

Abstract
Strategies employed by pathogenic enteric bacteria, such as Shigella, to subvert the host adaptive immunity are not well defined. Impairment of T lymphocyte chemotaxis by blockage of polarised edge formation has been reported upon Shigella infection. However, the functional impact of Shigella on T lymphocytes remains to be determined. Here, we show that Shigella modulates CD4+ T cell F‐actin dynamics and increases cell cortical stiffness. The scanning ability of T lymphocytes when encountering antigen‐presenting cells (APC) is subsequently impaired resulting in decreased cell–cell contacts (or conjugates) between the two cell types, as compared with non‐infected T cells. In addition, the few conjugates established between the invaded T cells and APCs display no polarised delivery and accumulation of the T cell receptor to the contact zone characterising canonical immunological synapses. This is most likely due to the targeting of intracellular vesicular trafficking by the bacterial type III secretion system (T3SS) effectors IpaJ and VirA. The collective impact of these cellular reshapings by Shigella eventually results in T cell activation dampening. Altogether, these results highlight the combined action of T3SS effectors leading to T cell defects upon Shigella infection.
Keywords: actin, cell polarisation, Golgi, immunological synapse, Rab proteins, Shigella, T cell activation, T3SS effectors, TCR, vesicular trafficking
IMPORTANCE
Considering the burden of diarrhoeal diseases in low‐ to middle‐income countries, understanding how the most prevalent pathogens, such as the Gram‐negative enteroinvasive bacterium Shigella causing bacillary dysentery, impact on the host immunity appears to be of importance. Although natural infection induces a poorly efficient short‐lasting protection, interactions of Shigella virulence effectors with host targets to subvert adaptive immunity are poorly documented. Our study provides new insights into the outcome of the direct targeting of T lymphocytes by Shigella. We indeed report that human T lymphocytes invaded by Shigella have a defect in both actin cytoskeleton dynamics and vesicular trafficking, resulting in the impairment of the immunological synapse formation. This is likely to impact on the priming of the specific immunity, which could represent an advantage for this bacterium with no animal reservoir to permit reinfection of its human host.
1. INTRODUCTION
The study of host–pathogen crosstalk has highlighted an immense number of strategies applied by microbes to subvert the host immune response. In the case of pathogenic enteric bacteria, such as Shigella, interactions of bacterial virulence effectors with host targets to subvert innate immunity have been extensively studied. Surprisingly, interference with the host adaptive immunity is far less documented, including the impact of bacterial pathogens in its priming (Pinaud, Sansonetti, & Phalipon, 2018). However, as reported for other pathogens, the establishment of a protective immunity to Shigella upon natural infection has been noted to arise only after multiple rounds of infection, is often short‐lived, and poorly effective in preventing reinfection, particularly in young children (Raqib et al., 2000; Raqib et al., 2002). Shigella is a facultative intracellular pathogen and the causative agent of shigellosis or bacillary dysentery. This acute rectocolitis is characterised by a large destruction of the colonic tissue due to the massive inflammation elicited upon infection. The invasive and pro‐inflammatory phenotype of Shigella relies on the presence of a type III secretion system (T3SS), leading to the rapid injection of several effector proteins into the host cell cytoplasm with dramatic impact on cellular functions (Killackey, Sorbara, & Girardin, 2016; Mattock & Blocker, 2017; Parsot, 2009). Some reports indicate that the characteristic acute inflammation induced upon Shigella infection may influence the outcome of adaptive immunity. Indeed, massive death of adaptive immune cells including lamina propria dendritic cells (DCs) and B and T lymphocytes has been observed in rectal biopsies of Shigella‐infected individuals, alongside with high levels of immunosuppressive cytokines (Islam, Azad, Bardhan, Raqib, & Islam, 1994; Mathan & Mathan, 1991). In a model of human intestinal xenograft, Shigella has been shown to downregulate the production of the chemokine CCL20 by infected enterocytes, resulting in a weak recruitment of DCs to the site of infection (Sperandio, Regnault, Guo, et al., 2008).
More recently, the direct targeting of host adaptive immune cells including DCs and B and T lymphocytes has been highlighted (Brunner, Samassa, Sansonetti, & Phalipon, 2019). Such interactions may take place in isolated lymphoid follicles associated to the colonic mucosa, in the lamina propria and also possibly within mesenteric lymph nodes. Several reports demonstrated that Shigella infection of human B and T lymphocytes results in critical dampening of immune cell function. Indeed, Shigella induces cell death in B lymphocytes, by various invasion dependent and independent mechanisms (Nothelfer et al., 2014). Shigella targeting of T lymphocytes on the other hand impairs their chemotaxis in vitro (Konradt et al., 2011), along with the alteration of their migration pattern in vivo (Salgado‐Pabon et al., 2013). Shigella‐induced impairment of T cell chemotaxis is due to the action of the T3SS effector IpgD, a phosphoinositide‐4‐phosphatase mediating the hydrolysis of phosphatidylinositol 4,5 biphosphate (PI[4,5]P2). The reduction of the PI(4,5)P2 pool at the plasma membrane consequently prevents the formation of the polarised edge that is necessary for the initiation of T cell migration (Konradt et al., 2011).
CD4+ T cell activation promoted upon infection leads to the induction of a pathogen‐specific T cell response, which is usually key for the efficiency of the protective immunity towards the pathogenic agent. This activation is achieved due to the remarkable capacity of these cells to deform and polarise upon recognition of an activating antigen‐presenting cell (APC), shifting from a migratory asymmetrical phenotype to a more symmetrical polarised phenotype, characterising the immunological synapse (IS). The IS is a complex macromolecular structure at the T cell–APC interface involving the T cell receptor (TCR) recognition of the cognate peptide ligand associated to the major histocompatibility complex on APCs, which results in the coordinated polarisation of receptors, adhesion molecules, kinases, signalling molecules, cytoskeletal elements, and organelles towards the contact area between the two cells (Alcover, Alarcón, & Di Bartolo, 2018; Brown et al., 2003; Dustin & Choudhuri, 2016; Hivroz & Saitakis, 2016). The IS is a highly dynamic platform, which requires two critical cellular processes to ensure its appropriate formation: actin cytoskeleton remodelling and polarised vesicular trafficking. Both are mandatory to bring all the necessary molecules, including TCR, to the contact zone between the two cells to coordinate the complex activation cascade in a timely manner. Noticeably, both have been shown to be the targets of Shigella T3SS effectors in intestinal epithelial cells (Burnaevskiy et al., 2013; Dong et al., 2012; Ferrari et al., 2019; Mounier et al., 2012). We thus hypothesised that Shigella could impact T cell activation by interfering with processes regulating cellular motility and IS formation. As a follow‐up with our previous studies (Konradt et al., 2011; Salgado‐Pabon et al., 2013), the analysis has been centred on Shigella‐invaded CD4+ T cells upon in vitro infection. Additionally, a classical in vitro assay for IS formation using human Jurkat T cells or primary lymphoblasts co‐cultured with superantigen‐pulsed Raji B cells as APCs has been performed for a step‐by‐step analysis of IS formation upon Shigella infection. Here, we report that Shigella, via its T3SS, acts at different stages of the molecular activation process to dampen efficient T cell activation.
2. RESULTS
2.1. Shigella induces cortical stiffness associated with increased F‐actin in human T lymphocytes
Actin cytoskeleton remodelling is an essential component of T cell motility (Roy & Burkhardt, 2018). The impact of Shigella on T cell cortical actin structure and dynamics remains unknown. We first investigated the T lymphocyte actin dynamics upon Shigella infection by quantifying F‐actin at the cell membrane by flow cytometry (Figure 1a) and confocal microscopy (Figure 1‐figure supplement 1). As compared to the non‐infected cells, wild‐type (WT) Shigella‐invaded cells displayed a twofold increase in their F‐actin content (Figure 1a, Figure 1‐figure supplement 1). In addition, the mxiD strain, which is T3SS defective and thus, non‐invasive, was used as a control to assess the contribution of effective T3SS effectors secretion in the phenotype observed with the invasive T3SS effective WT strain. Notably, infection of Jurkat cells with the mxiD mutant did not result in increased F‐actin content, indicating the involvement of a T3SS effector‐dependent mechanism (Figure 1a).
Figure 1.
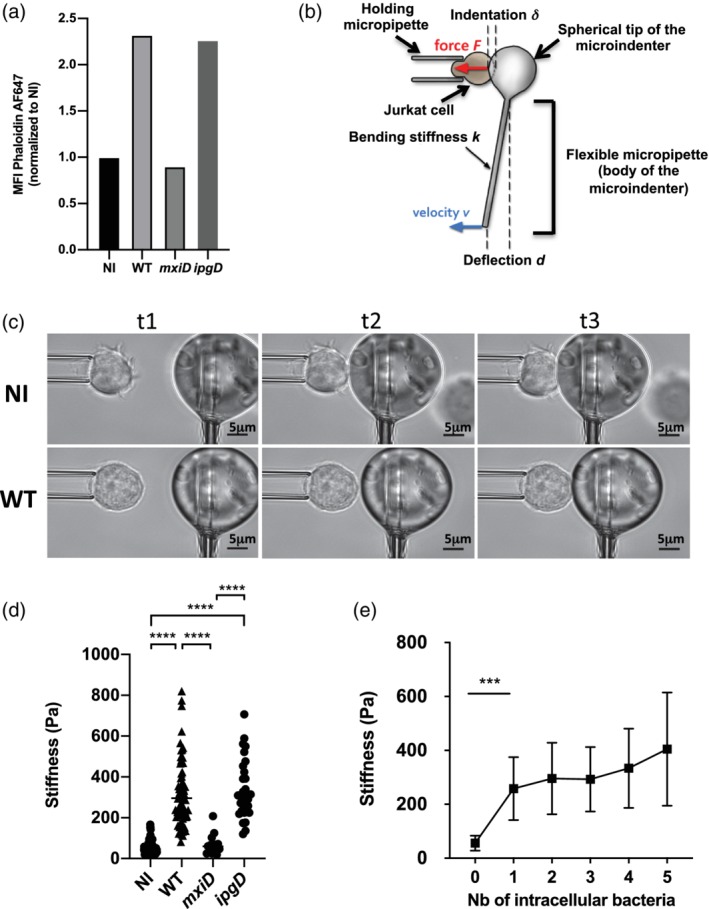
Increased stiffness associated with increased filamentous actin in Shigella‐invaded Jurkat cells. (a) F‐actin content measurement. Cells were either non‐infected (NI) or infected with WT‐Shigella or the mxiD or ipgD mutants. Cells were fixed and permeabilised prior to staining with fluorescent phalloidin whose intensity was analysed by flow cytometry. Representative of three independent experiments. (b) Principle of the profile microindentation setup. A stiff micropipette holds a cell. A flexible micropipette has a glass sphere at its tip and is used as a microindenter, with its base translated towards the cell at a constant velocity (v). Upon contact with the cell, the glass bead induces a deformation of the cell (refers as the indentation δ) allowing calculation of the cell stiffness. (c) Representative snapshots to illustrate the microindentation experiment at three randomly chosen time points (referred as t1, t2, and t3). Representative NI cell (upper panel) and wild‐type (WT) Shigella‐invaded cell (bottom panel) are represented. (d) Stiffness measurements in NI, mxiD‐infected cells, WT‐ or ipgD‐invaded cells (three independent experiments, except for the mxiD strain, two independent experiments). (e) Impact of the number of intracellular WT Shigella on cell stiffness from six independent experiments. (a,d,e) M ± SD, ***p < .001, ****p < .0001 (Kruskal–Wallis and Tukey's multiple comparisons test)
Because the actomyosin cytoskeleton is a major regulator of cortical stiffness (Thauland, Hu, Bruce, & Butte, 2017), we next sought to investigate cellular stiffness upon bacterial invasion. For this, we utilised microindentation, which served as a physical measurement reflecting cell cortical stiffness. In brief, individual Jurkat cells were subjected to a continuously increasing, compressive force applied by a glass bead, which is attached to a flexible glass microfibre (Figure 1b). This force promotes the glass bead to progressively touch—indented—the cell surface resulting in a local deformation of the cell, which can be quantified. The degree of deformation is inversely correlated with the cellular stiffness. WT Shigella‐invaded Jurkat cells displayed a characteristic round morphology with undetectable membrane protrusions, as opposed to non‐infected cells (Figure 1c). Microindentation showed that stiffness of WT Shigella‐invaded cells was sixfold higher than that of non‐invaded ones, whereas that of the mxiD‐infected Jurkat cells was similar to that of non‐infected cells (Figure 1d). Of note, one single intracellular bacterium was sufficient to account for the drastic increase in cell stiffness, whereas no significant further increase was observed in cells harbouring two to five intracellular bacteria (Figure 1e), showing that stiffness is independent from the number of intracellular bacteria. In contrast, T cells infected with the Shigella mutant strain ipgD displayed the same F‐actin content (Figure 1a) and stiffness (Figure 1a) as WT Shigella‐invaded cells.
