

Abstract
Inactivating variants in the centrosomal CEP78 gene have been found in cone‐rod dystrophy with hearing loss (CRDHL), a particular phenotype distinct from Usher syndrome. Here, we identified and functionally characterized the first CEP78 missense variant c.449T>C, p.(Leu150Ser) in three CRDHL families. The variant was found in a biallelic state in two Belgian families and in a compound heterozygous state—in trans with c.1462‐1G>T—in a third German family. Haplotype reconstruction showed a founder effect. Homology modeling revealed a detrimental effect of p.(Leu150Ser) on protein stability, which was corroborated in patients' fibroblasts. Elongated primary cilia without clear ultrastructural abnormalities in sperm or nasal brushes suggest impaired cilia assembly. Two affected males from different families displayed sperm abnormalities causing infertility. One of these is a heterozygous carrier of a complex allele in SPAG17, a ciliary gene previously associated with autosomal recessive male infertility. Taken together, our data indicate that a missense founder allele in CEP78 underlies the same sensorineural CRDHL phenotype previously associated with inactivating variants. Interestingly, the CEP78 phenotype has been possibly expanded with male infertility. Finally, CEP78 loss‐of‐function variants may have an underestimated role in misdiagnosed Usher syndrome, with or without sperm abnormalities.
Keywords: CEP78, cilia, cone‐rod dystrophy with hearing loss (CRDHL), founder, male infertility, missense
1. INTRODUCTION
Inherited retinal diseases (IRDs) are a major cause of blindness worldwide. They combine a group of genetic eye disorders with a complex phenotypic spectrum caused by progressive degeneration of rod and cone photoreceptors and/or the retinal pigment epithelium (Berger, Kloeckener‐Gruissem, & Neidhardt, 2010). Tremendous genetic heterogeneity has been demonstrated by variants identified in over 270 genes (RetNet—Retinal Information Network: https://sph.uth.edu/retnet; Daiger et al., 1998), allowing a molecular diagnosis in up to 60% of cases (Lee & Garg, 2015). A subset of IRDs, so‐called retinal ciliopathies, is caused by mutations in genes involved in cilia biogenesis or function and is usually associated with complex phenotypes (Hildebrandt, Benzing, Katsanis, Schwartz, & Hildebrandt, 2011). Well‐described ciliopathies including IRD are Usher syndrome (USH; MIM# 276900) and Bardet‐Biedl syndrome (BBS; MIM# 209900), characterized by multiorgan defects (Koenig, 2003). Due to their large clinical heterogeneity with overlapping features, the establishment of clinical diagnosis for ciliopathies is often difficult, and in some cases, the phenotype results from multiallelic defects (Reiter & Leroux, 2017; Shaheen et al., 2016).
Biallelic inactivating variants in CEP78, encoding the Centrosomal Protein 78 (MIM# 617110), have been linked to autosomal recessive cone‐rod dystrophy with hearing loss (CRDHL; MIM# 617236; Namburi et al., 2016; Nikopoulos et al., 2016). The unique combination of CRD and sensorineural hearing impairment was stated to be distinct from the well‐known Usher syndrome, characterized by recessively inherited retinitis pigmentosa (RP) and congenital mild to severe hearing impairment or complete sensorineural deafness. Up to now, seven different truncating mutations have been reported in CEP78, segregating in CRDHL families of different origins (Fu et al., 2016; Namburi et al., 2016; Nikopoulos et al., 2016). Furthermore, a homozygous CEP78‐truncating variant was found in a family with reported nonsyndromic RP (MIM# 268000; de Castro‐Miró et al., 2016). So far, only frameshift and splice site variants have been identified, with the latter all displaying exon skipping at the RNA level (Fu et al., 2016; Namburi et al., 2016; Nikopoulos et al., 2016). In addition, a homozygous deletion‐inversion‐deletion overlapping CEP78 has recently been described in a CRDHL case (Sanchis‐Juan et al., 2018). Several functional studies at the protein level point to a loss‐of‐function (LOF) effect with decreased amounts of protein, correct subcellular localization, and importantly, elongated primary cilia in patients' fibroblasts, a feature that has been observed in other ciliopathies as well (Mokrzan, Lewis, & Mykytyn, 2007; Nikopoulos et al., 2016).
CEP78 (UniProt: Q5JTW2, 4 isoforms reported—http://www.uniprot.org) is a component of the centrosome and localizes to the mature centrioles (Brunk et al., 2016). The human full‐length protein of 722 amino acids (Q5JTW2‐2) contains an N‐terminal leucine‐rich repeat (LRR) domain with five consecutive LRR domains, and a C‐terminal coiled‐coil domain (Brunk et al., 2016; Hossain, Javadi Esfehani, Das, & Tsang, 2017). Centrioles are the main components of centrosomes, key microtubule‐organizing centers in eukaryotic cells, with the mother centriole acting as the basal body during cilia formation (Gönczy & Hatzopoulos, 2019). Centrioles duplicate only once per cell cycle, and anomalies in their structure or number are associated with several diseases including cancer and developmental disorders such as ciliopathies (Gönczy, 2015; Nigg & Raff, 2009). Interestingly, the dysregulation of CEP78 has already been associated with prostate and colorectal cancers (Nesslinger et al., 2007; Zhang et al., 2016). Recently, CEP78 has been suggested as a promising molecular biomarker in thyroid carcinoma (Hammad et al., 2019).
Here, we functionally studied the first CEP78 missense variant, which we identified in homozygous and compound heterozygous states in three unrelated CRDHL families with and without male infertility. We assessed other potential genetic causes underlying male infertility, performed haplotype reconstruction for the missense variant in the three families, evaluated protein modeling, and studied induced cilia in patients' fibroblasts.
2. MATERIALS AND METHODS
2.1. Ethics statement
This study was conducted following the tenets of the Declaration of Helsinki and ethical approval was given by the local ethics committee (Ghent University Hospital, EC UZG 2017/1540; Lausanne University Hospital, Protocol 09/14, Technical University Munich 4360/13). All individuals involved gave their informed consent before inclusion in this study.
2.2. Phenotypic evaluation
All affected individuals were subjected to a hearing assessment and to detailed ophthalmologic evaluation including best‐corrected visual acuity measurement, fundoscopy, visual field assessment, both infrared and blue light reflectance and autofluorescence imaging, spectral‐domain optical coherence tomography (SD‐OCT), and electroretinography (ERG).
Because of a presumed syndromic ciliary phenotype, body mass index (BMI) was calculated (family F1), the Sniffin' sticks smell test (families F1 and F3) was used to estimate olfactory function and a saccharin test to assess nasal mucociliary clearance (F1; Hummel, Sekinger, Wolf, Pauli, & Kobal, 1997; Sherly & Prathibha, 2014). In addition, semen evaluation was performed for the male probands from F1 (subject III:2) and F2 (subject II:2) and F3 (subject II:1).
2.3. Whole exome sequencing and variant validation
Genomic DNA (gDNA) was extracted from leukocytes according to the manufacturer's guidelines. Whole exome sequencing (WES) was performed for two individuals from F1 (III:2 and III:5) using SureSelectXT human All Exon V6 enrichment (Agilent) and NextSeq500 sequencing technology (Illumina). Read mapping and variant calling were performed using the CLC Genomics Workbench (hg19 human reference genome, v. 7.5.4, Qiagen). Duo exome sequencing was performed for both affected individuals from family F3 (II:1 and II:2) using SureSelectXT human All Exon V5 enrichment (Agilent) and HighSeq2500 (Illumina) for sequencing. Reads were aligned to the UCSC human reference assembly (hg19—https://genome.ucsc.edu) with BWA (v.0.5.8). Single‐nucleotide variants and small insertions and deletions were detected with SAMtools (v.0.1.7). Copy number variations (CNVs) were detected with ExomeDepth (Plagnol et al., 2012) and Pindel (Ye, Schulz, Long, Apweiler, & Ning, 2009).
Sanger sequencing of the coding exons and intron‐exon boundaries of CEP78 (NM_001098802.2; F2, II:2) and SPAG17 (sperm associated antigen 17; NM_206996.2; MIM# 616554) was performed using an ABI 3730xl DNA Analyzer (Applied Biosystems) with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Primer sequences are listed in Tables S1 and S2.
