

Abstract
Background and Aims
Patients with liver disease acquire complex changes in their hemostatic system, which results in a fragile rebalanced status. The status of the fibrinolytic system is controversial, as is the role of fibrinolytic dysfunction in bleeding and thrombosis in patients with cirrhosis. Here, we aimed to determine fibrinolytic status and its relationship with outcome in acutely ill patients with cirrhosis.
Approach and Results
We assessed plasma fibrinolytic potential in a large cohort of patients with acutely decompensated cirrhosis (AD, n = 52) or acute‐on‐chronic liver failure (ACLF, n = 57). Compared with 40 healthy volunteers, median clot lysis times (CLTs) were shorter in patients with AD but comparable to controls in patients with ACLF. However, the variability in CLTs in patients was much larger than in healthy controls, and in both patient groups, a proportion of patients had clearly prolonged or shortened CLTs. The variability in CLTs in patients was not readily explained by variations in plasma levels of key fibrinolytic proteins. However, CLTs were clearly related to clinical characteristics, with longer CLTs in patients with sepsis and patients with any organ failure (as defined by the European Foundation for the Study of Chronic Liver Disease organ failure scores). CLTs were not different between patients that did or did not experience bleeding or a thrombotic event during follow‐up. Baseline CLTs were substantially longer in patients that died within 30 days of admission.
Conclusions
Our study demonstrates a mixed fibrinolytic phenotype in acutely ill patients with cirrhosis with baseline hypofibrinolysis associated with sepsis, organ failure, and short‐term mortality. These associations may be explained by defective clearance of intraorgan microthrombi that have been proposed to drive organ failure.
Abbreviations
- ACLF
acute‐on‐chronic liver failure
- AD
acute decompensation
- CLT
clot lysis time
- MELD
Model for End‐Stage Liver Disease
- PAI‐1
plasminogen activator inhibitor type 1
- SOFA
Sequential Organ Failure Assessment
- TAFI
thrombin activatable fibrinolysis inhibitor
- tPA
tissue‐type plasminogen activator
Patients with cirrhosis frequently have complex changes in their hemostatic system. The net effect of these changes is a hemostatic system that is in a “rebalanced” status,1 although notable hyper‐ and hypocoagulable features may be present.2, 3, 4 Although there is increasing consensus that a decreased platelet count in patients with cirrhosis is (in part) balanced by highly elevated plasma levels of von Willebrand factor5 and that the thrombin‐generating capacity is normal to increased compared with healthy individuals,6, 7 there is continuing controversy on the status of the fibrinolytic system.8 Using various approaches, it has been demonstrated that some patients with cirrhosis are in a hyperfibrinolytic state, which is ascribed to complex changes in plasma levels of fibrinolytic proteins, with an important role for elevated levels of tissue‐type plasminogen activator (tPA).9, 10, 11 However, it has also been argued that the fibrinolytic system is in balance in these patients because of the concomitant decrease of antifibrinolytics (antiplasmin, thrombin activatable fibrinolysis inhibitor [TAFI]) and the profibrinolytic plasminogen.12 However, this fibrinolytic balance may be easily disturbed, and whether the balance remains intact in patients with severe disease is questionable.
Patients with acute decompensation of cirrhosis (AD) or acute‐on‐chronic liver failure (ACLF) have progressive changes in their hemostatic system2, 13, 14 and are at particular risk for bleeding and thrombotic complications.15 Patients with ACLF are characterized by development of organ failure and high short‐term mortality.16, 17, 18 An intense systemic inflammatory response that is often complicated (or precipitated) by infection is one of the hallmarks of ACLF.19 Despite recent advances in supportive care, ACLF remains associated with high mortality and resource use, including blood and blood products.20
We have studied fibrinolytic status in patients with AD and ACLF and showed that although plasma fibrinolytic potential was similar in patients with AD and ACLF compared with healthy controls, individual patients showed accelerated as well as inhibited clot lysis.2 We have also demonstrated that patients with ACLF had decreased lysis of whole blood clots, assessed by thromboelastometry, in comparison with patients with AD, 72 hours after hospital admission.14 Interestingly, it has been well established that patients with sepsis and patients with acute liver failure, both of which are inflammatory syndromes that are frequently complicated by multiple organ failure, frequently have inhibited fibrinolysis,21, 22 which may contribute to organ failure by intraorgan thrombotic events.23, 24 The role of sepsis and multiorgan failure on the fibrinolytic status of acutely ill patients with cirrhosis has not yet been established.
With the aim to provide a better clinical approach to hemostasis management of acutely ill patients with cirrhosis, we sought to describe the fibrinolytic profile in a large cohort of patients from two expert clinical centers. In addition, we assessed laboratory and clinical determinants of the fibrinolytic status with particular emphasis on sepsis and multiorgan failure.
Patients and Methods
Between February 2018 and September 2018, adult patients with AD (n = 52) or ACLF (n = 58) consecutively admitted to King’s College Hospital London (United Kingdom) and Hospital Clínic Barcelona (Spain) who gave written informed consent were included in this study. The National Research Ethics Service (NRES) Committee London, Westminster (12/LO/1417) and the Medical Ethical Committee Hospital Clínic Barcelona (2017/0948) approved the study protocol, which was in accordance with the Helsinki Declaration of 1975. Informed consent or assent was obtained from participants or their personal consultees. Exclusion criteria for this study were acute liver failure, known congenital coagulation disorders, the use of anticoagulants or platelet function inhibitors, pregnancy, human immunodeficiency virus positivity, extrahepatic malignancy, and hepatocellular carcinoma outside the Milan criteria. A Strengthening the Reporting of Observational Studies in Epidemiology diagram outlining details of patient inclusion is given in Supporting Fig. S1. Cirrhosis was defined by the presence of two or more of i) histological evidence of cirrhosis on liver biopsy, ii) laboratory abnormalities consistent with cirrhosis, or iii) radiological findings consistent with cirrhosis and portal hypertension. AD of chronic liver disease and ACLF were defined and graded according to the number of organ failures in concordance with criteria reported in the CANONIC study.16 Patients were followed up with until 30 days after discharge, death, or transplant, whatever happened first. Healthy controls aged >18 years (n = 40) were enrolled at King’s College Hospital (n = 20) and Hospital Clínic Barcelona (n = 20) to establish reference values for the various laboratory tests performed. Healthy controls provided informed consent with the protocol approved by the NRES Committee London, Westminster (12/LO/1417) and the Medical Ethical Committee Hospital Clínic Barcelona (2017/0948). Exclusion criteria for healthy controls were body mass index below 18 or above 28, pregnancy or active breastfeeding, a personal history of thrombotic or liver disease, untreated medical conditions, chronic medical conditions requiring regular primary or secondary care review, or current use of anticoagulants, platelet function inhibitors, or oral contraceptives.
Data Collection
We collected baseline data on patient demographics, comorbidities, biochemistry, and illness severity scores. The severity of liver disease was evaluated with Sequential Organ Failure Assessment (SOFA), CLIF‐AD, CLIF‐ACLF, Model for End‐Stage Liver Disease (MELD), and Child‐Turcotte‐Pugh scores. Sepsis syndrome was defined according to the Sepsis‐3 guidelines. Data on hemorrhagic or thrombotic events and on transfusion requirements were collected throughout hospitalization. Bleeding events were defined according to the following criteria: fatal bleeding, symptomatic bleeding in a critical area or organ, and/or bleeding causing a fall in hemoglobin level of ≥2 g/L or leading to transfusion ≥2 units of packed red cells.
Blood Samples
Blood samples were collected in sodium citrate‐containing vacutainer tubes (0.129 M) from an arterial line, from a central venous catheter, or by standard peripheral venous phlebotomy within the first 2 days of admission or after the development of ACLF. Samples were obtained before the administration of blood products, anticoagulants, or platelet function inhibitors. Within 2 hours after the blood draw, the sample was centrifuged at 2,000g and 10,000g for 10 minutes at ambient temperature. Plasma was stored at −80°C until it was used for analyses.
Clot Lysis Assay
Lysis of a tissue factor–induced clot by exogenous tPA was studied by monitoring changes in turbidity during clot formation and subsequent lysis as described.25 In short, 50 μL plasma was pipetted in a 96‐well microtiter plate. Subsequently, 50 μL of a mixture containing phospholipid vesicles (40% l‐α‐dioleoylphosphatidylcholine, 20% l‐α‐dioleoylphosphatidylserine, and 40% l‐α‐dioleoylphosphatidylethanolamine, final concentration 10 μM), tPA (final concentration 56 ng/mL), tissue factor (Innovin [Siemens Healthcare Diagnostics, Marburg, Germany] final dilution 1:1,000), and CaCl2 (final concentration 17 mM), diluted in N‐2‐hydroxyethylpiperazine‐N′‐2‐ethanesulfonic acid (HEPES) buffer (25 mM HEPES, 137 mM NaCl, 3.5 mM KCl, 3 mM CaCl2, 0.1% bovine serum albumin, pH 7.4), was added using a multichannel pipette. After thorough mixing, the plate was incubated at 37°C in a Spectramax 340 kinetic microplate reader (Molecular Devices, San Jose, CA, USA), and the optical density at 340 nm was monitored every 20 seconds, resulting in a clot lysis turbidity profile. The clot lysis time (CLT) was derived from this clot lysis profile and defined as the time (minutes) from the midpoint of the clear to maximum turbid transition, representing clot formation, to the midpoint of the maximum turbid to clear transition, representing the lysis of the clot.
Plasma Protein Levels
Plasminogen levels were measured on an automated coagulation analyzer (ACL TOP 300) with reagents and protocols from the manufacturer (Werfen, Breda, the Netherlands). Plasma levels of tPA, plasminogen activator inhibitor type 1 (PAI‐1), and TAFI were determined using commercially available enzyme‐linked immunosorbent assays: Zymutest tPA (Hyphen Biomed, Neuville‐sur‐Oise, France), PAI‐1 DuoSet (R&D systems, Minneapolis, MN), and Zymutest Activatable TAFI (Hyphen Biomed).
Statistical Analyses
Statistical analyses were performed using SPSS Statistics, version 23 (IBM, Inc., Chicago, IL) and GraphPad Prism (San Diego, CA). Data were presented as means with standard deviation or medians and interquartile ranges for continuous variables as appropriate and as percentages for categorical variables. Means of two groups were compared by Student t test or Mann‐Whitney U test as appropriate. Multiple groups were compared using one‐way analysis of variance (with the Bonferroni posttest) or Kruskal‐Wallis H test (with Dunn’s posttest) as appropriate. Pearson’s correlation coefficient was used to assess correlation between variables. Receiver operating characteristic (ROC) curves and their corresponding areas under the curve (AUC) were used to assess the performance of the MELD, SOFA, and Child‐Turcotte‐Pugh score relative to the CLT in predicting outcome. Logistic regression models were used to assess the relation of CLT to outcome with adjustment for MELD, SOFA, or Child‐Turcotte‐Pugh score. P value < 0.05 was considered significant.
Results
Patient Characteristics
We have determined CLTs and plasma levels of tPA, PAI‐1, plasminogen, and TAFI in 52 adult patients with AD cirrhosis and 57 adult patients with ACLF and compared these levels with plasma concentrations in 40 healthy controls. Patient demographics and clinical and laboratory data are presented in Table 1. Healthy controls were younger than patients (35 [28‐43] years, and 45% were male). Among patients with ACLF, 27 (47%) had ACLF‐1, 13 (23%) had ACLF‐2, and 17 (30%) had ACLF‐3 at inclusion.
Table 1.
Demographic and Laboratory Data of the Study Population
| Variable | AD | ACLF |
|---|---|---|
| n = 52 | n = 57 | |
| Age, years | 58 (50‐67) | 59 (49‐66) |
| Etiology | ||
| Alcohol | 32 | 33 |
| Viral | 0 | 11 |
| NASH | 10 | 6 |
| Biliary | 3 | 2 |
| Other | 7 | 5 |
| Male | 29 | 40 |
| SOFA score | 4 (3‐6) | 8 (6‐11) |
| CLIF‐SOFA score | 60 (47‐108) | 87 (74‐97) |
| CLIF‐AD score | 51 (42‐57) | |
| CLIF‐ACLF score | 53 (45‐60) | |
| MELD | 15 (11‐21) | 27 (23‐35) |
| Child‐Turcotte‐Pugh, points | 9 (7‐10) | 10 (8‐12) |
| Medication on admission | ||
| antibiotics | 25 | 32 |
| Betablocker | 22 | 36 |
| Antiviral | 0 | 6 |
| Lactulose | 21 | 19 |
| Rifaximin | 4 | 14 |
| Ascites | ||
| No | 9 | 2 |
| Minimal | 18 | 25 |
| Moderate | 22 | 18 |
| Severe | 3 | 12 |
| Reason(s) for decompensation | ||
| (Suspected) infection | 10 | 24 |
| Ascites | 23 | 17 |
| Encepalopathy | 7 | 12 |
| Multiorgan failure | 0 | 2 |
| Variceal bleeding | 6 | 4 |
| Alcoholic hepatitis | 5 | 5 |
| Other | 1 | 2 |
| Hemoglobin, g/dL | 80 (86‐110) | 85 (77‐99) |
| Na, mmol/L | 136 (132‐139) | 135 (132‐141) |
| Urea, mmol/L | 5 (3‐8) | 8 (4‐9) |
| Creatinine, µmol/L | 73 (54‐109) | 211 (134‐282) |
| Bilirubin, µmol/L | 39 (28‐101) | 100 (40‐381) |
| Gamma glutamyl transpeptidase, IU/L | 77 (43‐145) | 70 (42‐158) |
| Alkaline phosphatase, IU/L | 120 (82‐181) | 116 (80‐142) |
| Aspartate aminotransferase, IU/L | 62 (44‐100) | 65 (40‐108) |
| Albumin, g/L | 29 (26‐33) | 29 (24‐33) |
| Platelets × 109/L | 88 (62‐127) | 70 (38‐107) |
| Fibrinogen, g/L | 2.2 (1.4‐2.9) | 1.8 (1.1‐2.5) |
| INR | 1.4 (1.3‐1.8) | 1.7 (1.4‐2.6) |
| APTT, seconds | 36 (30‐43) | 41 (33‐56) |
Abbreviations: APTT, activated partial thromboplastin time; INR, international normalized ratio; NASH, nonalcoholic steatohepatitis.
Plasma Fibrinolytic Profile in Patients With AD and ACLF
We studied plasma fibrinolytic potential in patients with AD and ACLF. CLT was significantly shorter in the AD cohort compared with the healthy controls and was similar between the ACLF cohort and healthy controls. CLT was significantly prolonged in the ACLF cohort compared with the AD cohort (Fig. 1). However, a large variability was observed in the CLT values, with a substantial proportion of patients that had CLTs shorter than the reference values of the healthy controls and a substantial proportion that had substantially prolonged CLTs. The spread in CLT values was particularly profound in patients with ACLF with some patients with very rapid clot lysis and some patients that showed no lysis at all during the time span of the experiment. CLTs were similar between patients with ACLF grades 1 and 2 but were substantially elevated in those with ACLF grade 3 (ACLF‐1 66 minutes [57‐82]; ACLF‐2 60 minutes [30‐86]; ACLF‐3 180 minutes [60‐180]).
Figure 1.
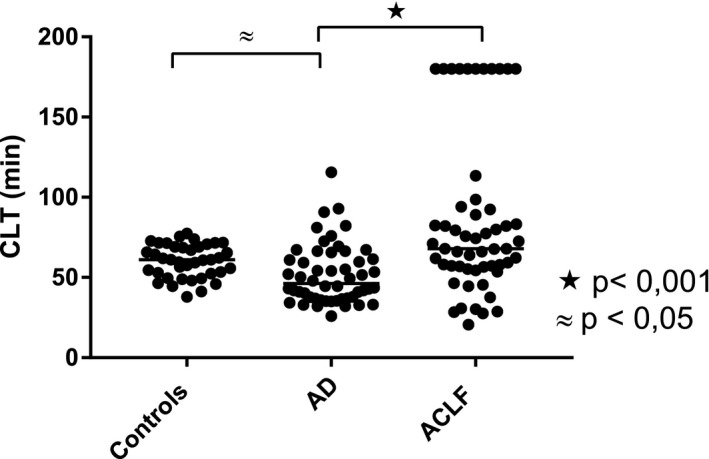
Plasma fibrinolytic potential in acutely ill patients with cirrhosis. CLTs were determined in patients with AD and ACLF and in a group of healthy volunteers. Horizontal lines indicate medians.
Plasma levels of PAI‐1, plasminogen, and TAFI are important determinants of CLT in healthy individuals.26 Fig. 2 shows levels of these analytes in our cohort. Plasma levels of PAI‐1 were substantially higher in patients compared with controls, but levels were comparable between patients with AD and ACLF. Plasminogen and TAFI levels were substantially lower in patients, with significantly lower levels in patients with ACLF compared with patients with AD. Within the ACLF cohort, there was a stepwise decrease in plasminogen and TAFI levels with increasing ACLF grade (ACLF‐1 36% [27‐56], ACLF‐2 34% [21‐43], ACLF‐3 18% [15‐22] for plasminogen, ACLF‐1 58% [31‐77], ACLF‐2 38% [21‐56], ACLF‐3 30% [17‐47] for TAFI).
Figure 2.
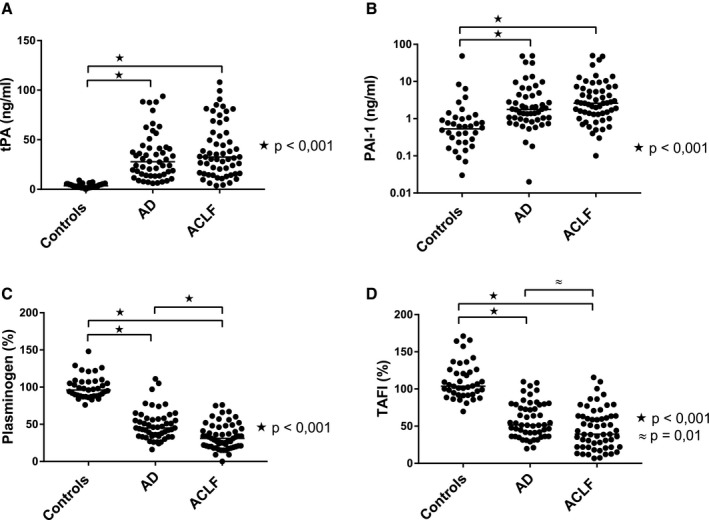
Plasma levels of fibrinolytic proteins. Plasma levels of (A) tPA, (B) PAI‐1, (C) plasminogen, and (D) TAFI were determined by enzyme‐linked immunosorbent assay or functional assay (for plasminogen) in patients with AD and ACLF, and in a group of healthy volunteers. Horizontal lines indicate medians.
Although tPA is not a determinant of CLT in healthy individuals, as exogenous tPA is added to the test reagent mixture, excessive tPA levels, as for example encountered during liver transplantation, appear to drive fibrinolysis in the assay. We therefore assessed tPA antigen levels, which reflect both free tPA and tPA‐PAI‐1 complexes, and found substantially increased levels in patients compared with controls, with no differences between patients with AD and ACLF.
In linear regression analyses, we found that plasma levels of plasminogen, TAFI, tPA, and PAI‐1 weakly correlated with CLTs, but only tPA levels showed correlations both in all patients combined and in AD and ACLF patients analyzed as separate cohorts (Table 2).
Table 2.
Correlations Between CLT and Plasma Levels of Individual Fibrinolytic Proteins
| Group | Plasminogen | TAFI | tPA | PAI‐1 |
|---|---|---|---|---|
| AD + ACLF | r 2 = 0.080 | r 2 = 0.001 | r 2 = 0.105 | r 2 = 0.062 |
| P = 0.003 | P = 0.699 | P = 0.0007 | P = 0.009 | |
| AD | r 2 = 0.009 | r 2 = 0.098 | r 2 = 0.104 | r 2 = 0.0009 |
| P = 0.488 | P = 0.023 | P = 0.019 | P = 0.824 | |
| ACLF | r 2 = 0.080 | r 2 = 0.009 | r 2 = 0.115 | r 2 = 0.156 |
| P = 0.036 | P = 0.478 | P = 0.011 | P = 0.002 |
Pearson correlation coefficients are shown with corresponding P values.
Clinical Determinants of CLT
As plasma levels of fibrinolytic proteins did not appear to explain why some patients were clearly hypofibrinolytic and others clearly hyperfibrinolytic, we assessed clinical features in relation to CLT. In all patients combined, CLT was significantly longer in patients with sepsis at the time of sampling compared with patients without sepsis (Fig. 3). In line with this, patients with sepsis had higher PAI‐1 (3.9 ng/mL [1.6‐9.6] vs. 1.7 ng/mL [0.8‐3.8], P < 0.01) and lower plasminogen (29% [18‐41] vs. 42% [31‐56], P < 0.01) and TAFI (39% [21‐62] vs. 51% [35‐78] P = 0.03) plasma levels compared with patients without sepsis. tPA levels were comparable between patients who were septic and patients who were nonseptic.
Figure 3.
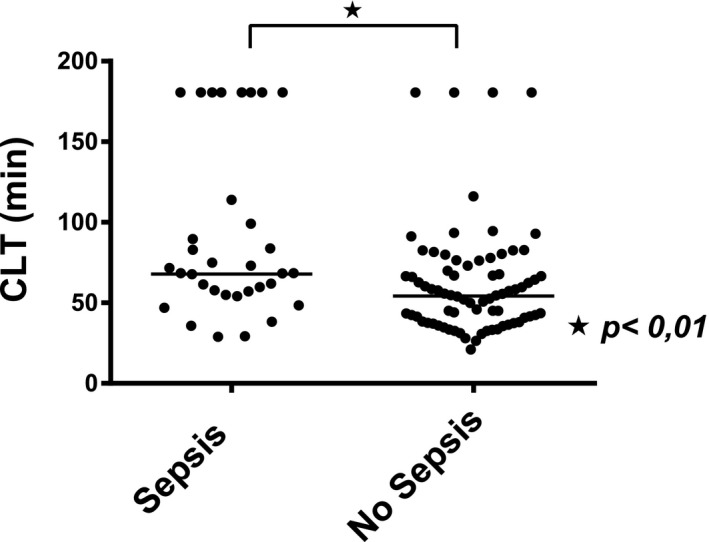
Fibrinolytic status in critically ill patients with cirrhosis stratified by sepsis status. CLT in patients with AD and ACLF combined are shown, stratified for presence or absence of sepsis. Horizontal lines indicate medians.
Because hypofibrinolysis has been proposed to contribute to organ failure in acutely ill patients by microvascular occlusion, we have explored the relation between fibrinolysis and the severity of organ failure. CLTs were higher in patients with SOFA scores above the median, and CLT were slightly higher in patients with CLIF‐AD and CLIF‐ACLF scores above the median (Table 3). In addition, when patients were stratified according to absence or presence of individual organ failure, CLTs were higher in patients with all individual organ failures, although the difference in CLT in patients with or without coagulation failure did not reach statistical significance. CLTs were similar when patients were stratified according to ascites severity (no ascites 54 min [36‐72], minimal ascites 57 min [44‐77], moderate ascites 61 min [43‐82], severe ascites 57 min [32‐94]).
Table 3.
CLT Stratified by SOFA, CLIF‐AD, CLIF‐ACLF, Child‐Turcotte‐Pugh Scores and by the Presence or Absence of Liver, Renal, Coagulation, Hemodynamic, Respiratory, and Neurological Failure
| SOFA (Median Value 6) | |||
| Low, n = 61 | High, n = 48 | P | |
| CLT, min | 54 (37‐66) | 68 (47‐130) | <0.01 |
| CLIF‐AD (Median Value 51) | |||
| Low, n = 25 | High, n = 27 | P | |
| CLT, min | 42 (37‐52) | 59 (35‐67) | ns |
| CLIF‐ACLF (Median Value 53) | |||
| Low, n = 32 | High, n = 25 | P | |
| CLT, min | 66 (56‐82) | 79 (55‐180) | ns |
| Child‐Turcotte‐Pugh (Median Value 9) | |||
| Low, n = 55 | High, n = 54 | P | |
| CLT, min | 57 (41‐67) | 59 (42‐93) | ns |
| Liver Failure (Bilirubin > 205 µmol/l) | |||
| No, n = 80 | Yes, n = 29 | P | |
| CLT, min | 57 (41‐72) | 79 (75‐113) | <0.01 |
| Renal Failure (Creatinine > 176 µmol/l) | |||
| No, n = 65 | Yes, n = 44 | P | |
| CLT, min | 50 (36‐67) | 67 (56‐92) | <0.001 |
| Coagulation Failure (INR > 2.5) | |||
| No, n = 90 | Yes, n = 19 | P | |
| CLT, min | 57 (41‐75) | 67 (36‐180) | ns |
| Hemodynamic Failure (Requirement for Vasoactive Support) | |||
| No, n = 88 | Yes, n = 21 | P | |
| CLT, min | 55 (39‐71) | 80 (55‐180) | <0.01 |
| Respiratory Failure (PaO2/FiO2 < 200) | |||
| No, n = 98 | Yes, n = 11 | P | |
| CLT, min | 55 (40‐72) | 180 (89‐180) | <0.001 |
| Neurologic Failure (Encephalopathy Moderate‐Severe) | |||
| No, n = 76 | Yes, n = 33 | P | |
| CLT, min | 56 (40‐72) | 65 (48‐115) | 0.02 |
Median and interquartile ranges are shown with corresponding P values.
Abbreviation: INR, international normalized ratio.
CLT in Relation to Clinical Events and Outcome
In all patients combined, 35 (24%) experienced a bleeding event during hospitalization (8 patients in the AD group and 27 patients in the ACLF group), and more than half (63%) of these events were related to portal hypertension (Table 4). Six thrombotic events were observed during hospitalization: 4 patients had a de novo portal vein thrombosis, 1 patient experienced thrombosis of a previously placed transjugular intrahepatic portosystemic shunt, and 1 patient had deep venous thrombosis.
Table 4.
Bleeding and Thrombotic Events During Hospitalization
| Variable | All | AD | ACLF |
|---|---|---|---|
| n = 109 | n = 52 | n = 57 | |
| Bleeding events, n (% patients) | 35 (24) | 8 (15) | 27 (47) |
| Related to PH, n (% bleeding events) | 22 (63) | ||
| UGIB | 3 | 14 | |
| LGIB | 2 | 3 | |
| Not related to PH, n (% bleeding events) | 13 (37) | ||
| Epistaxis | 2 | 2 | |
| Soft tissue | 1 | ||
| Punctures | 1 | 2 | |
| Hematoma after procedure | 2 | ||
| Hematuria | 3 | ||
| Thrombotic events, n (% patients) | 6 (4) | 2 (4) | 4 (7) |
| Portal vein thrombosis | 2 | 2 | |
| TIPS thrombosis | 1 | ||
| Left upper arm DVT | 1 |
Abbreviations: DVT, deep vein thrombosis; LGIB, lower gastrointestinal bleeding; PH, portal hypertension; TIPS, transjugular intrahepatic portosystemic shunt; UGIB, upper gastrointestinal bleeding.
CLTs were similar in those patients who bled compared with those who did not, 54 minutes (42‐83) versus 61 minutes (41‐78), P = 0.7, and there were no differences in CLT between patients with bleeding related to portal hypertension or bleeding from other causes, 59 minutes (41‐78) versus 56 minutes (44‐95), P = 0.5. CLTs were similar in patients who had thrombotic complications compared with those who did not, 66 minutes (49‐105) versus 57 minutes (41‐78), P = 0.4.
Seventeen patients (15 ACLF and 2 AD) died within 30 days after hospital admission. Patients who died had significantly longer CLTs compared with survivors, 101 minutes (57‐180) versus 57 minutes (40‐72), P < 0.001 (Fig. 4). In logistic regression analyses, CLT remained longer in those who died compared with survivors when adjusted for Child‐Turcotte‐Pugh (P = 0.002) or MELD (P = 0.004) score. However, when CLT was adjusted for SOFA score, the difference between those who died and survivors was no longer significant (P = 0.118). The performance of the CLT in relation to mortality prediction assessed using ROC analyses was inferior to that of commonly used clinical scores. The AUC was 0.772 (95% confidence interval: 0.631‐0.914) for CLT, 0.849 (0.733‐0.965) for the Child‐Turcotte‐Pugh score, 0.886 (0.791‐0.980) for the MELD score, and 0.913 (0.826‐1.00) for the SOFA score.
Figure 4.
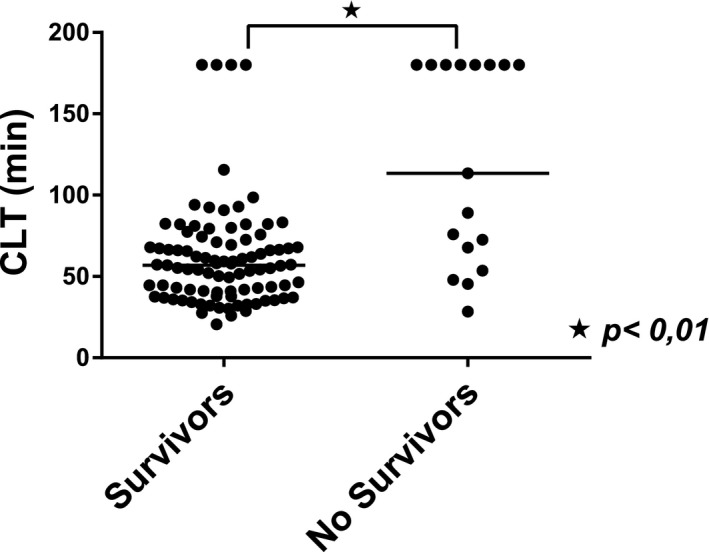
Fibrinolytic status in critically ill patients with cirrhosis stratified by 30‐day survival. CLT in patients with AD and ACLF combined are shown, stratified by 30‐day survivors and those who died within 30 days after admission. Horizontal lines indicate medians.
Discussion
In the present study, we found a mixed fibrinolytic profile of acutely ill patients with cirrhosis, with some patients who were evidently hyperfibrinolytic and some patients with profound hypofibrinolysis. Plasma levels of individual fibrinolytic proteins did not clearly identify those patients with hypofibrinolysis or hyperfibrinolysis, but the presence of sepsis and organ failure was clearly associated with hypofibrinolysis. Finally, we found marked baseline hypofibrinolysis in patients that died within 30 days of admission, which appeared independent of severity of liver disease.
We have studied plasma fibrinolytic potential using an assay that is associated with risk of venous and arterial thrombosis in the general population,27 and it is tempting to speculate that those patients with cirrhosis that are hypofibrinolytic using this test are at risk for thrombotic complications as well. These thrombotic events not only encompass deep vein thrombosis but possibly also portal vein thrombosis and intraorgan microthrombosis. Such microthrombi could contribute to hepatic and extrahepatic organ failure and could explain the relation between hypofibrinolysis and short‐term mortality in patients with AD and ACLF. Indeed, intraorgan microthrombosis is thought to contribute to multiple organ dysfunction syndrome in patients with sepsis without an underlying liver disease, and a hypofibrinolytic state is linked to intraorgan microthrombosis in these patients.28, 29 There is continuing interest in the use of anticoagulant agents, including heparin, antithrombin concentrate, tissue factor pathway inhibitor, activated protein C, and recombinant thrombomodulin in the treatment of sepsis,30, 31 and all these agents are thought to be beneficial at least in part by reduction of intraorgan thrombosis. Although it is conceivable that such agents might benefit patients with ACLF as well, the use of anticoagulant agents is complicated by the extensive hemostatic changes in these patients.2, 13, 14 On the other hand, patients with sepsis without underlying liver disease may also have profound hemostatic changes, and it is incompletely defined what proportion of hemostatic changes in patients with ACLF is related to their underlying liver disease relative to the hemostatic changes induced by excessive inflammation and sepsis.
Our study cohort, and particularly those patients with ACLF, showed large variability in CLTs, which makes the fibrinolytic status of patients with ACLF hardly predictable at an individual level. The heterogeneity underlying AD and ACLF could partially explain this finding. The trigger for decompensation, etiology of the liver disease, extent of liver dysfunction, and involvement of extrahepatic organ failure vary widely between patients and may affect the fibrinolytic status. In addition, the concurrence of sepsis, which frequently complicates the clinical course of patients with ACLF, may also contribute to the variability in fibrinolytic status.32 The inflammation‐induced impairment of fibrinolysis is a well‐established consequence of sepsis and is among other factors related to the excessive release of PAI‐1 by endothelial cells, which is at least partly related to production of TNFα.29, 33, 34 Indeed, in our cohort, PAI‐1 levels and CLTs were significantly higher in patients with sepsis compared with those patients without sepsis. As the inflammation‐induced impairment of fibrinolysis plays a pivotal role in multiple organ dysfunction by promoting microvascular fibrin deposition in sepsis‐associated disseminated intravascular coagulation in patients without underlying liver disease,28 it seems plausible that a similar mechanism acts in patients with ACLF. Indeed, the functional characteristics of organ failure associated with sepsis in patients without underlying liver disease are very close to the sequence of events leading to ACLF in cirrhosis. For example, the severity of systemic inflammation in ACLF is similar to that of patients with sepsis.35 Interestingly, fibrinolytic capacity did not differ between patients stratified according to ascites severity, although ascites is an established source of fibrinolytic activity.36 Indeed, D‐dimer levels, which were reported in our cohort37 and reflect in vivo fibrinolytic activity, progressively increased with increasing severity of ascites (data not shown). Even though the presence of ascites does not appear to alter plasma fibrinolytic potential, it may be that patients with ascites do have altered local fibrinolytic activity, and this possibility requires additional study.
Our present data are in line with another study from our group that showed increased short‐term mortality in those critically ill patients with cirrhosis with a relative hypofibrinolytic status as assessed by thromboelastometry at 72 hours after admission.14 Given the large variability in the fibrinolytic status in AD and ACLF and its potential significance, it would be useful to have a diagnostic global fibrinolytic test as predictor of outcome and possibly to guide prohemostatic and antifibrinolytic therapy in these patients. Although thromboelastometry is widely available for clinical use, it is limited by the low sensitivity compared with the global tests of fibrinolysis performed in a research setting (such as the CLT). Efforts to bring such tests into clinical practice would be desired, for example, by adapting the test for use on automated coagulation analyzers used in the diagnostic laboratory. It has to be noted, however, that AD and ACLF are very dynamic syndromes, and it is not unlikely that fibrinolytic potential changes substantially over time. Theoretically, patients may shift from a very hyperfibrinolytic to a very hypofibrinolytic phenotype, for example, when sepsis develops during the course of their decompensating event. Future studies are required to study the dynamics of fibrinolytic potential over time and its consequences for the use of fibrinolytic capacity in clinical management and prediction.
In summary, our study demonstrates a mixed fibrinolytic phenotype in acutely ill patients with cirrhosis with hypofibrinolysis associated with sepsis, organ failure, and short‐term mortality. Efforts to implement sensitive fibrinolytic assays into clinical practice will assist in personalized management of fibrinolytic dysfunction and will aid outcome prediction of these patients.
Supporting information
Acknowledgment
A.B. acknowledges Dr. Graciela Martinez‐Pallí and colleagues of the digestive disease anesthesia section of Hospital Clínic Barcelona for taking over clinical duties during the period when this research was performed in the University Medical Center Groningen in the Netherlands.
Supported by departmental funds from Ton Lisman.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate co‐first authorship.
- 1. Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood 2010;116:878‐885. [DOI] [PubMed] [Google Scholar]
- 2. Fisher C, Patel VC, Stoy SH, Singanayagam A, Adelmeijer J, Wendon J, et al. Balanced haemostasis with both hypo‐ and hyper‐coagulable features in critically ill patients with acute‐on‐chronic‐liver failure. J Crit Care 2017;43:54‐60. [DOI] [PubMed] [Google Scholar]
- 3. Lisman T, Violi F. Cirrhosis as a risk factor for venous thrombosis. Thromb Haemost 2017;117:3‐5. [DOI] [PubMed] [Google Scholar]
- 4. Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost 2017;1:150‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006;44:53‐61. [DOI] [PubMed] [Google Scholar]
- 6. Bos S, van den Boom B, Kamphuisen PW, Adelmeijer J, Blokzijl H, Schreuder T, et al. Haemostatic profiles are similar across all aetiologies of cirrhosis. Thromb Haemost 2019;119:246‐253. [DOI] [PubMed] [Google Scholar]
- 7. Tripodi A, Salerno F, Chantarangkul V, Clerici M, Cazzaniga M, Primignani M, et al. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005;41(3):553‐558. [DOI] [PubMed] [Google Scholar]
- 8. Leebeek FW, Rijken DC. The fibrinolytic status in liver diseases. Semin Thromb Hemost 2015;41:474‐480. [DOI] [PubMed] [Google Scholar]
- 9. Leebeek FW, Kluft C, Knot EA, de Maat MP, Wilson JH. A shift in balance between profibrinolytic and antifibrinolytic factors causes enhanced fibrinolysis in cirrhosis. Gastroenterology 1991;101:1382‐1390. [DOI] [PubMed] [Google Scholar]
- 10. Rijken DC, Kock EL, Guimarães AH, Talens S, Darwish Murad S, Janssen HL, et al. Evidence for an enhanced fibrinolytic capacity in cirrhosis as measured with two different global fibrinolysis tests. J Thromb Haemost 2012;10:2116‐2122. [DOI] [PubMed] [Google Scholar]
- 11. Goodpasture EW. Fibrinolysis in chronic hepatic insufficiency. Bull Johns Hopkins Hosp 1914;25:330‐336. [Google Scholar]
- 12. Lisman T, Leebeek FW, Mosnier LO, Bouma BN, Meijers JC, Janssen HL, et al. Thrombin‐activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology 2001;121:131‐139. [DOI] [PubMed] [Google Scholar]
- 13. Premkumar M, Saxena P, Rangegowda D, Baweja S, Mirza R, Jain P, et al. Coagulation failure is associated with bleeding events and clinical outcome during systemic inflammatory response and sepsis in acute‐on‐chronic liver failure: an observational cohort study. Liver Int 2019;39:694‐704. [DOI] [PubMed] [Google Scholar]
- 14. Blasi A, Calvo A, Prado V, Reverter E, Reverter JC, Hernández‐Tejero M, et al. Coagulation failure in patients with acute‐on‐chronic liver failure and decompensated cirrhosis: beyond the international normalized ratio. Hepatology 2018;68:2325‐2337. [DOI] [PubMed] [Google Scholar]
- 15. Lisman T, Bernal W. Management of hemostatic disorders in patients with advanced liver disease admitted to an intensive care unit. Transfus Med Rev 2017;31:245‐251. [DOI] [PubMed] [Google Scholar]
- 16. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al.; CANONIC Study Investigators of the EASL‐CLIF Consortium . Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013;144:1426‐1437. [DOI] [PubMed] [Google Scholar]
- 17. Mahmud N, Kaplan DE, Taddei TH, Goldberg DS. Incidence and mortality of acute‐on‐chronic liver failure using two definitions in patients with compensated cirrhosis. Hepatology 2019;69:2150‐2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hernaez R, Kramer JR, Liu Y, Tansel A, Natarajan Y, Hussain KB, et al. Prevalence and short‐term mortality of acute‐on‐chronic liver failure: a national cohort study from the USA. J Hepatol 2019;70:639‐647. [DOI] [PubMed] [Google Scholar]
- 19. Hernaez R, Solà E, Moreau R, Ginès P. Acute‐on‐chronic liver failure: an update. Gut 2017;66:541‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng YL, Chang CH, Chen WT, Tsai MH, Lee WC, Tu KH, et al. Prognostic factors and treatment effect of standard‐volume plasma exchange for acute and acute‐on‐chronic liver failure: a single‐center retrospective study. Transfus Apher Sci 2018;57:537‐543. [DOI] [PubMed] [Google Scholar]
- 21. Semeraro F, Colucci M, Caironi P, Masson S, Ammollo CT, Teli R, et al. Platelet drop and fibrinolytic shutdown in patients with sepsis. Crit Care Med 2018;46:e221‐e228. [DOI] [PubMed] [Google Scholar]
- 22. Lisman T, Bakhtiari K, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT. Intact thrombin generation and decreased fibrinolytic capacity in patients with acute liver injury or acute liver failure. J Thromb Haemost 2012;10:1312‐1319. [DOI] [PubMed] [Google Scholar]
- 23. Levi M, van der Poll T. Coagulation and sepsis. Thromb Res 2017;149:38‐44. [DOI] [PubMed] [Google Scholar]
- 24. Lisman T, Stravitz RT. Rebalanced hemostasis in patients with acute liver failure. Semin Thromb Hemost 2015;41:468‐473. [DOI] [PubMed] [Google Scholar]
- 25. Meltzer ME, Lisman T, Doggen CJ, de Groot PG, Rosendaal FR. Synergistic effects of hypofibrinolysis and genetic and acquired risk factors on the risk of a first venous thrombosis. PLoS Med 2008;5:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meltzer ME, Lisman T, de Groot PG, Meijers JC, le Cessie S, Doggen CJ, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI‐1. Blood 2010;116:113‐121. [DOI] [PubMed] [Google Scholar]
- 27. Lisman T. Decreased plasma fibrinolytic potential as a risk for venous and arterial thrombosis. Semin Thromb Hemost 2017;43:178‐184. [DOI] [PubMed] [Google Scholar]
- 28. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis, thrombosis and organ dysfunction. Thromb Res 2012;129:290‐295. [DOI] [PubMed] [Google Scholar]
- 29. Gando S. Role of fibrinolysis in sepsis. Semin Thromb Hemost 2013;39:392‐399. [DOI] [PubMed] [Google Scholar]
- 30. Iba T, Levy JH, Raj A, Warkentin TE. Advance in the management of sepsis‐induced coagulopathy and disseminated intravascular coagulation. J Clin Med 2019;8:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Umemura Y, Yamakawa K, Ogura H, Yuhara H, Fujimi S. Efficacy and safety of anticoagulant therapy in three specific populations with sepsis: a meta‐analysis of randomized controlled trials. J Thromb Haemost 2016;14:518‐530. [DOI] [PubMed] [Google Scholar]
- 32. Foreman MG, Mannino DM, Moss M. Cirrhosis as a risk factor for sepsis and death: analysis of the national hospital discharge survey. Chest 2003;124:1016‐1020. [DOI] [PubMed] [Google Scholar]
- 33. van Deventer SJ, Buller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood 1990;76:2520‐2526. [PubMed] [Google Scholar]
- 34. van der Poll T, Coyle SM, Levi M, Jansen PM, Dentener M, Barbosa K, et al. Effect of a recombinant dimeric tumor necrosis factor receptor on inflammatory responses to intravenous endotoxin in normal humans. Blood 1997;89:3727‐3734. [PubMed] [Google Scholar]
- 35. Clària J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al. Systemic inflammation in decompensated cirrhosis: characterization and role in acute‐on‐chronic liver failure. Hepatology 2016;64:1249‐1264. [DOI] [PubMed] [Google Scholar]
- 36. Agarwal S, Joyner KA Jr, Swaim MW. Ascites fluid as a possible origin for hyperfibrinolysis in advanced liver disease. Am J Gastroenterol 2000;95:3218‐3224. [DOI] [PubMed] [Google Scholar]
- 37. