

Abstract
Trifarotene is a new drug with retinoic acid receptor activity and selectivity for retinoic acid receptor‐γ. The reported studies aimed at assessing the clinical pharmacology and safety of trifarotene. The clinical pharmacology of topical trifarotene up to 100 µg/g was extensively investigated through 2 maximal usage pharmacokinetic trials (MUsT) conducted in adult (≥18 years) and pediatric patients (9‐17 years) with moderate to severe acne and two studies conducted in healthy volunteers: 1 thorough QTC study and 1 drug‐drug interaction study with concomitantly administered oral levonorgestrel (0.15 mg)/ethinyl estradiol (0.03 mg). Safety assessments included adverse event reporting and assessment of erythema, scaling, dryness, and stinging/burning using a scale from 0 = none to 4 = severe, as well as the evaluation of the systemic safety of trifarotene through routine laboratory testing. Systemic absorption of trifarotene was generally unquantifiable in the target population, especially when applied at 50 µg/g. QTC investigations did not show any risk of cardiovascular health issues; trifarotene did not reduce the systemic exposure to oral contraceptives such as levonorgestrel/ethinyl estradiol. Safety analyses did not show local or systemic safety concerns with trifarotene up 100 µg/g, a dose twice as high as the intended market dose. Results showed that trifarotene 50 µg/g cream is well tolerated and safe, even when applied under maximized conditions in adults and pediatric acne patients presenting with severe acne. Daily use of trifarotene 50 µg/g cream was not associated with cardiovascular effects and did not result in drug‐drug interaction in women of childbearing potential using oral contraception.
Keywords: drug‐drug interaction, MUsT, pharmacokinetics, TQC study, trifarotene
Acne vulgaris is a common chronic skin disease of the face, neck, upper chest, and back.1, 2 Retinoids play a central role in the treatment of acne due to their ability to reduce inflammation and to normalize the desquamation of follicular epithelium, leading to the elimination of comedones and the inhibition of new microcomedone formation.3 Trifarotene is a new chemical entity (Galderma Research and Development LLC, Fort Worth, Texas) recently approved in the United States (label: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211527s000lbl.pdf). In primary pharmacology studies, trifarotene showed retinoic acid receptor (RAR) activity and a RARγ selectivity. RARγ is the receptor subtype present in keratinocytes and is recognized to be the most relevant in acne over RARα and RARβ. Moreover, trifarotene modulates retinoid target genes (differentiation and inflammatory processes) in immortalized keratinocytes and reconstructed epidermis.4 Trifarotene 50 µg/g cream was developed for the treatment of acne vulgaris on large areas of the face and/or trunk in patients 9 years of age and older.
Systemic exposure to topically applied trifarotene up to 100 µg/g was investigated through 4 clinical studies: 2 studies were maximal usage pharmacokinetic (PK) trials (MUsT5, 6) conducted in subjects with acne vulgaris. The 2 other studies were conducted in healthy volunteers: 1 clinical drug‐drug interaction (DDI) study and 1 thorough QTC (TQT7) study to assess the potential for cardiac repolarization delays. Considering the pharmacologic class of trifarotene (retinoids) and the class‐associated potential teratogenicity, and because trifarotene is intended to be used by women of childbearing potential, a DDI study was conducted to assess the potential trifarotene effects on contraceptive steroids as recommended by European Medicines Agency guidelines.8
The nonclinical development of trifarotene was performed in agreement with International Conference on Harmonization guideline M3(R2) for a chronic clinical indication. For reproductive and development toxicity, reproductive and development toxicity studies in rats, embryo‐fetal toxicity in rats and rabbits, and pre‐ and post natal development toxicity in rats were performed.
It is worth noting that preclinical investigations indicated that trifarotene had a large safety margin for teratogenic effects and no in vitro DDI potential; nonetheless, a clinical DDI study was conducted to provide clear guidance in terms of risk mitigation measures by specifically addressing possible interactions affecting the efficacy of levonorgestrel/ethinyl estradiol (LNG/EE, Portia, Teva Pharmaceuticals USA, Inc, Forest, Virginia), a contraceptive steroid. LNG/EE is a well‐known hormonal oral contraceptive that has been on the market since the early 1970s. Ethinyl estradiol is metabolized to various hydroxylated metabolites and their glucuronide and sulfate conjugates via cytochrome P450 (CYP) 3A4 and CYP2C9.9 Compounds that induce CYP3A4 have been shown to reduce the systemic exposure to oral contraceptives such as LNG/EE, with associated risks of contraceptive failure.10 The purpose of the QTC study was to assess the potential of trifarotene to prolong ventricular repolarization.11, 12
Trifarotene 50 µg/g cream once daily was selected as the final concentration and dose regimen for the topical treatment of acne vulgaris of the face and trunk based on results from phase 2 studies completed during the development program. Although both doses of 50 µg/g and 100 µg/g of trifarotene provide a positive benefit‐risk ratio in the treatment of acne vulgaris, the better safety and local tolerability profile of trifarotene 50 µg/g cream supported the use of this dose in the phase 3 studies. Phase 3 studies demonstrated that trifarotene 50 µg/g cream provides significant and clinically meaningful benefit to subjects 9 years of age and older with acne vulgaris of the face and trunk.
Methods
All studies were conducted between 2012 and 2017 in accordance with the Declaration of Helsinki, Good Clinical Practice, and other relevant regulatory guidelines. Study protocols/amendments were approved by appropriate institutional review boards/independent ethics committees. All participants and/or their legal representatives, provided written informed consent prior to study enrollment.