Altogether, these results show that Shigella, via its T3SS effectors, but independently of IpgD, increases T cell F‐actin content associated with an increase of T cell cortical stiffness.
2.2. Shigella alters TCR endocytosis and recycling in human T lymphocytes
Along with actin cytoskeleton dynamics, intracellular vesicular trafficking is a central player of IS assembly and maintenance and is required for T cell activation. In particular, Golgi and recycling endosome reorientation towards the interface is one of the hallmarks of IS formation, enabling the polarised delivery of TCR and signalling molecules (such as Lck and LAT) at the contact site (Alcover & Thoulouze, 2010; Depoil et al., 2005; Kupfer & Dennert, 1984; Pais‐Correia, Thoulouze, & Alcover, 2007). Shigella invasion of epithelial cells is known to induce Golgi fragmentation, vesicular trafficking arrest, and reorganisation of the endocytic recycling compartment (Burnaevskiy et al., 2013; Dong et al., 2012; Ferrari et al., 2019; Mounier et al., 2012). We thus analysed these subcellular structures in WT Shigella‐invaded Jurkat T cells using anti‐GM130 and an antibody recognising the small GTPase Rab11, which associates primarily with recycling endosomes and regulates the recycling of endocytosed proteins (Takahashi et al., 2012). We observed the disassembly of the Golgi compartment in the invaded cells in contrast to non‐infected ones (Figure 2a). In addition, in the invaded cells, the perinuclear pool of the small GTPase Rab11 was dispersed throughout the cell (Figure 2a).
Figure 2.
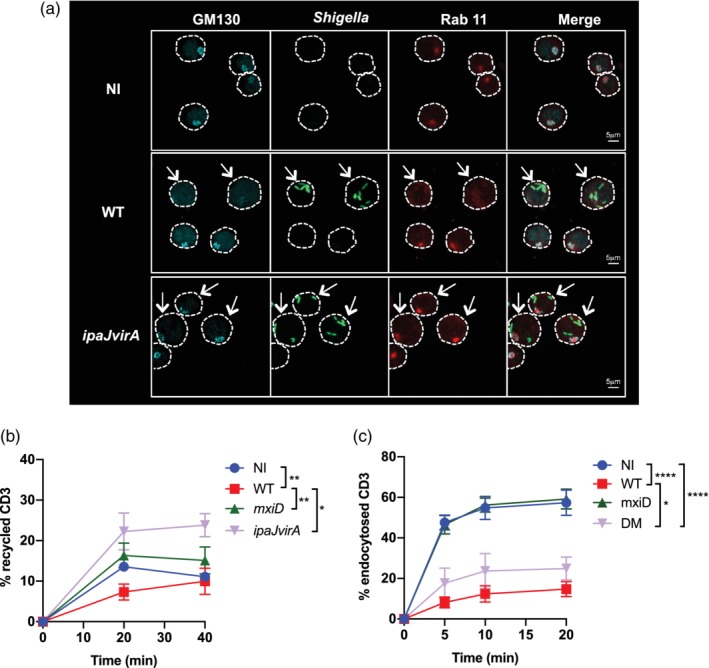
Vesicular trafficking is modulated by T3SS effectors in Shigella‐invaded Jurkat cells. (a) Jurkat cells were infected with GFP wild‐type (WT) Shigella or the ipaJvirA mutant for 30 min followed by 1 h30 incubation with gentamicin to kill extracellular bacteria. Cells were fixed, permeabilised, and then labelled with anti‐GM130 and anti‐Rab11 mAbs. Images are representative of three independent experiments. The cells are delineated by a dotted line. The arrows indicate the invaded cells. (b–c) Flow cytometry analysis on 20,000 cells per time point of T cell receptor (TCR; CD3) recycling (b) or TCR (CD3) endocytosis (c) in Jurkat cells non‐infected or infected with WT Shigella, or the mutant strains mxiD or ipaJvirA. Data are presented as the percentage of internalised receptors that have recycled to the cell surface (b) or as the percentage of internalised TCR (c) and are from five independent experiments. (b–c) M ± SD, *p < .05, **p < .005, ***p < .0005, ****p < .0001 (Tukey's multiple comparisons test). NI, non‐infected
Previous studies showed that the T3SS Shigella effectors, VirA and IpaJ, block global host cell secretion, recycling, and receptor‐mediated endocytosis in intestinal epithelial cells (Burnaevskiy et al., 2013; Dong et al., 2012; Ferrari et al., 2019). VirA acts as a GAP of Rab1 GTPase, which regulates endoplasmic reticulum‐to‐Golgi transport, and has a GAP activity towards other RabGTPases in vitro (Dong et al., 2012). IpaJ is a cysteine protease that cleaves the myristoylated glycine of Arf1 GTPase, a key player in intra‐Golgi and retrograde endoplasmic reticulum‐to‐Golgi transport and in the maintenance of Golgi structure (Burnaevskiy et al., 2013). Thus, we tested a Shigella ipaJvirA‐deficient mutant in our settings. We observed that Jurkat cells invaded with the ipaJvirA mutant strain displayed an intact Golgi structure and an intact Rab11 compartment (as observed in 92.2% of cells), similar to that of non‐infected cells (99.6%; Figure 2a). This observation stands in striking contrast to WT Shigella‐invaded cells, as 10.6% of cells only showed intact GM130+ and Rab11+ compartments (Figure 2a and details in material and methods).
TCR trafficking between the plasma membrane and the endocytic compartment is dependent on Rab11 (Finetti et al., 2015; Kumar, Ferez, & Swamy, 2011; Liu, Rhodes, Wiest, & Vignali, 2000). Because WT Shigella‐invaded T cells displayed an atypical Rab11 staining, we performed time course analyses to follow TCR recycling by using an anti‐CD3ε antibody in Jurkat infected with WT Shigella or the mxiD mutant strain. TCR recycling was measured as percentage of endocytosed TCR that recycled back to the plasma membrane. TCR recycling was significantly reduced in WT Shigella‐invaded Jurkat cells, but not in the mxiD mutant‐infected cells (Figure 2b). By performing a kinetic of TCR internalisation (TCR endocytosis rate), we observed that after 20 min of incubation, almost 60% of the TCR was endocytosed in non‐infected cells and mxiD mutant strain‐infected cells, in contrast to only 15% in WT Shigella‐invaded Jurkat cells (Figure 2c). The defect in TCR recycling was fully reverted in ipaJvirA‐invaded Jurkat cells (Figure 2b), whereas the decrease of its endocytosis rate was only partially restored (Figure 2c). In parallel, the recycling and endocytosis of transferrin (Tf), a classical marker of endocytic trafficking whose intracellular pool is also polarised towards the contact site during IS formation, were also affected in WT Shigella‐invaded Jurkat cells due to the concerted action of VirA and IpaJ effectors (Figure 2‐figure supplements 1A,B), as previously reported in epithelial cells (Ferrari et al., 2019).
Altogether, these results show that similar to epithelial cells, Shigella disrupts the Golgi apparatus via IpaJ and VirA effectors in T lymphocytes. In addition, a novel key role for these two effectors is revealed with the disruption of the Rab11 compartment and TCR recycling, along with their contribution to the decreased TCR endocytosis rate.
2.3. Shigella impairs the scanning capacity of CD4+ T lymphocytes and prevents the formation of canonical IS
As previously stated, IS formation is dependent on actin cytoskeleton dynamics in two ways: first, by promoting T cell mobility necessary for APC scanning and second, by mediating the spatiotemporal organisation of IS components at the contact site, which conditions subsequent signalling cascade (Roy & Burkhardt, 2018). In addition, intracellular vesicular trafficking is a central player of IS assembly and maintenance and is required for T cell activation (Alcover & Alarcon, 2000; Onnis, Finetti, & Baldari, 2016). Considering the impact of Shigella on these two critical pathways presented above, we thus investigated the formation of ISs upon T cell infection. First, we analysed the dynamics of T cell scanning by performing time‐lapse video microscopy of TCR‐CD3ζ‐GFP expressing Jurkat cells infected with DsRed‐expressing Shigella co‐cultured with fluorescently labelled SAg‐Raji cells. As already shown in Figure 1c, WT Shigella‐invaded Jurkat cells displayed a round phenotype with undetectable membrane protrusions, appeared less motile, as previously reported (Konradt et al., 2011), and less efficient in scanning SAg‐Raji cells when compared with non‐infected cells (Figure 3‐video supplement 1). As a consequence, the percentage of conjugates formed by WT Shigella‐invaded T cells was significantly lower than that measured in the absence of infection (Figure 3A). Furthermore, the few cell–cell contacts formed between SAg‐Raji cells and WT Shigella‐invaded Jurkat cells failed to induce the polarised delivery and accumulation of TCR‐CD3ζ complexes at the contact zone characterising canonical IS formation (Alcover, Alarcon, & Di Bartolo, 2017; Blanchard, Di Bartolo, & Hivroz, 2002; Krummel, Sjaastad, Wulfing, & Davis, 2000), in striking contrast to non‐infected T cells (Figure 3‐video supplement 1).
Figure 3.
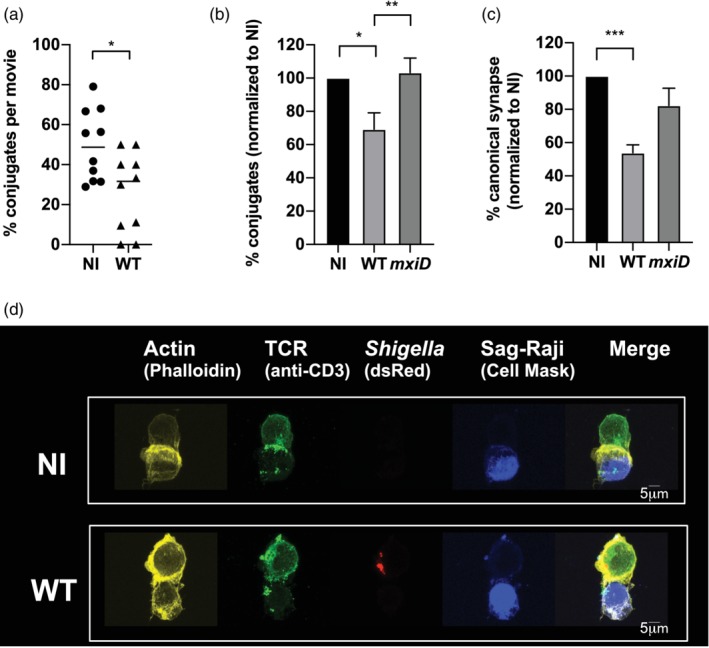
Shigella prevents human Jurkat T cells to form canonical synapses. Jurkat cells were infected with wild‐type (WT) Shigella or the mxiD mutant for 30 min at a multiplicity of infection of 20 followed by 1 h30 incubation with gentamicin to kill extracellular bacteria. Infected and non‐infected (NI) Jurkat cells were incubated with Raji cells pulsed with SAg (SAg‐Raji). (a) ζ‐GFP Jurkat cells were infected with DsRed fluorescent bacteria and incubated with fluorescently labelled SAg‐Raji. Cells were placed on an imaging chamber coated with matrigel matrix and time lapses were acquired with 20‐s frame duration. Ten videos were analysed per condition with about 10–20 cells analysed per movie. *p = .0336. (b) Quantification of the percentage of conjugates (CD3+CD19+) by flow cytometry from six independent experiments. **p = .0022 (c) Quantification of the percentage of canonical synapses (T cell receptor [TCR] and F‐actin accumulation at the interface) following ImageStream acquisition from three independent experiments (strategy detailed in Figure 3‐figure supplement 1). For (b) and (c), 20,000 cells were acquired. (d) Conjugates formed between non‐infected (upper panel) or wild‐type (WT) Shigella‐invaded (bottom panel) Jurkat cells and SAg‐Raji cells, analysed by confocal microscopy. Images show a medial optical section of a representative conjugate. Cells were fixed and stained with fluorescent phalloidin and anti‐CD3 mAb (TCR labeling) without cell permeabilization to detect only surface TCRs. M ± SD; (a) *p < .05 (Mann–Whitney); (b–c) *p < .05, **p < .005, ***p < .005 (Kruskal–Wallis)
To further confirm these results, we performed flow cytometric quantification of Jurkat‐SAg‐Raji cell conjugates forming in a similar co‐culture set‐up. The percentage of CD3+CD19+ Jurkat‐SAg‐Raji cell conjugates was shown to decrease by 30% with Jurkat cells infected with WT Shigella as compared with non‐infected ones (Figure 3b). This process was dependent on Shigella invasion as the non‐invasive Shigella mutant strain mxiD with defective T3SS displayed similar degree of conjugates formation as non‐infected controls (Figure 3b).
We then quantified the formation of canonical ISs, that is, cell–cell contacts characterised by an accumulation of TCR‐CD3 and F‐actin at the contact site, using ImageStream technology that combines flow cytometry and epifluorescence microscopy (Figure 3‐figure supplement 1). We observed that canonical ISs formation was reduced by 50% with WT Shigella‐infected T cells, as compared with non‐infected cells (Figure 3c). Similarly, WT Shigella‐infected T cells displayed significantly reduced capacity to form ISs formation as compared with mxiD mutant‐infected Jurkat cells (Figure 3c). Subsequent confocal microscopy analysis focusing on WT Shigella‐invaded Jurkat cells confirmed the decreased TCR‐CD3 complexes recruitment to the area of cell contact and a lack of F‐actin enrichment (Figure 3d). In comparison, canonical IS formation was observed in the absence of infection (Figure 3d).
Of note, WT Shigella‐invaded human primary lymphoblasts also failed to form canonical ISs (Figure 3‐figure supplement 2).
Altogether, these results demonstrate that Shigella reduces the scanning ability of T cells towards APCs and, upon cell contact, disrupts the formation of canonical ISs.
2.4. Shigella prevents human T cell activation
One of the main outcome of IS formation is the induction of T cell activation (Fooksman et al., 2010). Following TCR triggering, the early activation events are characterised by a cascade of tyrosine phosphorylation following TCR triggering, eventually leading to cytokines secretion. We thus assessed the consequences of altered IS formation induced by Shigella on the TCR downstream signalling pathways. The first actors of the tyrosine‐phosphorylation cascade, namely Lck, Zap70, and LAT, were analysed by immunoblotting on cell lysates using specific antibodies (Figure 4a,b). As expected, in the non‐infected condition, the increase of specific tyrosine phosphorylation of Lck, Zap70, and LAT was observed upon incubation of Jurkat cells with SAg‐Raji cells, as compared with the incubation with non‐pulsed Raji cells (Figure 4a). Similarly, this phosphorylation was observed in Jurkat cells infected with the non‐invasive mxiD mutant strain but not in WT Shigella‐invaded cells (Figure 4a). Quantification of the ratio of activated phosphorylated proteins P‐Lck, P‐Zap70, and P‐LAT out of the total amount of each respective protein suggests that WT Shigella invasion almost completely abolishes cell activation (Figure 4b).
Figure 4.
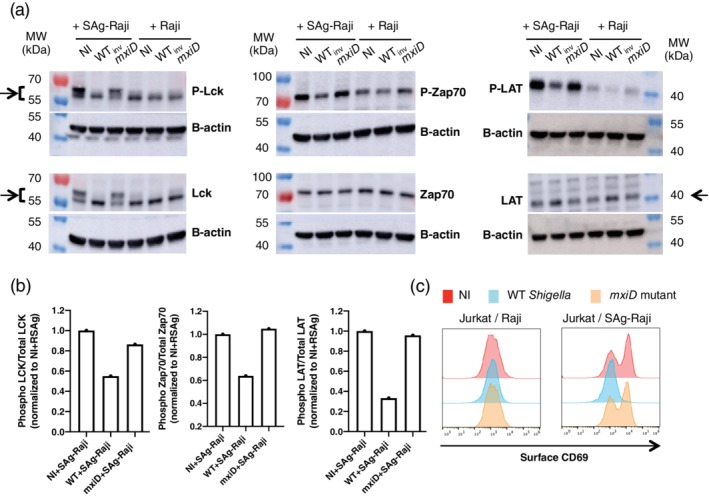
Shigella prevents human Jurkat T cells activation. Jurkat cells were infected with wild‐type (WT) Shigella or the mxiD mutant for 30 min at a multiplicity of infection of 20 followed by 1 h30 gentamicin to kill extracellular bacteria. (a) Following sorting of Shigella‐invaded Jurkat cells, these cells were then incubated with Raji or SAg‐Raji cells for 20 min and lysed. Supernatants were analysed by immunoblotting using individually specific antibodies against Lck, ZAP70, and LAT and their phosphorylated forms. β‐Actin immunoblotting used as a loading control for each blot is shown. The arrows indicate the bands used for quantification in Figure 4b. (b) Ratio of phosphorylated protein over total for Zap70, Lck, and LAT when Jurkat cells were incubated with SAg‐Raji cells. One representative experiment is shown. (c) Jurkat cells were stimulated with Raji cells pulsed (SAg‐Raji) or not for 2h30 min. Cells were fixed and stained with anti‐CD69 mAb. Surface expression of CD69 on live cells from one representative experiment out of three is shown. NI, non‐infected
In line with this, WT Shigella‐invaded Jurkat cells also failed to induce the surface expression of CD69, a classical marker of T cell activation, as compared with both non‐infected and mxiD‐infected Jurkat cells (Figure 4c).
Altogether, these results demonstrate that Shigella drastically impairs T cell activation.
3. DISCUSSION
Our study provides new insights into the outcome of the direct targeting of T lymphocytes by Shigella. We have previously reported that Shigella impairs the migratory competence of T lymphocytes by suppressing cell polarisation and, more globally, disrupts T cell dynamics in vivo (Konradt et al., 2011; Salgado‐Pabon et al., 2013). Here, we show that Shigella further impairs T cell functionality by preventing T cell activation via perturbation of canonical IS formation.
Among others, the canonical IS is dependent on two important cellular events, which are the rearrangement of F‐actin and intracellular trafficking, resulting in the polarisation of key molecules, including the TCR, to the contact zone of T lymphocyte and APC.
We report that Shigella promotes an increase in the F‐actin content in the invaded T cells, which coincides with a significant increase in cellular stiffness. Because cellular stiffness is related to a densely polymerised array of actin filaments and associated crosslinking proteins just beneath the plasma membrane (Gilden & Krummel, 2010), we propose that the increased cellular stiffness is related to the higher F‐actin content. Interestingly, Shigella mediates this process with such efficiency that a single intracellular bacterium is sufficient to promote the maximal level of stiffness measured. This suggests that this phenomenon relies on a bacterial‐induced mechanism rather than a passive consequence of multiple intracellular bacteria overwhelming the T cell limited cytoplasm space. Interestingly, cytoskeletal stiffness was recently shown to be correlated with IS formation and TCR stimulation (Thauland et al., 2017). Particularly, naïve T cells have a stiffer cortical actin cytoskeleton and form smaller IS as compared with effector T cells, evoking that, prior to the encounter with the APC, T cell stiffness can predict the morphology of the IS to be formed. Our results suggest that Shigella induces a sort of naïve T cell‐like cortical actin cytoskeleton stiffness that eventually renders T cells less responsive towards APCs. In regard to the T3SS effectors accounting for the increase in both F‐actin content and stiffness, we showed that IpgD (Figure 1d), IpaJ, and VirA (Figure 1‐figure supplement 2B) are not involved. For IpgD, this is consistent with the inactivation of the ezrin, radixin, and moesin proteins (that are key in linking the cortical actin cytoskeleton and the plasma membrane; Barret, Roy, Montcurrier, Mangeat, & Niggli, 2000; Kikuchi et al., 2002) it mediates, which is expected to induce relaxation of the cell membrane. Other T3SS effectors, which are key in modulating actin cytoskeleton in epithelial cells, such as IcsB, IpgB1, and IpgB2 (Liu et al., 2018; Mattock & Blocker, 2017; Valencia‐Gallardo, Carayol, & Tran Van Nhieu, 2015) were tested for modulation of F‐actin content, and none were found to be implicated in the F‐actin increase observed with WT Shigella (data not shown). These findings suggest the existence of additional mechanism(s) involved in actin dynamics remodelling and cell stiffness, triggered by a yet to be identified T3SS effector, or a combination of effectors.
Besides cellular stiffness, we show that Shigella targets the second core cellular component required for canonical IS formation. Indeed, Shigella induces fragmentation of the Golgi apparatus and the disruption of the Rab11 compartment in invaded T cells by the combined action of the T3SS effectors IpaJ and VirA. In link with the Rab11 compartment, these effectors are shown to disrupt TCR trafficking. Whereas the role of these effectors in the disruption of the Golgi in epithelial cells was known (Burnaevskiy et al., 2013), their role on the Rab11 compartment was not reported as yet. As initially reported in vitro, VirA binds the Rab protein Rab1, but it was further shown to target additional Rabs including Rab11, as well as Rab22 and Rab35, which are involved in endosomal trafficking (Dong et al., 2012). This is consistent with the recently proposed role of VirA and IpaJ to be involved in global alteration of vesicular trafficking leading to a profound defect in the effective subcellular localisation of many proteins (Ferrari et al., 2019). In epithelial cells, T3SS‐mediated targeting of the Golgi apparatus is one of the reported strategies utilised by several enteric pathogens to counteract host cell functions. For instance, in the case of Salmonella, the intracellular multiplication of bacteria implies their close association with the Golgi network via the effector SseG (Ramsden, Holden, & Mota, 2007; Salcedo & Holden, 2003). On the opposite, the EPEC and EHEC effector EspG induces Golgi fragmentation, therefore disrupting the secretory pathway, similar to what is observed with Shigella (Clements et al., 2011).
The consequences of Shigella‐mediated increase in F‐actin content and cellular stiffness along with impairment of vesicular trafficking in invaded T cells are likely responsible for the limitation in the formation of canonical IS while encountering with APCs and for the subsequent blockage of T cell activation we observed. Of note, besides the invaded T lymphocytes, it is likely that T lymphocytes targeting via the injection of T3SS effectors not resulting in bacterial invasion (Pinaud et al., 2017) further does its part in the prevention of T cell activation. The role in conjugates formation of some T3SS effectors known to mediate actin cytoskeleton rearrangements, such as IpgD (Konradt et al., 2011) tested for T cell stiffness, IpgB1B2, IcsB, and IcsA (Liu et al., 2018; Mattock & Blocker, 2017; Valencia‐Gallardo et al., 2015) was assessed by using Shigella strains defective for their expression. None of them had a detectable impact on the number of conjugates formed as compared with WT Shigella (data not shown). For T3SS effectors responsible for alteration of vesicular trafficking, ipaJvirA‐invaded Jurkat T cells, despite proficient TCR recycling, did not form more conjugates than WT Shigella‐invaded cells (Figure 3 figure supplement 3). Together, these findings indicate for additional effectors to be involved in Shigella‐induced impairment of IS formation—probably via a combined action of several effectors—highlighting the multiple levels of targeting mechanisms applied by Shigella to coordinate the shutdown of this complex cell activation system. Our findings are in accordance with the modulation of host innate immune responses exerted by Shigella and more generally by enteroinvasive bacteria such as Yersinia, Salmonella, EPEC/EHEC, which has been shown to occur with multiple host proteins being targeted by several T3SS effectors in a given cellular pathway (Pinaud et al., 2018).
So far, only a few T cell invading bacterial pathogens have been reported to impair IS architecture and subsequent T cell activation. Particularly, in Bordetella pertussis infection, the effector CyA interacts with the integrin LFA‐1 in its active conformation and induces its mislocalisation from the IS in a cAMP‐ and protein kinase A‐dependent manner to prevent T cell activation (Paccani et al., 2008; Paccani, Finetti, & Davi, 2011). Alternatively, in Yersinia infection, the bacterial phosphatase YopH targets SLP76 and LAT to block T cell activation (Gerke, Falkow, & Chien, 2005; Yao, Mecsas, Healy, Falkow & Chien 1999). Targeting of DCs as APCs to suppress T cell activation has also been reported (Adkins, Schulz, Borgmann, Autenrieth, & Gröbner, 2008; Autenrieth et al., 2007; Lindner et al., 2007). Noteworthy, T cell ability to capture bacterial antigens directly from an infected DC has been highlighted (Cruz‐Adalia et al., 2017). The ability of these so‐called trans‐infected T cells to subsequently form IS was shown to be maintained, ensuring cell activation and proliferation (Cruz‐Adalia et al., 2017).
To our knowledge, this study is the first to report the dual targeting of actin dynamics and intracellular trafficking during IS formation by a pathogenic bacterium. A similar action was previously reported only for the human immunodeficiency virus (HIV)‐1 retrovirus, suggesting that intracellular lymphotropic pathogens, viruses, and bacteria have evolved common strategies to impair CD4+ T cell functions. In the case of HIV‐1, two distinct mechanisms regulated by two distinct viral effectors were shown to impact IS formation and to shape a virally induced T cell activation state independent of antigen recognition. First, the Nef accessory viral protein, which interferes with endosomal vesicular trafficking and interacts with regulators of actin polymerisation, induces Lck and TCR accumulation in an endosomal compartment, and alters TCR vesicular trafficking, resulting in the decrease of early signalling events (Thoulouze et al., 2006) Second, the HIV envelope protein gp120, which alters the kinetics of TCR recruitment to the IS and prolongs TCR‐induced activation, leads to increased CD69 upregulation and increased IL‐2 secretion. Thereby, HIV‐1 likely promotes T cell survival to ensure HIV intracellular replication (Deng et al., 2016).
Considering that there is no animal reservoir for Shigella, infection of humans is key for the survival of this bacterium. Its ability to prevent the priming of proper immune response might be critical to permit re‐infection of the same host multiple times and to drive appearance of asymptomatic carriers excreting bacteria that are still infectious. Establishment of long‐term carriers probably reflects a particular balance between Shigella‐mediated impairment of host immune responses and control of infection by the host to counteract pathogenicity without killing the bacteria (Ghosh, Pazhani, Niyogi, Nataro, & Ramamurthy, 2014; Haider, Huq, Samadi, & Ahmad, 1985; Levine, Dupont, Khodabandelou, & Hornick, 1973). By providing a better understanding of the crosstalk between Shigella and host cells that are key in the priming of specific immunity, these findings give hints for the development of an efficient vaccine that might contribute to the battle against antimicrobial resistance (Jansen, Knirsch, & Anderson, 2018; Lipsitch & Siber, 2016).
4. EXPERIMENTAL PROCEDURES
4.1. Bacterial strains and culture
The Shigella flexneri serotype 5a strains and their derivative mutants used in this study are the following: WT (M90T), mxiD, ipgD, and ipaJvirA transformed or not with plasmids coding for DsRed (Sörensen et al., 2003) or GFP (pFPV 25.1) (Jaumouillé, Francetic, Sansonetti, & Tran Van Nhieu, 2008; Valdivia, Hromockyj, Monack, Ramakrishnan, & Falkow, 1996). Bacteria were grown at 37°C on tryptic soy broth (TSB; Becton, Dickinson) agar plates supplemented or not with 100‐μg/ml ampicillin, 30‐μg/ml kanamycin, and congo red (0.1%). Congo red‐positive colonies corresponding to T3SS‐active bacteria were selected to be grown overnight in TSB supplemented or not with 30 μg/ml of ampicillin at 30°C. Subcultures (1:50) were performed in antibiotic‐free TSB until early exponential phase was reached (OD 1.2).
4.2. Superantigens
Toxin shock syndrome toxin 1 (TSST‐1) and staphylococcal enterotoxins A, C1, C3, and E (SEA, SEC‐1, SEC‐3, and SEE, respectively; Toxin Biotechnology, FL, USA) were used according to the manufacturer's instructions. For binding and activation assays of human TCR subsets, the following superantigens were used: TSST‐1 to recognise the Vβ2.1 subset; SEA for Vβ1.1/5.3/6.3/6.4/6.9/7.3/7.4/9.1/23.1; and SEC‐1, SEC‐3, and SEE for Vβ3.2/6.46.9.12/15.1, Vβ5.1/12, and Vβ 5.1/6.1/6.3/8 subsets, respectively (Fraser & Proft, 2008; Toxin Technology).
4.3. Cell lines and primary T lymphoblasts
The human Jurkat T (clone E6.1, ATCC TIB‐152) and the Raji cell lines (Burkit lymphoma B cell line, major histocompatibility complex class II positive, ATCC clone CCL‐86) were used. The Jurkat cell line expressing a GFP‐tagged TCRζ (ζ‐GFP) was kindly provided by Claire Hivroz. Cells were maintained at a concentration of 5 × 105 cells/ml in RPMI1640 Glutamax (Gibco) supplemented with 10% heat‐inactivated fetal bovine serum (FBS; Biowest) and cultured at 37°C in a 5% CO2 incubator. Passages were performed three times a week, by a threefold dilution in a fresh medium. SAg‐specific lymphoblasts from human peripheral blood mononuclear cells (PBMC) were obtained as previously described (Das et al., 2004). Briefly, human PBMCs were isolated from healthy donors using a Ficoll Paque Plus gradient separation (GE Healthcare) following centrifugation at 800 g without brake at room temperature (RT) for 30 min. SAg‐specific T cells were then expanded by culturing PBMCs (106 cells/ml) in RPMI1640 with Glutamax (Invitrogen) supplemented with 10% FBS, non‐essential AA (Gibco), sodium pyruvate (Gibco), Penicillin/Streptomycin (Gibco), and IL‐7 (5 ng/ml; Biolegend) in the presence of 1 μg/ml of each superantigen SEA, SEC‐1, SEC‐3, SEE, and TSST‐1 (Toxin Technology, FL, USA) at 37°C in 5% CO2 incubator. After 3 days, cells were adjusted to a concentration of 5 × 105 cells/ml in the same medium, without superantigens, supplemented with human recombinant IL‐2 at 50 IU/ml, and cultured for 8–10 days before experiments were performed.
4.4. T cell infection
Jurkat T cells or T cell blasts were cultured in 96 round‐bottom well plates (Corning) at a concentration of 1.5 × 106 cells per well in RPMI1640 with Glutamax (Invitrogen). Bacterial subcultures were diluted in this culture medium to a multiplicity of infection of 20 and centrifuged onto the cells at 300 g for 5 min at 37°C. Infected T cells were incubated at 37°C in a 5% CO2 incubator for 30 min, followed by a 60‐ or 90‐min incubation with gentamicin at a final concentration of 50 μg/ml to kill extracellular bacteria, prior to performing different assays as described below.
4.5. Conjugate formation assay
For IS formation, the protocol is based on a classical assay previously reported for IS formation in vitro (Roumier et al., 2001). In brief, Raji cells used as APC were preincubated at 37°C for 30 min with either 10 μg/ml of SEE (Toxin Technology, FL, USA) for conjugate formation with Jurkat cells, or with a mix of SAgs composed of SEA, SEC1, SEC3, SEE, and TSST‐1 (10 μg/ml each) for conjugate formation with T lymphoblasts. Conjugates were formed in suspension by incubating SAg‐pulsed Raji cells with Jurkat cells or T blasts, previously infected as described above at 37°C for 20 min, prior to fixation for flow cytometry analysis and immunofluorescence microscopy or lysis for western blotting. Non‐infected cells were used as control. For confocal microscopy experiments, Raji cells were preincubated either with CellTrace Far Red DDAO‐SE or CellTracker Blue CDMC dyes (ThermoFisher) according to the manufacturer's instructions. For flow cytometry experiments, conjugates were stained with anti‐human‐CD3‐FITC (Clone OKT3, ThermoFisher) and anti‐human‐CD19‐APC (Clone HIB19, Invitrogen). Acquisition was performed on a FACSCantoII flow cytometer (BD Bioscience) equipped with 405‐, 488‐, and 633‐nm lasers, or an AttuneNxT flow Cytometer (ThermoFisher) equipped with 405‐, 488‐, 561‐, and 638‐nm lasers. The software Flowjo version 10.4.2 was used for subsequent analysis.