Human Genome Variation Society nomenclature (http://varnomen.hgvs.org) was used for variant notation, with the A of the initiation codon ATG as +1. The functional impact of identified sequence variants was assessed trough in silico predictions in Alamut Visual (v.2.8.1), Alamut Batch (v.1.9), and dbNSFP (v.3.4a), including missense prediction tools, splice prediction tools, physicochemical distance (Grantham score), evolutionary conservation, location in protein domains, presence in dbSNP (build 151—https://www.ncbi.nlm.nih.gov/SNP), MetaDome (https://stuart.radboudumc.nl/metadome/—v.1.0.1), Exome Variant Server from the NHLBI Exome Sequencing Project (ESP—http://evs.gs.washington.edu/EVS), ExAC (http://exac.broadinstitute.org/), and gnomAD (http://gnomad.broadinstitute.org/; Liu, Wu, Li, & Boerwinkle, 2016; Wiel et al., 2019).
Multiplex polymerase chain reactions (PCRs) on the Y‐chromosome (AZFa, AZFb, and AZFc) and for the SRY and USP9Y (DFFRY) genes were performed to evaluate the presence of Y‐microdeletions in F1, III:2 (Krausz, Hoefsloot, Simoni, Tüttelmann, & European Academy of Andrology, & European Molecular Genetics Quality Network, 2014).
2.4. Haplotype analysis
In total, 11 microsatellite markers (Genome data viewer—https://www.ncbi.nlm.nih.gov/genome/gdv) and 15 single‐nucleotide polymorphisms (SNPs; dbSNP, build 151) were selected for haplotype reconstruction in the area flanking CEP78. SNPs were Sanger sequenced according to the standard procedures (see above). For microsatellite fragment size determination, PCR products were mixed with an internal GeneScan size standard (ROX‐500, Applied Biosystems) and Hi‐Di formamide (Applied Biosystems), and were then loaded on the ABI 3730xl DNA Analyzer. Fragment sizes were calculated using the GeneMapper software (v. 5.0, Applied Biosystems). PCR primers were designed with Primer3Plus (http://www.bioinformatics.nl/primer3plus) and sequences can be found in Table S3 (Untergasser et al., 2007).
2.5. CNV analysis for SPAG17
CNV analysis was performed for the SPAG17 gene by quantitative PCR (qPCR). qPCR assays were designed per coding exon, as previously described (D'haene, Vandesompele, & Hellemans, 2010). Assays for each gDNA sample were prepared using the SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad Laboratories) and were subsequently run in triplicate on a LightCycler 480 System (Roche). Data were analyzed with qbase+ software (v. 3.1, Biogazelle). qPCR primer design was done using Primer3Plus or PrimerXL, and ZNF80 and GPR15 were used as reference genes (Lefever et al., 2017; Untergasser et al., 2007). Primer sequences can be found in Table S4.
2.6. Homology modeling and FoldX calculations for p.(Leu150Ser)
Amino acid residues 42‐199 of CEP78 were used as input sequences for an HHPRED structure prediction (https://toolkit.tuebingen.mpg.de/#/tools/hhpred; Söding, Biegert, & Lupas, 2005). Based on the HHPRED output, structures with PDB code 5IRN, 1Z7X, and 1IO0 were chosen as best nonredundant templates for CEP78 modeling. The corresponding HHPRED alignments of CEP78 with these templates were used as input alignment for modeling of the CEP78 fragment using the YASARA structure homology modeling engine (http://yasara.org/products.htm#structure; Krieger & Vriend, 2015). The figure of the model was generated using UCSF chimera (Pettersen et al., 2004). For FoldX mutant stability calculations on the three models and on the three template structures, we used the BuildModel command after structure optimization with the RepairPDB command (Guerois, Nielsen, & Serrano, 2002).
2.7. Immunoblotting on fibroblasts
Skin biopsies, taken from the inner aspect of the arm, were obtained from patients with biallelic CEP78 variants (F1, III:2 and III:5; F3, II:1) and heterozygous carriers (F1, II:2 and II:8). Starting from the skin biopsy, a fibroblast culture was established in Dulbecco's minimal essential medium (MEM) with phenol red (Life Technologies) supplemented with 10% fetal bovine serum (Labconsult), 1% kanamycin (Life Technologies), 1% penicillin‐streptomycin (Life Technologies), and 1% MEM nonessential amino acids (Life Technologies). To perform immunoblotting of CEP78, total protein from cultured fibroblasts was extracted with RIPA Buffer (Sigma‐Aldrich) from a full T75 flask. Lysis buffer included protease inhibitory cocktail (Roche Diagnostics), phosphatase inhibitory cocktail 2, and phosphatase inhibitory cocktail 3 (Sigma‐Aldrich). After centrifugation and reduction with 1 M DTT (Sigma‐Aldrich), protein lysates were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (NuPAGE™ 4–12% Bis‐Tris Protein Gels, ThermoFisher Scientific) with a ladder (Precision Plus Protein All Blue Standards, Bio‐Rad Laboratories). Proteins were transferred to a nitrocellulose membrane using the iBlot 2 Dry Blotting System (Thermo Fisher Scientific). Membranes were blocked 2 hours room temperature in 2% bovine serum albumin (BSA; Sigma‐Aldrich), incubated with primary antibodies against CEP78, overnight at 4°C (1:1000, A301‐800A, Bethyl) or β‐tubulin, 1 hour room temperature (1:2500, ab6046, Abcam), and subsequently incubated with horseradish‐peroxidase‐conjugated secondary antibody, 2 hours room temperature (1:2500, 7074S, Cell Signaling Technologies). Membranes were scanned with an Amersham Imager 680 system (GE Healthcare Life Sciences), and CEP78 signal intensity quantitation was achieved using ImageJ (NIH, v. 1.50i) and normalized to the amount of β‐tubulin. Graphs display data points of three independent experiments and the statistical analysis was performed in GraphPad (v. 5.04) using the Mann–Whitney test.
2.8. Immunocytochemistry on fibroblasts
Fibroblasts of controls and affected individuals and heterozygous carriers of F1 were grown on glass chamber slides and starved in serum‐free medium for 48 hr when confluent. Cells were fixed with 3.7% paraformaldehyde, permeabilized with 0.2% Triton X‐100, blocked in 10% BSA in phosphate‐buffered saline, and incubated overnight at 4°C with primary antibodies against TAPT1 (1:500, Sigma, HPA042567) and α‐acetylated tubulin (1:1000, Sigma, T6793). The TAPT1 antibody is marking the base of the cilium, whereas the acetylated tubulin antibody is marking the ciliary axoneme (Symoens et al., 2015). After washing, cells were incubated for 3 hr with secondary antibodies at room temperature (1:750, Life Technologies, donkey anti‐mouse Alexa Fluor 488 and donkey anti‐rabbit Alexa Fluor 594) and mounted with coverslips (Vectashield Mounting Medium with 4′,6‐diamidino‐2‐phenylindole). Images were captured with a Zeiss AxioObserved‐Z1 microscope. Unbiased counting of at least 100 events per sample (975 events in affected individuals, 405 in carriers, and 1,211 in controls) was performed. Cilia length was measured using segmented lines in ImageJ and independently evaluated by two investigators. The statistical analysis was performed in GraphPad using one‐way ANOVA (Kruskal–Wallis test).
2.9. Expression analysis and splicing assessment on fibroblasts
For quantitative reverse‐transcription PCR (qRT‐PCR), total RNA was extracted from cultured fibroblasts with the RNeasy Kit (Qiagen), treated with RNase‐Free DNase (Qiagen), and complementary DNA (cDNA) was synthesized with the iScript cDNA Synthesis Kit (Bio‐Rad Laboratories; CEP78) or the SuperScript VILO cDNA Synthesis Kit (Invitrogen) (SPAG17). For each cDNA sample, assays were prepared with the addition of SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad Laboratories) and were subsequently run in triplicate on a Roche LightCycler 480 System. Data were analyzed with qbase+ (v. 3.1; Biogazelle) and normalized to the HMBS, SDHA, and YWHAZ genes. For nonquantitative RT‐PCR, cDNA underwent standard PCR and was then loaded on the Fragment Analyzer 5300 (Agilent). No cycloheximide (CHX) treatment has been performed before RNA extraction. Primers were designed using Primer3plus or PrimerXL (http://www.primerxl.org/) and sequences are listed in Table S5.
