

Abstract
Androgen deprivation therapy (ADT) is first‐line palliative treatment in androgen receptor‐positive (AR+) salivary duct carcinoma (SDC), and response rates are 17.6–50.0%. We investigated potential primary ADT resistance mechanisms for their predictive value of clinical benefit from ADT in a cohort of recurrent/metastatic SDC patients receiving palliative ADT (n = 30). We examined mRNA expression of androgen receptor (AR), AR splice variant‐7, intratumoral androgen synthesis enzyme‐encoding genes AKR1C3, CYP17A1, SRD5A1 and SRD5A2, AR protein expression, ERBB2 (HER2) gene amplification and DNA mutations in driver genes. Furthermore, functional AR pathway activity was determined using a previously reported Bayesian model which infers pathway activity from AR target gene expression levels. SRD5A1 expression levels and AR pathway activity scores were significantly higher in patients with clinical benefit from ADT compared to those without benefit. Survival analysis showed a trend toward a longer median progression‐free survival for patients with high SRD5A1 expression levels and high AR pathway activity scores. The AR pathway activity analysis, and not SRD5A1 expression, also showed a trend toward better disease‐free survival in an independent cohort of locally advanced SDC patients receiving adjuvant ADT (n = 14) after surgical tumor resection, and in most cases a neck dissection (13/14 patients) and postoperative radiotherapy (13/14 patients). In conclusion, we are the first to describe that AR pathway activity may predict clinical benefit from ADT in SDC patients, but validation in a prospective study is needed.
Keywords: salivary gland neoplasms, salivary duct carcinoma, androgen receptor antagonists, drug resistance, neoplasm, computational biology
Short abstract
What's new?
Androgen deprivation therapy (ADT) is a leading treatment strategy in the palliative care of patients with androgen receptor (AR)‐positive salivary duct carcinoma (SDC). However, while as many as half of patients may respond to ADT, resistance frequently emerges, undermining its use. In this investigation of primary ADT resistance mechanisms, expression of the androgen synthesis enzyme‐encoding gene SRD5A1 and functional activity of the AR pathway were found to predict clinical benefit from ADT in SDC patients. High AR pathway activity scores were further linked to improved disease‐free survival in SDC patients with locally advanced disease who received adjuvant ADT.
Abbreviations
- ADT
androgen deprivation therapy
- AKR1C3
aldo‐keto reductase family 1 member C3
- AR
androgen receptor
- ASCO
American Society of Clinical Oncology
- CAP
College of American Pathologists
- CI
confidence interval
- CRPC
castration‐resistant prostate cancer
- CYP17A1
cytochrome P450 17A1
- DFS
disease‐free survival
- FFPE
formalin‐fixed paraffin‐embedded
- FISH
fluorescent in situ hybridization
- H&E
hematoxylin and eosin
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- IQR
interquartile range
- LA
locally advanced
- OS
overall survival
- PFS
progression‐free survival
- R/M
recurrent/metastatic
- ROC
receiver operating characteristic
- SDC
salivary duct carcinoma
- smMIP
single‐molecule molecular inversion probe
- SRD5A1/2
steroid 5 alpha‐reductase 1/2
Introduction
Salivary duct carcinoma (SDC) is an aggressive subtype of salivary gland cancer, which is often androgen receptor (AR) positive (66.7–96.4%).1, 2, 3 Primary treatment consists of a tumor resection, most often in combination with a neck dissection and postoperative radiotherapy. Despite this extensive treatment, the 3‐year disease‐free survival (DFS) rate is only 27.7% in locally advanced patients.4 In patients with recurrent and/or metastatic (R/M) SDC, androgen deprivation therapy (ADT) is often used as first‐line palliative treatment. In retrospective studies, ADT has shown response rates of 17.6–50.0% and an OS of 17 months compared to 5 months in a best supportive care cohort.5, 6 A recent prospective phase 2 trial in Japan showed a response rate of 41.7%, median progression‐free survival (PFS) of 8.8 months and median OS of 30.5 months.7 Because of the efficacy of ADT in R/M SDC patients, we evaluated ADT as adjuvant treatment in 22 patients with locally advanced (LA) AR‐positive SDC. Multivariable Cox regression analysis showed a significantly improved DFS (hazard ratio 0.14, 95% CI 0.03–0.75, p = 0.022) and OS (hazard ratio 0.06, 95% CI 0.01–0.76, p = 0.030) compared to 111 controls who did not receive adjuvant ADT.4
Besides ADT, other treatment options are available for patients with R/M SDC. In the case of ERBB2 (HER2) gene amplification (29.4–46.4%),1, 2 patients can be treated with docetaxel plus trastuzumab, showing an overall response rate of 70.2% and median PFS of 8.9 months.8 Double HER2 blockade with docetaxel–trastuzumab–pertuzumab or in second‐line with the antibody‐drug conjugate trastuzumab‐emtansine also showed promising results.9, 10, 11 Finally, the high frequency (61.3%) of oncogenic driver gene mutations offers personalized treatment options.12
Despite the efficacy of ADT in the palliative and adjuvant setting, ADT is only effective in a subgroup of patients and little is known about primary resistance mechanisms. Although AR expression, determined by immunohistochemistry, is a hallmark of SDC, intratumoral and intertumoral variation of AR expression is frequently observed.13 Therefore, variation in AR mRNA and protein levels may cause variable responses. Furthermore, AR‐V7, an AR splice variant that lacks the ligand‐binding domain and is constitutively active, may cause ADT resistance. In prostate cancer AR‐V7 expression is 20‐fold higher in castration‐resistant prostate cancer (CRPC) compared to hormone‐naïve prostate cancer, though in SDC the presence of AR‐V7 has also been shown in hormone‐naïve tumors.14, 15 Another ADT resistance mechanism described in CRPC is increased expression of genes involved in intratumoral androgen synthesis.16 Key enzymes involved in the conversion of androgen precursors, such as dehydroepiandrosterone into dihydrotestosterone are aldo‐keto reductase family 1 member C3 (AKR1C3), cytochrome P450 17A1 (CYP17A1), steroid 5 alpha‐reductase 1 (SRD5A1) and SRD5A2. Finally, a low‐active or inactive AR signal transduction pathway, in which androgen stimulation does not result in (full) AR transcriptional activity, may cause primary ADT resistance, simply because no effective AR signaling is present. AR pathway activity can be quantified by a recently developed and validated method, in which expression of AR target genes is measured and subsequently converted into a pathway activity score (ranging between 0 and 100) using a Bayesian computational model.17, 18 Besides, these AR‐related mechanisms, primary ADT resistance may be caused by activity of other tumor‐driving pathways, for instance induced by ERBB2 gene amplification or other tumor‐driving gene mutations. The aim of our study was to assess these potential primary ADT resistance mechanisms in a cohort of R/M SDC patients receiving palliative ADT and a cohort of LA SDC patients receiving adjuvant ADT. For those factors that differed significantly between R/M SDC patients with and without clinical benefit from ADT, the optimal cut‐off value and survival differences were assessed. Subsequently, this cut‐off value was used to evaluate DFS differences in the LA cohort.
