

Abstract
Tropifexor (LJN452) is a potent, orally available, non–bile acid farnesoid X receptor agonist under clinical development for chronic liver diseases. Here, we present results from a first‐in‐human study of tropifexor following single‐ and multiple‐ascending doses (SAD/MAD) and food effect substudy in healthy volunteers. The SAD study included 6 fasted cohorts receiving 10‐ to 3000‐µg tropifexor or placebo and 1 cohort receiving 300‐µg tropifexor with a high‐fat meal. The MAD study included 4 lean cohorts receiving 10 to 100 µg and 1 obese cohort receiving 30‐µg once‐daily doses or placebo for 14 days. Pharmacodynamic assessment of fibroblast growth factor 19 and fasting plasma lipids was performed after dosing. Overall, 95 volunteers received at least 1 tropifexor or placebo dose. Tropifexor was well tolerated up to 3000 µg and 100 µg in the SAD and MAD studies, respectively; however, 2 subjects discontinued the MAD study due to asymptomatic elevation of liver transaminases. At single doses, tropifexor showed a moderate rate of absorption (median time to maximum concentration, 4 hours), dose‐proportional increases in exposure, and elimination half‐life of 13.5 to 21.9 hours. When taken with food, tropifexor exposure increased by ∼60%. With multiple dosing, steady state was reached on day 4 with <2‐fold accumulation. Single and multiple doses showed dose‐dependent increases in fibroblast growth factor 19. No changes in serum lipids were observed in tropifexor‐ vs placebo‐treated obese subjects. In conclusion, tropifexor was well tolerated, had a pharmacokinetic profile suitable for once‐daily dosing and showed dose‐dependent target engagement without altering plasma lipids in healthy volunteers.
Keywords: farnesoid X receptor, fibroblast growth factor 19, low‐density lipoprotein cholesterol, pharmacodynamics, pharmacokinetics, tropifexor
The farnesoid X receptor (FXR; NR1H4) is a nuclear receptor activated by bile acids.1, 2, 3 FXR is expressed abundantly in the liver, intestine, and kidney, all of which play a role in bile acid metabolism.4, 5 FXR controls a sensitive negative feedback loop affecting various aspects of bile acid metabolism,6 both directly in target organs and indirectly via fibroblast growth factor 19 (FGF19), a naturally occurring hormone released from enterocytes in response to physiological FXR agonism.7 Furthermore, FXR plays an essential role in various aspects of cholesterol, triglyceride, and carbohydrate metabolism, primarily in the liver.8, 9, 10
FXR agonists are under investigation as therapies for chronic liver diseases such as nonalcoholic steatohepatitis (NASH) and primary biliary cholangitis (PBC). The bile acid–derived FXR agonist obeticholic acid (OCA) was provisionally approved by the US Food and Drug Administration and the European Medicines Agency in 2016 for patients with PBC who are not responsive to or intolerant of the standard of care, ursodeoxycholic acid.11, 12, 13, 14 Ursodeoxycholic acid is associated with an incomplete biochemical response in up to 40% of patients with PBC.15 In phase 2 and phase 3 trials with OCA in PBC, dose‐dependent increases in the incidence and severity of pruritus were observed.13, 14 Currently, there are no approved therapies for NASH; however, clinical validation for the potential use of FXR agonists in NASH was obtained from phase 2 trials with OCA.16, 17 In the latter of these trials (Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non‐cirrhotic, Non‐alcoholic Steatohepatitis [FLINT]), OCA led to improvements in key histologic features of NASH, including fibrosis, but did not show complete NASH resolution.17 Furthermore, pruritus was observed in 23% of patients, and significant increases in total and low‐density lipoprotein (LDL) cholesterol and a modest decrease in high‐density lipoprotein (HDL) cholesterol were observed in OCA‐treated vs placebo‐treated patients. In light of these limitations of this bile acid–derived FXR agonist, the development of more efficacious, non–bile acid FXR agonists with improved pharmacokinetics (PK) and an improved adverse event profile remains a key unmet need in the management of these chronic liver diseases.
Tropifexor is a highly potent, orally available, non–bile acid FXR agonist that has shown effective target engagement in cell‐based assays and in vivo experiments in rodents.18 Its potency has been attributed to the inclusion of a bicyclic nortropine‐substituted benzothiazole carboxylic acid.18 Recently, preclinical validation of tropifexor was performed in various animal models of cholestasis and NASH, demonstrating effective reduction in various disease parameters including fibrosis.19, 20 Here, we present results from a phase 1 study of the safety, tolerability, PK, and pharmacodynamics (PD) of tropifexor. In addition to lean, healthy volunteers, a cohort of obese volunteers was also included to investigate PK and PD in individuals resembling those with nonalcoholic fatty liver disease/NASH.21, 22
Methods
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The study was performed at the Buffalo Clinical Research Center, LLC, and the protocol was reviewed and approved by an institutional review board (Integreview, Austin, Texas).
This was a first‐in‐human study and was conducted in 2 parts: (1) a randomized, double‐blind, placebo‐controlled single‐ascending‐dose (SAD) part with an additional food effect cohort in lean healthy subjects and (2) a randomized, double‐blind, placebo‐controlled multiple‐ascending‐dose (MAD) part in lean and obese healthy subjects.
Study Population
Male and female healthy volunteers aged 18 to 65 years who weighed ≥50 kg and who provided written informed consent were included. The following sitting vital sign ranges were required for inclusion: (1) oral body temperature, 35.0 to 37.5°C; (2) systolic blood pressure, 90 to 140 mmHg (lean subjects) or 90 to 150 mmHg (obese subjects); (3) diastolic blood pressure, 50 to 90 mmHg; and (4) pulse rate, 40 to 90 beats per minute. The required body mass index (BMI) ranges for inclusion were 18 to 30 kg/m2 and 35 to 45 kg/m2 for lean and obese subjects, respectively. Key exclusion criteria included women of childbearing potential; any surgical or medical condition that could significantly alter the absorption, metabolism, distribution, and excretion of the study drug; medical history and/or clinical or laboratory evidence of liver disease or liver injury as indicated by abnormal liver tests, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), γ‐glutamyl transferase (GGT), alkaline phosphatase, or serum bilirubin levels exceeding 1× the upper limit of normal (ULN) for lean subjects and 2× ULN for obese subjects; or a positive hepatitis B surface antigen or hepatitis C test result.
Study Design
In the SAD part of the study, 8 subjects per cohort were randomized to receive a single dose of tropifexor (10, 30, 100, 300, 1000, or 3000 µg) or placebo in a 3:1 ratio (6 active:2 placebo). The study included a 27‐day screening period, baseline evaluation for eligibility on day −2, and a meal test to determine the physiological change in FGF19 preceded by a ≥10‐hour overnight fast on day −1. This was followed by a single oral dose of tropifexor on day 1 and an end‐of‐study (EOS) visit on day 8. All safety and tolerability data up to 72 hours after dosing for at least 6 subjects within a cohort were reviewed and assessed as acceptable before escalating the dose. The study also explored the effect of food in the 300‐µg cohort where subjects underwent a second dosing period following a 7‐day washout period and received a high‐fat meal immediately prior to a single dose of 300‐µg tropifexor (Figure S1).
In the MAD part of the study, 8 subjects per cohort were randomized to receive ascending daily doses of tropifexor (10, 30, 60, or 100 µg) or placebo in a 3:1 ratio (6 active:2 placebo). The study comprised a 27‐day screening period, baseline evaluation for eligibility on day −2, a meal test to measure the physiological change in FGF19 preceded by a ≥10‐hour overnight fast on day −1, treatment period from days 1 to 15 with a second meal test on day 14, safety follow‐up visits on days 21 and 42, and an EOS visit on day 70. All safety and tolerability assessments up to 8 days after the last dose (day 21) for at least 6 subjects within a cohort were reviewed and assessed as acceptable before dose escalation. The MAD study was repeated in 11 obese subjects randomized in an approximately 2:1 ratio (7 active:4 placebo) to receive 30 µg of tropifexor or placebo once daily (qd) for 14 days. All study timelines were similar to that of the study in lean subjects, except for the EOS visit, which was on day 21 in obese subjects.
Bioanalytical Methods for PK Samples
Tropifexor and CKS577 and their internal standards (IS) [13C2H2 34S]tropifexor and [13C2H2 34S]CKS577 were supplied by Novartis (Basel, Switzerland). Chromatographic separation was performed using Shimadzu LC‐30AD pumps (Columbia, Maryland) on Quattro 3 C8 analytical column (2.1 × 50 mm, American Chromatography Supplies, Vineland, New Jersey) at 40°C. The mobile phases were water (A) and acetonitrile (B) containing 0.1% formic acid. The gradient elution conditions were 0.0 to 0.03 minutes at 40% B; 0.03 to 1.50 minutes from 40% to 75% B; 1.50 to 1.80 minutes at 75% B; 1.80 to 1.90 minutes from 75% to 40% B. The flow rate was 0.4 mL/min, and the run time was 2.5 minutes per injection. A Triple Quad 6500 mass spectrometer (AB Sciex, Framingham, Massachusetts) coupled with an electrospray ionization ion source was used to monitor the mass transitions. The system was operated in positive mode, and multiple reaction monitoring mass transitions were m/z 604.2/228.0 and m/z 609.2/227.9 for tropifexor and its IS, respectively, and m/z 780.3/603.9 and m/z 785.0/608.9 for CKS577 and its IS, respectively. A 50‐µL volume of human plasma was added to an Ostro 96‐well plate (Waters, Milford, Massachusetts), which was placed on top of a 1‐mL 96‐well collection plate. To each well, a 300‐µL aliquot of the IS working solution (each at 3 ng/mL in acetonitrile/ethanol/formic acid, 90/10/1, v/v/v) was added except for the control blanks. The combo plate was vortexed and centrifuged at approximately 1200 rpm (262 × g) for about 10 minutes. A 50‐µL volume of human urine was added to a 1‐mL 96 square‐well plate. To each well, a 300‐µL aliquot of the IS working solution (3 ng/mL each in acetonitrile/ethanol/formic acid, 90/10/1, v/v/v) was added, and the plate was vortexed and centrifuged as above. Thereafter, 10 µL of the above extracts was injected onto the liquid chromatography with tandem mass spectrometry (LC‐MS/MS) system.
The lower limit of quantification (LLOQ) was 20 pg/mL in plasma and 100 pg/mL in urine for both tropifexor and CKS577, respectively, whereas the upper limit of quantification (ULOQ) was 20 ng/mL in plasma and 100 ng/mL in urine, respectively. The overall coefficient of variance percentage (%CV) and bias percentage of calibration standards were 9.5 and −3.6 to 2.0 for plasma tropifexor, 9.8 and −1.9 to 3.5 for plasma CKS577, 6.7 and −1.5 to 1.0 for urine tropifexor, and 7.2 and −2.0 to 3.0 for urine CKS577, respectively. The overall %CV and bias percentage quality controls were 10.8 and 0.8 to 5.0 for plasma tropifexor, 8.2 and 1.8 to 3.5 for plasma CKS577, 8.5 and 1.3 to 4.1 for urine tropifexor, and 15.4 and −2.1 to 8.0 for urine CKS577, respectively.
PK Assessments
During the SAD study, blood samples were collected for PK assessment of tropifexor before dosing (0 hours) and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, and 72 hours after dosing. Urine samples were collected in 12‐hour batches starting at dosing and ending 72 hours after dosing. During the MAD study, blood samples were collected before dosing (0 hours) and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 hours after dosing on days 1 and 13 and before dosing (0 hours) on days 4, 5, 6, 7, and 10. Urine samples were collected in 12‐hour batches from postdose on day 1 to predose on day 2 and from postdose on day 13 to predose on day 14.
The following PK parameters were evaluated for tropifexor and CKS577: for the SAD part, the observed maximum plasma concentration following drug administration (Cmax); time to reach Cmax (tmax); area under the plasma concentration–time curve from time 0 to infinity (AUCinf); terminal elimination half‐life (t1/2); apparent systemic clearance from plasma. In the MAD part, steady‐state exposure was calculated as AUC in 1 dose interval (AUCtau). In addition, Cmax, tmax, and accumulation ratio (Racc = AUCtau,Day13/AUCtau,Day 1) were estimated. For urine PK, the amount of tropifexor or CKS577 excreted into the urine from time 0 to time t, where t is a defined time point after administration (Ae0‐t), clearance, and percent recovery of tropifexor or CKS577 were estimated. These parameters were derived using the actual recorded sampling times and noncompartmental analysis with Phoenix WinNonlin, version 6.3 (Certara, Princeton, New Jersey).
Bioanalytical Methods for PD Samples
Quantitative determination of FGF19 in human serum was performed at WuXi AppTec (Shanghai, China) using a commercial human FGF19 immunoassay (R&D Systems, Minneapolis, Minnesota) as previously described.23 Internal validation of the assay characteristics demonstrated an LLOQ of 27.20 pg/mL and a ULOQ of 1096.3 pg/mL. Of 2370 study samples analyzed for FGF19, 30 (1.3%) were found to be below the LLOQ of the assay. Samples were diluted as necessary to allow quantitation, and no assay values were above the ULOQ following appropriate dilution. The interrun accuracy ranged from 88.9% to 105.8%, and the interrun precision ranged from 5.4% to 9.3% (data not shown).
Quantitative determination of 7α‐hydroxy‐4‐cholesten‐3‐one (C4) in human serum samples was by performed at WuXi AppTec (Shanghai, China) by means of LC‐MS/MS. Briefly, human serum samples (sample volume, 80 µL) were precipitated by acetonitrile/ammonium sulfate, the extracts evaporated, and the reconstituted samples analyzed by LC‐MS/MS. Analysis was performed by reversed‐phase liquid chromatography, with separation on a ZORBAX C18 (Agilent Technologies, Santa Clara, California), 3.5 µm, 100 × 2.1 mm column at 55°C at a flow rate of 700 µL/min with an 11.0‐minute run time. The analyte was ionized by atmospheric pressure chemical ionization in positive ion mode on a 5500 triple‐quad (AB Sciex, Framingham, Massachusetts). The reference standard (C27H44O2 7α‐hydroxy‐4‐cholesten‐3‐one) and IS (C27H37D7O2 7α‐hydroxy‐4‐cholesten‐3‐one‐d7) (Toronto Research Chemicals Inc., Toronto, Canada) was diluted in human serum purchased from Bioreclamation IVT (Westbury, New York). Validation of the assay characteristics demonstrated an LLOQ of 3.90 ng/mL and a ULOQ of 1250 ng/mL. Across 22 accepted runs performed on 21 different days, the variability of the calibration parameters (mean ± standard deviation) was as follows: slope 0.01968 ± 0.004109; intercept 0.01181 ± 0.02236, and coefficient of determination r2 0.9962 ± 0.0021. Overall, a total of 1211 samples were analyzed in a total of 22 accepted runs. C4 levels ranged from 3.92 ng/mL to 190 ng/mL, with 125 samples falling below the LLOQ. Seven samples had abnormal IS peak area responses and were repeated.
PD Assessments
For PD analyses, plasma concentrations of the biomarkers of FXR target engagement FGF19 and C4 were evaluated at various time points after dosing in the SAD and MAD studies. In the SAD study, FGF19 was analyzed at 0 hours on day –2; at 0, 1, 2, 3, 4, 6, 8, and 12 hours on day –1 (following a high‐fat meal challenge) or at 0 hours on day –1 (in the pilot food effect cohort); and at 0, 1, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours after dosing (with a standard breakfast given at 4 hours after dosing). In the MAD study, FGF19 was analyzed at 0 hours on day –2; at 0, 1, 2, 3, 4, 6, 8, and 12 hours on day –1 (following a high‐fat meal challenge); and at 0, 1, 2, 3, 4, 6, 8, 12, and 24 hours after dosing on days 1 and 13 (with a standard breakfast given at 4 hours after dosing). The LLOQ and ULOQ for FGF19 were 27.2 pg/mL and 1096.3 pg/mL, respectively; samples with values above the ULOQ were diluted and retested.
C4 concentration was measured at 0, 1, 2, 3, 4, 6, 8, 12, and 24 hours after dosing on days 1 and 13 in lean subjects and on days 1 and 14 in obese subjects. The LLOQ and ULOQ for C4 were 3.9 ng/mL and 1250 ng/mL using 80 µL of human serum, respectively. The PD analysis set included all subjects with available PD data, who received at least 1 dose of the study drug or matching placebo and had no protocol deviations.
Secondary Assessments
Serum levels of total, LDL, and HDL cholesterol and triglycerides were analyzed at baseline (day –1) and at 1, 2, 3, 4, 6, 8, and 12 hours after dosing at 10, 30, 100, 300 µg (fed and fasted condition), 1000 and 3000 µg tropifexor or placebo in the SAD study. In the MAD study, all lipid assessments were made at baseline (day –1) and at 0 hours before dosing on days 1, 13, and 14.
Safety and Tolerability Assessments
All adverse events (AEs) and serious AEs (SAEs), with their severity and relationship to the study drug, were recorded as part of the safety assessment. In addition, standard hematology and blood chemistry tests were performed, including serum biomarkers of liver function, such as ALT and AST. The safety analysis set included all subjects who received at least 1 dose of the study drug or matching placebo.