2.10. Electron microscopy on nasal brush and spermatids
Biopsies were obtained from the nasal (inferior turbinate) mucosa of a homozygous individual (F1, III:2) and a heterozygous carrier (F1, II:8). Spermatid electron microscopy (EM) pictures were obtained from a semen sample from F2, II:2. Biopsies were immersed in 2.5% glutaraldehyde and processed for ultrastructural analysis, according to Shahana et al. (2005). Ultrathin sections were examined at a final magnification of ×60,000 or ×85,000. In each specimen, an analysis of at least 50 transverse ciliary sections of different cells was required to study the internal axonemal structure. For the spermatids, ultrathin sections of a gold interference color were cut using an ultramicrotome (Leica EM UC6), followed by a poststaining in a Leica EM AC20 for 40 min in uranyl acetate at 20°C and for 10 min in lead stain at 20°C. Sections were collected on formvar‐coated copper slot grids. Grids were viewed with a JEM 1400plus transmission electron microscope (JEOL, Tokyo, Japan) operating at 80 kV. A qualitative evaluation of general ciliary sections was performed as described (Lucas, Paff, Goggin, & Haarman, 2016).
3. RESULTS
3.1. Clinical findings in individuals with CRDHL from three families
The families included in this study are of Belgian (F1 and F2) and German origin (F3). All patients included were clinically diagnosed with CRDHL. Ophthalmic images and more detailed clinical features are provided in Figure 1 and Table 1, respectively.
Figure 1.
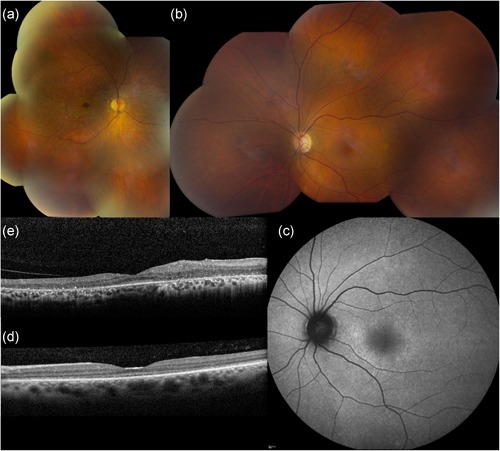
Representative ophthalmological pictures of individuals carrying c.449T>C p.(Leu150Ser) in CEP78 (F1, III:2 and III:5). Clockwise from top left: (a) Composite fundus image of OD of male patient aged 53 (III:2). Severe reduction in vascular caliber, small white dots from the macula up to the midperiphery, relatively better preservation of fovea as well as peripheral retina, and spicular intraretinal pigmentation in nasal midperiphery. (b) Composite fundus picture of OS of female patient aged 47 (III:5). Virtually normal fundus except for mild reduction in vascular caliber. Abnormality of superonasal vessels to optic disc is composite artifact. (c) Blue light autofluorescence image of OS of III:5. Diffuse, mottled hyperautofluorescence is more pronounced in circle around central macula. (d) SD‐OCT of central macula of OS of III:5. Thinning of foveal area with considerable loss of definition of outer retinal layers (RPE and PRs). (e) SD‐OCT of central macula of OD of III:2. Thinning of outer retinal layers representing RPE and PRs, which are relatively better preserved in foveal area. Other retinal layers seem rather disorganized in the temporal macula. OD, oculus dexter (right eye); OS, oculus sinister (left eye); SD‐OCT, spectral‐domain optical coherence tomography; PR, photoreceptor; RPE, retinal pigment epithelium
Table 1.
Overview of clinical findings in CEP78‐associated CRDHL
| Age/Sex | Age of onset, years | BCVA (OD/OS) | Goldmann visual fields | Fundus imaging | BAF | OCT | ERG | Hearing impairment | Other findings | |
|---|---|---|---|---|---|---|---|---|---|---|
| F1: III:2 | 56/M | 17 | CF at 50 cm/HM at 1 m (at age 53) | ODS: only preserved inferior caecocentral island with object V4 of Goldmann with severely reduced sensitivity | ODS: severe reduction in vascular caliber, small white dots from the macula up to the midperiphery, relatively better preservation of fovea as well as peripheral retina, and spicular intraretinal pigmentation in nasal midperiphery | ODS: diffuse, mottled mixed hypo‐ and limited hyperautofluorescence more pronounced in macular area; better preserved autofluorescence in foveal area immediately surrounded by hypoautofluorescent ring | ODS: thinning of outer retinal layers representing RPE and PRs, which are relatively better preserved in foveal area | ODS: severely reduced rod‐responses to one‐fourth of normal amplitude and delayed peak time. Absent cone‐specific responses | Sensorineural hearing loss for high tones | Photophobia and acquired achromatopsia |
| Diabetes mellitus type 2 | ||||||||||
| Morbid obesity | ||||||||||
| Oligoasthenoteratospermia | ||||||||||
| F1: III:5 | 50/F | 15 | CF at 70 cm/CF at 20 cm (at age 48) | ODS: large central, absolute scotoma including blind spot, and pericentral visual field; normal peripheral limits with object V4 | ODS: fundus virtually normal, except for mild reduction in vascular caliber | ODS: diffuse, mottled hyperautofluorescence is more pronounced in circle around central macula | ODS: thinning of foveal area with considerable loss of definition of outer retinal layers | ODS: moderately reduced rod‐responses to half of normal amplitude and delayed peak time; absent cone‐specific responses. | Sensorineural hearing loss for high tones | Photophobia and acquired achromatopsia |
| Morbid obesity | ||||||||||
| F2: II2 | 57/M | 20 | HM at 1 m/HM at 1 m | ODS: moderately constricted peripheral limits; extensive central, absolute scotoma | ODS: limited outer retinal atrophy in macula up to area nasal to optic disc; severely reduced vascular caliber | ODS: diffuse hypo‐ and hyperautofluorescence; hyperautofluorescent ring around central macula; mottled hypoautofluorescence of retina nasal to optic disc | ODS: thinning of outer retinal layers representing RPE and PRs, which are relatively better preserved in foveal area | ODS: severely reduced rod‐responses to one‐tenth of normal amplitude and delayed peak time. Absent cone‐specific responses | Sensorineural hearing loss for high tones | Photophobia and acquired achromatopsia |
| F3: II:1 | 29/M | 6 | 0.2/0.2 | Normal peripheral limits; central, relative doughnut scotoma with small, spared central area | ODS: normal | ODS: mottled hyper‐ and hypoautofluorescence in macular area | ODS: granular aspect of outer retinal layers; disrupted outer retinal layer in fovea | ODS: mildly reduced rod‐specific responses; moderately reduced cone‐specific responses | Sensorineural hearing loss for high tones | Photophobia |
| Acquired, severe color vision deficiency | ||||||||||
| Asthenoteratozoospermia | ||||||||||
| F3: II:2 | 26/M | 6 | 0.63/0.63 | Normal peripheral limits; central, relative doughnut scotoma with small, spared central area | ODS: normal | ODS: mottled hyper‐ and hypoautofluorescence in macular area | ODS: granular aspect of outer retinal layers; disrupted outer retinal layer in fovea | ODS: normal rod‐specific responses; moderately reduced cone‐specific responses | Sensorineural hearing loss for high tones | Photophobia |
| Normal color vision | ||||||||||
| Diabetes type 1 |
Abbreviations: BAF, blue light autofluorescence imaging; BCVA, best‐corrected visual acuity; CF, counting fingers; ERG, full‐field flash electroretinography; F, female; HM, hand movements; M, male; OD, oculus dexter (right eye); ODS, oculus dexter et sinister (right and left eye); OS, oculus sinister (left eye); SD‐OCT, spectral‐domain optical coherence tomography.
The ocular phenotype of the affected siblings in F1 has previously been clinically described in part by us before the identification of CEP78 as CRDHL disease gene (Witters, De Zaeytijd, Leys, & Leroy, 2004). In F1, a syndromic phenotype was first suspected due to the presence of several ciliopathy‐like characteristics. BMI calculation showed obesity class II or III in affected individuals and obesity class I or II in their parents (Table S6). Several family members reported respiratory problems. However, smell and saccharin tests showed only mild abnormalities (Table S7). Importantly, the male proband of F1 (III:2) displays infertility, which was supported by his semen evaluation showing oligoasthenoteratospermia (Table S8). In addition, diabetes type 2 has been diagnosed for several members of F1 (II:8, III:2 and III:4) and a history of colon cancer has been reported in the family branch of II:8 (three out of four sisters).
The presence of male infertility in F1 prompted us to evaluate male fertility in other families. Hence, we assessed the semen quality of the male proband of F2 (II:2); however, no abnormalities were observed (Table S8). Furthermore, there are no additional features that support a syndromic ciliopathy in F2. Interestingly, additional clinical features were found in F3: male infertility, more specifically asthenoteratozoospermia in F3 (II:1; Table S8), and diabetes type 1 in F3 (II:2). A smell test performed in these individuals did not show any abnormalities (Table S7).
3.2. Identification of the first CEP78 missense variant in three CRDHL families
WES in two affected individuals of F1 (III:2 and III:5) revealed a novel homozygous missense variant in CEP78, c.449T>C, p.(Leu150Ser) (NM_001098802.1; Figures 2 and 3). In another Belgian family with a CRDHL phenotype (F2, II:2), targeted analysis of CEP78 revealed the same homozygous missense variant. WES in two siblings from a third family with CRDHL (F3: II:1 and II:2) showed presumed compound heterozygosity for p.(Leu150Ser) and a novel splice site variant, c.1462‐1G>T. This substitution is located in the acceptor splice site of intron 12 and is predicted to cause skipping of exon 13, resulting in a frameshift. Aberrant splicing was confirmed using RT‐PCR (Figure S1). The variants segregate with disease in the three families proving the biallelic localization of the identified variants (Figure 2a).
Figure 2.
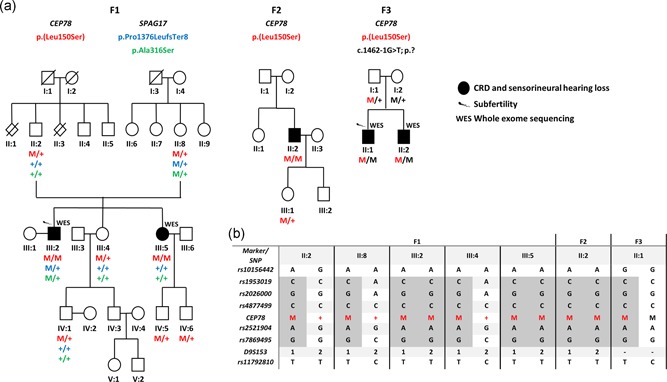
Pedigrees segregating the CEP78 and SPAG17 variants, and haplotype reconstruction for the c.449T>C/p.(Leu150Ser) variant in CEP78. (a) F3, II:1 and II:2 are compound heterozygous for c.[449T>C]; [1462‐1G>T] p.[Leu150Ser];[?]. In F1, two variants p.(Pro1376LeufsTer8) and p.(Ala316Ser) were identified in SPAG17, occurring however in cis. (b) Haplotype reconstruction highlighted p.(Leu150Ser) in CEP78 as a founder allele. Complete haplotype is available in Table S10. M, mutant allele; +, wild‐type allele
Figure 3.
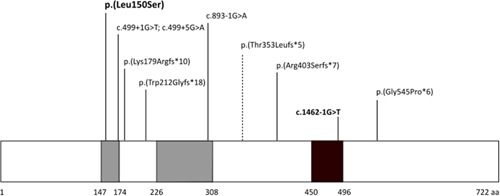
Location of known CEP78 variants. Schematic diagram of the CEP78 protein showing the location of the previously described truncating variants and the novel variants reported in this study (in bold). The previously published variant causing RP is indicated with a dashed line. CEP78 includes multiple leucine‐rich repeats (LRR, in gray) located at the N‐terminal, and a coiled‐coil domain at the C‐terminal (in black). The missense change described in this study is located in one of the LRRs. The splice site substitution is predicted to cause skipping of exon 13. This figure has been adapted from material from Brunk et al. (2016). RP, retinitis pigmentosa
The missense variant changes an extremely conserved amino acid position and is predicted to affect protein function according to several in silico prediction tools (Figure S2 and Table S9). This change is a known variant (rs761661253) and has an overall allele frequency of 0.0000964 in ExAC and of 0.00004967 in gnomAD with no homozygotes observed in both.
Haplotype analysis with 11 microsatellite markers and 15 SNPs in F1 and F2 revealed a common haplotype of 1.7‐2.3 Mb, which is suggestive for a founder effect (Figure 2b, Table S10). One sibling of F3 has been subsequently included in the haplotype reconstruction.
3.3. Assessment of other potential genetic causes of male infertility
The proband of F1 (III:2) suffers from male infertility (Table S8). To assess a potential distinct genetic cause, we first evaluated the occurrence of microdeletions of the Y chromosome, which was negative (data not shown; Flannigan & Schlegel, 2017). Second, we assessed the presence of (likely) pathogenic variants in known and candidate genes for male infertility in the WES data (Table S11). From the filtered variants, two heterozygous variants were found in the SPAG17 gene: c.4127del, p.(Pro1376LeufsTer8), and c.946G>T p.(Ala316Ser) (NM_206996.2). Both variants are predicted to be pathogenic (Table S11). Segregation analysis showed that the two variants occur in cis and are maternal (F1, II:8; Figure 2a). Expression analysis of SPAG17 messenger RNA (mRNA) in fibroblasts of the heterozygous F1, II:8 showed decreased expression compared to controls (Figure S3). This can likely be attributed to nonsense‐mediated decay (NMD) resulting from the frameshift variant c.4127del, p.(Pro1376LeufsTer8). So far, a homozygous missense variant in SPAG17 c.4343G>A p.(Arg1448Gln) has been reported in one patient from a consanguineous family, putting forward SPAG17 as candidate gene for recessive male infertility (Figure S4; Xu et al., 2018). No second (likely) pathogenic SPAG17 variant could be identified however in F1 (III:2) by sequencing of all exons that had coverage below 20x in the WES data, by sequencing of the untranslated regions (UTRs) and by CNV analysis of all exons (data not shown). Deep intronic variants or noncoding variants in regulatory regions cannot be excluded by this assessment.
As the proband of F3 (II:1) displays male infertility as well, his WES data were revisited, but no (likely) pathogenic variants were identified. No Y‐deletion test was available for F3 (II:1).
3.4. Homology modeling and FoldX calculations of p.(Leu150Ser)
The N‐terminal domain of CEP78 is predicted to contain multiple LRRs, some of which have been shown to be involved in protein–protein interactions (Matsushima et al., 2005). Leu150 is the first leucine residue of an LRR and the LRR seems to fit the consensus sequence of RNAse inhibitor‐like (RI‐like) class leucine rich repeats (Matsushima et al., 2005). Variants of the first leucine to proline, phenylalanine, or histidine in the repeats of four other LRR proteins have been found to be disruptive and to underlie different diseases, including congenital stationary night blindness (NYX; MIM# 300278), hereditary bleeding disorder (GP1BA; MIM# 606672), and immunodysregulation (CARMIL2; MIM# 610859; Matsushima et al., 2005; Sorte et al., 2016). We used homology modeling and FoldX calculations to predict whether the p.(Leu150Ser) variant can similarly affect the structure or stability of CEP78.
HHPRED searches confirmed that LRR containing structures are the best templates for homology modeling of the CEP78 N‐terminal region (Söding et al., 2005). In the alignments, Leu150 indeed aligns with the first leucine of an LRR of the templates. We used the best three HHPRED hits as template for modeling of the CEP78 N‐terminal region: NOD2 (PDB code: 5IRN), Tropomodulin C20 domain (PDB code: 1IO0), and RNAse inhibitor (PDB code: 1Z7X, chain W). All these structures have RI class LRRs. Three separate models were built based on these three templates.
The model based on the RNAse inhibitor (PDB code1Z7X) template can be seen in Figure 4a. A very similar arrangement is found in both other models. Leu150 is packed between six other leucine residues in the hydrophobic core of the LRR fold, and replacing Leu150 to serine disturbs this packing and introduces a serine in a very hydrophobic environment. This is most likely detrimental for the protein folding. We used FoldX to predict the effect of p.(Leu150Ser) on protein stability in the three models (Guerois et al., 2002). The variant is predicted to have a very destabilizing effect, with a ΔΔG of >4.5 kcal/mol in FoldX for all three models. Similarly, mutation of the aligning leucine residue to serine in the three template structures (5IRN: L965; 1IO0: L282; 1Z7X: L371) is predicted to have a very disruptive effect with a ΔΔG of >4.5 kcal/mol in FoldX for all three structures. This suggests that the p.(Leu150Ser) variant in CEP78, and similar variants of the first leucine to serine in other LRR proteins, severely affect protein stability.
Figure 4.
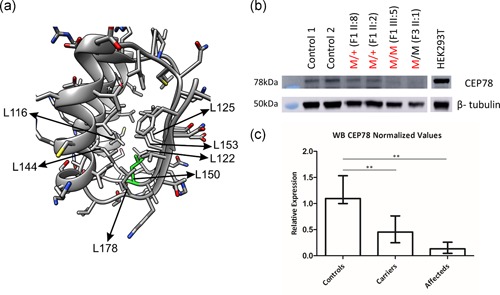
(a) Structural modeling of the p.(Leu150Ser) CEP78 missense variant. Residues 106‐184 of the model of CEP78 N‐terminal domain (V42‐K198) based on an RNAse inhibitor structure were used as a template. In this model, Leu150 (L150, green) is surrounded by six other leucine residues (indicated with arrows). (b) Immunoblotting of endogenous CEP78 in primary fibroblasts. Since the p.(Leu150Ser) change is predicted to affect protein stability, protein levels of CEP78 were evaluated in available primary fibroblasts of F1 (II:2, II:8 and III:5) and F3 (II:1). Undetectable protein or strongly reduced CEP78 levels has been detected in the homozygous/compound heterozygous and heterozygous individuals, respectively. (c) Relative amounts of CEP78 were calculated on three independent western blots. Compared to controls, the amount of CEP78 is reduced in heterozygous carriers (p = .0075) and nearly undetectable in homozygous/compound heterozygous patients (p = .0048). M, mutation (in black: c.1462‐1G>T; in red: c.449T>C); +, wild type
3.5. Missense variant p.(Leu150Ser) causes decreased CEP78 protein levels in patients' fibroblasts
Since the result of the homology protein modeling is indicating a strong effect of p.(Leu150Ser) on protein stability, immunoblot of CEP78 was performed using fibroblast protein extracts from a homozygous (F1, III:5) and compound heterozygous patient (F3, II:1), from two p.(Leu150Ser) heterozygous carriers (F1, II:2 and II:8), and from two unrelated controls. Compared to controls, the amount of CEP78 is significantly reduced in heterozygous carriers (p = .0075) and nearly undetectable in homozygous/compound heterozygous patients (p = .0048; Figures 4b,c, S5, and S6). Subsequent mRNA expression analysis of CEP78 showed no decreased expression in homozygous individual F1, III:5, confirming that the effect of the variant is probably restricted to the protein level. Compound heterozygous individual F3, II:1 shows only a subtly decreased CEP78 expression that can be attributed to the splicing variant (Figure S7).
3.6. Ciliary assessment in patients' fibroblasts, nasal brush, and sperm
Biallelic truncating variants in CEP78 have previously been associated with elongated primary cilia (Nikopoulos et al., 2016). To evaluate the ciliary length in individuals biallelic or monoallelic for p.(Leu150Ser), immunostaining against α‐acetylated tubulin and TAPT1 was performed on fibroblast cultures. Subsequent cilia length assessment in members of F1 revealed that induced primary cilia are significantly longer in fibroblasts from homozygous and heterozygous individuals than in those from control cells (p < 1.0 x 10−4; Figures 5a and S8 and Table S12).
Figure 5.
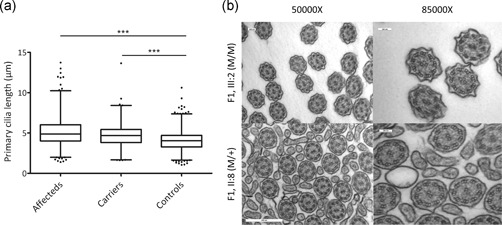
(a) Analysis of ciliary length. Fibroblasts from homozygous (F1, III:2 and III:5) and heterozygous (F1, II:2 and II:8) individuals show significantly longer primary cilia than those from unaffected control individuals (p < .001). (b) EM cilia. Ciliary ultrastructure from nasal brush samples obtained from one affected individual (F1, III:2) and one heterozygous carrier (F1, II:8), presents the conventional 9 + 2 microtubule doublet configuration. Scale bars 100 or 500 nm. EM, electron microscopy
Transmission EM of cross sections of ciliated nasal epithelium cells showed the conventional “9 + 2” microtubule doublet configuration with the presence of dynein arms in both a homozygous individual (F1, III:2) and a heterozygous carrier (F1, II:8) (Figure 5b). Transmission EM was also performed on a sperm sample obtained from F2, II:2, but no relevant anomalies of the main structures were identified (Figure S9). Overall, no clear ultrastructural abnormalities of cilia were observed both in nasal brush and sperm.
4. DISCUSSION
Several studies have linked CEP78‐truncating variants with a unique CRDHL phenotype and, so far, all described variants are frameshift or splice site sequence variants with a presumed LOF effect. Recently, a homozygous structural variant affecting CEP78, that is, a deletion‐inversion‐deletion, has also been described in a patient with CRDHL (Table S13; Fu et al., 2016; Namburi et al., 2016; Nikopoulos et al., 2016; Sanchis‐Juan et al., 2018; de Castro‐Miró et al., 2016). Here, we report the first missense variant causing CRDHL, c.449T>C, p.(Leu150Ser), expanding the CEP78 molecular spectrum. The founder effect of the variant identified in three unrelated families of Belgian and German origin may indicate a potentially higher prevalence of this variant in other cases of Northwestern origin with a recognizable CRDHL phenotype. Indeed, based on a clinical diagnosis of CRDHL, targeted testing of CEP78 has revealed this pathogenic variant in F2, II:2.
Other features that may advance the differential diagnosis between the CRDHL phenotype and Usher syndrome are extra‐sensorial, syndromic symptoms such as male infertility (F1 and F3). To assess if other causes for male infertility are present in F1 and F3, WES data of F1, III:2 and F3, II:1 were filtered for variants in reported male infertility genes (Table S11). This revealed two novel heterozygous variants in SPAG17 in F1, III:2, which proved to occur in cis. A decreased expression of SPAG17 could be shown in fibroblasts of F1, II:8. No additional (likely) pathogenic variants could be found in the male infertility gene panel and so far only one homozygous missense variant has been reported in two twin brothers from a consanguineous family with severe asthenozoospermia (Xu et al., 2018). In parallel, homozygous variants in SPAG17 and WDR35 were described in a patient with multiple brain and skeletal anomalies, with a presumed combinatorial effect on the ciliary phenotype (Córdova‐Fletes et al., 2018). Furthermore, a SPAG17 missense variant has been associated with primary ciliary dyskinesia (Andjelkovic et al., 2018). SPAG17 encodes a protein present in the axoneme central pair complex of motile cilia and flagella (Goduti & Smith, 2012; Mitchell & Smith, 2009). Conditional knockout mouse models support a critical role for SPAG17 in the function and structure of cilia, showing that SPAG17 is essential not only for sperm motility but also for normal manchette structure, protein transport, and formation of the sperm head and flagellum (Kazarian et al., 2018; Teves et al., 2015, 2013). Despite the role of SPAG17 in sperm biology and its previous potential disease associations, the role of the SPAG17 complex allele in the pathogenesis of the male infertility of F1, III:2 is currently unclear. Given the co‐occurrence of male infertility in two affected individuals (F1, III:2 and F3, II:1) of two unrelated families and the absence of other (likely) pathogenic variants in male infertility genes in both cases, we can speculate about a primary role of the CEP78 genotype in the pathogenesis of the male infertility in F1 and F3.
Apart from the genetic data, this study provides other lines of evidence that support the pathogenicity of p.(Leu150Ser). First, amino acid conservation and in silico predictions indicate a disease‐causing effect. Next, homology modeling and FoldX calculation for this substitution in the CEP78 protein model point to a detrimental effect on protein folding and stability due to an amino acid change in an extremely conserved domain, probably involved in protein–protein interactions (Matsushima et al., 2005). Subsequently, expression studies on patient material showed a reduced expression restricted to the protein level and correlated with the zygosity of the missense variant. In the compound heterozygous individual, a subtly decreased mRNA expression level was observed while a clearly lower protein level was seen, which can likely be attributed to NMD caused by the novel splice site variant. Overall, the strong predicted effect of the missense variant on protein stability is suggestive for an LOF mechanism, similar to the already described CEP78 variants.
The ciliary consequences of the missense variant are recapitulating the effect already described for truncating CEP78 variants and for other proteins involved in ciliogenesis (Nikopoulos et al., 2016; Wang et al., 2013). Specifically, we reported elongated primary cilia in affected fibroblasts, that is, a difference of ~1 μm between patient and control cells, which is comparable with what has been reported by Nikopoulos et al. (2016). No clear ultrastructural defects could be demonstrated in nasal brushes and semen sample however. Remarkably, heterozygous carriers of the missense variant displayed an intermediate cellular phenotype, a feature that has already been described both in ciliary and nonciliary contexts (Fog et al., 2018; Noone et al., 2004). Downstream analysis of the Sonic Hedgehog (SHH) signaling pathway was not informative under the given experimental setup (data not shown). Phenotypes typically associated with defective SHH signaling, such as polydactyly and central nervous system developmental anomalies, were not observed however (Reiter & Leroux, 2017).
CEP78 has been described as an interactor of polo‐like kinase 4 (PLK4) where colocalization of CEP78 and PLK4 is required for PLK4‐mediated centriole overduplication (Brunk et al., 2016). PLK4 is the major serine/threonine kinase in centrioles and recent evidence pointed out that PLK4‐mediated phosphorylation of CCP110 (often called CP110), a well‐described regulator of ciliogenesis, is an essential step for centriole assembly (Habedanck, Stierhof, Wilkinson, & Nigg, 2005; Kleylein‐Sohn et al., 2007; M. Lee, Seo, Chang, Hwang, & Rhee, 2017). Interestingly, CEP78 directly interacts with viral protein R binding protein (DCAF1, also known as VPRBP), a component of an E3 ubiquitin ligase, and restrains the transfer of ubiquitin from the complex to CCP110, thus influencing the cellular processes mediated by CCP110, an endogenous inhibitor of ciliogenesis (Figure S10; Hossain et al., 2017). A more recent study, in the context of HIV‐1 infection, reinforced the idea that CEP78 counterbalances the effects of VPRBP on CCP110 (Hossain, Ferreira Barbosa, Cohen, & Tsang, 2018). Although the exact mechanism needs to be proven, it is tempting to speculate about dysregulated processes controlled by CCP110, cilia elongation processes in primis, as a result from a lower amount of CEP78 protein (Schmidt et al., 2009; Walentek et al., 2016).
In conclusion, we found the first CEP78 missense variant to underlie the same sensorial CRDHL phenotype as previously associated with inactivating variants, expanding the molecular spectrum. Due to a founder effect, we may anticipate a higher prevalence in other cases of Northwestern origin with a recognizable CRDHL phenotype. Apart from the sensorial phenotype, we reported male infertility in CEP78 missense cases. Finally, CEP78 LOF variants may have an underestimated role in the pathogenesis of atypical or misdiagnosed Usher syndrome with or without sperm abnormalities.
CONFLICT OF INTERESTS
F. C. is a cofounder of pxlence.
Supporting information
Supporting information
ACKNOWLEDGMENTS
This study was supported by the Ghent University Special Research Fund (BOF15/GOA/011) to E. D. B., by Hercules Foundation AUGE/13/023 to E. D. B., by the Ghent University Hospital Innovation Fund NucleUZ (E. D. B. and F. C.), by the Swiss National Science Foundation (grant #176097 to C. R.), by the German Bundesministerium für Bildung und Forschung (BMBF) through the German Network for Mitochondrial Diseases (mitoNET, 01GM1113C and 01GM1906A to T. K.), by the Independent Research Fund Denmark (grant #8020‐00162B to P. F. and L. B. P.), and by the Carlsberg Foundation (#CF18‐0294 to L. B. P.). We gratefully acknowledge the families who participated in this study and Caroline Van Cauwenbergh and Maria E. Teves for their help. We would like to thank Dr. Francesc Garcia‐Gonzalo (Autonomous University of Madrid, Madrid, Spain) for the gift of puromycin‐sensitive hTERT‐RPE1 cells. G. A. is a PhD fellow (11B4818N; 11B4820N), and E. D. B. (1802215N; 1802220N) and B. P. L. (1803816N) are Senior Clinical Investigators of the Research Foundation—Flanders (FWO). E. D. B. and B. P. L. are members of ERN‐EYE.
Ascari G, Peelman F, Farinelli P, et al. Functional characterization of the first missense variant in CEP78, a founder allele associated with cone‐rod dystrophy, hearing loss, and reduced male fertility. Human Mutation. 2020;41:998–1011. 10.1002/humu.23993
Elfride De Baere and Frauke Coppieters contributed equally to this study.
Contributor Information
Elfride De Baere, Email: elfride.debaere@ugent.be.
Frauke Coppieters, Email: frauke.coppieters@ugent.be.
DATA AVAILABILITY STATEMENT
Data available on request, but may be subjected to restrictions due to privacy/ethical restrictions. The variants reported in this paper have been deposited in the Leiden Open Variation Database (LOVD—https://databases.lovd.nl/shared/genes) under variant IDs 0000603156, 0000603158, 0000603157, 0000603159, and 0000603160.