Study Design
Maximal Usage PK trials
The two MUsT (EUDRACT 2012–000521047 and IND 111091) were multicenter, randomized, double‐blind, 2‐parallel‐group studies conducted in patients with acne to assess trifarotene plasma concentrations after 29 daily topical applications of trifarotene 50 µg/g or 100 µg/g under maximal use conditions. The adult MUsT (MUsT 1) was conducted in Germany, Hungary, and the United States. The pediatric MUsT (MUsT 2) was conducted in the United States.
In MUsT 1, patients were aged 18 to 34 years, with an acne severity grade of 4 (severe acne) on the 0 to 4 grade Investigator's Global Assessment (IGA, graded from 0 = none to 4 = severe), and at least 30 noninflammatory and 40 inflammatory lesions on the face at Screening and Baseline. In MUsT 2, participants were aged 10 to 17 years; subjects up to 11 years were required to have an IGA ≥3 (moderate to severe acne), while patients aged 12 to 17 years had to have an IGA = 4 and at least 40 noninflammatory and 25 inflammatory lesions on the face.
Once‐daily applications of trifarotene 50‐ or 100‐µg/g cream for 29 days were performed by a qualified person. To ensure maximal conditions of use, patients were treated in all zones potentially affected by acne, such as the face, shoulders, upper back, and upper chest, regardless of acne severity at baseline. In MUsT 1, the average daily dose of trifarotene cream was 2 g; in MUsT 2, it was calculated in milligrams per kilogram of body weight on a subject‐specific basis ranging from 1.1 to 2 g.
TQT study
This was a randomized, double‐blind, vehicle and moxifloxacin (positive) control, parallel‐group study to assess effects of supratherapeutic topical exposure to trifarotene on ventricular repolarization conducted at one study site in France (EUDRACT 2012‐001979‐37 and IND 111091). Participants were healthy subjects aged 18 to 65 years, with a normal 12‐lead electrocardiogram and without specific contraindication to moxifloxacin. The study was designed in accordance with the International Conference on Harmonization E14 guideline.13 Trifarotene 100‐µg/g gel was used in this study to obtain a higher exposure than that achievable with the to‐be‐marketed formulation. Subjects were treated once daily (twice on day 14) for 15 days with either 12 g of trifarotene 100‐µg/g gel (supratherapeutic dose) or with trifarotene vehicle. Both treatments were combined with the appropriate control on day 15 (placebo or moxifloxacin). There were 3 treatment groups in this study: trifarotene vehicle + moxifloxacin capsules, trifarotene vehicle + placebo capsules, and trifarotene + placebo capsules. Twelve‐lead electrocardiograms extracted from Holter over a 24‐hour period with time‐matched PK sampling were performed on day 15. In addition to PK parameters on day 15, the pharmacodynamic relationship between the duration of the QT/QTc intervals and the trifarotene plasma concentrations was assessed.
DDI study
This was an open‐label study conducted to assess the potential for trifarotene 100‐µg/g cream to reduce the systemic exposure of coadministered oral contraceptives (LNG 0.15 mg/EE 0.03 mg). LNG and EE were selected because they are frequently used in combined oral contraceptives and they are extensively metabolized by the CYP enzymes, including CYP3A4 and CYP2C9 which contribute to the direct metabolism of trifarotene. The PK profile of the oral contraceptive combination (LNG/EE) was assessed before (day 1) and after (day 18) repeated topical applications of trifarotene once daily for 14 days (started on day 5). Blood samples for PK assessments for LNG and EE were collected at day 1 and day 18 visits; before dosing; and at 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 48, and 72 hours after dosing. In addition, the 72‐hour complete PK profile of trifarotene was assessed at the end of the treatment period (day 18) at the same time points, to confirm exposure to trifarotene.
Participants were healthy females aged 18 to 35 years, not breastfeeding, and not pregnant or trying to become pregnant. Subjects received multiple daily applications of 2 g of trifarotene cream 100 µg/g to be exposed to plasma concentrations close to the highest trifarotene levels observed in the MUsT studies.
Pharmacokinetic/Bioanalysis Evaluations
Complete PK profiles were obtained at different study time periods, and trifarotene plasma concentrations were determined using validated methods of liquid chromatography coupled with tandem mass spectrometry. The lower limit of quantification (LLOQ) were 5 pg/mL for trifarotene, 5 pg/mL for EE, and 100 pg/mL for LNG. If sufficient plasma concentrations were above LLOQ, PK parameters were calculated using a noncompartmental analysis approach (Phoenix WinNonlin, validated version 6.3). The following PK parameters were determined for each subject: maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), residual drug concentration (Ctrough), area under the plasma concentration–time curve calculated from time 0 to the time corresponding to the last quantifiable concentration and over 24 hours (AUC0‐t, AUC0‐24h), elimination rate constant value (kel), AUC calculated from time 0 and extrapolated to infinity (AUC0‐inf), and half‐life (t1/2). Of note, when the acceptability criteria for kel were not met (ie, at least 3 data points log‐linear terminal phase, excluding Cmax and a coefficient of regression ≥0.98), t1/2 was not reported.
Safety Assessments
Safety assessments included adverse event (AE) reporting, assessment of local tolerance, including erythema, scaling, dryness, and stinging/burning using a scale from 0 = none to 4 = severe, and routine laboratory testing.