4.6. T cell activation assays
Jurkat cells were left non‐infected or infected, as already described. Then, 3 × 105 cells were left untreated or stimulated with phorbol‐12‐myristate‐13‐acetate (25 ng/ml; PMA) and calcium ionophore A23187 (1 μg/ml; PMA/iono) or with 3 × 105 Raji cells prepulsed with SEE (10 μg/ml) at 37°C in a 5% CO2 incubator for 2.5 hr. Following fixation with 4% PFA, cells were stained with anti‐human CD3‐PEeF610 (eBioscience) and anti‐CD69 PerCP‐eFluor710 (eBioscience). Viability was assessed by using LiveDead fixable violet (ThermoFisher) or 4,6‐diamidino‐2‐phenylindole (DAPI).
4.7. Imaging flow cytometry acquisition and data analysis
Conjugates were acquired on the ImageStream X Mk I (Amnis, Merck‐Millipore) equipped with dual camera and 405‐, 488‐, 561‐, and 633‐nm excitation lasers. Acquisition was performed at 40× magnification (0.75 NA). The following channels were used: channels 1 and 9 for brightfield, channel 2 for CD19‐AF488, channel 4 for CD3‐PEeF610, channel 7 for 4,6‐diamidino‐2‐phenylindole, and channel 11 for Phalloidin AF647. Analysis was performed using the IDEAS software (version 6.2, Amnis). Masks (areas of interest) and features (calculations made from masks) were generated to quantify the percentage of canonical synapses formed. The detailed gating strategy is described in Figure 3‐figure supplement 1.
4.8. Confocal microscopy acquisition
T cells (Jurkat or SAg‐T blasts) alone or in conjugates with Raji cells were seeded onto coverslips precoated with poly‐L‐lysine (Sigma) prior to fixation with 4% PFA for 10 min at RT. After three washings with PBS, the coverslips were then incubated with NH4Cl for 10 min to remove residual PFA. TCR and F‐actin were stained with anti‐CD3 Ab (clone UCHT1, Biolegend) and Phalloidin AF647 (ThermoFisher, A22287), respectively, followed by staining with secondary antibodies for 45 min at RT. Depending on the Abs, several protocols were used for intracellular staining. For mouse anti‐human Lck, permeabilisation with PBS supplemented with 0.05% saponin was performed for 15 min at RT, followed by incubation with the Ab diluted in the same buffer for 1 hr. For rabbit‐anti human‐GM130 (Abcam, ab52649) and mouse‐anti‐Rab11 (BD Bioscience, 610,657), permeabilisation was done with PBS supplemented with 0.1% Triton. Confocal microscopy was performed on a SP5 (Leica) or on an Opterra (Bruker) using either a 63× objective or a 100× objective (NA 1.4). Z‐stack acquisitions of optical sections were performed at 0.3 increments for analysis. Images were analysed using Fiji analysis software (Schindelin et al., 2012).
4.9. Object detection and cell segmentation
Automated image analysis was performed with Fiji (Schindelin et al., 2012) via a Jython script, available upon reasonable request. First, the Golgi was detected using the LoG detector of TrackMate (Tinevez, Perry, & Schindelin, 2017). The Golgi channel was first projected along the Z axis using maximum intensity projection and then filtered with a median or radius 8. This gives a processed image where the Golgi appears as clean blobs, amenable to detection with the LoG detector, tuned for a radius of 2 μm and a quality threshold of 14. The Rab11 pool was detected the same way, changing the quality threshold to a value of 40. Bacteria were also detected the same way, changing the quality threshold to a value of 2.
Cells were segmented on the maximum intensity projection image filtered by a Gaussian of sigma = 2 μm. Segmentation was done using intensity thresholding, with the Triangle algorithm for automatic threshold determination, followed by a watershed transformation on the resulting binary image, to separate touching cells.
Each cell region of interest is then inspected one by one and classified as follows: non‐infected cells or infected cells (with or without Golgi, and with or without Rab11 pool). The total count of each class was then exported to a CSV file, aggregating results from several images, later used for subsequent analysis.
4.10. Live imaging and cell‐to‐cell contact analysis
Jurkat cells expressing TCR fused to GFP were infected, as described in the T cell infection section above, washed, and re‐suspended at a concentration of 1 × 106 cells/ml in RPMI1640 without phenol red containing 10% FBS. Cells were then mixed (ratio 1:1) with Raji cells previously stained with Cell Tracker Blue CMHC Dye (ThermoFisher) and pulsed with SEE SAg, as described in the conjugates formation section above. Cells were immediately placed in a 35‐mm imaging μ‐dish (ibidi, Germany) coated with Matrigel matrix (Corning). The dish and image acquisition chamber were preheated and kept at 37°C and 5% CO2 during the duration of the acquisition. Time‐lapse images were acquired on Zeiss Definite Focus microscope using a 20× objective (LD Achroplan) with 20‐s frame duration. TL Halogen lamp was used as a light source. The following excitation/emission filters were used: Zeiss FS01, modified emission (Blue), Zeiss FS38 HE (GFP), Semrock HcRed (Texas Red), and TL Brightfield. The resulting movies were then analysed with a custom image analysis tools, implemented as a Fiji (Schindelin et al., 2012) plugin and a MATLAB (v2018a, The MathWorks, Natick, USA) script, described briefly here. The cell‐to‐cell contact analyser is a TrackMate (Tinevez et al., 2017) extension that produces tracks following cell‐to‐cell contacts over time, from their appearance to their disappearance. Contacts are detected on a contact image produced as follows. The user specifies a threshold value for each channel T1 and T2 that separates the background from the cell image. The contact image is then produced from the following expression: C = (I1‐T1) × (I2‐T2); where I1 and I2 are respectively the image in channel for T‐cells and AP cells, filtered by a Gaussian of σ = 1.0 pixels and dilated using the grayscale morphology dilation operation with a radius of 3 pixels. C is clipped to 0 for negative values. On the image C, contacts between T cells and AP cells appear as bright and smooth blobs over 0 background. They are segmented in distinct objects by simple thresholding above 0. The resulting objects are tracked using TrackMate, with the LAP tracker using a maximal linking distance of 5 μm, a gap closing of maximum 2 frames and a maximal gap closing distance of 5 μm. T cells and AP cells are tracked separately with TrackMate, using the LoG detector configured with a radius of 7.5 μm and a threshold depending of the movie image quality for detection, and the LAP tracker using a maximal linking distance of 11 μm, a gap closing of maximum 2 frames, and a maximal gap closing distance of 11 μm. The resulting tracks were inspected and curated manually again using TrackMate.
In a second step performed in MATLAB, the contact tracks are matched to a T cell track and an AP cell track, by searching the cell track with the minimal distance from contact to cell, averaged over all the common time points of the track. T cells infected by Shigella are identified by measuring the mean intensity in the DsRed channel within the cell radius. The resulting track trio are used to measure the number of tracks with or without T cell infected Shigella and the contact duration. The cell‐to‐cell contact analyser is an open source, and the code and binaries are available on GitHub (https://github.com/imagopole/CellContactAnalyzer).
4.11. Measurement of T cell stiffness
Cells were put into a Petri dish (Fluorodish, WPI, Sarasota, FL, USA) in a 4‐ml medium and placed on the stage of an inverted microscope (TE300, Nikon Instruments, Tokyo, Japan) equipped with a 100× oil immersion, 1.3 NA objective (Nikon Instruments) on an air suspension Table (CVI Melles Griot, Netherlands). Images were acquired using a Flash 4.0 CMOS camera (Hamamatsu Photonics, Hamamatsu City, Japan) controlled by a code implemented in MATLAB (Mathworks, Natick, MA, USA) and the MicroManager software (Edelstein et al., 2014). A custom‐made heating plate was used to keep cells at 37°C. The experiments were performed under brightfield illumination with brief use of standard fluorescence illumination between acquisitions to monitor the number of bacteria in the analysed Jurkat cells. Micropipettes used to hold Jurkat cells were prepared as described previously by others (Basu & Huse, 2017; Guillou et al., 2016; Guillou, Dahl, J‐MG, et al., 2016; Sawicka et al., 2017). Microindenters were prepared from the bead micropipettes, as described previously (Guillou, Babataheri, et al., 2016). Profile microindentation and quantification of cell stiffness were previously described (Guillou, Babataheri, et al., 2016).
4.12. TCR recycling and endocytosis and vesicular compartment trafficking assay
The protocols used were adapted from Mounier et al. (Mounier et al., 2012), Finetti et al. (Finetti et al., 2014), and Patrussi and Baldari (Patrussi & Baldari, 2017). Briefly, Jurkat cells were infected with Shigella at a multiplicity of infection of 20 for 30 min at 37°C followed by 1‐hr incubation with gentamicin at 50 μg/ml for 1 hr in RPMI1640, 1% BSA. For analysis of TCR recycling, Shigella‐infected Jurkat cells were incubated on ice for 30 min with saturated concentration of anti‐CD3 Ab (clone UCHT1, Biolegend) corresponding to the binding step, followed by washing with cold PBS, and transferred at 37°C for 40 min for internalisation of TCR‐Ab complexes. For Tf recycling experiments, infection of cells was performed in the presence of human Tf coupled to Alexa647 (TfAF647) at a concentration of 0.5 μg/ml. For both Tf and TCR recycling analysis, cells were then treated with a stripping solution (100‐mM NaCl, 100‐mM Glycine, pH 2.5) for 1–3 min at 4°C for TfR and at RT for TCR to remove residual membrane‐associated TfAF647 or anti‐CD3 Ab. After washing with PBS, cells were incubated at 37°C and a time course was performed to assess recycling of anti‐CD3 Ab‐TCR‐ or TfAF647‐TfR complexes. For TCR recycling, TCR‐Ab complexes that had recycled back to the cell surface were quantified by flow cytometry upon incubation with non‐permeabilised cells with a fluorescently labelled secondary antibody. Data were presented as the percentage of internalised receptors that have recycled back to the plasma membrane, as previously described (Margadant, Kreft, de Groot, Norman, & Sonnenberg, 2012). Tf recycling time course was performed in the presence of 50 μg/ml of holotrasferrin and represented as the percentage of the remaining intracellular TfAF647 fluorescence at each time point (100% being the amount at the first time point of the kinetics).
For endocytosis assays, cells were infected as described before, incubated on ice for 30 min with saturating concentration of anti‐CD3‐PeCy5 (clone UCHT1, eBioscience) or TfAF647 to allow for surface binding. After washing with ice‐cold PBS, a time course was performed by incubating cells at 37°C for different time points to allow endocytosis of anti‐CD3‐receptor or TfAF647‐TfR complexes, followed by a wash step with the stripping solution in order to remove residual membrane associated TfAF647 or anti‐CD3. The total internalised fluorescent CD3 or TfAF647 was quantified by flow cytometry. Results are presented as the percentage of internalised anti‐CD3 receptor or as the total internal TfAF647.
4.13. Western blotting
Following conjugate formation, cells were centrifuged (300 g, RT) and lysed, and whole cell lysates were analysed with anti‐Lck (3A5, Santa Cruz), anti‐phospho‐Src (for phospho‐Lck, Y416, 2101S, Cell Signalling), anti‐LAT (05–561, Merck Millipore), anti‐phospho‐LAT (Y191, 3,584 T, Cell Signalling),anti‐Zap70 (Y319, Syk [Y352], 2701S, Cell Signalling), and anti‐phospho‐Zap70 (2701S, Cell signalling). These antibodies were used separately. Each blot was then stained with an anti‐Β‐actin (ab8227, Abcam), following stripping for the anti‐LAT and anti‐phospho‐LAT, with a molecular weight (37 kDa) similar to the one of Β‐actin (42 kDa).
4.14. Data presentation and statistical analysis
Prism 6.0 (GraphPad Software) was used for graphs and statistical analyses. Means and standard deviations are represented. Mann–Whitney test was used to compare two groups. Kruskal–Wallis or Tukey's multiple comparisons tests were used to compare more than two groups. The Illustrator CS5 software (Adobe) was used to assemble figures.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
Supporting information
Video S1 supporting Information
Video S2 supporting Information
Figure S1 Shigella infection increases F‐actin content in Jurkat T cells. F‐actin content measurements. Following infection, cells were fixed and stained with fluorescent phalloidin whose intensity was quantified by confocal microscopy. Data from 3 independent experiments. Mean +/− SD, **** p < 0.0001 (Mann–Whitney).
Figure S2‐figure supplement 1: Tf vesicular trafficking is modulated by T3SS effectors in Shigella‐invaded Jurkat cells. Flow cytometry analysis of Tf recycling (A) or endocytosis (B) in Jurkat cells non‐infected or infected with Shigella WT or the mxiD or ipaJvirA mutant strains as previously described. (A) To assess for Tf recycling, Jurkat cells were infected with WT Shigella or the ipaJvirA mutant in the presence of fluorescently labelled TfAF647, prior to incubation with gentamicin containing media to kill extracellular bacteria. Following the removal of surface‐attached TfA647 by acid stripping, a Tf recycling time‐course was performed in the presence of non‐fluorescent holotransferrin. Data are shown as the percentage of the remaining intracellular TfA647 fluorescence at each time point. (B) For Tf endocytosis assays, cells were infected with WT Shigella and the mxiD or ipaJvirA mutants, prior to incubation on ice for 30 min with TfAF647. Cells were then incubated at 37°C and the rate of Tf endocytosis was measured over time following acid stripping. Data are presented as mean fluorescent intensity (MFI) of the intracellular TfAF647 pool and are representative of three individual experiments.
Figure S3‐figure supplement 1: ImageStream gating strategy. (1) Unfocused cells were excluded by using the gradient RMS feature, which measures the quality of an image by detecting important changes of pixel values in the image. (2) In order to discriminate between single cells and doublets, the brightfield Area versus Aspect ratio (i.e., minor axis divided by the major axis) for the focused cells was plotted. Indeed, as opposed to single cells, which have intermediate Area values and a high Aspect Ratio, doublets have a higher area value and a smaller aspect ratio. (3) Doublets of CD3+ and CD19+ cells were identified by plotting the CD3 vs CD19 intensity. (4) In order to focus on doublet with only one CD19+ cell, the aspect ratio intensity feature was used. This feature corresponds to the minor axis intensity divided by the major axis intensity. Doublets with high aspect ratio intensity for CD19 (i.e., close to 1) were considered as doublet of interest. (5) Doublets with high intensity of CD3 were identified by plotting the max pixel feature versus the area. Indeed, the max pixel feature is used to estimate the true peak fluorescence activity. Concerning CD3, a high max pixel intensity value is expected at the interface when a canonical synapse is formed. (6) Similarly, actin clustering in the IS mask was identified by plotting the max pixel intensity of actin versus the area in order to formally identify canonical ISs. BF: Brightfield.
ACKNOWLEDGEMENTS
We thank Claire Hivroz (Institut Curie, France) for kindly providing us with ζ‐GFP Jurkat cells and for helpful advices about lymphoblast experiments, Andres Alcover and Philippe Bousso (Institut Pasteur, Paris) for the kind gift of reagents and fruitful discussions. Many thanks to our PMM colleagues, Ellen Arena, Claude Parsot, Giulia Nigro, Laurie Pinaud, Ilia Belotserkovsky, and Nathalie Sauvonnet, for helpful discussions and constant support, with a particular thanks to Giulia for careful reading of the manuscript. We thank Ronan Thibaut (Institut Pasteur, Paris) for his help on some experimental set ups. We are grateful to the CRT Technology Cores (Institut Pasteur, Paris) for the use of their technical facilities and in particular to Pierre‐Henri Commère for cell sorting. We acknowledge the help of Emma Spanjaard from the Image Analysis Hub of the Institut Pasteur for the live image analysis. This work was supported by the French Ministry of Higher Education and Research (FS), the French Medical Research Foundation FS (FDT20170437005) and MF (SPF20121226366), the European Research Council (ERC) Grants 232798 and 339579 (PJS), the French Government's Investissement d'Avenir Program, Laboratoire d'Excellence “Integrative Biology of Emerging Infectious Diseases” (grant ANR‐10‐LABX‐62‐IBEID), Agency of Science, Technology and Research (A*STAR), Singapore (THT), INSIS‐CNRS through the call Ingénierie translationnelle 2017” (JH), Ecole polytechnique (JH), and LabeX LaSIPS (ANR‐10‐LABX‐0040‐LaSIPS) managed by the French National Research Agency under the “Investissements d'avenir” program (ANR‐11‐IDEX‐0003‐02) (JH).
Samassa F, Ferrari ML, Husson J, et al. Shigella impairs human T lymphocyte responsiveness by hijacking actin cytoskeleton dynamics and T cell receptor vesicular trafficking. Cellular Microbiology. 2020;22:e13166 10.1111/cmi.13166
Funding information Agency of Science, Technology and Research (A*STAR), Singapore; Ecole polytechnique; INSIS‐CNRS Ingénierie translationnelle 2017; LabeX LaSIPS (ANR‐10‐LABX‐0040‐LaSIPS), Grant/Award Number: ANR‐10‐LABX‐0040‐LaSIPS; Ministère de l'Education Nationale, de l'Enseignement Superieur et de la Recherche; the European Research Council, Grant/Award Numbers: 232798, 339579; the French Government's Investissement d'Avenir Program, Laboratoire d'Excellence “Integrative Biology of Emerging Infectious Diseases”, Grant/Award Number: ANR‐10‐LABX‐62‐IBEID; the French Medical Research Foundation, Grant/Award Numbers: FDT20170437005, SPF20121226366; the French National Research Agency under the “Investissements d'avenir” program (ANR‐11‐IDEX‐0003‐02)
REFERENCES
- Adkins, I. , Schulz, S. , Borgmann, S. , Autenrieth, I. B. , & Gröbner, S. (2008). Differential roles of Yersinia outer protein P‐mediated inhibition of nuclear factor‐kappa B in the induction of cell death in dendritic cells and macrophages. Journal of Medical Microbiology, 57(2), 139–144. 10.1099/jmm.0.47437-0 [DOI] [PubMed] [Google Scholar]
- Alcover, A. , & Alarcon, B. (2000). Internalization and intracellular fate of TCR‐CD3 complexes. Critical Reviews in Immunology, 20(4), 325–346. http://www.ncbi.nlm.nih.gov/pubmed/11100805 [PubMed] [Google Scholar]
- Alcover, A. , Alarcon, B. , & Di Bartolo, V. (2017). Cell biology of T cell receptor expression and regulation. Annual Review of Immunology, 36, 103–125. 10.1146/annurev-immunol-042617-053429 [DOI] [PubMed] [Google Scholar]
- Alcover, A. , Alarcón, B. , & Di Bartolo, V. (2018). Cell biology of T cell receptor expression and regulation. Annual Review of Immunology, 36(1), 103–125. 10.1146/annurev-immunol-042617-053429 [DOI] [PubMed] [Google Scholar]
- Alcover, A. , & Thoulouze, M. I. (2010). Vesicle traffic to the immunological synapse: A multifunctional process targeted by lymphotropic viruses. Current Topics in Microbiology and Immunology, 340, 191–207. 10.1007/978-3-642-03858-7_10 [DOI] [PubMed] [Google Scholar]
- Autenrieth, S. E. , Soldanova, I. , Rösemann, R. , Gunst, D. , Zahir, N. , Kracht, M. , … Autenrieth, I. B. (2007). Yersinia enterocolitica YopP inhibits MAP kinase‐mediated antigen uptake in dendritic cells. Cellular Microbiology, 9(2), 425–437. 10.1111/j.1462-5822.2006.00800.x [DOI] [PubMed] [Google Scholar]
- Barret, C. , Roy, C. , Montcurrier, P. , Mangeat, P. , & Niggli, V. (2000). Mutagenesis of the phosphatidylinositol 4,5‐bisphosphate (PIP2) binding site in the NH2‐terminal domain of ezrin correlates with its altered cellular distribution. The Journal of Cell Biology, 151, 1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, R. , & Huse, M. (2017). Mechanical communication at the immunological synapse. Trends in Cell Biology, 27(4), 241–254. 10.1016/j.tcb.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard, N. , Di Bartolo, V. , & Hivroz, C. (2002). In the immune synapse, ZAP‐70 controls T cell polarization and recruitment of signaling proteins but not formation of the synaptic pattern. Immunity, 17, 389–399. [DOI] [PubMed] [Google Scholar]
- Brown, M. J. , Nijhara, R. , Hallam, J. A. , Gignac, M. , Yamada, K. M. , Erlandsen, S. L. , … Shaw, S. (2003). Chemokine stimulation of human peripheral blood T lymphocytes induces rapid dephosphorylation of ERM proteins, which facilitates loss of microvilli and polarization. Blood, 102, 3890–3899. [DOI] [PubMed] [Google Scholar]
- Brunner, K. , Samassa, F. , Sansonetti, P. J. , & Phalipon, A. (2019). Shigella‐mediated immunosuppression in the human gut: Subversion extends from innate to adaptive immune responses. Human Vaccines & Immunotherapeutics, 15, 1317–1325. 10.1080/21645515.2019.1594132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnaevskiy, N. , Fox, T. G. , Plymire, D. A. , Ertelt, J. M. , Weigele, B. A. , Selyunin, A. S. , … Alto, N. M. (2013). Proteolytic elimination of N‐myristoyl modifications by the Shigella virulence factor IpaJ. Nature, 496(7443), 106–109. 10.1038/nature12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements, A. , Smollett, K. , Lee, S. F. , Hartland, E. L. , Lowe, M. , & Frankel, G. (2011). EspG of enteropathogenic and enterohemorrhagic E. coli binds the Golgi matrix protein GM130 and disrupts the Golgi structure and function. Cellular Microbiology, 13(9), 1429–1439. 10.1111/j.1462-5822.2011.01631.x [DOI] [PubMed] [Google Scholar]
- Cruz‐Adalia, A. , Ramirez‐Santiago, G. , Osuna‐Pérez, J. , Torres‐Torresano, M. , Zorita, V. , Martínez‐Riaño, A. , … Veiga, E. (2017). Conventional CD4 + T cells present bacterial antigens to induce cytotoxic and memory CD8 + T cell responses. Nature Communications, 8, 1591 10.1038/s41467-017-01661-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, V. , Nal, B. , Dujeancourt, A. , Thoulouze, M. I. , Galli, T. , Roux, P. , … Alcover, A. (2004). Activation‐induced polarized recycling targets T cell antigen receptors to the immunological synapse; involvement of SNARE complexes. Immunity, 20(5), 577–588. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15142526 [DOI] [PubMed] [Google Scholar]
- Deng, J. , Mitsuki, Y. , Shen, G. , Ray, J. C. , Cicala, C. , Arthos, J. , … Hioe, C. E. (2016). HIV envelope gp120 alters T cell receptor mobilization in the immunological synapse of uninfected CD4 T cells and augments T cell activation. Kirchhoff F, Ed. J Virol., 90(23), 10513–10526. 10.1128/JVI.01532-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depoil, D. , Zaru, R. , Guiraud, M. , Chauveau, A. , Harriague, J. , Bismuth, G. , … Valitutti, S. (2005). Immunological synapses are versatile structures enabling selective T cell polarization. Immunity, 22(2), 185–194. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15723807 [DOI] [PubMed] [Google Scholar]
- Dong, N. , Zhu, Y. , Lu, Q. , Hu, L. , Zheng, Y. , & Shao, F. (2012). Structurally distinct bacterial TBC‐like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell, 150(5), 1029–1041. 10.1016/j.cell.2012.06.050 [DOI] [PubMed] [Google Scholar]
- Dustin, M. L. , & Choudhuri, K. (2016). Signaling and polarized communication across the T cell immunological synapse. Annual Review of Cell and Developmental Biology, 32(1), 303–325. 10.1146/annurev-cellbio-100814-125330 [DOI] [PubMed] [Google Scholar]
- Edelstein, A. D. , Tsuchida, M. A. , Amodaj, N. , Pinkard, H. , Vale, R. D. , & Stuurman, N. (2014). Advanced methods of microscope control using μManager software. Journal of Biological Methods, 1(2), 10 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, M. L. , Malardé, V. , Grassart, A. , Salavessa, L. , Nigro, G. , Descorps‐Declere, S. , … Sauvonnet, N. (2019). Shigella promotes major alteration of gut epithelial physiology and tissue invasion by shutting off host intracellular transport. Proc Natl Acad Sci. June, 116, 13582–13591. 10.1073/pnas.1902922116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finetti, F. , Patrussi, L. , Galgano, D. , Cassioli, C. , Perinetti, G. , Pazour, G. J. , & Baldari, C. T. (2015). The small GTPase Rab8 interacts with VAMP‐3 to regulate the delivery of recycling T‐cell receptors to the immune synapse. Journal of Cell Science, 128(14), 2541–2552. 10.1242/jcs.171652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finetti, F. , Patrussi, L. , Masi, G. , Onnis, A. , Galgano, D. , Lucherini, O. M. , … Baldari, C. T. (2014). Specific recycling receptors are targeted to the immune synapse by the intraflagellar transport system. Journal of Cell Science, 127(Pt 9), 1924–1937. 10.1242/jcs.139337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooksman, D. R. , Vardhana, S. , Vasiliver‐Shamis, G. , Liese, J. , Blair, D. A. , Waite, J. , … Dustin, M. L. (2010). Functional anatomy of T cell activation and synapse formation. Annual Review of Immunology, 28, 79–105. 10.1146/annurev-immunol-030409-101308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, J. D. , & Proft, T. (2008). The bacterial superantigen and superantigen‐like proteins. Immunological Reviews, 225(1), 226–243. 10.1111/j.1600-065X.2008.00681.x [DOI] [PubMed] [Google Scholar]
- Gerke, C. , Falkow, S. , & Chien, Y. (2005). The adaptor molecules LAT and SLP‐76 are specifically targeted by Yersinia to inhibit T cell activation. The Journal of Experimental Medicine, 201(3), 361–371. 10.1084/jem.20041120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, S. , Pazhani, G. P. , Niyogi, S. K. , Nataro, J. P. , & Ramamurthy, T. (2014). Genetic characterization of Shigella spp. isolated from diarrhoeal and asymptomatic children. Journal of Medical Microbiology, 63, 903–910. 10.1099/jmm.0.070912-0 [DOI] [PubMed] [Google Scholar]
- Gilden, J. , & Krummel, M. F. (2010). Control of cortical rigidity by the cytoskeleton: Emerging roles for septins. Cytoskeleton, 67(8), 477–486. 10.1002/cm.20461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou, L. , Babataheri, A. , Saitakis, M. , Bohineust, A. , Dogniaux, S. , Hivroz, C. , … Husson, J. (2016). T‐lymphocyte passive deformation is controlled by unfolding of membrane surface reservoirs. Molecular Biology of the Cell, 27(22), 3574–3582. 10.1091/mbc.E16-06-0414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou, L. , Dahl, J. B. , Lin, J. G. , Barakat, A. I. , Husson, J. , Muller, S. J. , & Kumar, S. (2016). Measuring cell viscoelastic properties using a microfluidic extensional flow device. Biophysical Journal, 111(9), 2039–2050. 10.1016/j.bpj.2016.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider, K. , Huq, M. I. , Samadi, A. R. , & Ahmad, K. (1985). Plasmid characterization of shigella spp. isolated from children with shigellosis and asymptomatic excretors. The Journal of Antimicrobial Chemotherapy, 16, 691–698. 10.1093/jac/16.6.691 [DOI] [PubMed] [Google Scholar]
- Hivroz, C. , & Saitakis, M. (2016). Biophysical aspects of T lymphocyte activation at the immune synapse. Frontiers in Immunology, 7, 46 10.3389/fimmu.2016.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, M. M. , Azad, A. K. , Bardhan, P. K. , Raqib, R. , & Islam, D. (1994). Pathology of shigellosis and its complications. Histopathology, 24, 65–71. 10.1111/j.1365-2559.1994.tb01272.x [DOI] [PubMed] [Google Scholar]
- Jansen, K. U. , Knirsch, C. , & Anderson, A. S. (2018). The role of vaccines in preventing bacterial antimicrobial resistance. Nature Medicine, 24, 10–19. 10.1038/nm.4465 [DOI] [PubMed] [Google Scholar]
- Jaumouillé, V. , Francetic, O. , Sansonetti, P. J. , & Tran Van Nhieu, G. (2008). Cytoplasmic targeting of IpaC to the bacterial pole directs polar type III secretion in Shigella. The EMBO Journal, 27, 447–457. 10.1038/sj.emboj.7601976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, S. , Hata, M. , Fukumoto, K. , Yamane, Y. , Matsui, T. , Tamura, A. , … Tsukita, S. (2002). Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nature Genetics, 31, 320–325. [DOI] [PubMed] [Google Scholar]
- Killackey, S. A. , Sorbara, M. T. , & Girardin, S. E. (2016). Cellular aspects of Shigella pathogenesis: Focus on the manipulation of host cell processes. Frontiers in Cellular and Infection Microbiology, 6, 38 10.3389/fcimb.2016.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradt, C. , Frigimelica, E. , Nothelfer, K. , Puhar, A. , Salgado‐Pabon, W. , di Bartolo, V. , … Phalipon, A. (2011). The Shigella flexneri type three secretion system effector IpgD inhibits T cell migration by manipulating host phosphoinositide metabolism. Cell Host & Microbe, 9(4), 263–272. 10.1016/j.chom.2011.03.010 [DOI] [PubMed] [Google Scholar]
- Krummel, M. F. , Sjaastad, M. D. , Wulfing, C. , & Davis, M. M. (2000). Differential clustering of CD4 and CD3zeta during T cell recognition. Science, 289(5483), 1349–1352. https://www.ncbi.nlm.nih.gov/pubmed/10958781 [DOI] [PubMed] [Google Scholar]
- Kumar, R. , Ferez, M. , Swamy, M. , Arechaga, I. , Rejas, M. T. , Valpuesta, J. M. , & van Santen, H. M. (2011). Increased sensitivity of antigen‐experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity, 35(3), 375–387. 10.1016/j.immuni.2011.08.010 [DOI] [PubMed] [Google Scholar]
- Kupfer, A. , & Dennert, G. (1984). Reorientation of the microtubule‐organizing center and the Golgi apparatus in cloned cytotoxic lymphocytes triggered by binding to lysable target cells. Journal of Immunology, 133(5), 2762–2766. http://www.ncbi.nlm.nih.gov/pubmed/6384372 [PubMed] [Google Scholar]
- Levine, M. M. , Dupont, H. L. , Khodabandelou, M. , & Hornick, R. B. (1973). Long‐term Shigella‐carrier state. The New England Journal of Medicine, 288(22), 1196–1171. [DOI] [PubMed] [Google Scholar]
- Lindner, I. , Torrvellas‐Garcia, J. , Kolonias, D. , Carlson, L. . M. , Tolba, K. . A. , Plano, G. . V. , & Lee, K. . P. (2007). Modulation and dendritic cell differentiation and fuction by YopJ of Yersinia pestis. European Journal of Immunology, 37(9), 2450–2462. 10.1002/eji.200635947 [DOI] [PubMed] [Google Scholar]
- Lipsitch, M. , & Siber, G. R. (2016). How can vaccines contribute to solving the antimicrobial resistance problem? Klugman R. John KPC, Ed. MBio., 7(3), e00428–e00416. 10.1128/mBio.00428-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Rhodes, M. , Wiest, D. L. , & Vignali, D. A. A. (2000). On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity, 13, 665–675. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Zhou, Y. , Peng, T. , Zhou, P. , Ding, X. , Li, Z. , … Shao, F. (2018). N ε‐fatty acylation of multiple membrane‐associated proteins by Shigella IcsB effector to modulate host function. Nature Microbiology, 3, 996–1009. 10.1038/s41564-018-0215-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margadant, C. , Kreft, M. , de Groot, D.‐J. , Norman, J. C. , & Sonnenberg, A. (2012). Distinct roles of Talin and kindlin in regulating integrin α5β1 function and trafficking. Current Biology, 22(17), 1554–1563. 10.1016/j.cub.2012.06.060 [DOI] [PubMed] [Google Scholar]
- Mathan, M. M. , & Mathan, V. I. (1991). Morphology of rectal mucosa with shigellosis. Reviews of Infectious Diseases, 13(Suppl 4), S314–S318. [DOI] [PubMed] [Google Scholar]
- Mattock, E. , & Blocker, A. J. (2017). How do the virulence factors of Shigella work together to cause disease? Frontiers in Cellular and Infection Microbiology, 7(64), 1–24. 10.3389/fcimb.2017.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier, J. , Boncompain, G. , Senerovic, L. , Lagache, T. , Chrétien, F. , Perez, F. , … Sauvonnet, N. (2012). Shigella effector IpaB‐induced cholesterol relocation disrupts the golgi complex and recycling network to inhibit host cell secretion. Cell Host & Microbe, 12(3), 381–389. 10.1016/j.chom.2012.07.010 [DOI] [PubMed] [Google Scholar]
- Nothelfer, K. , Arena, E. T. , Pinaud, L. , Neunlist, M. , Mozeleski, B. , Belotserkovsky, I. , … Phalipon, A. (2014). B lymphocytes undergo TLR2‐dependent apoptosis upon Shigella infection. The Journal of Experimental Medicine, 211(6), 1215–1229. 10.1084/jem.20130914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onnis, A. , Finetti, F. , & Baldari, C. T. (2016). Vesicular trafficking to the immune synapse: How to assemble receptor‐tailored pathways from a basic building set. Frontiers in Immunology, 7, 50 10.3389/fimmu.2016.00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccani, S. R. , Finetti, F. , Davi, M. , Patrussi, L. , D'Elios, M. M. , Ladant, D. , & Baldari, C. T. (2011). The Bordetella pertussis adenylate cyclase toxin binds to T cells via LFA‐1 and induces its disengagement from the immune synapse. J Exp Med, 208(6), 1317–1330. 10.1084/jem.20101558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccani, S. R. , Molin, F. D. , Benagiano, M. , Ladant, D. , D'Elios, M. M. , Montecucco, C. , & Baldari, C. T. (2008). Suppression of T‐lymphocyte activation and chemotaxis by the adenylate cyclase toxin of Bordetella pertussis. Infection and Immunity, 76(7), 2822–2832. 10.1128/IAI.00200-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais‐Correia, A. M. , Thoulouze, M. I. , & Alcover, A. (2007). T cell polarization and the formation of immunological synapses: From antigen recognition to virus spread. Current Immunology Reviews, 3, 170–188. 10.2174/157339507781483513 [DOI] [Google Scholar]
- Parsot, C. (2009). Shigella type III secretion effectors: How, where, when, for what purposes? Current Opinion in Microbiology, 12, 110–116. 10.1016/j.mib.2008.12.002 [DOI] [PubMed] [Google Scholar]
- Patrussi, L. , & Baldari, C. T. (2017). Analysis of TCR/CD3 recycling at the immune synapse. Methods in Molecular Biology, 1584, 143–155. 10.1007/978-1-4939-6881-7_10 [DOI] [PubMed] [Google Scholar]
- Pinaud, L. , Samassa, F. , Porat, Z. , Ferrari, M. L. , Belotserkovsky, I. , Parsot, C. , … Phalipon, A. (2017). Injection of T3SS effectors not resulting in invasion is the main targeting mechanism of Shigella toward human lymphocytes. Proceedings of the National Academy of Sciences, 114(37), 9954–9959. 10.1073/pnas.1707098114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinaud, L. , Sansonetti, P. J. , & Phalipon, A. (2018). Host cell targeting by enteropathogenic bacteria T3SS effectors. Trends in Microbiology, 26(4), 266–283. 10.1016/j.tim.2018.01.010 [DOI] [PubMed] [Google Scholar]
- Ramsden, A. E. , Holden, D. W. , & Mota, L. J. (2007). Membrane dynamics and spatial distribution of salmonella‐containing vacuoles. Trends in Microbiology, 15(11), 516–524. 10.1016/j.tim.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Raqib, R. , Mia, S. M. S. , Qadri, F. , Alam, T. I. , Alam, N. H. , Chowdhury, A. K. , … Andersson, J. (2000). Innate immune responses in children and adults with shigellosis. Infection and Immunity, 68(6), 3620–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raqib, R. , Qadri, F. , SarkEr, P. , Mia, S. M. S. , Sansonnetti, P. J. , Albert, M. J. , & Andersson, J. (2002). Delayed and reduced adaptive humoral immune responses in children with shigellosis compared with in adults. Scandinavian Journal of Immunology, 55(4), 414–423. 10.1046/j.1365-3083.2002.01079.x [DOI] [PubMed] [Google Scholar]
- Roumier, A. , Olivo‐Marin, J. C. , Arpin, M. , Michel, F. , Martin, M. , Mangeat, P. , … Alcover, A. (2001). The membrane‐microfilament linker ezrin is involved in the formation of the immunological synapse and in T cell activation. Immunity, 15(5), 715–728. http://www.ncbi.nlm.nih.gov/pubmed/11728334 [DOI] [PubMed] [Google Scholar]
- Roy, N. H. , & Burkhardt, J. K. (2018). The actin cytoskeleton: A mechanical intermediate for signal integration at the immunological synapse. Frontiers in Cell and Development Biology, 6(September), 116 10.3389/fcell.2018.00116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo, S. P. , & Holden, D. W. (2003). SseG, a virulence protein that targets salmonella to the Golgi network. The EMBO Journal, 22(19), 5003–5014. 10.1093/emboj/cdg517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado‐Pabon, W. , Celli, S. , Arena, E. T. , Nothelfer, K. , Roux, P. , Sellge, G. , … Phalipon, A. (2013). Shigella impairs T lymphocyte dynamics in vivo. Proceedings of the National Academy of Sciences, 110(12), 4458–4463. 10.1073/pnas.1300981110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka, A. , Babataheri, A. , Dogniaux, S. , Barakat, A. I. , Gonzalez‐Rodriguez, D. , Hivroz, C. , & Husson, J. (2017). Micropipette force probe to quantify single‐cell force generation: Application to T‐cell activation. Théry M, Ed. Mol Biol Cell., 28(23), 3229–3239. 10.1091/mbc.e17-06-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , … Cardona, A. (2012). Fiji: An open‐source platform for biological‐image analysis. Nature Methods, 9(7), 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sörensen, M. , Lippuner, C. , Kaiser, T. , Mißlitz, A. , Aebischer, T. , & Bumann, D. (2003). Rapidly maturing red fluorescent protein variants with strongly enhanced brightness in bacteria. FEBS Letters, 552, 110–114. 10.1016/S0014-5793(03)00856-1 [DOI] [PubMed] [Google Scholar]
- Sperandio, B. , Regnault, B. , Guo, J. , Zhang, Z. , Stanley, S. L., Jr , Sansonetti, P. J. , & Pédron, T. (2008). Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. The Journal of Experimental Medicine, 205(5), 1121–1132. 10.1084/jem.20071698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S. , Kubo, K. , Waguri, S. , Yabashi, A. , Shin, H. W. , Katoh, Y. , & Nakayama, K. (2012). Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. Journal of Cell Science, 125(17), 4049–4057. 10.1242/jcs.102913 [DOI] [PubMed] [Google Scholar]
- Thauland, T. J. , Hu, K. H. , Bruce, M. A. , & Butte, M. J. (2017). Cytoskeletal adaptivity regulates T cell receptor signaling. Science Signaling, 10(469), 1–11. 10.1126/scisignal.aah3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoulouze, M. I. , Sol‐Foulon, N. , Blanchet, F. , Dautry‐Varsat, A. , Schwartz, O. , & Alcover, A. (2006). Human immunodeficiency virus type‐1 infection impairs the formation of the immunological synapse. Immunity, 24(5), 547–561. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16713973 [DOI] [PubMed] [Google Scholar]
- Tinevez, J. Y. , Perry, N. , Schindelin, J. , Hoopes, G. M. , Reynolds, G. D. , Laplantine, E. , … Eliceiri K. W. (2017). TrackMate: An open and extensible platform for single‐particle tracking. Methods, 115, 80–90. 10.1016/j.ymeth.2016.09.016 [DOI] [PubMed] [Google Scholar]
- Valdivia, R. H. , Hromockyj, A. E. , Monack, D. , Ramakrishnan, L. , & Falkow, S. (1996). Applications for green fluorescent protein (GFP) in the study of host‐pathogen interactions. Gene, 173, 47–52. 10.1016/0378-1119(95)00706-7 [DOI] [PubMed] [Google Scholar]
- Valencia‐Gallardo, C. M. , Carayol, N. , & Tran Van Nhieu, G. (2015). Cytoskeletal mechanics during Shigella invasion and dissemination in epithelial cells. Cellular Microbiology, 17, 174–182. 10.1111/cmi.12400 [DOI] [PubMed] [Google Scholar]
- Yao, T. , Mecsas, J. , Healy, J. I. , Falkow, S. , & Chien, Y. (1999). Suppression of T and B lymphocyte activation by a Yersinia pseudotuberculosis virulence factor, Yoph. The Journal of Experimental Medicine, 190(9), 1343–1350. 10.1084/jem.190.9.1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 supporting Information
Video S2 supporting Information
Figure S1 Shigella infection increases F‐actin content in Jurkat T cells. F‐actin content measurements. Following infection, cells were fixed and stained with fluorescent phalloidin whose intensity was quantified by confocal microscopy. Data from 3 independent experiments. Mean +/− SD, **** p < 0.0001 (Mann–Whitney).
Figure S2‐figure supplement 1: Tf vesicular trafficking is modulated by T3SS effectors in Shigella‐invaded Jurkat cells. Flow cytometry analysis of Tf recycling (A) or endocytosis (B) in Jurkat cells non‐infected or infected with Shigella WT or the mxiD or ipaJvirA mutant strains as previously described. (A) To assess for Tf recycling, Jurkat cells were infected with WT Shigella or the ipaJvirA mutant in the presence of fluorescently labelled TfAF647, prior to incubation with gentamicin containing media to kill extracellular bacteria. Following the removal of surface‐attached TfA647 by acid stripping, a Tf recycling time‐course was performed in the presence of non‐fluorescent holotransferrin. Data are shown as the percentage of the remaining intracellular TfA647 fluorescence at each time point. (B) For Tf endocytosis assays, cells were infected with WT Shigella and the mxiD or ipaJvirA mutants, prior to incubation on ice for 30 min with TfAF647. Cells were then incubated at 37°C and the rate of Tf endocytosis was measured over time following acid stripping. Data are presented as mean fluorescent intensity (MFI) of the intracellular TfAF647 pool and are representative of three individual experiments.
Figure S3‐figure supplement 1: ImageStream gating strategy. (1) Unfocused cells were excluded by using the gradient RMS feature, which measures the quality of an image by detecting important changes of pixel values in the image. (2) In order to discriminate between single cells and doublets, the brightfield Area versus Aspect ratio (i.e., minor axis divided by the major axis) for the focused cells was plotted. Indeed, as opposed to single cells, which have intermediate Area values and a high Aspect Ratio, doublets have a higher area value and a smaller aspect ratio. (3) Doublets of CD3+ and CD19+ cells were identified by plotting the CD3 vs CD19 intensity. (4) In order to focus on doublet with only one CD19+ cell, the aspect ratio intensity feature was used. This feature corresponds to the minor axis intensity divided by the major axis intensity. Doublets with high aspect ratio intensity for CD19 (i.e., close to 1) were considered as doublet of interest. (5) Doublets with high intensity of CD3 were identified by plotting the max pixel feature versus the area. Indeed, the max pixel feature is used to estimate the true peak fluorescence activity. Concerning CD3, a high max pixel intensity value is expected at the interface when a canonical synapse is formed. (6) Similarly, actin clustering in the IS mask was identified by plotting the max pixel intensity of actin versus the area in order to formally identify canonical ISs. BF: Brightfield.