Statistical Analysis
Descriptive statistical analyses were performed for PK parameters of tropifexor and CKS577 and for PD end points FGF19, C4, total bile acids, triglycerides, and cholesterol using SAS software (SAS Institute, Cary, North Carolina) procedures.
For the food effect part, PK parameters Cmax and AUCinf were log‐transformed and analyzed using a linear mixed‐effects model. The model included a fixed effect for treatment (tropifexor fed and tropifexor fasted) and a random effect for subject.
The Wilcoxon rank‐sum test was performed to compare the median difference of changes from baseline in FGF19 and C4 concentrations between each tropifexor dose and the pooled placebo group. The Hodges‐Lehmann estimate and 95% confidence interval for the median difference also were provided.
The log‐transformed ratio to baseline for triglycerides was analyzed using a linear mixed‐effects model with fixed effects for log‐transformed baseline, treatment, time, and treatment by time interaction. A first‐order autoregressive covariance structure was used to account for the correlation among multiple measurements from the same subject. Similarly, for cholesterol data, absolute change from baseline was analyzed using a linear mixed‐effects model for repeated measurements. The model included baseline, treatment, time, and treatment by time interaction as fixed effects with an unstructured covariance matrix.
For PK, concentrations below the LLOQ were considered as 0 in summary statistics and for PK parameter calculations. For PD end points FGF19 and C4, values below LLOQ were replaced by LLOQ/2 in summary statistics, analyses and for PD parameter calculations.
Results
Disposition and Demographics
In total, 95 subjects were enrolled in this study and received at least 1 dose of tropifexor or matching placebo. In all, 47 healthy subjects were enrolled in 6 cohorts in the SAD study, all of whom completed the study as per protocol. Six of 8 subjects who received the 300 µg dose of tropifexor in the SAD study, received a second 300 µg dose after a high‐fat meal, and all 6 (4 on tropifexor treatment and 2 on placebo) completed the study as per protocol. A total of 37 subjects were enrolled in the MAD study involving lean volunteers; of these, 34 completed the study as per protocol. In the portion of the MAD study involving obese subjects, 11 enrolled subjects completed the study as per protocol.
Demographics and baseline characteristics were similar across treatment groups from both SAD and MAD studies (Table 1); most treatment groups comprised predominantly male subjects. In the SAD study, median age ranged between 30.5 and 41.5 years and 24 to 44 years in the placebo and tropifexor groups, respectively. In the MAD study, median age range was 40 to 41 years in the placebo group and 26.5 to 40 years in the tropifexor groups. All treatment groups in the SAD study were comparable with regard to BMI. In the MAD study with lean subjects, the median BMI (29.15 kg/m2) of the 60‐µg treatment group was slightly higher than that of other treatment groups, while it was comparable among obese subjects receiving placebo (42 kg/m2) and 30 µg tropifexor (41 kg/m2).
Table 1.
Demographics and Baseline Characteristics
| Pooled Placebo | Tropifexor Dose (µg) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 300 | |||||||||
| Fasted | Fed | 10 | 30 | 100 | Fasted | Fed | 1000 | 3000 | |
| SAD | n = 12 | n = 2 | n = 5 | n = 6 | n = 6 | n = 6 | n = 4 | n = 6 | (n = 6) |
| Age, y, median (range) | 41.5 (23‐55) | 30.5 (23‐38) | 44.0 (20‐53) | 41.5 (23‐58) | 28.0 (22‐64) | 24.5 (21‐39) | 29.5 (21‐39) | 26.0 (22‐30) | 24.0 (19‐53) |
| Males, n (%) | 11 (91.7) | 2 (100) | 4 (80.0) | 6 (100) | 5 (83.3) | 5 (83.3) | 3 (75.0) | 6 (100) | 6 (100) |
| White, n (%) | 5 (41.7) | 1 (50.0) | 2 (40.0) | 3 (50.0) | 3 (50.0) | 4 (66.7) | 2 (50.0) | 0 (0.0) | 4 (66.7) |
| Black, n (%) | 7 (58.3) | 1 (50.0) | 3 (60.0) | 3 (50.0) | 3 (50.0) | 2 (33.3) | 2 (50.0) | 5 (83.3)a | 2 (33.3) |
| BMI, kg/m2, median (range) | 25.95 (22.7‐29.9) | 25.3 (25.2‐ 25.4) | 25.3 (19.8‐29.3) | 27.25 (23.2‐28.4) | 25.75 (21.2‐28.6) | 25.3 (20.0‐29.0) | 26.55 (22.4‐29.0) | 25.8 (22.6‐29.8) | 24.35 (20.0‐27.8) |
| Placebo | Tropifexor Dose | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 30 µg qd | |||||||||
| MAD | Lean | Obese | 10 µg qd | Lean | Obese | 60 µg qd | 100 µg qd | ||
| Lean Subjects | n = 10 | n = 4 | n = 9 | n = 6 | n = 7 | n = 6 | n = 6 | ||
| Age, y, median (range) | 40.0 (23‐59) | 41.0 (21‐63) | 31.0 (21‐62) | 40.0 (24‐54) | 29.0 (23‐52) | 26.5 (23‐40) | 35.0 (31‐54) | ||
| Males, n (%) | 10 (100) | 2 (50.0) | 8 (88.9) | 5 (83.3) | 5 (71.4) | 6 (100) | 4 (66.7) | ||
| White, n (%) | 6 (60.0) | 1 (25.0) | 5 (55.6) | 4 (66.7) | 3 (42.9) | 2 (33.3) | 1 (16.7) | ||
| Black, n (%) | 3 (30.0)a | 3 (75.0) | 3 (33.3)b | 2 (33.3) | 3 (42.9)a | 4 (66.7) | 4 (66.7)a | ||
| BMI, kg/m2, median (range) | 25.95 (21.7‐29.4) | 41.95 (38.6‐44.7) | 25.6 (24.0‐29.8) | 22.45 (19.5‐29.0) | 41.0 (37.0‐44.1) | 29.15 (22.3‐30.0) | 25.65 (23.9‐ 28.2) | ||
BMI, body mass index; MAD, multiple ascending dose; qd, once daily; SAD, single ascending dose; SD, standard deviation.
One subject in the 1000‐µg SAD cohort, 1 subject in the MAD placebo group, 1 subject in the 100‐µg MAD cohort, and 1 subject in the 30‐µg MAD obese cohort were Asians.
One subject in the 10‐µg MAD cohort was Native American.
Safety and Tolerability of Tropifexor
Tropifexor generally was well tolerated in both the SAD and the MAD studies, with most subjects completing the studies as planned. Tropifexor was well tolerated up to 3000 µg in the SAD study without any SAEs. In the MAD study, tropifexor was well tolerated up to 100 µg qd for 14 days. Three subjects were discontinued from the study, all in the MAD phase. One subject in the 60 µg qd cohort was lost to follow‐up a month after completing dosing; 1 subject in the 100 µg tropifexor qd cohort was discontinued per protocol at day 7 due to an elevated ALT level >5× ULN but <8× ULN; and 1 subject in the 100 µg tropifexor qd cohort was discontinued on day 9 owing to an abnormal ALT level of >3× ULN but <5× ULN.
With regard to overall safety, 69 AEs were reported in 39 of 95 subjects in both parts of the study (Table 2). Four SAEs were reported in 3 subjects, of which 1 was assessed as drug related. The single SAE related to study drug (ALT >5× ULN but <8× ULN at 100 µg qd of tropifexor) was similar to a study drug–related AE of increase in ALT >3× ULN but <5× ULN at 100 µg qd of tropifexor; ALT and AST were also increased by >1× ULN but <3× ULN in 1 additional subject during the dosing period. In all 3 cases, the subjects remained asymptomatic throughout the study and showed no meaningful changes in markers of liver damage such as total bilirubin, alkaline phosphatase, or GGT. In addition, during an extended follow‐up period, creatine kinase and lactate dehydrogenase were found to be elevated in 1 subject following physical injury. No drug‐exacerbated pruritus was reported at any dose either in the SAD or the MAD parts of the study.
Table 2.
Summary of Treatment‐Emergent Adverse Events (≥20% in Any Group)
| Placebo | Tropifexor (µg) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 300 | |||||||||
| Fasted | Fed | 10 | 30 | 100 | Fasted | Fed | 1000 | 3000 | |
| SAD | n = 12 | n = 2 | n = 5 | n = 6 | n = 6 | n = 6 | n = 4 | n = 6 | n = 6 |
| Any AE | … | 1 (50.0) | 2 (40.0) | 2 (33.3) | 2 (33.3) | 2 (33.3) | … | 1 (16.7) | 2 (33.3) |
| Abdominal pain | … | … | 1 (20.0) | … | … | … | … | … | … |
| Vomiting | … | … | … | … | … | … | … | … | 2 (33.3) |
| Muscle spasms | … | … | 1 (20.0) | … | … | … | … | … | … |
| Headache | … | … | 2 (40.0) | … | 1 (16.7) | 1 (16.7) | … | … | … |
| Macular rash | … | 1 (50.0) | … | … | … | … | … | … | … |
| Placebo | Tropifexor | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 30 µg qd | |||||||||
| Lean | Obese | 10 µg qd | Lean | Obese | 60 µg qd | 100 µg qd | |||
| MAD | n = 10 | n = 4 | n = 9 | n = 6 | n = 7 | n = 6 | n = 6 | ||
| Any AE | 6 (60.0) | 2 (50.0) | 4 (44.4) | 4 (66.7) | 4 (57.1) | 2 (33.3) | 5 (83.3) | ||
| Dyspepsia | … | … | … | 1 (16.7) | … | 1 (16.7) | 2 (33.3) | ||
| ALT increased | … | … | 1 (11.1) | – | … | … | 2 (33.3) | ||
| Dizziness | … | … | … | 2 (33.3) | … | … | 1 (16.7) | ||
| Headache | 2 (20.0) | … | 3 (33.3) | 1 (16.7) | … | 1 (16.7) | 1 (16.7) | ||
| Diarrhea | … | 2 (50.0) | … | 1 (14.3) | |||||
| Nausea | … | 1 (25.0) | … | 1 (14.3) | |||||
| Vomiting | … | 1 (25.0) | … | … | |||||
AE, adverse event; ALT, alanine aminotransferase; MAD, multiple ascending dose; qd, once daily; SAD, single ascending dose; qd, once daily.
PK Results
Following oral administration, peak tropifexor concentrations were achieved at a median tmax of 4 hours (Figure 1A). The mean t1/2 ranged from 13.5 to 21.9 hours (Table 3). The Cmax and exposure (AUCinf) appeared to increase dose proportionally from 10 µg to 3000 µg. Moderate to high intersubject variability was observed for PK parameters (%CV up to 71% for Cmax and 52% for AUCinf).
Figure 1.
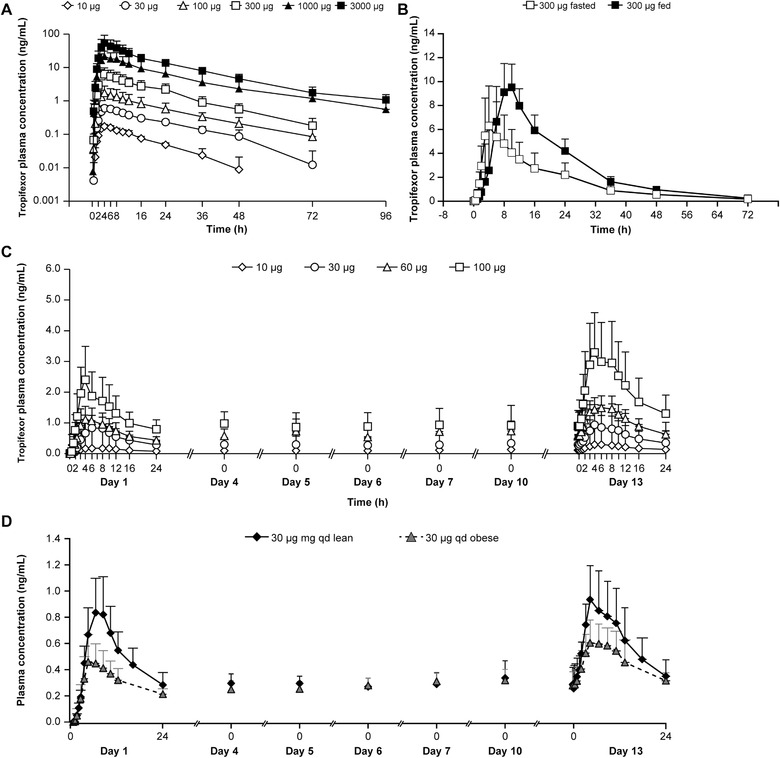
Pharmacokinetic profile of tropifexor following single and multiple ascending doses (PK analysis set). (A) Mean plasma concentration–time profiles following single oral doses of tropifexor under fasted conditions (semilogarithmic scale). (B) Mean plasma concentration–time profiles following a single oral dose of 300 µg under fed and fasted conditions (linear scale). (C) Mean plasma concentration–time profiles following multiple ascending doses of tropifexor for 14 days in lean healthy volunteers (linear scale). (D) Mean plasma concentration–time profiles following a multiple dose of tropifexor 30‐µg qd in lean and obese healthy volunteers for 14 days (linear scale). Data are represented as mean ± standard error.
Table 3.
Summary of Plasma PK Parameters of Tropifexor Following Single and Multiple Ascending Doses (PK Analysis Set)
| SAD, Mean (SD) | PK Parameters | ||||||
|---|---|---|---|---|---|---|---|
| Dose (µg) | n | AUCinf (ng • h/mL) | Cmax (ng/mL) | tmax (h)a | t1/2 (h) | CL/F (mL/h) | |
| 10 | 4 | 3.39 (0.333) | 0.19 (0.031)b | 4.0 (3.0‐6.0)b | 14.9 (3.22) | 2980 (324) | |
| 30 | 5 | 13.4 (1.16) | 0.63 (0.108)b | 4.0 (3.0‐6.0)b | 13.7 (4.74) | 2260 (202) | |
| 100 | 5 | 37.9 (17.2) | 1.7 (1.04)b | 4.0 (4.00‐4.03)b | 13.5 (3.36) | 3080 (1250) | |
| 300 | 6 | 123 (56.4) | 6.4 (3.24) | 4.0 (3.0‐8.0) | 15.3 (5.55) | 3210 (2300) | |
| 300 (fed) | 4 | 203 (52.8) | 10.4 (2.11) | 9.0 (8.0‐12.0) | 12.0 (1.41) | 1550 (364) | |
| 1000 | 5 | 482 (250) | 22.1 (9.37)b | 4.0 (3.0‐8.0)b | 21.9 (10.5) | 2430 (917) | |
| 3000 | 6 | 933 (361) | 54.2 (38.2) | 4.0 (4.0‐6.0) | 16.5 (3.53) | 3630 (1350) | |
| MAD, Mean (SD) | PK Parameters | ||||||
|---|---|---|---|---|---|---|---|
| qd Dose (µg) | n | AUC0‐24h (ng • h/mL) | Cmax (ng/mL) | tmax (h)a | CLss/F (mL/h) | Racc | |
| 10 | Day 1 | 9 | 2.88 (1.27) | 0.212 (0.109) | 4.0 (4.0‐10.0) | … | … |
| Day 13 | 9 | 4.88 (1.47) | 0.319 (0.104) | 4.0 (4.0‐6.0) | 2210 (604) | 1.87 (0.609) | |
| 30 | Day 1 | 6 | 11.7 (3.31) | 0.894 (0.305) | 6.0 (6.0‐8.0) | … | … |
| Day 13 | 6 | 14.0 (4.37) | 0.943 (0.260) | 4.0 (4.0‐10.0) | 2380 (989) | 1.21 (0.278) | |
| 30 (obese) | Day 1 | 7 | 7.19 (2.13) | 0.492 (0.174) | 4.0 (4.0‐8.0) | … | … |
| Day 14 | 7 | 10.4 (2.92)c | 0.650 (0.180) | 6.0 (4.0‐10.0) | 3140 (1170)c | 1.59 (0.693)c | |
| 60 | Day 1 | 6 | 16.2 (5.44) | 1.22 (0.475) | 4.0 (3.0‐6.0) | … | … |
| Day 13 | 6 | 25.3 (6.40) | 1.61 (0.331) | 4.0 (3.0‐6.0) | 2560 (882) | 1.66 (0.560) | |
| 100 | Day 1 | 6 | 30.3 (12.4) | 2.40 (1.09) | 4.0 (4.0‐4.0) | … | … |
| Day 13 | 4 | 50.2 (21.9) | 3.47 (1.38) | 4.0 (3.0‐8.0) | 2600 (1890) | 1.41 (0.287) | |
AUCinf, area under the plasma concentration‐time curve from time 0 to infinity; AUCtau, area under the plasma concentration–time curve from time 0 to the end of the dosing interval tau; AUC0‐24 h, area under the plasma concentration‐time curve from time 0 to 24 h; Cmax, observed maximum plasma concentration following drug administration; CL/F, apparent systemic (or total body) clearance from plasma following extravascular administration; CLss/F, apparent systemic clearance from plasma observed during a dosing interval at steady state following extravascular administration; MAD, multiple ascending dose; PK, pharmacokinetic; qd, once daily; Racc, accumulation ratio; SAD, single ascending dose; SD, standard deviation; tmax, time to reach Cmax; t1/2, terminal elimination half‐life.
tmax is represented as median and range.