Statistical Analysis
Pharmacokinetic parameters were analyzed descriptively using SAS software (version 9.2 or above). In the MUsTs, an analysis of variance was performed on systemic PK parameters to estimate time and dose effect. The following PK parameters were analyzed: Ctrough, AUC0‐24h, and Cmax using SAS Proc GLM. The 90% confidence intervals (CIs) for the pairwise differences between times or groups were calculated.
In the TQT study, a scatter plot of Fridericia's corrected QT interval (QTcF) changes from baseline adjusted for the mean placebo value at the corresponding time point vs logarithmically transformed trifarotene concentrations was provided. A Spearman correlation coefficient was calculated.
In the DDI study, ratios of the geometric least square (LS) means (day 18/day 1) and their 90%CIs for 2 PK parameters (AUC0‐t, Cmax) of LNG and EE were analyzed. There was DDI if the test (LNG/EE + trifarotene) to reference (LNG/EE alone) 90%CIs for the geometric LS mean ratios did not fall fully inside the accepted 80% to 125% range.
Results
MUsT 1: Adults
Thirty‐nine adult patients, with a mean age of 21 ± 4 years at baseline and with severe acne received trifarotene 50 µg/g cream (N = 21; 19 completed the study) or trifarotene 100‐µg/g cream (N = 18; 17 completed the study). See Table 1 for details.
Table 1.
Baseline Characteristics for MUsT Studies (Adult and Pediatric)
| Trifarotene 50 µg/g Cream | Trifarotene 100‐µg/g Cream | TOTAL | ||
|---|---|---|---|---|
| MUsT 1: Adult | (N = 21) | (N = 18) | (N = 39) | |
| Age, y | Mean ± SD | 20.81 ± 3.44 | 21.78 ± 4.25 | 21.26 ± 3.82 |
| Median (min‐max) | 19.00 (18‐31) | 20.00 (18‐34) | 20.00 (18‐34) | |
| Sex, n (%) | Male | 5 (23.8) | 4 (22.2) | 9 (23.1) |
| Female | 16 (76.2) | 14 (77.8) | 30 (76.9) | |
| Race, n (%) | White | 21 (100.0) | 18 (100.0) | 39 (100.0) |
| IGA score | 4 (severe) | 21 (100.0) | 18 (100.0) | 39 (100.0) |
| Inflammatory lesion counts | Mean ± SD | 62.0 ± 26.2 | 50.6 ± 9.1 | … |
| Median (min‐max) | 53.0 (40.0‐139.0) | 49.0 (40.0‐69.0) | … | |
| Noninflammatory lesion counts | Mean ± SD | 65.6 ± 31.3 | 50.6 ± 18.8 | 59.3 ± 16.88 |
| Median (min‐max) | 53.0 (32.0‐166.0) | 47.0 (33.0‐100.0) | 61.0 (35‐114) |
| Trifarotene 50 µg/g cream | Trifarotene 100‐µg/g cream | TOTAL | ||
|---|---|---|---|---|
| MUsT 2: Pediatric | (N = 18) | (N = 17) | (N = 35) | |
| Age, y | N | 18 | 17 | 35 |
| Mean ± SD | 14.9 ± 2.11 | 14.8 ± 1.79 | 14.8 ± 1.93 | |
| Median (min‐max) | 15.5 (10‐17) | 15.0 (12‐17) | 15.0 (10‐17) | |
| Age group, n (%) | 9‐11 years | 2 (11.1) | 0 | 2 (5.7) |
| 12‐17 years | 16 (88.9) | 17 (100.0) | 33 (94.3) | |
| Sex, n (%) | Male | 12 (66.7) | 13 (76.5) | 25 (71.4) |
| Female | 6 (33.3) | 4 (23.5) | 10 (28.6) | |
| Race, n (%) | White | 14 (77.8) | 14 (82.4) | 28 (80.0) |
| Black or African American | 3 (16.7) | 2 (11.8) | 5 (14.3) | |
| Other | 1 (5.6) | 1 (5.9) | 2 (5.7) | |
| IGA score, n (%) | 3 (moderate) | 2 (11.1) | 0 | 2 (5.7) |
| 4 (severe) | 16 (88.9) | 17 (100.0) | 33 (94.3) | |
| Inflammatory lesion counts | N | 16 | 17 | 33 |
| Mean ± SD | 35.6 ± 6.95 | 38.9 ± 10.56 | 37.3 ± 9.02 | |
| Median (min‐max) | 35.0 (26‐52) | 34.0 (27‐58) | 35.0 (26‐58) | |
| Noninflammatory lesion counts | N | 16 | 17 | 33 |
| Mean ± SD | 53.1 ± 12.14 | 65.1 ± 18.92 | 59.3 ± 16.88 | |
| Median (min‐max) | 48.0 (35‐77) | 63.0 (38‐114) | 61.0 (35‐114) |
IGA, Investigator's Global Assessment; max, maximum; min, minimum; SD, standard deviation.
