐sized tumor cells. The lack of c‐MYC/8q24 rearrangements, as well as cyclin D1 and MYC expression in the tumor cells allowed for a diagnosis of DLBCL. The MYC protein was detected only in case 15. A FISH study of tumor tissue in this case revealed additional signal at the c‐MYC/8q24 gene locus and the lack of c‐MYC/8q24 rearrangements. The detection of EBV in tumor cells allowed us to classify this case as EBV‐positive DLBCL, NOS.
Table 2.
Pathological findings from 18 DLBCL cases
| Case no. | CD20 | HGAL (>30%) | CD10 (>30%) | BCL6 (>30%) | MuM1 (>30%) | BCL2 (>50%) | MYC (>40%) | EBV | Subtype according to Hans algorithm | DLBCL type |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | + | + | − | − | − | GCB | DLBCL, NOS |
| 2 | + | − | − | + | + | − | − | − | Non‐GCB | DLBCL, NOS |
| 3 | + | − | − | − | + | − | − | − | Non‐GCB | DLBCL, NOS |
| 4 | + | − | − | − | + | − | − | − | Non‐GCB | DLBCL, NOS |
| 5 | + | − | − | + | + | + | − | − | Non‐GCB | DLBCL, NOS |
| 6 | + | − | − | + | + | − | NA | NA | Non‐GCB | DLBCL |
| 7 | + | NA | NA | NA | NA | NA | NA | NA | NA | DLBCL |
| 8 | + | − | − | − | + | − | − | − | Non‐GCB | DLBCL, NOS |
| 9 | + | − | − | + | + | + | − | − | Non‐GCB | DLBCL, NOS |
| 10 | + | − | − | − | + | − | − | − | Non‐GCB | DLBCL, NOS |
| 11 | + | NA | NA | NA | NA | NA | NA | NA | NA | DLBCL |
| 12 | + | NA | NA | NA | NA | NA | NA | NA | NA | DLBCL |
| 13 | + | NA | − | + | − | + | − | NA | GCB | DLBCL |
| 14 | + | − | − | + | − | + | − | − | GCB | DLBCL, NOS |
| 15 | + | + | − | − | + | + | + | + | Non‐GCB | EBV‐positive DLBCL, NOS; monomorphic |
| 16a | + | NA | NA | NA | NA | NA | NA | + | NA | EBV‐positive DLBCL, NOS; polymorphic |
| 17a | + | NA | NA | NA | NA | NA | NA | − | NA | THRLBCL |
| 18a | + | NA | NA | NA | NA | NA | NA | − | NA | THRLBCL |
Abbreviations: −, negative; +, positive; DLBCL, diffuse large B‐cell lymphoma; DLBCL, NOS, diffuse large B‐cell lymphoma, not otherwise specified; EBV, Epstein‐Barr virus; GCB, germinal center; NA, not available; THRLBCL, T‐cell/histiocyte‐rich large B‐cell lymphoma.
Cases 16‐18 had a limited number of scattered tumor cells, making it difficult to calculate the percentage of positively stained cells.
Of 18 cases of DLBCLs, in three cases there were no samples for IHC study (cases 7, 11, and 12), another three cases had a limited number of scattered large cells (cases 16‐18), which interfered with calculating the percentage of positively stained tumor cells. The remaining 12 cases of DLBCLs, according to the Hans algorithm,5 were subdivided into the germinal center B‐cell (GCB) subtype for three cases and the non‐GCB subtype for nine cases. The HGAL staining results matched those predicted by the Hans model in nine of the 11 tested cases.
According to the 2016 WHO classification, the 13 cases were classified as: DLBCL, NOS (nine cases), EBV‐positive DLBCL, NOS (two cases), and THRLBCL (two cases). Five cases of DLBCLs were not re‐classified because their EBV status was unknown.