Represents mean (SD) of 5 subjects in 10 µg cohort, 6 subjects in 30 µg cohort, 6 subjects in 100 µg cohort, and 6 subjects in 1000 µg cohort.
Represents mean (SD) of 6 subjects.
The metabolite CKS577 was detected in plasma only at tropifexor doses of 100 µg or higher. Peak concentrations were detected at approximately 4 hours following dosing with tropifexor. At the 1000 and 3000 µg doses, the mean t1/2 of CKS577 was 17.0 hours and 14.4 hours, respectively (Table S1). The systemic exposure ratio (CKS577/tropifexor) was <3% based on Cmax or AUClast.
In urine, tropifexor concentrations were below the LLOQ for most subjects, except for 1 subject in the 1000‐µg cohort and 6 subjects in the 3000‐µg cohort. Excretion of tropifexor in urine was low, with mean fraction of dose excreted at <0.015% and the renal clearance of 0.174 mL/h and 0.244 mL/h at 1000 µg and 3000 µg doses, respectively. Thus, renal clearance was <0.01% of the total plasma clearance at these doses. The mean fraction dose of CKS577 excreted in urine was ∼0.2% at 1000 µg and 3000 µg doses (Table S2).
Analysis of data from 4 subjects who received 300 µg of tropifexor with a high‐fat meal showed that tmax was delayed from 4.0 to 9.0 hours; Cmax and AUCinf increased by 55% and 63%, respectively, while t1/2 remained comparable (12 hours fasted vs 15.3 hours fed; Table 3, Figure 1B). Consistent with the PK profile of tropifexor, the median tmax of its metabolite CKS577 increased from 4 to 11 hours, and Cmax and AUCinf increased by about 54% and 68%, respectively (Table S1).
The mean plasma concentration–time profiles of tropifexor following repeat dose administration are shown in Figure 1C. After once‐daily oral dosing for 13 days, time to reach peak concentration at day 13 across all doses (10‐100 µg) was similar to that on day 1 with a median tmax of 4 hours (range, 3‐10 hours; Table 3), suggesting no potential changes in absorption mechanisms with multiple dosing. Steady state was reached by day 4 as trough levels of tropifexor were comparable from day 4 and onwards up to day 13. Consistent with the observed t1/2, the once‐daily dosing resulted in an accumulation ratio (Racc) of <2‐fold (1.21‐1.87).
Plasma CKS577 was detected only in 60 µg and 100 µg cohorts, except for 1 subject in the 10 µg cohort (Table S1), where CKS577 levels also were measurable. The median CKS577 tmax was 6 to 8 hours with a low CKS577 exposure (<3% of the parent tropifexor exposure) on day 13.
Urine tropifexor and CKS577 concentrations were below the LLOQ for most subjects across cohorts, suggesting that negligible fractions of the dose (<1% for tropifexor and <0.4% for CKS577) were excreted in the urine.
In obese healthy subjects who received 30 µg qd tropifexor for 13 days, tmax was similar on days 1 and 13, with a median tmax of 6 hours (range, 4‐10 hours), which was in general comparable to that in lean subjects (Table 3). Steady state was reached on day 4, and the accumulation ratio was <2‐fold. The overall mean exposure of tropifexor in obese subjects appeared to be slightly lower than that observed in lean subjects based on the limited sample size (Figure 1D).
PD Results
On the day before dosing (day –1), baseline and 24‐hour serum concentration–time profiles of FGF19 did not differ across treatment groups in the SAD part of the study. Serum FGF19 concentrations increased in response to feeding. In the pooled placebo subjects, FGF19 increased from a fasted median Cmax of 152 pg/mL to a median Cmax of 319 pg/mL on day −1 and 362 pg/mL on day 1, with a median maximal change from baseline in response to feeding of 177 pg/mL. In the SAD part of the study, drug‐associated elevations in mean FGF19 were observed, with a peak concentration achieved at approximately 6 hours after dosing across dose groups (Figure 2A). A dose‐dependent increase in FGF19 concentrations was observed up to 1000 µg of tropifexor. Median FGF19 Cmax ranged from 416 pg/mL to 1820 pg/mL with doses up to 3000 µg. In the MAD part of the study, day −1 baseline and 24‐hour serum concentration–time profiles of FGF19 did not differ across treatment groups. In the pooled placebo subjects, FGF19 increased from a fasted median of 69.5 pg/mL to a median Cmax of 299 pg/mL on day −1 and 353 pg/mL and 371 pg/mL on days 1 and 13, respectively, with a median maximal change from baseline in response to a meal of 191 pg/mL on day 1 and 240 pg/mL on day 13. A dose‐dependent increase in serum FGF19 was observed in the MAD part of the study, similar to that observed in the SAD study (Figure 2B). Day 13 trends were similar to those on day 1, with FGF19 Cmax peaking at approximately 6 hours after dosing for all groups. The median maximal change in FGF19 level from baseline was similar on day 1 and day 13 at dose levels of up to 60 µg of tropifexor (Table 4). Furthermore, the median maximal changes in FGF19 Cmax levels were comparable on days 1 and 13 (Table 4). The median change in FGF19 concentration from baseline to 6 hours after dosing was significantly higher (P < .05) relative to placebo in the 30, 100, 300, 1000, and 3000 µg SAD cohorts; the 30, 60, and 100 µg MAD lean cohorts on day 1; and the 30‐ and 100 µg MAD lean cohorts on day 13.
Figure 2.
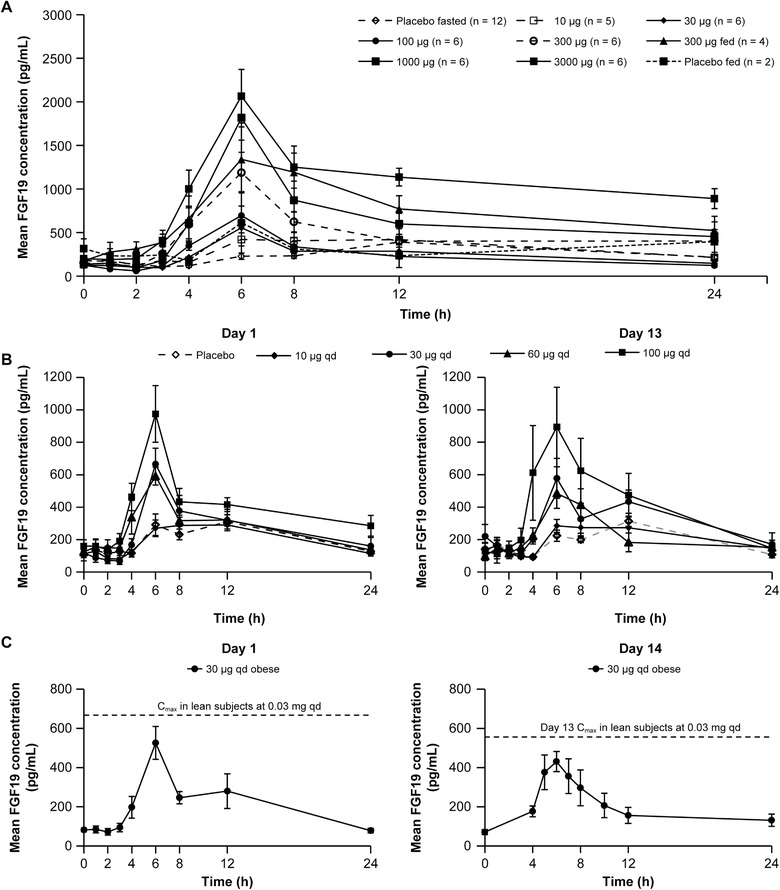
Pharmacodynamic effect of single and multiple ascending doses of tropifexor in lean or obese healthy volunteers (PD analysis set). (A) Mean fibroblast growth factor 19 (FGF19) concentration‐time profile following single ascending doses of tropifexor in fasted subjects and following a single oral dose of 300 µg under fed condition. (B) Day 1 and day 13 mean FGF19 concentration‐time profile in lean volunteers receiving multiple ascending doses of tropifexor. (C) Day 1 and Day 14 mean FGF19 concentration‐time profile in obese healthy volunteers receiving a once‐daily dose of 30 µg tropifexor for 14 days, in comparison with lean healthy volunteers. Data are represented as mean ± standard error.
Table 4.
Summary of PD Parameters of FGF19 Following Single and Multiple Ascending Doses
| n | AUC0‐24 h (pg • h/mL) | Cmax (pg/mL) | Maximum Change From Baseline (pg/mL) | Difference vs Placebo (pg/mL) | ||
|---|---|---|---|---|---|---|
| SAD | Median (Range) Mean (SD) | Median (95%CI) | ||||
| Dose (µg) | 6‐h After Dose | |||||
| 10 | 5 |
1250 (1040‐1410) 1230 (138) |
438 (337‐773) 489 (168) |
265 (156‐665) 313 (205) |
196.5 (−0.7 to 384.0) | |
| 30 | 6 |
980 (733‐2450) 1260 (662) |
416 (267‐1520) 609 (479) |
312 (168‐1330) 484 (438) |
235.4 (60.4‐676.0) | |
| 100 | 6 |
1450 (1020‐2130) 1540 (498) |
673 (325‐991) 704 (252) |
607 (291‐795) 578 (213) |
534.6 (320.0‐790.0) | |
| 300 | 6 |
2460 (782‐4900) 2660 (1500) |
1440 (447‐1730) 1210 (535) |
1250 (353‐1470) 1060 (485) |
1120.0 (405.2‐1463.0) | |
| 300 (fed) | 4 |
3090 (1720‐5690) 3400 (1910) |
1140 (769‐2380) 1350 (726) |
976 (567‐2200) 1180 (728) |
||
| 1000 | 6 |
3410 (1730‐4440) 3220 (985) |
1820 (1030‐2570) 1820 (630) |
1700 (963‐2430) 1680 (597) |
1679.0 (1058.0‐2244.0) | |
| 3000 | 6 |
4240 (2530‐6800) 4470 (1750) |
1750 (1400‐3250) 2070 (752) |
1570 (1260‐2950) 1860 (682) |
1554.0 (1290.3‐2563.0) | |
| MAD | Median (Range) Mean (SD) | Median (95%CI) | ||||
|---|---|---|---|---|---|---|
| qd Dose (µg) | 6‐h After Dose | |||||
| 10 | Day 1 | 9 |
737 (361‐1470) 806 (352) |
360 (215‐605) 380 (126) |
270 (7.00‐471) 267 (151) |
2.4 (−163.5 to 135.7) |
| Day 13 | 9 |
883 (615‐1030) 860 (151) |
405 (177‐613) 375 (126) |
245 (−62.0 to 538) 262 (158) |
81.35 (−55.6 to 187.3) | |
| 30 | Day 1 | 6 |
1000 (737‐2060) 1210 (529) |
626 (469‐1070) 691 (238) |
595 (207‐892) 574 (244) |
392.8 (113.8‐623.6) |
| Day 13 | 6 |
1330 (602‐2070) 1380 (557) |
649 (262‐1040) 607 (290) |
538 (248‐737) 490 (197) |
357.0 (140.8‐552.7) | |
| 30 (obese)a | Day 1 | 7 |
1040 (435‐1860) 1110 (495) |
518 (178‐918) 551 (242) |
460 (104‐845) 469 (237) |
299.70 (–37.5 to 649.3) |
| Day 14 | 7 |
1050 (648‐1960) 1180 (447) |
479 (226‐877) 531 (238) |
418 (152‐752) 449 (217) |
117.05 (–285.7 to 304.10) | |
| 60 | Day 1 | 6 |
1370 (983‐3140) 1610 (780) |
581 (462‐847) 592 (137) |
456 (306‐706) 469 (138) |
323.2 (131.8‐490.6) |
| Day 13 | 6 |
1140 (640‐2090) 1280 (612) |
597 (235‐829) 573 (230) |
475 (131‐735) 450 (258) |
237.5 (9.2‐529.4) | |
| 100 | Day 1 | 6 |
2320 (1310‐3190) 2240 (725) |
936 (428‐1660) 981 (424) |
731 (350‐1480) 821 (422) |
541.55 (254.80‐1094.0) |
| Day 13 | 4 |
2290 (743‐4000) 2330 (1470) |
1150 (309‐1470) 1020 (502) |
1050 (231‐1290) 907 (469) |
671.65 (144.3‐1179.6) | |
AUC0‐24h, area under the plasma concentration–time curve from time 0 to 24 h; Cmax, observed maximum plasma concentration following drug administration; CI, confidence interval; FGF19, fibroblast growth factor 19; MAD, multiple ascending dose; PD, pharmacodynamic; qd, once daily; SAD, single ascending dose.
Bolded values indicate P < .05 vs placebo by Wilcoxon rank‐sum test.
Day 14 assessment was performed in obese subjects instead of Day 13.
Similar results were observed in obese subjects receiving 30‐µg qd doses for 14 days (Table 4). Nevertheless, median FGF19 AUC0‐24h and Cmax and change in concentration from baseline relative to placebo in obese subjects were numerically lower than those in lean subjects at the same dose on both days 1 and 13/14, except for median AUC0‐24h on day 1. In the obese subjects, the median difference in change from baseline FGF19 relative to placebo was numerically higher but not significantly different (P > .05), on both day 1 and day 14 (Figure 2C and Table 4).
Concomitant with the elevation in postdose FGF19 levels, there was a dose‐dependent decline in the levels of 7‐ α‐hydroxy‐4‐cholesten‐3‐one (C4), an intermediate in the bile acid biosynthesis pathway. Following multiple dosing, the median change from baseline in C4 with the 30‐µg dose between lean and obese subjects was similar on day 1 (−18.3 and −21.4 pg/mL) and day 13/14 (−17.0 and −17.1 pg/mL; Table S3).
An approximately linear PK‐PD relationship was found between tropifexor exposure and FGF19 exposure as shown by scatter plots of tropifexor Cmax vs FGF19 Cmax (SAD: if tropifexor Cmax ≤40 ng/mL) and tropifexor AUC0‐24h vs FGF19 AUC0‐24h in both SAD and MAD studies (Figure 3).
Figure 3.
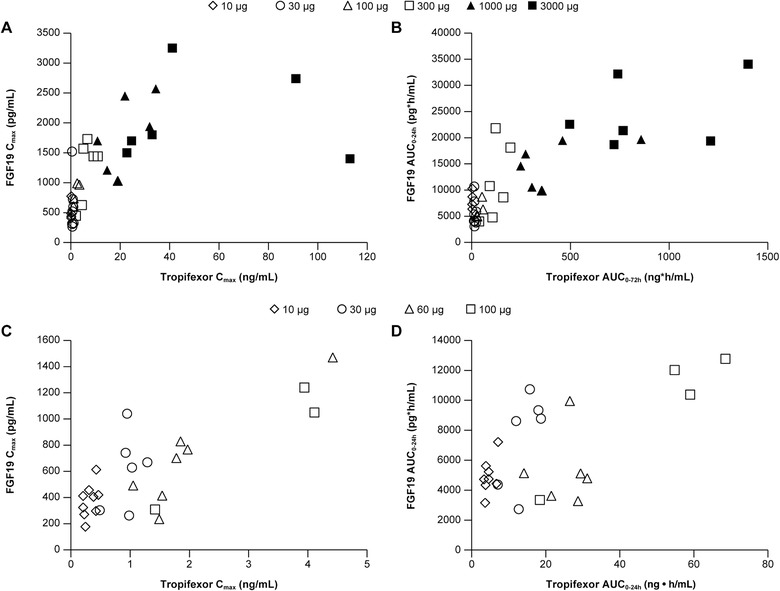
Pharmacokinetic‐pharmacodynamic relationship. Scatter plots of (A) FGF19 observed maximum plasma concentration following drug administration (Cmax) vs tropifexor Cmax and (B) FGF area under the plasma concentration‐time curve from time 0 to 24 h (AUC0‐24h) vs tropifexor AUC0‐24h following single ascending doses of tropifexor in fasted subjects. Scatter plots of (C) FGF Cmax vs tropifexor Cmax and (D) FGF AUC0‐24h vs tropifexor AUC0‐24h following multiple ascending doses of tropifexor in lean healthy subjects.
Blood Chemistry Panel and Serum Biomarkers
With regard to triglycerides and total, HDL, and LDL cholesterol, no clinically significant changes were observed from baseline compared to placebo in the SAD and MAD studies.
In obese healthy volunteers, no clinically significant differences were observed in baseline‐normalized triglycerides and total, LDL, and HDL cholesterol between tropifexor‐treated and placebo‐treated groups on day 14 before the last dose (Figure 4).
Figure 4.