A descriptive summary of PK results on day 1, day 15, and day 29 is presented in Table 2. Mean plasma concentrations over 24 hours and 72 hours are presented in Figure 1A‐B. After 29 applications of trifarotene 50 µg/g cream, trifarotene plasma concentrations were quantifiable in 7 of 19 (37%) patients, with a Cmax ranging from 5 to 10 pg/mL and an AUC0‐24h from 75 to 104 pg • h/mL. In the trifarotene 100‐µg/g cream group, trifarotene plasma concentrations were quantifiable in 11 of 18 (61%) patients, with a Cmax of 11 ± 8 pg/mL and an AUC0‐24h of 119 ± 53 pg • h/mL (mean ± SD). Tmax averaged approximately 4 hours in both the 50‐ and 100‐µg/g groups, suggesting a rapid absorption of trifarotene through the skin. T1/2 could be determined in only 3 subjects in the 100‐µg/g group and ranged from 2 to 9 hours.
Table 2.
Descriptive Summary of Trifarotene PK Parameters in Adult Population (MUsT 1) and Pediatric Population (MUsT 2)
| Trifarotene 50 µg/g | Trifarotene 100 µg/g | ||||||
|---|---|---|---|---|---|---|---|
| MUsT 1: Adult | Cmax (pg/mL) | tmax (h) | AUC0‐24h (pg • h/mL) | Cmax (pg/mL) | tmax (h) | AUC0‐24h (pg • h/mL) | |
| Day 1 | Mean ± SD | – | – | – | – | – | – |
| N (N quantifiable) | 19 (0) | 19 (0) | 19 (0) | 18 (2) | 18 (2) | 18 (2) | |
| Min‐max | <5 | – | – | <5 ‐ 7 | 4 ‐ 4 | 80 ‐ 83 | |
| CV (%) | – | – | – | – | – | – | |
| Day 15 | Mean ± SD | – | – | – | 10 ±10 | 4 ± 2 | 113 ± 88 |
| N (N quantifiable) | 19 (2) | 19 (2) | 19 (2) | 18 (9) | 18 (9) | 18 (9) | |
| Min‐max | <5‐8 | 4‐6 | 86‐100 | <5‐49 | 2‐8 | 79‐456 | |
| CV (%) | – | – | – | 110 | 37 | 78 | |
| Day 29 | Mean ± SD | – | – | – | 11 ± 8 | 4 ± 1 | 119 ± 53 |
| N (N quantifiable) | 19 (7) | 19 (7) | 19 (7) | 18 (11) | 18 (11) | 18 (11) | |
| Min‐max | <5‐10 | 3.9‐4.0 | 75‐104 | <5‐31 | 4‐8 | 79‐244 | |
| CV (%) | – | – | – | 71 | 28 | 45 | |
| Trifarotene 50 µg/g Cream | Trifarotene 100‐µg/g Cream | ||||||
|---|---|---|---|---|---|---|---|
| MUsT 2: Pediatric | Cmax (pg/mL) | tmax (h) | AUC0‐24h (pg • h/mL) | Cmax (pg/mL) | tmax (h) | AUC0‐24h (pg • h/mL) | |
| Day 1 | Mean ± SD | – | – | – | – | – | – |
| N (N quantifiable) | 18 (0) | – | 18 (0) | 17 (2) | 2 | 17 (2) | |
| Min‐max | – | – | – | 5‐7 | 4‐4 | 68‐83 | |
| CV (%) | – | – | – | – | – | – | |
| Day 15 | Mean ± SD | – | – | – | 18 ± 17 | 3.6 ± 0.9 | 163 ± 123 |
| N (N quantifiable) | 14 (0) | – | 14 (0) | 13 (9) | 9 | 13 (9) | |
| Min‐max | – | – | – | 5 ‐ 52 | 2 ‐ 4 | 68 ‐ 416 | |
| CV (%) | – | – | – | 95 | 24 | 75 | |
| Day 29 | Mean ± SD | – | – | – | 12 ± 12 | 3.5 ± 1.3 | 137 ± 119 |
| N (N quantifiable) | 17 (3) | 17 (3) | 17 (3) | 16 (11) | 16 (11) | 16 (11) | |
| Min‐max | 7‐9 | 4‐4 | 89‐106 | 5‐52 | 2‐6 | 68‐547 | |
| CV (%) | – | – | – | 98 | 37 | 87 | |
AUC0‐24h, area under the concentration‐time curve from predose through 24 hours after dosing; Cmax, maximum plasma concentration; CV, coefficient of variation; min, minimum; max, maximum; PK, pharmacokinetics; SD, standard deviation; tmax, time at which Cmax occurs; –, nonreportable (ie, when strictly <50% of the data are quantifiable).
For descriptive statistics calculations, data <5 pg/mL were replaced by the lower limit of quantification for Cmax (ie, 5 pg/mL) and by the lowest AUC value of the study (ie, 79 pg • h/mL for the trifarotene 100‐µg/g cream AUC0‐24h).
Figure 1.