Based on the IPI, six patients were assigned to low and low/intermediate risk groups, while 12 patients were assigned to intermediate/high and high risk groups. Extranodal DLBCL lesions were detected in 14 patients, which most commonly involved the lungs (six patients) and stomach (two patients). Baseline clinical characteristics, treatments, and outcomes for the 18 pSS‐related DLBCLs are shown in Table 3. The average follow‐up period after the diagnosis of DLBCL was 44 months (range: 0‐200 months). The OS of the patients after the lymphoma diagnosis is shown in Figure 1. The median OS after the lymphoma diagnosis was 3 months (range: 0‐212 months), and the 5‐year OS rate after the lymphoma diagnosis was 37.5%.
Table 3.
Clinical characteristics, treatments, and outcomes for the 18 pSS‐related DLBCLs
| Case no. | Age (y) at DLBCL diagnosis | Time (y) from the onset of SS symptoms to the diagnosis of DLBCL | IPI | Lymphoma location | DLBCL treatment | Outcome/DLBCL evolution/survival from DLBCL diagnosis (mo) |
|---|---|---|---|---|---|---|
| 1 | 52 | 32 | Low/intermediate | Liver | R‐CHOP × 6 | Alive/CR/87 |
| 2 | 59 | 19 | Intermediate/high | Bone marrow, buccal mucosa, lymph nodes | NA | Dead/NA/3 |
| 3 | 53 | 18 | Low/intermediate | Lymph nodes | CHOP × 6 | Death from colon cancer/CR/172 |
| 4 | 48 | 38 | Low | Lymph nodes | R‐CHOP × 6 | Dead/CR/151 |
| 5 | 83 | 22 | Intermediate/high | Lymph nodes | NA | Dead/DP/12 |
| 6 | 59 | 13 | Low | Stomach | R‐CHOP × 6 | Alive/CR/67 |
| 7 | 47 | 26 | High | Brain, lung, lymph nodes | high doses of glucocorticosteroids | Death from cerebral edema/ postmortem diagnosis / 0 |
| 8 | 51 | 22 | High | Parotid gland, lung, lymph nodes | NA | Dead/NA/3 |
| 9 | 39 | 6 | High | Lung, stomach, lymph nodes | R‐CHOP × 2 | Dead/NA/3 |
| 10 | 62 | 15 | High | Lung, lymph nodes | NA | Dead/NA/2 |
| 11 | 45 | 22 | Intermediate/high | Lung, spleen, lymph nodes | NA | Dead/NA/2 |
| 12 | 70 | 18 | High | Lung, lymph nodes | CHOP × 3 | Death from acute pulmonary embolism/CR/3 |
| 13 | 73 | 18 | High | Retroperitoneal tumor with infiltration of the diaphragm crus, left psoas major muscle, and left kidney sinus | CHOP × 1 | Dead/NA/2 |
| 14 | 73 | 24 | Intermediate/high | Parotid gland, lymph nodes | R‐CHOP × 3 | Alive/continuing therapy/4 |
| 15 | 61 | 20 | Intermediate/high | Left mandibular fossa spreading anteriorly to the parotid area | R‐CHOP × 1 | Dead/NA/1 |
| 16 | 42 | 21 | Low/intermediate | Lymph nodes | VR‐CAP × 6/CR, relapse after 4 mo, ICE × 3 | Dead/DP/21 |
| 17 | 43 | 22 | Low | Amygdala, lymph nodes | CHOP × 6 | Alive/CR/200 |
| 18 | 57 | 14 | Intermediate/high | Spleen, posterolateral pharyngeal wall, lymph nodes | R‐CHOP × 6 | Alive/CR/55 |
Abbreviations: CR, complete remission; DLBCL, diffuse large B‐cell lymphoma; DP, disease progression; EBV, Epstein‐Barr virus; ICE, ifosfamide, carboplatin, etoposide; IPI, International Prognostic Index; NA, not available; pSS, primary Sjögren's syndrome; R‐CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; VR‐CAP, bortezomib, rituximab, cyclophosphamide, doxorubicin, prednisone.
Figure 1.
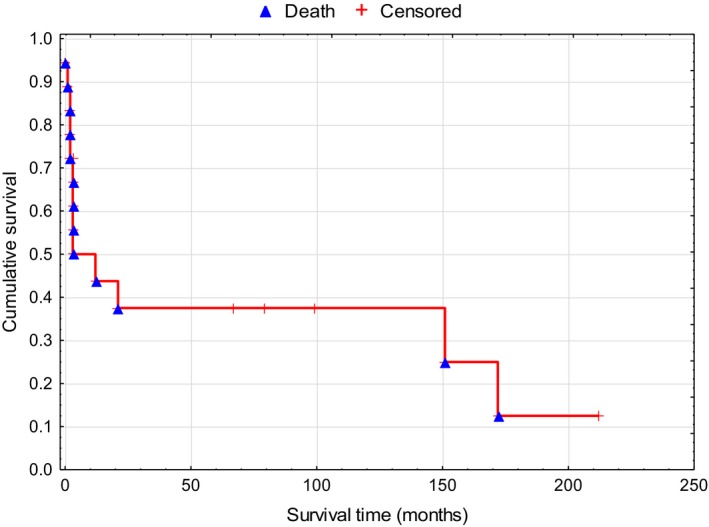
Overall survival among the 18 patients with primary Sjögren's syndrome and diffuse large B‐cell lymphoma
4. DISCUSSION
The main predictors of lymphoma development in patients with pSS are permanently enlarged salivary glands,8, 9, 10 palpable purpura,9, 10, 11 lymphadenopathies,8, 10 cryoglobulinaemia,10, 12 low complement levels (especially C4),9, 10, 11, 13 a monoclonal component in the serum or urine,8 and lymphopenia.11, 13 However, these studies evaluated risk factors for the development of the entire group of non‐Hodgkin's lymphomas, and did not indicate which predictors could lead to the development of a specific variant of non‐Hodgkin's lymphoma. Given that the predominant variant of non‐Hodgkin's lymphoma in patients with pSS is marginal zone B‐cell lymphoma (MZL),14 these risk factors presumably predispose patients to the development of MZL. To the best of our knowledge, only the study by Baimpa et al determined that the presence of lymphocytopenia (<1000 cells/µL) at the pSS diagnosis was a risk factor for the development of DLBCL.15 In our cohort of patients with pSS‐related DLBCLs, lymphopenia was also detected in 43% of the tested patients (6/13) at the time of their pSS diagnosis.
After reviewing the relevant literature, we only identified 117 cases of SS‐related DLBCL that have been described in case reports or small case series.2, 11, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 This very small sample size is likely related to the low prevalence of pSS in the general population, which is estimated to be 0.2%–1.4%,33 combined with the estimated incidence of DLBCL, which is 7/100 000 population.34 For example, Chiu et al identified only nine cases of DLBCL in 16 396 patients with pSS,31 while Smedby et al conducted a pooled analysis using the InterLymph Consortium database and identified only eight cases of DLBCL in 8176 patients with pSS.2 An analysis of the literature, as well as our results, indicate that DLBCL is the predominant variant among large B‐cell lymphomas in patients with pSS, and only sporadic cases have involved other specific entities of large B‐cell lymphomas in patients with pSS: one case of THRLBCL,25 two cases of primary cutaneous DLBCL, leg type,13, 17 one case of primary DLBCL of the central nervous system,20 one case of intravascular large B‐cell lymphoma,24 one case of lymphomatoid granulomatosis,32 and three cases of EBV‐positive DLBCL, NOS.26, 28, 32
According to Vasaitis, EBV was detected in the lymphoma tissue in 22% of investigated cases of pSS‐related DLBCLs.32 In our cohort, EBV was detected in the DLBCL tissue for 15.4% of the cases. Baecklund et al analyzed a large group of patients with RA and DLBCL, and detected EBV in 8.6% of the RA‐related DLBCL cases.35 The results of these studies and our data indicate that the prevalence of EBV‐positive DLBCL, NOS is higher among DLBCLs in patients with autoimmune diseases, relative to that in the general population.3 This suggests that the EBV may play a role in the pathogenesis of DLBCLs associated with autoimmune diseases.