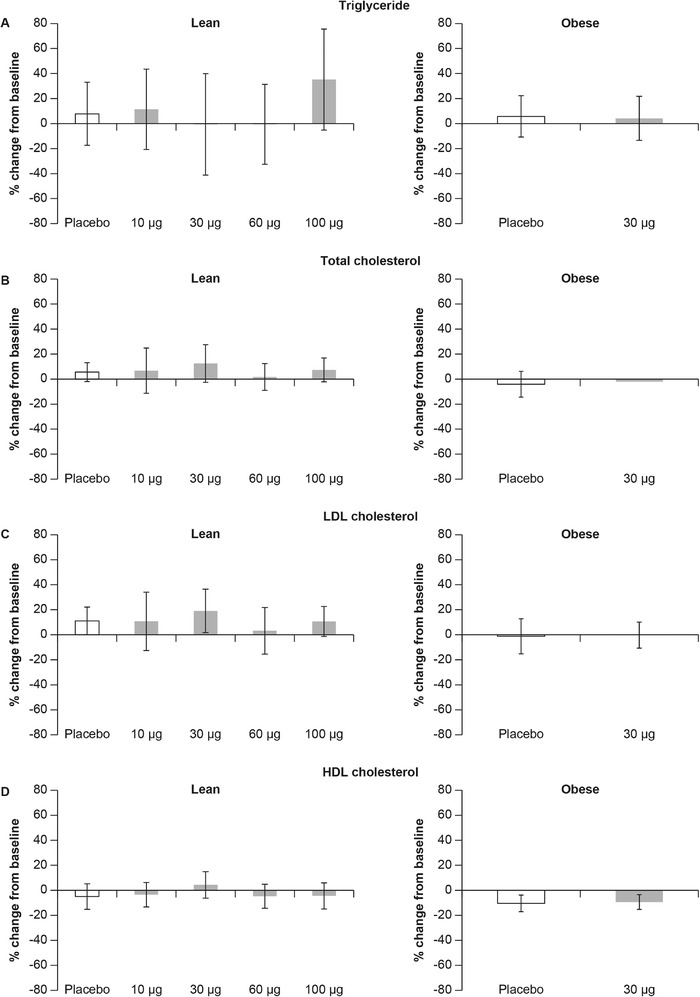
Effect of tropifexor on lipids. Day 14 percent change from baseline in levels of (A) triglycerides, (B) total cholesterol, (C) low‐density lipoprotein (LDL) cholesterol, and (D) high‐density lipoprotein (HDL) cholesterol following multiple doses of tropifexor for 14 days in lean and obese healthy volunteers. Data are represented as mean ± standard deviation.
No statistically significant reduction in fasting total serum bile acids were observed up to day 4, 72 hours after dosing in SAD. During this part of the study, the placebo adjusted ratio to baseline ranged at day 2 from 0.929 to 1.211 for 30 µg and 100 µg tropifexor respectively; at day 3 from 0.953 to 1.461 for 30 µg and 10‐µg tropifexor, respectively; and at day 4 from 1.099 to 1.589 for 1000 µg and 300 µg tropifexor, respectively. Similarly, no significant reduction in fasting total serum bile acids was observed at day 15 in MAD. At this time point, the placebo adjusted ratio to baseline ranged from 0.554 to 1.699 for 60 µg and 30 µg tropifexor, respectively, in the lean cohorts and 1.117 for 30 µg tropifexor in the obese cohort.
Discussion
This first‐in‐human study was performed based on promising preclinical validation results of tropifexor in various animal models of NASH and cholestasis19, 20 and acceptable preclinical safety assessments, to evaluate the safety, tolerability, PK, and PD of tropifexor following single‐ascending doses in the fasted or fed state and multiple‐ascending doses in lean and obese volunteers. A double‐blind placebo arm was included to provide unbiased collection of safety and tolerability data, and the effect of food was explored to gain an early understanding of impact of food on PK and PD effect of tropifexor.
Approximately dose‐proportional increases in both Cmax and AUC were observed following tropifexor single doses up to 3000 µg and multiple doses up to 100 µg. Consistent with the observed t1/2 (range, 13.5‐21.9 hours), steady‐state tropifexor levels were reached by day 4 with <2‐fold accumulation following a once‐daily dosing regimen. Moderate‐to‐slow absorption (median tmax of ∼4 hours) was observed following single or multiple oral dosing, possibly due to the low aqueous solubility of tropifexor.18 Overall, tropifexor demonstrated pharmacokinetic properties that would support a once‐daily dosing regimen in future clinical trials.
Tropifexor is likely a Biopharmaceutics Classification System Class II compound due to its low solubility and high permeability.18 As anticipated, when tropifexor was taken with a high‐fat meal, there was an increase of ∼60% in the systemic exposure of tropifexor and its metabolite CKS577. Although there was a trend toward an increase in the median AUC0‐24 for FGF19 in the fed state, the overall median Cmax and median maximal change from baseline were comparable during the food effect study (Table 4). The potential for formulation optimization to mitigate the effect of food on the pharmacokinetics and pharmacodynamics of tropifexor will be addressed in future studies.
Results from earlier in vitro metabolism studies showed that CKS577 (acyl glucuronide) was detected in the hepatocytes of all animal species incubated with high concentrations of 14C‐tropifexor and appeared to be the primary metabolite in human hepatocytes (data not shown). This indicated that glucuronidation contributed to the metabolism pathway for tropifexor. CKS577 is at least 10‐fold less potent than tropifexor on FXR and only partially active (∼58%) on cellular assays for FXR activity (data not shown).
In both the SAD and MAD parts of the study, the exposure of CKS577 in plasma was minimal (<3% as compared to the parent compound). The low exposure of this metabolite in human subjects and its relatively weak potency as an FXR agonist compared to its parent tropifexor, suggests CKS577 will have no meaningful contribution to the overall observed pharmacodynamic effects, as well as a low likelihood that tropifexor‐related AEs would be caused by this metabolite.
Increases in FGF19 exposures were observed with single doses of tropifexor from 10 µg to 1000 µg for median Cmax or to 3000 µg for median AUC0‐24 and with daily dosing of tropifexor for 13 days from 10 to 100 µg, thereby confirming FXR target engagement in the intestine.24 Moreover, a near linear exposure‐response relationship was observed following single or multiple dosing of tropifexor (Figure 3).
Because a high percentage of patients with NASH are obese,16, 17 and bile acid FXR agonists have been associated with an increase in LDL cholesterol, a cohort of obese (median BMI, 41 kg/m2) healthy volunteers was included in the MAD part of the study to evaluate PD effects on cholesterol. The mean steady‐state exposure (AUC and Cmax) in obese subjects appeared to be slightly lower than that in lean subjects. Similarly, the median steady‐state FGF19 exposure (day 14) in obese subjects was also slightly lower compared to lean subjects on day 13/14 (Table 3). In this cohort, no adverse changes were noted in the cholesterol profile in the active group when compared to those receiving placebo. However, given the low number of subjects, a further population PK/PD analysis with more robust clinical data is warranted.
In transgenic mice overexpressing human FGF19, prolonged exposure to constantly very high circulating levels of FGF19 (mean serum concentration of up to 77.7 ng/mL) throughout early life has been associated with the development of hepatocellular carcinoma.25, 26 Multiple dosing of tropifexor resulted in dose‐dependent, yet transient, elevations of FGF19. At the highest dose tested in the MAD part of the study (100 µg), the median maximum change from baseline of 1050 pg/mL was approximately 4.4‐fold of that seen in response to a meal, indicating that tropifexor treatment is unlikely to yield the sustained, very high FGF19 levels observed in the rodent transgenic models cited above.25, 26 Furthermore, numerous studies suggest that FXR suppresses tumorigenesis in target tissues.27, 28, 29
C4 is a key intermediate in the bile acid biosynthesis pathway and a biomarker of FXR target engagement in the liver.30 Dose‐dependent decreases in C4 levels from baseline were observed up to the 3000‐µg dose in the SAD study and with 30‐ and 100‐µg doses until day 15 in the MAD study. However, C4 levels were below LLOQ at most doses, a finding that may be attributed to the low resting levels of C4 in healthy volunteers.
Tropifexor was well tolerated at single doses up to and including 3000 µg and in multiple doses up to 60 µg, with a low incidence of AEs. The main dose‐limiting factor in this healthy volunteer population was an increase in ALT levels in the absence of meaningful changes in markers of liver damage such as GGT and bilirubin. These increases in ALT levels may represent an adaptation response to FXR agonist–mediated reduction in hepatic cholesterol synthesis that maintains cellular homeostasis in the context of decreased demand for bile acid production.