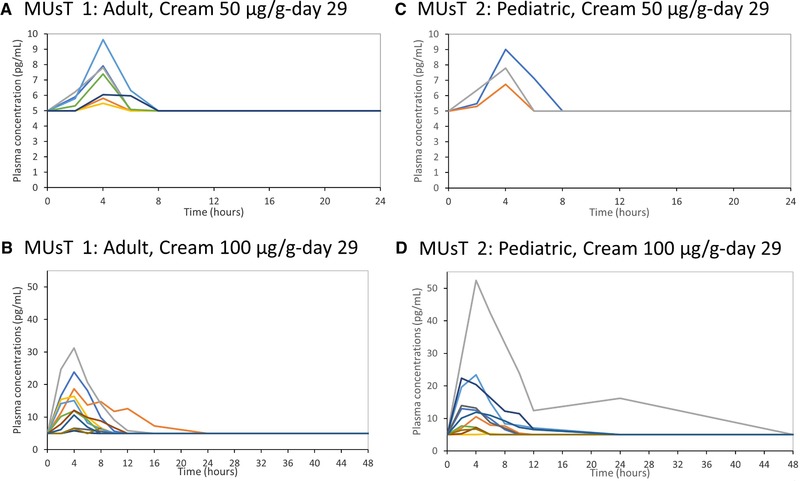
Adult MUsT (MUsT 1): Individual plasma profiles on day 29 for (A) trifarotene 50 µg/g cream (7/19 quantifiable subjects) and (B) trifarotene 100‐µg/g cream (11/18 quantifiable subjects). Pediatric MUsT (MUsT 2): Individual plasma profiles on day 29 for (C) trifarotene 50 µg/g cream (3/17 quantifiable subjects) and (C) trifarotene 100‐µg/g cream (11/16 quantifiable subjects). Note the 72‐hour time point was excluded from the figure because all plasma concentrations were nonquantifiable. Nonquantifiable data were replaced by the LLOQ (ie, 5 pg/mL). LLOQ, lower level of quantification; MUsT, maximal usage pharmacokinetic trial.
Due to the low number of quantifiable plasma concentrations, no formal statistical analysis to assess the time and dose effects could be performed. In the 100‐µg/g group the arithmetic mean values at day 15 and day 29 in 11 of 18 (61%) patients were comparable for Cmax (10 ± 10 and 11 ± 8 pg/mL, respectively) and AUC0‐24h (113 ± 88 and 119 ± 53 pg • h/mL, respectively), suggesting that steady state was reached on day 15. Similar results were obtained with geometric mean values comparison for both Cmax and AUC0‐24h.
Overall, there were no serious AEs, and no AEs led to discontinuation. The percentages of subjects with at least 1 AE were 88.9% for the trifarotene 100‐µg/g group and 66.7% for the trifarotene 50 µg/g group. Most of the related AEs were dermatological (either erythema or skin irritation), and only 1 of these was of severe intensity. The worst scores for erythema, scaling, dryness, and stinging/burning were higher in the trifarotene 100‐µg/g group than in the 50 µg/g group over the treatment period, indicating that the 100‐µg/g cream was less well tolerated than the 50 µg/g cream. No clinically significant changes in hematology or biochemistry were observed.
MUsT 2
Thirty‐five pediatric patients with a mean age of 15 ± 2 years at baseline and moderate to severe acne applied trifarotene 50 µg/g cream (N = 18; 17 completed the study) or trifarotene 100‐µg/g cream (N = 17; 16 completed the study). Detailed demographic data are provided in Table 1.
A descriptive summary of PK results on day 1, day 15, and day 29 is presented in Table 2. Mean plasma profiles are presented in Figure 1C‐D. After 29 applications of trifarotene 50 µg/g cream under maximal use conditions, trifarotene plasma concentrations were quantifiable in 3 of 17 (18%) patients aged 12 to 17 years, with Cmax ranging from 7 to 9 pg/mL and AUC0‐24h from 89 to 106 pg • h/mL. In the trifarotene 100‐µg/g group, trifarotene plasma concentrations were quantifiable in 11 of 16 (69%) in patients aged 10 to 17 years, with a Cmax of 12 ± 12 pg/mL and an AUC0‐24h of 137 ± 119 pg • h/mL (mean ± SD). A peak plasma concentration (tmax) was observed approximately 4 hours after application for both groups, suggesting rapid absorption of trifarotene through the skin. Due to the lack of a distinct elimination phase, t1/2 was determined in only 2 patients treated with trifarotene 100‐µg/g cream (3 hours for both patients).
The time effect was assessed on the trifarotene 100‐µg/g cream–treated group only. The mean geometric ratio of AUC0‐24h on day 29 vs AUC0‐24h on day 15 was 0.94 (90%CI, 0.7‐1.3) providing evidence for steady‐state achievement by day 15. A dose effect of trifarotene could not be assessed due to the low number of quantifiable plasma concentrations in the 50 µg/g group; however, a higher systemic exposure was observed with trifarotene 100 µg/g.
Overall, 8 subjects experienced 13 treatment‐emergent adverse events (TEAEs). Of these, 4 subjects experienced 6 related TEAEs of skin exfoliation, erythema, and skin irritation. Worst postbaseline local tolerability scores of erythema, scaling, dryness, and stinging/burning were mild in the majority of subjects. A total of 8 subjects who received trifarotene 100‐µg/g cream, required dose modification due to skin irritations. The mean duration of dose modification was 5.5 days, ranging from 2 to 15 days.
No adverse event led to study discontinuation. No clinically significant changes in hematology or biochemistry were observed.
TQT Study
One hundred eighty adult subjects were randomized to treatment, 60 in each treatment group. Demographic and baseline data are detailed in Table 3. Peak plasma concentrations are detailed in Table 4.
Table 3.