In our study, the DLBCL patients were younger at their diagnosis (median age: 55 years), relative to other studies (median age: 67 years).11, 32 Interestingly, pSS‐related DLBCLs tend to arise in individuals with longstanding pSS, and Theander et al reported that the median time from the onset of sicca symptoms to the DLBCL diagnosis was 17 years, and the median time from the pSS diagnosis until DLBCL development was 10 years.11 Vasaitis reported similar results, with a median time of 18.5 years from the onset of sicca symptoms to the DLBCL diagnosis and a median time of 9 years from the pSS diagnosis until DLBCL development.32 Similarly, we observed that the median time from the initial symptoms of pSS to the DLBCL diagnosis was 20.5 years, and the median time from the pSS diagnosis until the DLBCL diagnosis was 14 years. Interestingly, Baecklund et al reported that the duration of RA before the diagnosis of DLBCL was also 20 years.35
The algorithm of Hans et al uses a combination of three immunostaining markers (CD10, BCL6, and MUM1) and allows for DLBCL to be classified into two subsets with different pathologies, treatment outcomes, and prognoses: the GCB subtype with a better prognosis and the non‐GCB subtype with a worse prognosis.5, 36, 37, 38 Unlike the findings of Vasaitis,32 who reported similar proportions of the non‐GCB (13 cases) and GCB (13 cases) subtypes among pSS‐related DLBCLs, our group observed a significant predominance of the non‐GCB subtype (9 cases), relative to the GCB subtype (three cases) of DLBCLs. Kojima et al have also reported that all nine of their patients with RA‐related DLBCLs had the non‐GCB subtype,39 which also agrees with the findings of Baecklund et al, who reported a statistically significant predominance of the non‐GCB subtype (70% of 139 RA‐related DLBCLs).35
However, comparison of this IHC model with the gold‐standard gene expression profiling revealed a 20% misclassification rate,5 has highlighted the need for additional IHC markers to increase the model's predictive value. Human germinal center‐associated lymphoma (HGAL) is a B‐cell specific marker the expression of which is observed in the cytoplasm of normal germinal center B cells and lymphomas of GCB derivation.40 Gualco et al reported that the inclusion of HGAL in the Hans immunohistological algorithm improved the detection of the GCB subtype of DLBCL.41 In our study, the HGAL staining results matched those predicted by the Hans model in 82% (9/11) of cases.
In the group of pSS‐related DLBCLs that were analyzed by Vasaitis, the overall median survivals were similar between the non‐GCB and GCB subtypes.32 However, given the small sample of patients with the GCB subtype, we could not assess the difference in survival between the non‐GCB and GCB subtypes. Nevertheless, the median survival time after the lymphoma diagnosis in our group was only 3 months, which was significantly shorter than the result from the Vasaitis cohort (6 years).32 The short survival in our cohort may be related to several reasons. First, many DLBCL patients were diagnosed in the early 2000s, when the therapeutic options did not include rituximab for DLBCL treatment. In these patients, the non‐GCB subtype of DLBCL according to the IHC‐based Hans algorithm was associated with significantly poorer OS.42 Second, approximately two‐thirds of our cases belonged to the medium/high and high risk groups based on the IPI, which have relatively poor outcomes. Third, most of the deaths in our cohort were observed during chemotherapy. Since chemotherapy was administered in an outpatient setting for most patients, we assume a lack of timely doctor‐patient feedback which could lead to an untimely initiation of the therapy for febrile neutropenia.
Rheumatologists treating pSS patients should remember that DLBCL can manifest under several clinical masks. For example, case 1 involved isolated hepatic lymphoma which could be misdiagnosed as hepatocellular carcinoma or liver metastasis. In other instances, the lymphoma manifested as peripheral paralysis of the facial nerve (case 15) or diabetes insipidus (case 7).