No meaningful changes in plasma lipid levels were observed compared to placebo with single or multiple dosing of tropifexor in either lean or obese healthy volunteers dosed for up to 2 weeks. In contrast, in healthy volunteer studies,31 OCA has been found to cause significant elevation in LDL and reduction in HDL cholesterol, and these findings are paralleled in patients with NASH.16, 17 Although the size of the obese cohort was chosen to detect an OCA‐like effect on plasma lipids, the effect of tropifexor on plasma lipids needs to be validated in larger cohorts with longer dosing regimens and in patients groups with metabolic parameters more closely aligned with NASH patients.
Pruritus has been a frequently reported AE in studies with the bile acid–derived FXR agonist OCA.14, 17 Pruritus is a well‐recognized underlying symptom in many patients with PBC14 and is also noted in placebo‐treated patients with NASH.17 However, the mechanism of pruritus induction by FXR agonists is not well understood. It may involve activation of the G‐protein coupled bile acid receptor TGR5,32,33 either directly or indirectly by a drug metabolite, or pruritus may reflect an alteration of the bile acid pool such that an endogenous pruritogenic bile acid or bile acid metabolite is produced. Tropifexor did not cause pruritus in healthy volunteers up to 2 weeks following daily dosing potentially owing to its high specificity, lack of activity on TGR5, shorter duration of FXR agonism in the liver, or non–bile acid–derived structure. However, these findings need to be validated in larger cohorts of patients treated with tropifexor for longer durations.
Conclusions
Results from this first‐in‐human study indicate that tropifexor is well tolerated at pharmacologically active doses and has an acceptable safety profile, with no drug‐induced pruritus and only mild and transient elevations in serum ALT. Furthermore, tropifexor has a PK profile favorable for once‐daily dosing; shows effective FXR target engagement in the intestine via transient and dose‐dependent elevations in FGF19; and causes no significant changes in total, HDL, and LDL cholesterol levels in lean or obese healthy volunteers up to 2 weeks following daily dosing.
Conflicts of Interest
M.K.B., J.C., S.D., S.V., J.Z., and L.B.K. are employees of Novartis Institutes for BioMedical Research. S.N. is an employee of Novartis Healthcare Pvt. Ltd. M.K.B., B.L., and L.B.K. are stockholders of Novartis. L.G. and K.D. have no financial interests to disclose. The study was funded by Novartis Institutes for Biomedical Research.
Funding
The study was funded by Novartis.
Data‐Sharing Statement
In accordance with Novartis policy on sharing of clinical trial data for phase 1 studies available at https://www.clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Novartis.aspx, data from this study have not been shared on a public repository.
Supporting information
Figure S1. Design of the first‐in‐human study. The study was conducted in 2 parts, SAD and MAD.
Figure S1. Design of the first‐in‐human study. The study was conducted in 2 parts, SAD and MAD.
Table S1. Summary of plasma PK parameters of CKS577 following single and multiple ascending doses of tropifexor.
Table S2. Summary of urine PK parameters of tropifexor and CKS577 following single ascending dose on day 1 and following multiple ascending doses on day 13.
Table S3. Summary of pharmacodynamic parameters of C4 on days 1 and 14 following multiple ascending doses in lean and obese healthy volunteers.
Acknowledgments
The authors thank Yiming Zhang for providing statistical inputs and for review of the manuscript draft and Prachiti Narvekar, Novartis Healthcare Pvt. Ltd., for medical writing support and editorial assistance.
Fellows of the American College of Clinical Pharmacology (FCP): None
References
- 1. Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284(5418):1362‐1365. [DOI] [PubMed] [Google Scholar]
- 2. Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science.1999;284(5418):1365‐1368. [DOI] [PubMed] [Google Scholar]
- 3. Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3(5):543‐553. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Kast‐Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem. 2003;278(1):104‐110. [DOI] [PubMed] [Google Scholar]
- 5. Lee H, Zhang Y, Lee FY, Nelson SF, Gonzalez FJ, Edwards PA. FXR regulates organic solute transporters alpha and beta in the adrenal gland, kidney, and intestine. J Lipid Res. 2006;47(1):201‐214. [DOI] [PubMed] [Google Scholar]
- 6. Zollner G, Marschall HU, Wagner M, Trauner M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm. 2006;3(3):231‐251. [DOI] [PubMed] [Google Scholar]
- 7. Kir S, Kliewer SA, Mangelsdorf DJ. Roles of FGF19 in liver metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:139‐144. [DOI] [PubMed] [Google Scholar]
- 8. Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol‐activated receptors LXR and FXR. Nat Rev Mol Cell Biol. 2012;13(4):213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731‐744. [DOI] [PubMed] [Google Scholar]
- 10. Lambert G, Amar MJ, Guo G, Brewer HB Jr, Gonzalez FJ, Sinal CJ. The farnesoid X‐receptor is an essential regulator of cholesterol homeostasis. J Biol Chem. 2003;278(4):2563‐2570. [DOI] [PubMed] [Google Scholar]
- 11. Poupon RE, Lindor KD, Cauch‐Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113(3):884‐890. [DOI] [PubMed] [Google Scholar]
- 12. Poupon RE, Lindor KD, Pares A, Chazouilleres O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol. 2003;39(1):12–16. [DOI] [PubMed] [Google Scholar]
- 13. Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148(4):751‐761.e8. [DOI] [PubMed] [Google Scholar]
- 14. Nevens F, Andreone P, Mazzella G, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631‐643. [DOI] [PubMed] [Google Scholar]
- 15. Angulo P, Jorgensen RA, Lindor KD. Incomplete response to ursodeoxycholic acid in primary biliary cirrhosis: is a double dosage worthwhile? Am J Gastroenterol. 2001;96(11):3152‐3157. [DOI] [PubMed] [Google Scholar]
- 16. Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(3):574‐582.e1. [DOI] [PubMed] [Google Scholar]
- 17. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet. 2015;385(9972):956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tully DC, Rucker PV, Chianelli D, et al. Discovery of tropifexor (LJN452), a highly potent non‐bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J Med Chem. 2017;60(24):9960‐9973. [DOI] [PubMed] [Google Scholar]
- 19. Hernandez ED, Zheng L, Kim Y, et al. Tropifexor‐mediated abrogation of steatohepatitis and fibrosis is associated with the antioxidative gene expression profile in rodents. Hepatol Commun. 2019;3(8):1085‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laffitte B, Young K, Joseph S, et al. A novel, highly potent, non‐bile acid FXR agonist for the treatment of NASH and cholestasis. Hepatol Int. 2016;10(S1):S97. [Google Scholar]
- 21. Sarwar R, Pierce N, Koppe S. Obesity and nonalcoholic fatty liver disease: current perspectives. Diabetes Metab Syndr Obes. 2018;11:533‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masuoka HC, Chalasani N Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals. Ann N Y Acad Sci. 2013;1281:106‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pournaras DJ, Glicksman C, Vincent RP, et al. The role of bile after Roux‐en‐Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology. 2012;153(8):3613‐3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217‐225. [DOI] [PubMed] [Google Scholar]
- 25. Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160(6):2295‐2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell. 2011;19(3):347‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Ge X, Heemstra LA, et al. Loss of FXR protects against diet‐induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol Endocrinol. 2012;26 (2):272‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo F, Xu Z, Zhang Y, et al. FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget. 2015;6(33):34606‐34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He J, Zhao K, Zheng L, et al. Upregulation of microRNA‐122 by farnesoid X receptor suppresses the growth of hepatocellular carcinoma cells. Mol Cancer. 2015;14:163‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keely SJ, Walters JR The farnesoid X receptor: good for BAD. Cell Mol Gastroenterol Hepatol. 2016;2(6):725‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pencek R, Marmon T, Roth JD, Liberman A, Hooshmand‐Rad R, Young MA. Effects of obeticholic acid on lipoprotein metabolism in healthy volunteers. Diabetes Obes Metab. 2016;18(9):936‐940. [DOI] [PubMed] [Google Scholar]
- 32. Alemi F, Kwon E, Poole DP, et al. The TGR5 receptor mediates bile acid‐induced itch and analgesia. J Clin Invest. 2013;123(4):1513‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lieu T, Jayaweera G, Zhao P, et al. The bile acid receptor TGR5 activates the TRPA1 channel to induce itch in mice. Gastroenterology. 2014;147(6):1417‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Design of the first‐in‐human study. The study was conducted in 2 parts, SAD and MAD.
Figure S1. Design of the first‐in‐human study. The study was conducted in 2 parts, SAD and MAD.
Table S1. Summary of plasma PK parameters of CKS577 following single and multiple ascending doses of tropifexor.
Table S2. Summary of urine PK parameters of tropifexor and CKS577 following single ascending dose on day 1 and following multiple ascending doses on day 13.
Table S3. Summary of pharmacodynamic parameters of C4 on days 1 and 14 following multiple ascending doses in lean and obese healthy volunteers.