REFERENCES
- Andjelkovic, M. , Minic, P. , Vreca, M. , Stojiljkovic, M. , Skakic, A. , Sovtic, A. , … Pavlovic, S. (2018). Genomic profiling supports the diagnosis of primary ciliary dyskinesia and reveals novel candidate genes and genetic variants. PLoS One, 13(10):e0205422 10.1371/journal.pone.0205422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, W. , Kloeckener‐Gruissem, B. , & Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Progress in Retinal and Eye Research, 29(5), 335–375. 10.1016/j.preteyeres.2010.03.004 [DOI] [PubMed] [Google Scholar]
- Brunk, K. , Zhu, M. , Bärenz, F. , Kratz, A. , Haselmann‐Weiss, U. , Antony, C. , & Hoffmann, I. (2016). Cep78 is a new centriolar protein involved in Plk4‐induced centriole overduplication. Journal of Cell Science, 129(14), 2713–2718. 10.1242/jcs.184093 [DOI] [PubMed] [Google Scholar]
- Córdova‐Fletes, C. , Becerra‐Solano, L. E. , Rangel‐Sosa, M. M. , Rivas‐Estilla, A. M. , Alberto Galán‐Huerta, K. , Ortiz‐López, R. , … García‐Ortiz, J. E. (2018). Uncommon runs of homozygosity disclose homozygous missense mutations in two ciliopathy‐related genes (SPAG17 and WDR35) in a patient with multiple brain and skeletal anomalies. European Journal of Medical Genetics, 61(3), 161–167. 10.1016/j.ejmg.2017.11.011 [DOI] [PubMed] [Google Scholar]
- Daiger, S. , Rossiter, B. , Greenberg, J. , Christoffels, A. , Hide, W. , P Daiger, S. , … Hide, W. (1998). Data services and software for identifying genes and mutations causing retinal degeneration. Investigative Ophthalmology and Visual Science, 39, S295 10.1002/acr.21596 [DOI] [Google Scholar]
- de Castro‐Miró, M. , Tonda, R. , Escudero‐ferruz, P. , Andre, R. , Mayor‐Lorenzo, A. , Castro, J. , … Gonzàlez‐Duarte, R. (2016). Novel candidate genes and a wide spectrum of structural and point mutations responsible for inherited retinal dystrophies revealed by exome sequencing. PLoS One, 11(12):e0168966 10.1371/journal.pone.0168966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'haene, B. , Vandesompele, J. , & Hellemans, J. (2010). Accurate and objective copy number profiling using real‐time quantitative PCR. Methods, 50(4), 262–270. 10.1016/j.ymeth.2009.12.007 [DOI] [PubMed] [Google Scholar]
- Flannigan, R. , & Schlegel, P. N. (2017). Genetic diagnostics of male infertility in clinical practice. Best Practice & Research. Clinical Obstetrics & Gynaecology, 44, 26–37. 10.1016/j.bpobgyn.2017.05.002 [DOI] [PubMed] [Google Scholar]
- Fog, C. K. , Zago, P. , Malini, E. , Solanko, L. M. , Peruzzo, P. , Bornaes, C. , … Kirkegaard, T. (2018). The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine, 38, 142–153. 10.1016/j.ebiom.2018.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Q. , Xu, M. , Chen, X. , Sheng, X. , Yuan, Z. , Liu, Y. , … Chen, R. (2016). CEP78 is mutated in a distinct type of Usher syndrome. Journal of Medical Genetics, 54, 190–195. 10.1136/jmedgenet-2016-104166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goduti, D. J. , & Smith, E. F. (2012). Analyses of functional domains within the PF6 protein of the central apparatus reveal a role for PF6 sub‐complex members in regulating flagellar beat frequency. Cytoskeleton, 69(3), 179–194. 10.1002/cm.21010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönczy, P. (2015). Centrosomes and cancer: Revisiting a long‐standing relationship. Nature Reviews Cancer, 15(11), 639–652. 10.1038/nrc3995 [DOI] [PubMed] [Google Scholar]
- Gönczy, P. , & Hatzopoulos, G. N. (2019). Centriole assembly at a glance. Journal of Cell Science, 132(4), jcs228833 10.1242/jcs.228833 [DOI] [PubMed] [Google Scholar]
- Guerois, R. , Nielsen, J. E. , & Serrano, L. (2002). Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. Journal of Molecular Biology, 320(2), 369–387. 10.1016/S0022-2836(02)00442-4 [DOI] [PubMed] [Google Scholar]
- Habedanck, R. , Stierhof, Y.‐D. , Wilkinson, C. J. , & Nigg, E. A. (2005). The Polo kinase Plk4 functions in centriole duplication. Nature Cell Biology, 7(11), 1140–1146. 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- Hammad, M. O. , Elabbasy, L. M. , Abd elghaffar, M. A. , Zaki, M. M. A. , Bazeed, F. B. , & Zahran, M. A. (2019). Significance of CEP78 and WDR62 gene expressions in differentiated thyroid carcinoma: Possible predictors of lateral lymph node metastasis. Asia‐Pacific Journal of Clinical Oncology, 15, e154–e161. 10.1111/ajco.13143 [DOI] [PubMed] [Google Scholar]
- Hildebrandt, F. , Benzing, T. , & Katsanis, N. (2011). Ciliopathies. The New England Journal of Medicine, 364(16), 1533–1543. 10.1056/NEJMra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain, D. , Ferreira Barbosa, J. A. , Cohen, É. A. , & Tsang, W. Y. (2018). HIV‐1 Vpr hijacks EDD‐DYRK2‐DDB1DCAF1 to disrupt centrosome homeostasis. The Journal of Biological Chemistry, 293(24), 9448–9460. 10.1074/jbc.RA117.001444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain, D. , Javadi Esfehani, Y. , Das, A. , & Tsang, W. Y. (2017). Cep78 controls centrosome homeostasis by inhibiting EDD‐DYRK2‐DDB1(Vpr)(BP). EMBO Reports, 18(4), 632–644. 10.15252/embr.201642377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel, T. , Sekinger, B. , Wolf, S. R. , Pauli, E. , & Kobal, G. (1997). 'Sniffin' sticks': Olfactory performance assessed by the combined testing of odor identification, odor discrimination and olfactory threshold. Chemical Senses, 22(1), 39–52. [DOI] [PubMed] [Google Scholar]
- Kazarian, E. , Son, H. , Sapao, P. , Li, W. , Zhang, Z. , Strauss, J. F. , & Teves, M. E. (2018). SPAG17 is required for male germ cell differentiation and fertility. International Journal of Molecular Sciences, 19(4), 1252 10.3390/ijms19041252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein‐Sohn, J. , Westendorf, J. , Le Clech, M. , Habedanck, R. , Stierhof, Y. D. , & Nigg, E. A. (2007). Plk4‐induced centriole biogenesis in human cells. Developmental Cell, 13(2), 190–202. 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Koenig, R. (2003). Bardet‐Biedl syndrome and Usher syndrome. Developments in Ophthalmology, 37, 126–140. [DOI] [PubMed] [Google Scholar]
- Krausz, C. , Hoefsloot, L. , Simoni, M. , & Tüttelmann, F. , European Academy of Andrology, & European Molecular Genetics Quality Network . (2014). EAA/EMQN best practice guidelines for molecular diagnosis of Y‐chromosomal microdeletions: State‐of‐the‐art 2013. Andrology, 2(1), 5–19. 10.1111/j.2047-2927.2013.00173.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger, E. , & Vriend, G. (2015). New ways to boost molecular dynamics simulations. Journal of Computational Chemistry, 36(13), 996–1007. 10.1002/jcc.23899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. , & Garg, S. (2015). Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genetics in Medicine, 17(4), 245–252. 10.1038/gim.2015.15 [DOI] [PubMed] [Google Scholar]
- Lee, M. , Seo, M. Y. , Chang, J. , Hwang, D. S. , & Rhee, K. (2017). PLK4 phosphorylation of CP110 is required for efficient centriole assembly. Cell Cycle, 16, 1225–1234. 10.1080/15384101.2017.1325555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefever, S. , Pattyn, F. , De Wilde, B. , Coppieters, F. , De Keulenaer, S. , Hellemans, J. , & Vandesompele, J. (2017). High‐throughput PCR assay design for targeted resequencing using primerXL. BMC Bioinformatics, 18(1), 400 10.1186/s12859-017-1809-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Wu, C. , Li, C. , & Boerwinkle, E. (2016). dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, J. S. , Paff, T. , Goggin, P. , & Haarman, E. (2016). Diagnostic methods in primary ciliary dyskinesia. Paediatric Respiratory Reviews, 18, 8–17. 10.1016/j.prrv.2015.07.017 [DOI] [PubMed] [Google Scholar]
- Matsushima, N. , Tachi, N. , Kuroki, Y. , Enkhbayar, P. , Osaki, M. , Kamiya, M. , & Kretsinger, R. H. (2005). Structural analysis of leucine‐rich‐repeat variants in proteins associated with human diseases. Cellular and Molecular Life Sciences, 62(23), 2771–2791. 10.1007/s00018-005-5187-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, D. R. , & Smith, B. (2009). Analysis of the central pair microtubule complex in Chlamydomonas reinhardtii. Methods in Cell Biology, 92, 197–213. 10.1016/S0091-679X(08)92013-6 [DOI] [PubMed] [Google Scholar]
- Mokrzan, E. M. , Lewis, J. S. , & Mykytyn, K. (2007). Differences in renal tubule primary cilia length in a mouse model of Bardet‐Biedl syndrome. Nephron. Experimental Nephrology, 106(3), e88–e96. 10.1159/000103021 [DOI] [PubMed] [Google Scholar]
- Namburi, P. , Ratnapriya, R. , Khateb, S. , Lazar, C. H. , Kinarty, Y. , Obolensky, A. , … Ben‐yosef, T. (2016). Bi‐allelic truncating mutations in CEP78, encoding centrosomal protein 78, cause cone‐rod degeneration with sensorineural hearing loss. The American Journal of Human Genetics, 99(3), 777–784. 10.1016/j.ajhg.2016.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesslinger, N. J. , Sahota, R. A. , Stone, B. , Johnson, K. , Chima, N. , King, C. , … Nelson, B. H. (2007). Standard treatments induce antigen‐specific immune responses in prostate cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 13(5), 1493–1502. 10.1158/1078-0432.CCR-06-1772 [DOI] [PubMed] [Google Scholar]
- Nigg, E. A. , & Raff, J. W. (2009). Centrioles, centrosomes, and cilia in health and disease. Cell, 139(4), 663–678. 10.1016/j.cell.2009.10.036 [DOI] [PubMed] [Google Scholar]
- Nikopoulos, K. , Farinelli, P. , Giangreco, B. , Tsika, C. , Royer‐Bertrand, B. , Mbefo, M. K. , … Rivolta, C. (2016). Mutations in CEP78 cause cone‐rod dystrophy and hearing loss associated with primary‐cilia defects. American Journal of Human Genetics, 99(3), 770–776. 10.1016/j.ajhg.2016.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noone, P. G. , Leigh, M. W. , Sannuti, A. , Minnix, S. L. , Carson, J. L. , Hazucha, M. , … Knowles, M. R. (2004). Primary ciliary dyskinesia: Diagnostic and phenotypic features. American Journal of Respiratory and Critical Care Medicine, 169(4), 459–467. 10.1164/rccm.200303-365OC [DOI] [PubMed] [Google Scholar]
- Pettersen, E. F. , Goddard, T. D. , Huang, C. C. , Couch, G. S. , Greenblatt, D. M. , Meng, E. C. , & Ferrin, T. E. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25, 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Plagnol, V. , Curtis, J. , Epstein, M. , Mok, K. Y. , Stebbings, E. , Grigoriadou, S. , … Nejentsev, S. (2012). A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics, 28(21), 2747–2754. 10.1093/bioinformatics/bts526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter, J. F. , & Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nature Reviews Molecular Cell Biology, 18(9), 533–547. 10.1038/nrm.2017.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis‐Juan, A. , Stephens, J. , French, C. E. , Gleadall, N. , Mégy, K. , Penkett, C. , … Carss, K. J. (2018). Complex structural variants in Mendelian disorders: Identification and breakpoint resolution using short‐ and long‐read genome sequencing. Genome Medicine, 10(1), 95 10.1186/s13073-018-0606-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, T. I. , Kleylein‐Sohn, J. , Westendorf, J. , Le Clech, M. , Lavoie, S. B. , Stierhof, Y.‐D. , & Nigg, E. A. (2009). Control of centriole length by CPAP and CP110. Current Biology, 19(12), 1005–1011. 10.1016/j.cub.2009.05.016 [DOI] [PubMed] [Google Scholar]
- Shahana, S. , Björnsson, E. , Lúdvíksdóttir, D. , Janson, C. , Nettelbladt, O. , Venge, P. , … BHR‐group (2005). Ultrastructure of bronchial biopsies from patients with allergic and non‐allergic asthma. Respiratory Medicine, 99(4), 429–443. 10.1016/j.rmed.2004.08.013 [DOI] [PubMed] [Google Scholar]
- Shaheen, R. , Szymanska, K. , Basu, B. , Patel, N. , Ewida, N. , Faqeih, E. , … Alkuraya, F. S. (2016). Characterizing the morbid genome of ciliopathies. Genome Biology, 17(1), 242 10.1186/s13059-016-1099-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherly, D. , & Prathibha, K. (2014). Measurement of nasal mucociliary clearance. Clinical Research in Pulmonology, 2(2), 1019. [Google Scholar]
- Söding, J. , Biegert, A. , & Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research, 33, W244–W248. 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorte, H. S. , Osnes, L. T. , Fevang, B. , Aukrust, P. , Erichsen, H. C. , Backe, P. H. , … Stray‐Pedersen, A. (2016). A potential founder variant in CARMIL2/RLTPR in three Norwegian families with warts, molluscum contagiosum, and T‐cell dysfunction. Molecular Genetics & Genomic Medicine, 4(6), 604–616. 10.1002/mgg3.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symoens, S. , Barnes, A. M. , Gistelinck, C. , Malfait, F. , Guillemyn, B. , Steyaert, W. , … Coucke, P. J. (2015). Genetic defects in TAPT1 disrupt ciliogenesis and cause a complex lethal osteochondrodysplasia. American Journal of Human Genetics, 97(4), 521–534. 10.1016/j.ajhg.2015.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves, M. E. , Sundaresan, G. , Cohen, D. J. , Hyzy, S. L. , Kajan, I. , Maczis, M. , … Strauss, J. F. (2015). Spag17 deficiency results in skeletal malformations and bone abnormalities. PLoS One, 10(5):e0125936 10.1371/journal.pone.0125936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves, M. E. , Zhang, Z. , Costanzo, R. M. , Henderson, S. C. , Corwin, F. D. , Zweit, J. , … Strauss, J. F. (2013). Sperm‐associated antigen‐17 gene is essential for motile cilia function and neonatal survival. American Journal of Respiratory Cell and Molecular Biology, 48(6), 765–772. 10.1165/rcmb.2012-0362OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser, A. , Nijveen, H. , Rao, X. , Bisseling, T. , Geurts, R. , & Leunissen, J. A. M. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Research, 35, W71–W74. 10.1093/nar/gkm306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walentek, P. , Quigley, I. K. , Sun, D. I. , Sajjan, U. K. , Kintner, C. , & Harland, R. M. (2016). Ciliary transcription factors and miRNAs precisely regulate Cp110 levels required for ciliary adhesions and ciliogenesis. Elife, 5:e17557 10.7554/eLife.17557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W.‐J. , Tay, H. G. , Soni, R. , Perumal, G. S. , Goll, M. G. , Macaluso, F. P. , … Tsou, M.‐F. B. (2013). CEP162 is an axoneme‐recognition protein promoting ciliary transition zone assembly at the cilia base. Nature Cell Biology, 15(6), 591–601. 10.1038/ncb2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel, L. , Baakman, C. , Gilissen, D. , Veltman, J. A. , Vriend, G. , & Gilissen, C. (2019). MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Human Mutation, 40, 1030–1038. 10.1002/humu.23798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witters, J. A. , De Zaeytijd, J. , Leys, M. , & Leroy, B. P. (2004). Progressive cone dystrophy and sensorineural hearing loss. Bulletin de la Societe Belge d'Ophtalmologie, 294, 35–42. [PubMed] [Google Scholar]
- Xu, X. , Sha, Y.‐W. , Mei, L.‐B. , Ji, Z.‐Y. , Qiu, P.‐P , Ji, H. , … Li, L. (2018). A familial study of twins with severe asthenozoospermia identified a homozygous SPAG17 mutation by whole‐exome sequencing. Clinical Genetics, 93(2), 345–349. 10.1111/cge.13059 [DOI] [PubMed] [Google Scholar]
- Ye, K. , Schulz, M. H. , Long, Q. , Apweiler, R. , & Ning, Z. (2009). Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired‐end short reads. Bioinformatics, 25(21), 2865–2871. 10.1093/bioinformatics/btp394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Duan, T. , Wang, L. , Tang, J. , Luo, R. , Zhang, R. , & Kang, T. (2016). Low expression of centrosomal protein 78 (CEP78) is associated with poor prognosis of colorectal cancer patients. Chinese Journal of Cancer, 35(1), 62 10.1186/s40880-016-0121-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
Data available on request, but may be subjected to restrictions due to privacy/ethical restrictions. The variants reported in this paper have been deposited in the Leiden Open Variation Database (LOVD—https://databases.lovd.nl/shared/genes) under variant IDs 0000603156, 0000603158, 0000603157, 0000603159, and 0000603160.