Methods
Patients
Clinicopathological characteristics and potential ADT resistance mechanisms were assessed in a cohort of R/M AR‐positive SDC patients receiving palliative ADT (n = 30) and a cohort of LA AR‐positive SDC patients receiving adjuvant ADT (n = 14) after surgical tumor resection, and in most cases a neck dissection (13/14 patients) and postoperative radiotherapy (13/14 patients). ADT consisted of bicalutamide or LHRH‐analog plus bicalutamide following shared decision making.5 Patients were treated in the Radboud university medical center, Nijmegen, the Netherlands, or received a second opinion in the Radboud university medical center and were treated under supervision elsewhere in the Netherlands.
Our study was approved by the local medical ethical committee (file code 2017‐3917). Clinical data were collected from the medical records. A no‐objection system was used for secondary use of human tissue and medical data in accordance with the code of conduct of the Federation of Dutch Medical Scientific Societies (Human tissue and medical research: Code of conduct for responsible use). For all patients who were alive at the start of our study, written informed consent was obtained.
Tissue
Formalin‐fixed paraffin‐embedded (FFPE) tissue samples of the primary tumor prior to treatment were collected (n = 36). If primary tumor material was unavailable, tumor material of locoregional lymph node metastases or distant metastases prior to treatment was used (n = 5 and n = 3, respectively). Nonstained FFPE sections were cut and used for DNA/RNA isolation, AR immunohistochemistry (IHC) and ERBB2 FISH. Before and after these sections, hematoxylin and eosin (H&E) stained sections were prepared for pathological confirmation of SDC and for tumor annotation. Median percentage of neoplastic cells in tissue sections used for further analysis was 70.0% (range 25.0–90.0%) in the R/M cohort and 70.0% (range 30.0–80.0%) in the LA cohort.
RNA isolation
For RNA extraction, three 10 μm sections were used. RNA was extracted from the marked tumor regions according to the manufacturer's protocol (Siemens, VERSANT® Tissue Preparation Reagents kit). RNA was eluted in 100 μl buffer according to the instructions. After DNase treatment (Siemens, Munich, Germany, VERSANT® Tissue Preparation Reagents kit) RNA was stored at −80°C. RNA concentration was measured using the Qubit® RNA HS Assay Kit (Thermo Fisher, Waltham, MA) on a Qubit® Fluorometer.
Reverse transcriptase and real‐time PCR analysis
One microgram of total DNase‐treated RNA was used for cDNA synthesis. Random‐primed cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA). Gene expression levels were determined by SYBR Green qPCR (Roche, Basel, Switzerland) using a LightCycler LC480 instrument (Roche). Relative gene expression levels of AR, AR‐V7, AKR1C3, CYP17A1, SRD5A1 and SRD5A2 were calculated using the ΔΔCt method using the hypoxanthine phosphoribosyltransferase 1 (HPRT1) gene for normalization. Primer sequences are listed in Supporting Information Table S1.
AR immunohistochemical staining
Androgen receptor expression was determined using the AR specific polyclonal antibody N‐20 (Santa Cruz Biotechnology, Santa Cruz, CA). FFPE sections were pretreated with citrate (pH 6.0) for 10 min in a pretreatment module. Immunostaining was carried out using a 1:200 dilution of the primary antibody, and staining was performed according to the Powervision method by Immunologic. The AR staining was scored considering the staining intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong) and the percentage of positive nuclei (0 = <10%, 1 = 10–30%, 2 = 30–70%, 3= >70%). The final staining score was recorded as the sum of the staining intensity and the staining extent.6
AR pathway analysis
To quantitatively assess the AR pathway activity a qPCR‐based AR pathway test was used, that was adapted from the previously described biologically validated AR pathway analysis method developed for Affymetrix U133Plus2.0 microarray.17, 18, 19 The qPCR AR pathway test was developed to allow the use of FFPE material and small sample inputs (Philips Molecular Pathway Diagnostics, Eindhoven, The Netherlands). For the qPCR‐based AR pathway test, the relevant AR target gene expression levels were measured on FFPE tissue‐derived total RNA using one‐step RT‐qPCR and a Bayesian pathway model was used to quantitatively assess pathway activity.18 For the target genes and reference genes, multiple diagnostic grade qPCR assays were developed according to standard procedures. A human AR‐positive prostate carcinoma cell line (LNCaP) with ground‐truth data with respect to AR pathway activity was used for calibration of the qPCR‐based Bayesian model, similar as described for the Affymetrix‐based model.18 Based on a probability score (p), the odds for AR pathway activity was provided by the Bayesian model (p/1 − p), and this was transformed into a (base 2) logarithmic scale and then normalized to scores ranging from 0 to 100, where 0 corresponds to the lowest and 100 corresponds to the highest odds in favor of an active AR pathway that a specific model can infer. For each analyzed sample, this normalized AR pathway activity score between 0 and 100 was calculated.
HER2 assessment
HER2 status was determined by ERBB2 fluorescent in situ hybridization (FISH) according to standard ISH protocol using the ERBB2 probe of Kreatech (KI‐10701, mapping 17q12). Scoring was performed according to breast cancer guidelines of the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP).20 In case of an inconclusive ISH results, HER2 IHC was determined using the polyclonal rabbit antihuman c‐erbB‐2 antibody of DAKO according to protocol.
DNA isolation and mutation analysis
For all patients in the R/M cohort, oncogenic driver gene mutations were assessed using single‐molecule molecular inversion probes (smMIPs) analysis and Next‐Generation Sequencing.21 DNA from FFPE tissue specimens was isolated as described and a validated 29‐gene panel was used to detect driver mutations (Supporting Information Table S2).
Statistical analysis
Clinical ADT response in the R/M cohort was classified into five categories according to RECIST criteria: complete response, partial response, stable disease longer than 6 months, stable disease shorter than 6 months and progressive disease.22 Subsequently, patients with a complete response, partial response or stable disease >6 months were classified as “clinical benefit” Gene expression levels and AR pathway activity scores in tumor tissue of patients with and without clinical benefit compared by a two‐tailed independent t‐test with equal variance assumed. Equal variance was confirmed by Levene's test for equality of variances. A log transformation, using the natural logarithms, was performed on gene expression data before t‐tests were conducted in order to satisfy the assumption of normal distribution. AR protein expression levels in tumor tissue of patients with and without clinical benefit were compared using the Mann–Whitney U test. HER2 status and the presence or the absence of a driver mutation in patients with and without clinical benefit were compared using Fisher's exact test. Values of p < 0.05 were considered statistically significant without correction for multiple testing, as these analyses are considered exploratory.