The mechanisms underlying the pathogenesis of pSS‐related DLBCL remain unclear, although the increased prevalence of the non‐GCB DLBCL subtype in our cohort is consistent with the hypothesis of chronic B‐cell stimulation and antigenic drive. However, given that a proportion of DLBCLs were of the GCB subtype, the malignant transformation is likely also related to other pathways. In conclusion, we found that pSS‐related DLBCLs arose relatively late in the course of pSS, with a median interval of 20.5 years between the onset of the pSS symptoms and the lymphoma diagnosis. The non‐GCB subtype of DLBCL was also prevalent in our cohort, and these characteristics seem similar to those of RA‐related DLBCL.
CONFLICT OF INTEREST
The authors declare they have no conflicts of interest regarding the publication of this study.
AUTHOR CONTRIBUTIONS
VRG acquired and analyzed the data, wrote the paper, and revised the paper and formulated conclusions with NAP and VIV. NAP analyzed the data. VIV acquired and analyzed the data. All authors approved the final manuscript and agree to be accountable for all aspects of the work.
COMPLIANCE WITH ETHICAL STANDARDS
Ethical approval for the study's retrospective protocol was obtained from the VA Nasonova Research Institute of Rheumatology Ethics Committee. Patients provided informed consent for the collection and analysis of their data and specimens.
ACKNOWLEDGEMENT
The manuscript was edited by Wiley Editing Services (https://wileyeditingservices.com/en/)
Gorodetskiy VR, Probatova NA, Vasilyev VI. Characteristics of diffuse large B‐cell lymphoma in patients with primary Sjögren's syndrome. Int J Rheum Dis. 2020;23:540–548. 10.1111/1756-185X.13800
REFERENCES
- 1. Anderson LA, Gadalla S, Morton LM, et al. Population‐based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int J Cancer. 2009;125:398‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smedby EK, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non‐Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood. 2008;111:4029‐4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Swerdlow S, Campo E, Harris N, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2017. ISBN‐13 (Print Book) 9789283244943. [Google Scholar]
- 4. Shiboski SC, Shiboski CH, Criswell L, et al. American college of rheumatology classification criteria for Sjogren's syndrome: a data‐driven, expert consensus approach in the Sjogren's international collaborative clinical alliance cohort. Arthritis Care Res (Hoboken). 2012;64:475‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275‐282. [DOI] [PubMed] [Google Scholar]
- 6. International Non‐Hodgkin's Lymphoma Prognostic Factors Project . A predictive model for aggressive non‐Hodgkin's lymphoma. N Engl J Med. 1993;329:987‐994. [DOI] [PubMed] [Google Scholar]
- 7. Cheson BD. Staging and response assessment in lymphomas: the new Lugano classification. Chin Clin Oncol. 2015;4:5. [DOI] [PubMed] [Google Scholar]
- 8. Anaya JM, McGuff HS, Banks PM, Talal N. Clinicopathological factors relating malignant lymphoma with Sjogren's syndrome. Semin Arthritis Rheum. 1996;25:337‐346. [DOI] [PubMed] [Google Scholar]
- 9. Ioannidis JP, Vassiliou VA, Moutsopoulos HM. Long‐term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjogren's syndrome. Arthritis Rheum. 2002;46:741‐747. [DOI] [PubMed] [Google Scholar]
- 10. Nishishinya MB, Pereda CA, Munoz‐Fernandez S, et al. Identification of lymphoma predictors in patients with primary Sjogren's syndrome: a systematic literature review and meta‐analysis. Rheumatol Int. 2015;35:17‐26. [DOI] [PubMed] [Google Scholar]