Baseline Characteristics for TQT Study
| Trifarotene Vehicle + Moxifloxacine | Trifarotene Vehicle + Placebo | Trifarotene + Placebo | Total | ||
|---|---|---|---|---|---|
| TQT | (N = 60) | (N = 60) | (N = 60) | (N = 180) | |
| Age, y | Mean ± SD | 38.72 ± 14.05 | 37.43 ± 13.59 | 37.00 ± 13.83 | 37.72 ± 13.77 |
| Median (min‐max) | 37.00 (19‐65) | 35.00 (19‐65) | 33.50 (19‐64) | 36.00 (19‐65) | |
| Sex, n (%) | Male | 32 (53.3) | 31 (51.7) | 33 (55.0) | 96 (53.3) |
| Female | 28 (46.7) | 29 (48.3) | 27 (45.0) | 84 (46.7) | |
| Race, n (%) | White | 56 (93.3) | 58 (96.7) | 57 (95.0) | 171 (95.0) |
| Black | 1 (1.7) | 1 (1.7) | 2 (3.3) | 4 (2.2) | |
| American Indian | 1 (1.7) | … | 1 (1.7) | 2 (1.1) | |
| Asian | 2 (3.3) | 1 (1.7) | … | 3 (1.7) |
TQT, thorough QTC.; min, minimum; max, maximum; SD, standard deviation.
Table 4.
Comparison of Trifarotene Maximum Plasma Exposure Across Studies Under Steady‐State Conditions in MUsT 1 and 2 and TQT Study
| Study | Population | N | Formulation | Daily Dose | Cmax (pg/mL), Mean ± SD (Min‐Max) |
|---|---|---|---|---|---|
| MUsT 1 | Acne adults | 19 | Cream 50 µg/g | 2 g | NR (<5‐8) |
| MUsT 2 | Acne adolescents | 17 | Cream 50 µg/g | Up to 2 g | NR (<5‐9) |
| TQT | Healthy subjects | 58 | Gel 100 µg/g | 12 g once daily on 6000 cm² (the day before ECG 12 g twice‐daily) | 33 ± 34 (<5‐187) |
max, maximum; min, minimum; MUsT, maximal usage pharmacokinetic trial; NR, nonreportable due to the high proportion of nonquantifiable data; SD, standard deviation; TQT, thorough QTC.
The mean trifarotene peak plasma concentration was 3‐fold higher (33 ± 34 pg/mL; range, <5‐187 pg/mL) than the highest Cmax obtained with trifarotene 50 µg/g cream under maximal use conditions, thus confirming that supratherapeutic conditions were achieved in the TQT study. Results of the primary end point, expressed as estimates of QTcF double‐delta (baseline‐adjusted, placebo‐subtracted) demonstrated over time that all upper bounds of the 90%CI were <10 milliseconds for all postdose time points for trifarotene. Likewise, the lower bound of the 97.5%CI for the QTcF double‐delta between moxifloxacin and placebo was >5 milliseconds for postdose time points.
No correlation was observed between logarithmically transformed plasma concentrations of trifarotene and placebo‐adjusted QTcF time‐matched changes from baseline (Figure 2). Overall, a supratherapeutic dose of trifarotene did not prolong the QTc interval. This was combined with an adequate response of moxifloxacin, confirming the sensitivity of the assay.
Figure 2.
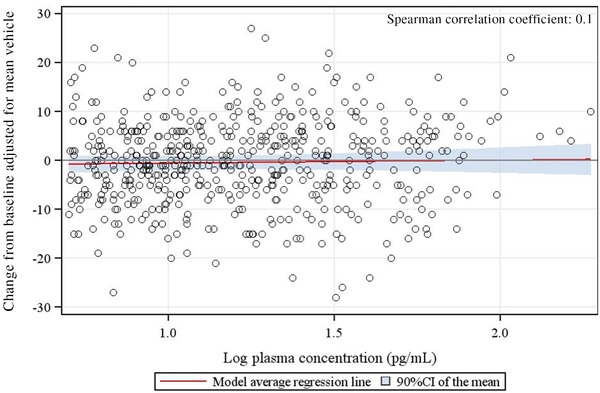
TQT study: Concentration response relationship.
Fifty‐one subjects reported trifarotene‐related moderate dermatologic TEAEs. Fifty subjects had erythema and scaling, and 1 had stinging/burning reported. One subject in the trifarotene + placebo group had 3 related AEs leading to discontinuation (severe erythema on right and left legs, moderate erythema on abdomen). All events of erythema were reported to have resolved on day 25. No TEAEs were reported in the vehicle + moxifloxacin or trifarotene vehicle + placebo groups. The most significant cutaneous irritation was reported in the trifarotene + placebo group, with the abdomen and back having the worst postbaseline scores compared to the legs. No clinically significant changes in hematology or biochemistry were observed.
DDI Study
Twenty‐four healthy female subjects were enrolled in the study. Two subjects were excluded from the PK analysis due to major protocol deviations. All subjects presented quantifiable EE and LNG plasma concentrations. Mean profiles of EE and LNG plasma concentrations at baseline and day 18 are presented in Figure 3A‐B. Repeated applications of trifarotene 100‐µg/g cream resulted in quantifiable systemic exposure in 6 of the 22 (27%) subjects, with Cmax ranging from 5 pg/mL to 11 pg/mL (Figure 4). An inferential analysis to determine DDI was performed for Cmax and AUC0‐t. The 90%CIs of the geometric LS mean ratios for these parameters fell within the acceptance range of bioequivalence (0.80‐1.25), indicating that trifarotene 100‐µg/g cream did not affect the circulating levels of LNG and EE (Table 5).
Figure 3.
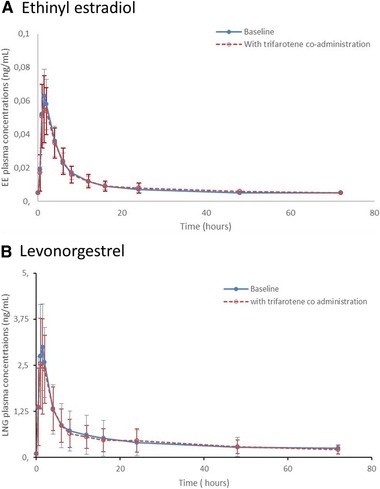
Drug‐drug interaction study: EE (A) and LNG (B) mean (SD) trifarotene plasma concentration profiles per treatment day (linear scale). EE, ethinyl estradiol; LNG, levonorgestrel; SD, standard deviation.
Figure 4.
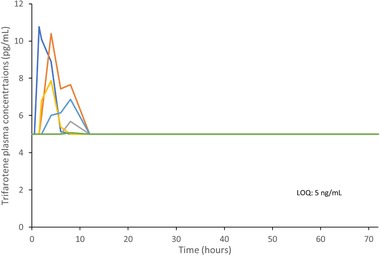
Drug‐drug interaction study: Trifarotene individual plasma concentrations vs time on day 18 (linear scale, LOQ: 5 pg/mL); 6 quantifiable subjects of 22; nonquantifiable data were replaced by the lower level of quantification (ie, 5 pg/mL). LOQ, level of quantification.
Table 5.
DDI Study, Inferential Analysis for EE and LNG Cmax and AUC0‐t
| Geometric Meana (Arithmetic Mean ± SD) | |||||
|---|---|---|---|---|---|
| Day 1 | Day 18 | Geometric Mean Ratio | 90%CI | ||
|
EE N = 22 |
Cmax (ng/mL) | 0.063 (0.065 ± 0.017) | 0.062 (0.063 ± 0.016) | 0.980 | (0.879‐1.094) |
| AUC0‐t (ng• h/mL) | 0.412 (0.427 ± 0.108) | 0.398 (0.418 ± 0.136) | 0.968 | (0.860‐1.090) | |
|
LNG N = 22 |
Cmax (ng/mL) | 2.951 (3.173 ± 1.306) | 2.781 (2.998 ± 1.177) | 0.943 | (0.864‐1.029) |
| AUC0‐t (ng • h/mL) | 25.20 (31.43 ± 25.66) | 25.02 (30.83 ± 19.3) | 0.993 | (0.878‐1.123) | |
AUC0‐t, area under the concentration‐time curve from T0 to the sampling time corresponding to the last quantifiable concentration; Cmax, maximum plasma concentration; CI, confidence interval; DDI, drug‐drug interaction; EE, ethinyl estradiol; LNG, levonorgestrel; SD, standard deviation.
Geometric means are based on least square means of Ln‐transformed values.
Overall, 37 AEs were reported by 19 (79.2%) subjects. The majority of AEs reported during the study were cutaneous in nature and were reported during the treatment period with trifarotene 100‐µg/g cream. The most common AE was reported as burning. TEAEs included 7 cutaneous events considered as being related to trifarotene 100‐µg/g cream and 2 systemic events considered as being related to the oral contraceptive (headache and dysmenorrhea). No serious AEs, AEs leading to discontinuation, or deaths were reported during the study. All of the treatment‐related AEs were of mild intensity, and they all resolved during the study (day 28). There were no safety signals in terms of laboratory parameters and vital signs. No clinically significant changes in hematology or biochemistry were observed. One subject became pregnant during the study. This subject was lost to follow‐up.
Discussion
Clinical pharmacology data demonstrate that trifarotene 50 µg/g cream, the to‐be‐marketed formulation, generates low systemic absorption when applied daily under maximal use conditions. With a trifarotene daily dose of 2 g, the mean Cmax in adults had a range of <5 to 8 pg/mL and in children a range of <5 to 9 pg/mL. In the TQT study of healthy subjects, 12 g once daily on 6000 cm2 resulted in a mean Cmax of 33 ± 34 pg/mL (<5‐187 pg/mL). Trifarotene plasma concentrations were close to or <5 pg/mL in most of the patients, including those with severe acne (IGA = 4). Quantifiable data obtained with trifarotene 100‐µg/g cream confirmed the absence of a sex or age effect on the systemic exposure to trifarotene. Moreover, patients with quantifiable trifarotene plasma concentrations in the higher dosage group indicated a trend of dose proportionality. Trifarotene was characterized by a short t1/2 ranging from 2 to 9 hours and without accumulation after repeated topical applications. Overall, the low systemic exposure levels and the rapid elimination of trifarotene from systemic circulation resulted in a good safety profile and prevented occurrence of systemic AEs.
Results from the TQT study showed that the mean Cmax with trifarotene 100‐µg/g cream was 3‐fold higher than the highest Cmax obtained with trifarotene 50 µg/g cream, thus confirming supratherapeutic conditions for the TQT study (Table 4). Moreover, trifarotene has no effect on ventricular repolarization.
Coadministration of trifarotene cream with LNG/EE did not result in any DDI indicating that no clinically meaningful DDIs may be expected when coadministrating trifarotene 50 µg/g cream and oral contraception (LNG/EE) in female acne subjects of childbearing potential.
Local and systemic safety evaluations did not lead to any notable safety concerns with trifarotene up 100 µg/g, a concentration twice as high as the to‐be‐marketed cream.