For those factors that differed significantly between patients with and without clinical benefit, receiver operating characteristic (ROC) curves were constructed to establish the optimal cut‐off value to predict clinical benefit. Subsequently, Kaplan–Meier survival curves were constructed for patients above and below the cut‐off value. The log‐rank test was used to compare survival data. Finally, Pearson correlations were calculated for AR and AR‐V7 gene expression levels, and for SRD5A1 gene expression levels and AR pathway activity scores. Analyses were performed using IBM SPSS version 25.0.
Results
Patient characteristics
In the R/M cohort, 30 patients (22 men, 8 women) who received palliative ADT were analyzed. Median age at the start of ADT was 62 years (range 36–79 years). Seven patients were treated with a LHRH‐analog plus bicalutamide, and 23 patients with bicalutamide monotherapy. Of these 30 patients, 5 had a partial response (16.7%), 4 had stable diseases for more than 6 months (13.3%), 3 had stable diseases for less than 6 months (10.0%) and 18 (60.0%) had progressive disease at first evaluation. Median FFPE tissue storage time was 49 months (range 7–195 months), and 23 samples were obtained from primary tumor, four from regional lymph nodes metastases and three from distant metastases. Further patient characteristics are listed in Table 1.
Table 1.
Patient and tumor characteristics of the recurrent/metastatic cohort, sorted by the AR pathway activity score
| Pt no. | Gender | Tumor tissue | Tumor % | Age tissue (mo.) | AR | AR‐V7 | AR IHC | AR pathway activity | AKR1C3 | SRD5A1 | SRD5A2 | HER2 status | Genetic alterations* | ADT | Clinical response | PFS (mo.) | OS (mo.) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | T | 70 | 46 | 0.278 | 0.005 | 5.0 | 33.1 | 0.072 | 0.637 | 0.001 | Pos | None | LHRH + bica | PD | 3 | 29+ |
| 2 | F | N | 60 | 45 | 0.030 | 0.000 | 4.0 | 33.9 | 0.712 | 0.865 | 0.000 | Neg | TP53 | Bica | PD | 2 | 5 |
| 3 | M | T | 50 | 136 | 0.260 | 0.001 | 6.0 | 34.8 | 2.099 | 3.117 | 0.001 | Pos | TP53 | Bica | PD | 3 | 9 |
| 4 | M | T | 60–70 | 133 | 0.345 | 0.004 | 5.0 | 36.1 | 0.061 | 1.741 | 0.008 | Pos | TP53 and ERBB2 | Bica | PD | 2 | 12 |
| 5 | F | T | 70 | 73 | 1.476 | 0.015 | 6.0 | 36.2 | 0.148 | 0.953 | 0.001 | Neg | PTEN and TP53 | Bica | PD | 3 | 7 |
| 6 | M | T | 25 | 50 | 1.097 | 0.008 | 5.0 | 38.9 | 0.075 | 0.933 | 0.003 | Neg | PTEN and TP53 | Bica | SD >6 mo. | 6 | 12 |
| 7 | F | N | 60–70 | 15 | 1.522 | 0.129 | 6.0 | 41.3 | 0.017 | 1.212 | 0.000 | Neg | None | LHRH + bica | PD | 9 | 14+ |
| 8 | M | T | 80 | 80 | 0.009 | 0.000 | 4.0 | 42.7 | 0.023 | 0.669 | 0.001 | Pos | TP53 | Bica | PD | 0 | 0 |
| 9 | M | T | 80 | 38 | 0.615 | 0.021 | 3.0 | 43.2 | 0.707 | 1.111 | 0.021 | Pos | None | Bica | PD | 2 | 34+ |
| 10 | M | T | 70 | 8 | 5.169 | 0.737 | 6.0 | 43.6 | 2.313 | 9.646 | 0.000 | Neg | None | Bica | PD | 2 | 3+ |
| 11 | F | M (epidural) | 30 | 7 | 0.063 | 0.001 | 0.0 | 43.7 | 0.323 | 0.674 | 0.001 | Pos | HRAS and PIK3CA | LHRH + bica | PD | 0 | 10 |
| 12 | M | T | 70 | 61 | 1.409 | 0.006 | 5.0 | 44.3 | 0.332 | 2.854 | 0.001 | Neg | AKT1 and BRAF | Bica | SD >6 mo. | 18 | 40+ |
| 13 | M | T | 70 | 66 | 1.251 | 0.006 | 6.0 | 45.3 | 0.093 | 3.622 | 0.000 | Pos | TP53 and ERBB2 | Bica | PR | 27 | 56+ |
| 14 | F | T | 70 | 195 | 3.227 | 0.454 | 6.0 | 45.4 | 0.732 | 5.540 | 0.001 | Neg | HRAS and PIK3CA | Bica | PD | 1 | 13 |
| 15 | F | T | 60 | 131 | 0.414 | 0.009 | 2.0 | 45.6 | 0.521 | 0.927 | 0.000 | Neg | None | LHRH + bica | PD | 2 | 5 |
| 16 | F | T | 40 | 48 | 0.082 | 0.001 | 2.0 | 46.6 | 0.065 | 1.206 | 0.001 | Neg | None | LHRH + bica | PD | 1 | 25 |
| 17 | M | T | 70 | 27 | 0.605 | 0.006 | 6.0 | 46.7 | 0.461 | 0.919 | 0.000 | Neg | None | Bica | SD <6 mo. | 5 | 12+ |
| 18 | M | T | 70 | 47 | 9.105 | 0.309 | 6.0 | 47.9 | 1.385 | 8.000 | 0.000 | Neg | BRAF | Bica | PR | 6 | 40+ |
| 19 | M | T | 30 | 13 | 5.046 | 0.081 | 6.0 | 48.8 | 0.732 | 5.429 | 0.007 | Neg | None | Bica | PD | 2 | 2+ |
| 20 | M | T | 30 | 25 | 1.598 | 0.038 | 6.0 | 49.4 | 0.395 | 8.877 | 0.003 | Neg | None | Bica | PD | 3 | 17 |
| 21 | M | M (liver) | 80 | 91 | – | – | 6.0 | 50.4 | – | – | – | Neg | TP53# | Bica | PD | 1 | 7 |
| 22 | M | T | 70 | 88 | 1.057 | 0.005 | 5.0 | 51.9 | 0.174 | 2.099 | 0.003 | Neg | TP53 | Bica | PD | 1 | 11 |
| 23 | M | T | 60 | 66 | 0.578 | 0.005 | 6.0 | 52.0 | 0.186 | 0.905 | 0.001 | Pos | TP53 | Bica | SD <6 mo. | 5 | 33 |