- 11. Theander E, Henriksson G, Ljungberg O, Mandl T, Manthorpe R, Jacobsson LT. Lymphoma and other malignancies in primary Sjogren's syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann Rheum Dis. 2006;65:796‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tzioufas AG, Boumba DS, Skopouli FN, Moutsopoulos HM. Mixed monoclonal cryoglobulinemia and monoclonal rheumatoid factor cross‐reactive idiotypes as predictive factors for the devolopment of lymphoma in primary Sjogren's syndrome. Arthritis Rheum. 1996;39:767‐772. [DOI] [PubMed] [Google Scholar]
- 13. Solans‐Laque R, Lopez‐Hernandez A, Bosch‐Gil JA, Palacios A, Campillo M, Vilardell‐Tarres M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjogren's syndrome. Semin Arthritis Rheum. 2011;41:415‐423. [DOI] [PubMed] [Google Scholar]
- 14. Royer B, Cazals‐Hatem D, Sibilia J, et al. Lymphomas in patients with Sjogren's syndrome are marginal zone B‐cell neoplasms, arise in diverse extranodal and nodal sites, and are not associated with viruses. Blood. 1997;90:766‐775. [PubMed] [Google Scholar]
- 15. Baimpa E, Dahabreh IJ, Voulgarelis M, Moutsopoulos HM. Hematologic manifestations and predictors of lymphoma development in primary Sjogren syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore). 2009;88:284‐293. [DOI] [PubMed] [Google Scholar]
- 16. Biasi D, Caramaschi P, Ambrosetti A, et al. Mucosa‐associated lymphoid tissue lymphoma of the salivary glands occurring in patients affected by Sjogren's syndrome: report of 6 cases. Acta Haematol. 2001;105:83‐88. [DOI] [PubMed] [Google Scholar]
- 17. Selva‐O'Callaghan A, Perez‐Lopez J, Solans‐Laque R, Lopez‐Peig C, Angel‐Bosch Gil J, Vilardell‐Tarres M. Primary cutaneous large B‐cell lymphoma of the legs in a patient with primary Sjogren's syndrome. Clin Exp Rheumatol. 2003;21:672. [PubMed] [Google Scholar]
- 18. Cortes M, Fernandes S, Teixeira V, Freitas LC. Upper gastrointestinal bleeding in a patient with Sjogren syndrome. Rev Esp Enferm Dig. 2016;108:576. [PubMed] [Google Scholar]
- 19. Kojima M, Tsukamoto N, Yokohama A, et al. B‐cell lymphoma associated with Sjogren's syndrome among Japanese patients: a clinicopathologic and immunohistochemical study of 15 cases. J Clin Exp Hematop. 2009;49:89‐95. [DOI] [PubMed] [Google Scholar]
- 20. Kinikli G, Erten S, Tanju S, Savas A, Kaygusuz G. Central nervous system lymphoma in a patient with Sjogren's syndrome and autoimmune thyroiditis (Hashimoto's thyroiditis). Clin Rheumatol. 2007;26:1377‐1379. [DOI] [PubMed] [Google Scholar]
- 21. Ramos‐Casals M, la Civita L, de Vita S, et al. Characterization of B cell lymphoma in patients with Sjogren's syndrome and hepatitis C virus infection. Arthritis Rheum. 2007;57:161‐170. [DOI] [PubMed] [Google Scholar]
- 22. Jia N, Tang FL. Characteristics of patients with primary Sjogren's syndrome and non‐Hodgkin's lymphoma: analysis of 9 cases. Zhonghua Yi Xue Za Zhi. 2009;89:2786‐2788. [in Chinese]. [PubMed] [Google Scholar]
- 23. Horvath IF, Szodoray P, Zeher M. Diffuse large B‐cell lymphoma as a sequela of Sjögren's syndrome: a case report. Open Autoimmun J. 2009;1:45‐49. [Google Scholar]
- 24. Zhang W, Feng S, Yan S, et al. Incidence of malignancy in primary Sjogren's syndrome in a Chinese cohort. Rheumatology (Oxford). 2010;49:571‐577. [DOI] [PubMed] [Google Scholar]
- 25. Voulgarelis M, Ziakas PD, Papageorgiou A, Baimpa E, Tzioufas AG, Moutsopoulos HM. Prognosis and outcome of non‐Hodgkin lymphoma in primary Sjogren syndrome. Medicine (Baltimore). 2012;91:1‐9. [DOI] [PubMed] [Google Scholar]
- 26. Kim CS, Choi YD, Choi JS, Bae EH, Ma SK, Kim SW. EBV‐positive diffuse large B‐cell lymphoma in a patient with primary Sjogren's syndrome and membranous glomerulonephritis. BMC Nephrol. 2012;13:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patel S, Kramer N, Cohen AJ, Rosenstein ED. Renal lymphoma: unusual lymphoproliferative manifestation of Sjogren's syndrome. J Rheumatol. 2013;40:102‐103. [DOI] [PubMed] [Google Scholar]
- 28. Strunk JE, Schuttler C, Ziebuhr J, et al. Epstein‐Barr virus‐induced secondary high‐grade transformation of Sjogren's syndrome‐related mucosa‐associated lymphoid tissue lymphoma. J Clin Oncol. 2013;31:e265‐268. [DOI] [PubMed] [Google Scholar]
- 29. Boussios S, Pentheroudakis G, Somarakis G, Markatseli TE, Drosos AA, Pavlidis N. Cancer diagnosis in a cohort of patients with Sjogren's syndrome and rheumatoid arthritis: a single‐center experience and review of the literature. Anticancer Res. 2014;34:6669‐6676. [PubMed] [Google Scholar]
- 30. Koga T, Mizokami A, Nakashima M, et al. Histological improvement in salivary gland along with effector memory Th17‐1 cell reduction in a primary Sjogren's syndrome patient with dermatomyositis and diffuse large B‐cell lymphoma by R‐CHOP therapy. Clin Immunol. 2016;165:35‐37. [DOI] [PubMed] [Google Scholar]
- 31. Chiu YH, Chung CH, Lin KT, et al. Predictable biomarkers of developing lymphoma in patients with Sjogren syndrome: a nationwide population‐based cohort study. Oncotarget. 2017;8:50098‐50108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vasaitis L. Lymphoma studies in patients with Sjögren's syndrome. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1331. 94 pp. Uppsala: Acta Universitatis. ISBN 978‐91‐554‐9912‐9. [Google Scholar]
- 33. Dafni UG, Tzioufas AG, Staikos P, Skopouli FN, Moutsopoulos HM. Prevalence of Sjogren's syndrome in a closed rural community. Ann Rheum Dis. 1997;56:521‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Y, Wang Y, Wang Z, Yi D, Ma S. Racial differences in three major NHL subtypes: descriptive epidemiology. Cancer Epidemiol. 2015;39:8‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baecklund E, Backlin C, Iliadou A, et al. Characteristics of diffuse large B cell lymphomas in rheumatoid arthritis. Arthritis Rheum. 2006;54:3774‐3781. [DOI] [PubMed] [Google Scholar]
- 36. Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature. 2000;403:503‐511. [DOI] [PubMed] [Google Scholar]
- 37. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large‐B‐cell lymphoma. N Engl J Med. 2002;346:1937‐1947. [DOI] [PubMed] [Google Scholar]
- 38. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression‐based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991‐9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kojima M, Itoh H, Shimizu K, et al. Malignant lymphoma in patients with systemic rheumatic disease (rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis, and dermatomyositis): a clinicopathologic study of 24 Japanese cases. Int J Surg Pathol. 2006;14:43‐48. [DOI] [PubMed] [Google Scholar]
- 40. Natkunam Y, Lossos IS, Taidi B, et al. Expression of the human germinal center‐associated lymphoma (HGAL) protein, a new marker of germinal center B‐cell derivation. Blood. 2005;105:3979‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gualco G, Bacchi LM, Domeny‐Duarte P, Natkunam Y, Bacchi CE. The contribution of HGAL/GCET2 in immunohistological algorithms: a comparative study in 424 cases of nodal diffuse large B‐cell lymphoma. Mod Pathol. 2012;25:1439‐1445. [DOI] [PubMed] [Google Scholar]
- 42. Salles G, de Jong D, Xie W, et al. Prognostic significance of immunohistochemical biomarkers in diffuse large B‐cell lymphoma: a study from the Lunenburg Lymphoma Biomarker Consortium. Blood. 2011;117:7070‐7078. [DOI] [PubMed] [Google Scholar]