Trifarotene represents a new generation of retinoids and is the first‐in‐class drug developed for the treatment of acne on both the face and trunk in patients 9 years and older. This is the first time that comprehensive data about the clinical pharmacology profile of trifarotene are provided. The present extensive clinical pharmacology investigations confirm that systemic absorption of trifarotene is low, especially when applied at 50 µg/g.
Conclusions
The present pharmacology studies confirm that trifarotene 50 µg/g cream is systemically well tolerated and safe when applied under maximized conditions in adults and pediatric acne patients, including patients with severe acne. Daily use of trifarotene 50 µg/g cream was not associated with cardiovascular effects and did not result in DDI in women of childbearing potential using contraception.
Conflicts of Interest
N.W., A.A.S., and M.G. are employees of Galderma R&D, LLC. USA. K.B. and M.P. were employees of Galderma R&D, France, at the time the studies were conducted. V.S. from Sanders Medical Writing and K.P.G., SMWS France, have no conflicts of interest to disclose
Funding
These clinical studies were sponsored by Galderma.
Author Contributions
N.W. provided clinical pharmacology expertise and designed the studies. K.B. performed the PK analysis of the clinical studies and contributed to the clinical design of QT/QTc and DDI studies. A.A. provided medical expertise contribution to the DDI study. M.P. provided guidance for the number of subjects to be included in the studies and performed the statistical analysis of the studies. M.G. provided medical expertise and contributed to the study design of the MUsT, DDI, and QT/QTc. K.P.G. provided writing and editorial assistance to the authors. All authors read and approved the final manuscript.
Acknowledgments
The authors acknowledge the writing assistance of Valerie Sanders of Sanders Medical Writing and Karl Patrick Göritz, Scientific and Medical Writing Services, France.
References
- 1. Oudenhoven MD, Kinney MA, McShane DB, Burkhart CN, Morrell DS. Adverse effects of acne medications: recognition and management. Am J Clin Dermatol. 2015;16(4):231‐242. [DOI] [PubMed] [Google Scholar]
- 2. Dreno B. What is new in the pathophysiology of acne, an overview. J Eur Acad Dermatol Venereol. 2017;31(suppl 5):8‐12. [DOI] [PubMed] [Google Scholar]
- 3. Eichenfield LF, Krakowski AC, Piggott C, et al. Evidence‐based recommendations for the diagnosis and treatment of pediatric acne. Pediatrics. 2013;131(suppl 3):S163‐S186. [DOI] [PubMed] [Google Scholar]
- 4. Aubert J, Piwnica D, Bertino B, et al. Nonclinical and human pharmacology of the potent and selective topical retinoic acid receptor‐gamma agonist trifarotene. Br J Dermatol. 2018;179(2):442‐456. [DOI] [PubMed] [Google Scholar]
- 5. Bashaw ED, Tran DC, Shukla CG, Liu X. Maximal usage trial: an overview of the design of systemic bioavailability trial for topical dermatological products. Ther Innov Regul Sci. 2015;49(1):108‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. FDA guidance: Maximal Usage Trials for Topically Applied Active Ingredients Being Considered for Inclusion in an Over‐The ‐ Counter Monograph: Study Elements and Considerations . https://www.fda.gov/media/125080/download Accessed May 2019.
- 7. Taubel J, Ferber G, Lorch U, Batchvarov V, Savelieva I, Camm AJ. Thorough QT study of the effect of oral moxifloxacin on QTc interval in the fed and fasted state in healthy Japanese and Caucasian subjects. Br J Clin Pharmacol. 2014;77(1):170‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. European Medicines Agency . Guideline on the investigation of drug interactions; CPMP/EWP/560/95/Rev. 1 Corr. 2**; Committee for Human Medicinal Products (CHMP). https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf. Published June 21, 2012. Accessed April 28, 2019.
- 9. Chang SY, Chen C, Yang Z, Rodrigues AD. Further assessment of 17alpha–ethinyl estradiol as an inhibitor of different human cytochrome P450 forms in vitro. Drug Metab Dispos. 2009;37(8):1667‐1675. [DOI] [PubMed] [Google Scholar]
- 10. Winkler J, Goldammer M, Ludwig M, Rohde B, Zurth C. Pharmacokinetic drug‐drug interaction between ethinyl estradiol and gestodene, administered as a transdermal fertility control patch, and two CYP3A4 inhibitors and a CYP3A4 substrate. Eur J Drug Metab Pharmacokinet. 2015;40(4):389‐399. [DOI] [PubMed] [Google Scholar]
- 11. Florian JA, Tornoe CW, Brundage R, Parekh A, Garnett CE. Population pharmacokinetic and concentration‐QTc models for moxifloxacin: pooled analysis of 20 thorough QT studies. J Clin Pharmacol. 2011;51(8):1152‐1162. [DOI] [PubMed] [Google Scholar]
- 12. Demolis JL, Kubitza D, Tenneze L, Funck‐Brentano C. Effect of a single oral dose of moxifloxacin (400 mg and 800 mg) on ventricular repolarization in healthy subjects. Clin Pharmacol Ther. 2000;68(6):658‐66. [DOI] [PubMed] [Google Scholar]
- 13. International Conference for Harmonisation . E14 Implementation Working Group ICH E14 Guideline: The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs questions & answers (R3). Published December 10, 2015. Accessed April 28, 2019.