| 24 | M | N | 80 | 7 | 4.666 | 0.196 | 5.5 | 52.7 | 0.993 | 0.827 | 0.013 | Pos | TP53 | Bica | PD | 2 | 6+ |
| 25 | M | T | 70 | 43 | 0.567 | 0.003 | 5.0 | 53.2 | 0.082 | 2.060 | 0.019 | Pos | None | LHRH + bica | SD >6 mo. | 9 | 18+ |
| 26 | M | T | 90 | 66 | 4.522 | 0.207 | 6.0 | 54.5 | 0.282 | 4.710 | 0.010 | Neg | PIK3CA | LHRH + bica | SD >6 mo. | 8+ | 13+ |
| 27 | M | T | 70 | 20 | 1.094 | 0.186 | 6.0 | 56.7 | 0.056 | 3.216 | 0.003 | Pos | None | Bica | PR | 5 | 18+ |
| 28 | M | T | 70 | 39 | 2.438 | 0.262 | 6.0 | 57.6 | 0.448 | 2.636 | 0.027 | Neg | HRAS and 2xPIK3CA | Bica | SD <6 mo. | 1 | 5 |
| 29 | M | M (skull base) | 90 | 154 | 3.287 | 0.068 | 6.0 | 61.8 | 9.590 | 33.988 | 0.004 | Neg | None | Bica | PR | 10 | 20 |
| 30 | M | N | 60 | 101 | 2.746 | 0.069 | 6.0 | 65.6 | 1.148 | 21.472 | 0.008 | Neg | HRAS and PIK3CA | Bica | PR | 14. | 44 |
Abbreviations: #, because of low DNA yield other mutations could have been missed; *, specific mutations are listed in Supporting Information Table S3; +, ongoing PFS/OS; ADT, androgen deprivation therapy; AKR1C3, aldo‐keto reductase family 1 member C3 gene expression; AR IHC, immunohistochemical androgen receptor expression; AR, androgen receptor gene expression; AR‐V7, androgen receptor splice variant 7 gene expression; bica, bicalutamide; F, female; LHRH, luteinizing hormone‐releasing hormone agonist; M, distant metastasis; M, male; mo., months; N, lymph node metastasis in neck; OS, overall survival; PD, progressive disease; PFS, progression‐free survival; PR, partial response; SD, stable disease; SRD5A, steroid 5 alpha‐reductase gene expression; T, primary tumor.
In the adjuvant ADT treated LA cohort, 14 patients (12 men, 2 women) were analyzed. Median age at the start of ADT was 61 years (range 36–84 years). Two patients were treated with an LHRH‐analog plus bicalutamide and 12 patients with bicalutamide monotherapy. Median FFPE tissue storage time was 13.5 months (range 3–108 months), and 13 samples were obtained from primary tumors and 1 from a regional lymph node metastasis. Further patient characteristics are listed in Table 2.
Table 2.
Patient and tumor characteristics of the locally advanced cohort, sorted by the AR pathway activity score
| Patient no. | Gender | Tumor tissue | Tumor % | Age tissue (mo.) | AR | AR‐V7 | AR IHC | AR pathway activity | AKR1C3 | SRD5A1 | SRD5A2 | HER2 status | ADT | DFS (mo.) | OS (mo.) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 31 | M | T | 70 | 18 | 1.187 | 0.042 | 6.0 | 37.9 | 0.121 | 0.883 | 0.003 | Neg | Bica | 18+ | 18+ |
| 32 | F | T | 60 | 6 | 4.045 | 0.812 | 6.0 | 42.9 | 0.351 | 3.364 | 0.005 | Neg | LHRH + bica | 5+ | 5+ |
| 33 | M | T | 40 | 108 | 1.014 | 0.045 | 6.0 | 44.4 | 0.007 | 0.164 | 0.002 | Neg | Bica | 17 | 52 |
| 34 | M | T | 70 | 12 | 0.886 | 0.006 | 6.0 | 45.3 | 0.883 | 1.050 | 0.028 | Pos | Bica | 10+ | 10+ |
| 35 | F | T | 80 | 3 | 4.441 | 0.237 | 5.5 | 46.0 | 0.829 | 7.062 | 0.046 | – | LHRH + bica | 2+ | 2+ |
| 36 | M | T | 70 | 3 | 4.056 | 0.162 | 4.5 | 48.4 | 0.829 | 1.117 | 0.003 | – | Bica | 3+ | 3+ |
| 37 | M | T | 30 | 13 | 5.046 | 0.081 | 6.0 | 48.8 | 0.732 | 5.429 | 0.007 | Neg | Bica | 6 | 8+ |
| 38 | M | N | 60 | 36 | 1.253 | 0.022 | 6.0 | 49.8 | 0.521 | 5.464 | 0.011 | Neg | Bica | 12 | 22 |
| 39 | M | T | 80 | 23 | 1.366 | 0.067 | 6.0 | 53.3 | 0.183 | 10.724 | 0.068 | Neg | Bica | 22 | 34+ |
| 40 | M | T | 40 | 10 | 2.235 | 0.084 | 6.0 | 54.0 | 1.357 | 6.635 | 0.002 | Neg | Bica | 11+ | 11+ |
| 41 | M | T | 70 | 18 | 1.886 | 0.116 | 6.0 | 54.4 | 0.369 | 1.905 | 0.012 | Pos | Bica | 15+ | 15+ |
| 42 | M | T | 50 | 30 | 1.663 | 0.048 | 6.0 | 58.6 | 0.532 | 22.098 | 0.052 | – | Bica | 25+ | 25+ |
| 43 | M | T | 70 | 9 | 6.755 | 0.147 | 6.0 | 59.6 | 0.737 | 46.340 | 0.004 | Neg | Bica | 11+ | 11+ |
| 44 | M | T | 70 | 14 | 0.657 | 0.020 | 4.5 | 61.6 | 0.198 | 7.835 | 0.222 | Pos | Bica | 15+ | 15+ |
Abbreviations: +, ongoing DFS/OS; ADT, androgen deprivation therapy; AKR1C3, aldo‐keto reductase family 1 member C3 gene expression; AR IHC, immunohistochemical androgen receptor expression; AR, androgen receptor gene expression; AR‐V7, androgen receptor splice variant 7 gene expression; bica, bicalutamide; DFS, disease‐free survival; F, female; M, male; mo., months; N, lymph node metastasis in neck; OS, overall survival; SRD5A, steroid 5 alpha‐reductase gene expression; T, primary tumor.
ADT resistance mechanisms
AR pathway activity scores and AR protein expression were determined in all patients in both cohorts. Insufficient RNA was available for gene expression analysis in one patient in the R/M cohort. Typical AR immunohistochemical staining expression patterns are shown in Figure 1. The difference in AR protein expression levels between patients with and without clinical benefit was not significant (median 6.0, interquartile range (IQR) 5.0–6.0 vs. 6.0, IQR 4.0–6.0, p = 0.29). Also, expression levels of AR (median 1.41 IQR 1.10–3.90 vs. 0.61, IQR 0.26–2.23, p = 0.054), AR‐V7 (median 0.068, IQR 0.006–0.20 vs. 0.008, IQR 0.002–0.12, p = 0.49), AKR1C3 (median 0.28, IQR 0.079–1.27 vs. 0.42, IQR 0.091–0.73, p = 0.79), SRD5A2 (median 0.003, IQR 0.0005–0.009 vs. 0.001, IQR 0.0003–0.006, p = 0.32), and CYP17A1 (negative in all tumors) mRNA were not significantly different between patients with and without clinical benefit. However, SRD5A1 mRNA expression levels were significantly higher (p = 0.008) in patients with clinical benefit (median 3.62, IQR 2.46–14.74) compared to those without (median 1.16, IQR 0.88–3.00). Moreover, the difference in AR pathway activity scores between patients with clinical benefit (median 53.2, IQR 44.8–59.3) and those without (median 45.4, IQR 38.7–49.9) was significant (p = 0.017). Box plots of ADT resistance mechanisms in patients with and without clinical benefit are shown in Supporting Information Figure S1. Although AR‐V7 expression levels were not significantly different in both groups, 41 of 43 evaluated patients in both cohorts (95.3%) had detectable AR‐V7 levels, and a significant correlation between AR and AR‐V7 expression levels was found (R 2 = 0.34, p < 0.001; Supporting Information Figure S2).
Figure 1.
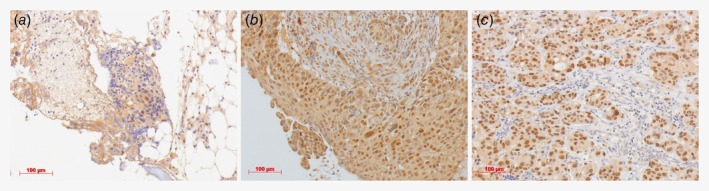
Androgen receptor (AR) immunohistochemical staining. Examples of a negative staining (score 0 + 0 = 0) in (a), moderate staining (score 2 + 2 = 4) in (b) and strong staining (score 3 + 3 = 6) in (c) are shown. The AR staining was scored as described in materials and methods. Images were taken at 200× magnification. [Color figure can be viewed at http://wileyonlinelibrary.com]
Prediction of clinical benefit from ADT using the AR pathway activity score
Patients with clinical benefit in the R/M cohort had a significantly higher AR pathway activity score compared to those without. A ROC‐curve of the AR pathway activity score to predict clinical benefit in the R/M cohort was constructed (Fig. 2 a). The area under the curve was 0.75 (95% CI 0.54–0.95, p = 0.035). The optimal cut‐off value for predicting clinical benefit was an AR pathway activity score of 52.9, which resulted in a sensitivity of 55.6%, a specificity of 95.2%, a positive predictive value of 83.3% and a negative predictive value of 83.3%. Using this cut‐off value, Kaplan–Meier survival curves were constructed for PFS (Fig. 3 a) and OS (Supporting Information Fig. S3 a) in the R/M cohort. The median PFS after treatment with palliative ADT was 2.9 months (95% CI 2.7–3.1 months) for patients with an inactive AR pathway and 9.9 months (95% CI 1.5–18.3 months) for patients with an active AR pathway (p = 0.13). The median OS was 25.9 months (95% CI 8.9–42.9 months) for patients with an inactive AR pathway and 43.6 months (95% CI 0.9–86.3 months) for patients with an active AR pathway (p = 0.39). Subsequently, this cut‐off value was applied to the LA cohort and the median DFS was 17.7 months (95% CI 8.4–27.0 months) for patients with an inactive AR pathway and 22.8 months (95% CI could not be calculated because only one patient had a recurrence) for patients with an active AR pathway (p = 0.061; Fig. 3 c). In the LA cohort, differences in OS were not calculated because of insufficient follow‐up.
Figure 2.
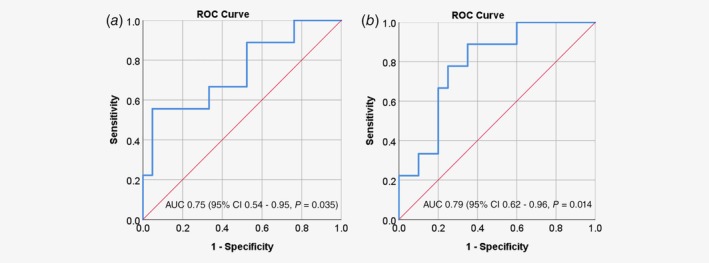
Receiver operating characteristic (ROC)‐curves describing the sensitivity and specificity to predict clinical benefit from androgen deprivation treatment in the R/M cohort. (a) ROC‐curve of androgen receptor pathway analysis. A cut‐off value of 52.9 was used for the subsequent survival analyses, which has a sensitivity of 0.556 and 1‐specificity of 0.048 in this cohort. (b) ROC‐curve of steroid 5 alpha‐reductase 1 (SRD5A1) gene expression levels. A cut‐off value of 2.75 was used, which has a sensitivity of 0.778 and one specificity of 0.250 in this cohort. AUC, area under the curve; CI, confidence interval. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
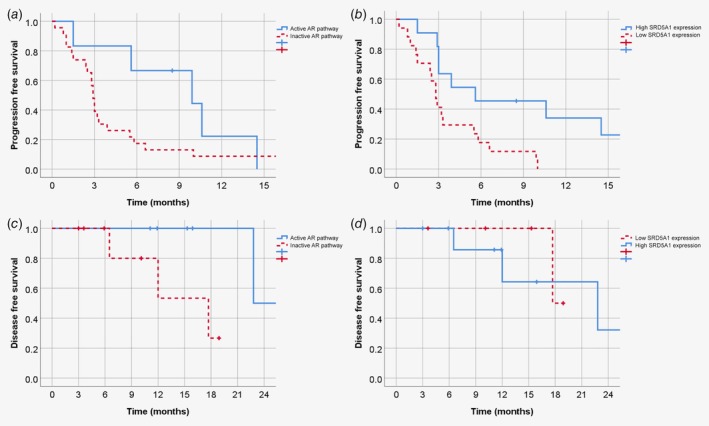
Kaplan–Meier survival curves. Progression‐free survival (PFS) after androgen deprivation therapy (ADT) in patients in the recurrent/metastatic (R/M) cohort for AR pathway activity score (p = 0.13) (a) and SRD5A1 expression (p = 0.008) (b). Disease‐free survival (DFS) after adjuvant ADT in patients in the locally advanced (LA) cohort for AR pathway activity (p = 0.061) (c) and SRD5A1 expression (p = 0.73) (d). [Color figure can be viewed at http://wileyonlinelibrary.com]
Prediction of clinical benefit from ADT using SRD5A1 mRNA expression
A significant correlation between SRD5A1 mRNA expression of the primary tumor tissue and clinical benefit was found. The area under the ROC curve for SRD5A1 in the R/M cohort (Fig. 2 b) was 0.79 (95% CI 0.62–0.96, p = 0.014). The optimal cut‐off value for predicting clinical benefit was an SRD5A1 expression of 2.75, which resulted in a sensitivity of 77.8%, a specificity of 75.0%, a positive predictive value of 58.3% and a negative predictive value of 88.2%. Using this cut‐off value, Kaplan–Meier survival curves were constructed for PFS (Fig. 3 b) and OS (Supporting Information Fig. S3 b) in the R/M cohort. The median PFS after treatment with palliative ADT was 2.8 months (95% CI 2.3–3.3 months) for patients with a low SRD5A1 expression and 5.6 months (95% CI 0.0–13.2 months) for patients with a high SRD5A1 expression (p = 0.008). The median OS was 24.2 months (95% CI 2.9–45.5 months) for patients with a low SRD5A1 expression and 46.3 months (95% CI 0.0–92.8 months) for patients with a high SRD5A1 expression (p = 0.069). Subsequently, this cut‐off value was applied to the LA cohort and the median DFS was 17.7 months (95% CI could not be calculated) for patients with a low SRD5A1 expression and 22.8 months (95% CI 6.3–39.3 months) for patients with a high SRD5A1 expression (p = 0.73; Fig. 3 d). In the LA cohort, differences in OS were not calculated because of insufficient follow‐up.
Combining the AR pathway activity score and SRD5A1 expression
Kaplan–Meier curves were constructed for PFS, OS (R/M cohort) and DFS (LA cohort) for patients with a high AR pathway activity score (>52.9) and high SRD5A1 expression (>2.75) vs. negative cases for both factors. Discrepant cases were left out. This resulted in a median PFS of 10.6 months (95% CI 3.1–18.1 months) and 2.8 months (95% CI 2.2–3.4 months, p = 0.005), a median OS of 46.3 months (95% CI 9.6–83.0 months) and 24.2 months (95% CI 20.7–27.7 months, p = 0.11), and a median DFS of 22.8 months (95% CI could not be calculated) and 17.7 months (95% CI could not be calculated, p = 0.32), respectively. Furthermore, a significant correlation between the AR pathway activity score and SRD5A1 expression factors was found (R 2 = 0.39, p < 0.001; Fig. 4).
Figure 4.
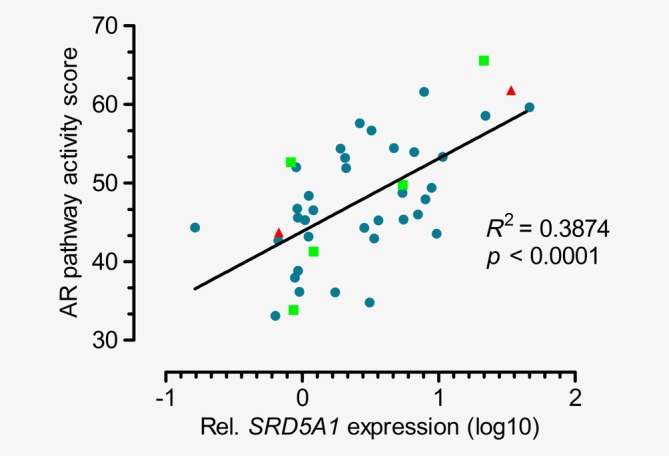
Correlation of relative SRD5A1 expression levels (normalized to HPRT1 housekeeping gene levels) and AR pathway activity scores measured in primary salivary duct carcinomas (blue dots, n = 36), regional lymph node metastases (green squares, n = 5) and distant metastases (red triangle, n = 2) of patients in the recurrent/metastatic cohort and locally advanced cohort. R‐squared and p values of the linear regression analysis are shown. [Color figure can be viewed at http://wileyonlinelibrary.com]
Prediction of clinical benefit in primary tumor vs. metastatic tissue
In the R/M cohort, primary tumor tissue was used in 23 patients, and tissue for regional lymph node metastases and distant metastases prior to ADT in 4 and 3 patients, respectively. SRD5A1 expression levels and AR pathway activity scores were not significantly different between primary tumor tissue, lymph node metastatic tissue and distant metastatic tissue (Supporting Information Fig. S4). However, the difference in SRD5A1 expression levels and AR pathway activity scores between patients with and without clinical benefit remained significant when the analyses were performed in metastatic tissue only (n = 7; SRD5A1 expression 27.73 (IQR 21.5– ‐) vs. 0.85 (IQR 0.71–1.13), p < 0.001 and median AR pathway activity 63.7 (IQR 61.8 – ‐) vs. 43.7 (IQR 37.6–51.5), p = 0.019, respectively. Supporting Information Fig. S5). Both ROC‐curves showed an area under the curve of 1 (95% CI 1–1, p = 0.064 and 95% CI 1–1, p = 0.053, respectively, Supporting Information Fig. S6). Median PFS was significantly longer for patients with high SRD5A1 expression levels (2.5 vs. 10.6 months, p = 0.049) and high AR pathway activity scores (2.5 vs. 10.6 months, p = 0.041). Median OS was not different for both factors. For primary tumor tissue (n = 23), the SRD5A1 expression levels and AR pathway activity scores were higher in patients with clinical benefit, but significance was lost (p = 0.57 and p = 0.20, respectively).
HER2 assessment
HER2 gene (ERBB2) amplification was determined in all patients in the R/M cohort and 10 of 14 patients in the LA cohort. Of these 40 patients, 14 patients (35.0%) had a tumor with an ERBB2 amplification (Tables 1 and 2). In the R/M cohort, no significant differences were found between the clinical benefit rate in patients with or without ERBB2 amplification (20.0% vs. 31.6%, p = 0.68) or OS (median 40.4 months, 95% CI 31.1–49.7 months vs. 22.4 months, 95% CI 17.6–27.2 months, p = 0.26).
Mutation analysis
Mutation analysis was performed in all 30 patients in the R/M cohort. In 12 tumors (40.0%), no driver mutations were found. In the other 18 tumors, mutations were detected in the TP53 (n = 11), PIK3CA (n = 5), HRAS (n = 4), PTEN (n = 2), BRAF (n = 2), AKT1 (n = 1) and ERBB2 (n = 2) genes (Table 1 and Supporting Information Table S3). In the R/M cohort, no significant differences were found between the clinical benefit rate in patients with or without the presence of one of these gene mutations (33.3% vs. 25.0%, p = 0.70) or OS (24.2 months, 95% CI 19.1–29.3 months vs. 43.6 months, 95% CI 36.5–50.7 months, p = 0.39).
Other palliative systemic treatments
Besides first‐line ADT, patients in the R/M cohort received other palliative systemic treatments after PD on ADT. An overview of the treatments is listed in Supporting Information Table S4. Based on the AR pathway activity score, patients with an inactive AR pathway received a mean number of 1.9 palliative systemic treatments vs. 1.5 in patients with an active AR pathway. Patients with an inactive AR pathway more often received second‐line ADT (37.5% vs. 16.7%), third‐line ADT (8.3% vs. 0.0%), chemo and/or targeted therapy (37.5% vs. 33.3%) and immunotherapy (4.2% vs. 0.0%).
Discussion
In our study, potential mechanisms of primary ADT resistance in SDC patients were investigated in order to predict clinical benefit from ADT. We are the first to describe that SRD5A1 expression levels and AR pathway activity scores were significantly higher in patients with clinical benefit compared to those without. Survival analysis in the R/M cohort revealed that median PFS was longer for patients with high SRD5A1 expression levels (2.8 vs. 5.6 months) and high AR pathway activity scores (2.9 vs. 9.9 months). Differences in OS were not found, but many patients received additional palliative systemic treatments, follow‐up was relatively short and the cohorts were relatively small. The threshold for elevated SRD5A1 expression and for high AR pathway activity was set (discovered) on the R/M cohort, and subsequently evaluated for differences in DFS in an independent cohort of locally advanced SDC patients receiving adjuvant ADT (n = 14). For the AR pathway activity analysis and not SRD5A1 expression, a trend toward a better DFS was found. Therefore, the AR pathway activity score is most promising as predictive factor for clinical benefit from ADT, but validation in a prospective study is needed before this method can be put into daily practice. Furthermore, it would be interesting to evaluate the prognostic value of AR‐pathway activity and SRD5A1 expression, as the survival differences may also be a result of a more indolent (well‐differentiated) tumor.
The predictive value of the AR pathway analysis may be further improved by using metastatic tissue only. In our study, only 7 of 30 samples in the R/M cohort were obtained from regional lymph node metastases or distant metastases prior to treatment. Nonetheless, the differences in SRD5A1 expression levels and AR pathway activity scores were still significant between patient with and without clinical benefit.
Combining SRD5A1 expression and AR pathway activity score showed a modest improvement in the prediction of clinical benefit in the R/M cohort, but worse prediction compared to the AR pathway activity score alone in the LA cohort. The most logical explanation for the lack of added value is that both biomarkers are either directly (the AR pathway activity score) or indirectly (SRD5A1 gene expression) associated with activity of the AR pathway, as SRD5A1 overexpression results in increased production of androgens in prostate cancer,23 resulting in AR pathway activation. Although intratumoral androgen synthesis has not yet been demonstrated in SDC, the former also may be an explanation for the positive correlation between SRD5A1 expression levels and AR pathway activity in SDC. More importantly, the level of SRD5A1 expression may have therapeutical consequences, as ADT response in SRD5A1 overexpressing tumors may be further enhanced by adding the SRD5A1 and SRD5A2 inhibitor dutasteride to the ADT regimen. In prostate cancer models, it was shown that dutasteride synergistically suppresses tumor cell proliferation when combined with enzalutamide (a new generation AR antagonist that is more potent than bicalutamide).24 Therefore, combining dutasteride with ADT is a rational approach for future clinical trials in AR‐positive SDC, especially in a translational setting in which the use of SRD5A1 expression levels and/or AR pathway activity scores as predictive biomarkers are confirmed, preferably on metastatic tissue.
The other potential primary resistance mechanisms were not significantly different between patients with and without clinical benefits from ADT and therefore cannot be used as predictive factors. However, some noteworthy findings were done. Although AR protein expression was in general high, one tumor lacked AR protein expression. This patient started palliative ADT based on an AR‐positive lung metastasis but had an AR negative epidural metastasis. This case shows the possible heterogeneity in AR expression in SDC metastases and could explain treatment resistance in such cases. Furthermore, we detected AR‐V7 mRNA expression in 95.3% of primary tumor samples, which is more frequent than the 37–70% of AR‐V7 positive SDC cases reported earlier.12, 15, 25 In CRPC, AR‐V7 expression explains at least in part the resistance to ADT,26 and AR‐V7 expression is elevated in response to ADT.14 We found AR‐V7 expression prior to ADT. Whether expression levels of AR or AR‐V7, or other genes, change during ADT in SDC remains to be investigated. Finally, genetic alterations such as ERBB2 amplification and functionally relevant mutations in oncogenes and tumor suppressor genes were found in 35.0 and 60.0% of patients, respectively, which is in accordance with the literature.12 This may cause ADT resistance by activating other tumor‐driving pathways, such as the PI3K‐AKT pathway, in the presence of an active AR pathway. It is therefore interesting to note that a positive HER2 status did not affect clinical benefit from ADT in AR and HER2 positive SDC patients, which is in line with data from other research groups.6, 7 Therefore, ADT seems to be a good treatment option in these patients, but the presence of other targetable genetic markers stresses the importance of prediction of clinical benefit in order to select the most suitable therapy. In our study, HER2 status was tested with FISH as first test according to ASCO‐CAP guideline.20 Because immunohistochemistry was only added in case of inconclusive results, we might have missed an occasional immunohistochemically 3+ case. In the most recent ASCO‐CAP guideline for HER2 testing in breast cancer, this has been addressed and the guideline has been adjusted accordingly.27
The positive results in our study are remarkable since our study has several limitations. First, the cohorts were relatively small, which is typical for a rare cancer but troublesome for elucidating predictive factors for clinical benefit. Second, not all patients received the same ADT, and compliance to ADT may differ, all of which could influence ADT response independent of AR pathway activity scores. Furthermore, for most patients, prediction of clinical benefit was based on primary tumor tissue samples, while treatment was installed when patients developed metastatic disease, with a varying time interval between primary tumor resection and development of metastases. It is well known that molecular characteristics frequently vary between primary tumors and metastases.18 For breast cancer, it was already shown that signaling pathway activity scores vary to a large extent between primary and distant metastases.28 In our study, the number of metastatic tissue samples, although small, was sufficient to detect significant differences between patients with and without clinical benefit. Finally, the quality of the tumor specimens varied. Some of the FFPE tumor specimens were old, up to more than 16 years, and the tumor percentage was low in some tumor specimens, resulting in “contamination” with benign salivary epithelial cells. Nonetheless, these tumor specimens endured RNA quality control and could still be used our analyses.
In conclusion, in our study, potential intrinsic ADT resistance mechanisms were studied in AR‐positive SDC patients that received ADT. We are the first to describe low SRD5A1 expression and low AR pathway activity scores as potential primary ADT resistance mechanisms in SDC patients. We speculate, based on our findings and studies in prostate cancer, that ADT response in SRD5A1 overexpressing tumors may be further enhanced by adding the SRD5A1 inhibitor dutasteride to the ADT regimen. The AR pathway activity score is a promising predictive biomarker for clinical benefit from ADT in SDC. Validation in a prospective study is needed, preferably in metastatic tissue samples.
Supporting information
Appendix S1: Supporting Information
Acknowledgements
The authors received no specific funding for this work.
Conflict of interest: DvS, HvZ, DK and AvdS are full employees of Philips Research. The other authors have no conflicts of interest to disclose related to this work. ML has been part of advisory boards on other subject for AstraZeneca, Bayer, Bristol‐Myers Squibb, Janssen Pharmaceuticals, MSD, and Roche.
Data availability
The data that support the findings of our study are available from the corresponding author upon reasonable request.
References
- 1. Boon E, Bel M, van Boxtel W, et al. A clinicopathological study and prognostic factor analysis of 177 salivary duct carcinoma patients from The Netherlands. Int J Cancer 2018;143:758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takase S, Kano S, Tada Y, et al. Biomarker immunoprofile in salivary duct carcinomas: clinicopathological and prognostic implications with evaluation of the revised classification. Oncotarget 2017;8:59023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams MD, Roberts D, Blumenschein GR Jr, et al. Differential expression of hormonal and growth factor receptors in salivary duct carcinomas: biologic significance and potential role in therapeutic stratification of patients. Am J Surg Pathol 2007;31:1645–52. [DOI] [PubMed] [Google Scholar]
- 4. van Boxtel W, Locati LD, van Engen‐van Grunsven ACH, et al. Adjuvant androgen deprivation therapy for poor‐risk, androgen receptor‐positive salivary duct carcinoma. Eur J Cancer 2019;110:62–70. [DOI] [PubMed] [Google Scholar]
- 5. Boon E, van Boxtel W, Buter J, et al. Androgen deprivation therapy for androgen receptor‐positive advanced salivary duct carcinoma: a nationwide case series of 35 patients in The Netherlands. Head Neck 2018;40:605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor‐positive salivary gland cancers. Head Neck 2016;38:724–31. [DOI] [PubMed] [Google Scholar]
- 7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor‐positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol 2018;29:979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takahashi H, Tada Y, Saotome T, et al. Phase II trial of Trastuzumab and docetaxel in patients with human epidermal growth factor receptor 2‐positive salivary duct carcinoma. J Clin Oncol 2018;37:125–34. [DOI] [PubMed] [Google Scholar]
- 9. van Boxtel W, Boon E, Weijs WLJ, et al. Combination of docetaxel, trastuzumab and pertuzumab or treatment with trastuzumab‐emtansine for metastatic salivary duct carcinoma. Oral Oncol 2017;72:198–200. [DOI] [PubMed] [Google Scholar]
- 10. Correa TS, Matos GDR, Segura M, et al. Second‐line treatment of HER2‐positive salivary gland tumor: ado‐Trastuzumab Emtansine (T‐DM1) after progression on Trastuzumab. Case Rep Oncol 2018;11:252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park JC, Ma TM, Rooper L, et al. Exceptional responses to pertuzumab, trastuzumab, and docetaxel in human epidermal growth factor receptor‐2 high expressing salivary duct carcinomas. Head Neck 2018;40:E100–6. [DOI] [PubMed] [Google Scholar]
- 12. Dalin MG, Desrichard A, Katabi N, et al. Comprehensive molecular characterization of salivary duct carcinoma reveals actionable targets and similarity to apocrine breast cancer. Clin Cancer Res 2016;22:4623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu B, Dogan S, Al Rasheed MRH, et al. Androgen receptor immunohistochemistry in salivary duct carcinoma: a retrospective study of 188 cases focusing on Tumoral heterogeneity and temporal concordance. Hum Pathol 2019;93:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu R, Dunn TA, Wei S, et al. Ligand‐independent androgen receptor variants derived from splicing of cryptic exons signify hormone‐refractory prostate cancer. Cancer Res 2009;69:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mitani Y, Rao PH, Maity SN, et al. Alterations associated with androgen receptor gene activation in salivary duct carcinoma of both sexes: potential therapeutic ramifications. Clin Cancer Res 2014;20:6570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cai C, Chen S, Ng P, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration‐resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011;71:6503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verhaegh W, van Ooijen H, Inda MA, et al. Selection of personalized patient therapy through the use of knowledge‐based computational models that identify tumor‐driving signal transduction pathways. Cancer Res 2014;74:2936–45. [DOI] [PubMed] [Google Scholar]
- 18. Stolpe AV, Holtzer L, van Ooijen H, et al. Enabling precision medicine by unravelling disease pathophysiology: quantifying signal transduction pathway activity across cell and tissue types. Sci Rep 2019;9:1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verhaegh W, Van de Stolpe A. Knowledge‐based computational models. Oncotarget 2014;5:5196–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013. [DOI] [PubMed] [Google Scholar]
- 21. Eijkelenboom A, Kamping EJ, Kastner‐van Raaij AW, et al. Reliable next‐generation sequencing of formalin‐fixed, paraffin‐embedded tissue using single molecule tags. J Mol Diagn 2016;18:851–63. [DOI] [PubMed] [Google Scholar]
- 22. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 23. Chang KH, Li R, Papari‐Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration‐resistant prostate cancer. Proc Natl Acad Sci USA 2011;108:13728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamid AR, Verhaegh GW, Smit FP, et al. Dutasteride and enzalutamide synergistically suppress prostate tumor cell proliferation. J Urol 2015;193:1023–9. [DOI] [PubMed] [Google Scholar]
- 25. Yang RK, Zhao P, Lu C, et al. Expression pattern of androgen receptor and AR‐V7 in androgen deprivation therapy naive salivary duct carcinomas. Hum Pathol 2018;84:173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cai C, He HH, Chen S, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine‐specific demethylase 1. Cancer Cell 2011;20:457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol 2018;36:2105–22. [DOI] [PubMed] [Google Scholar]
- 28. Anja Van De Stolpe AB, Moelans C, Inda MA, et al. Abstract 3690: measuring functional signal transduction pathway activity on breast cancer tissue samples to determine intra‐tumor heterogeneity and heterogeneity between primary and metastatic tumors. AACR Annual Meeting 2018; April 14–18, 2018; Chicago, IL. Cancer Res 2018;78:Abstract:3690. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of our study are available from the corresponding author upon reasonable request.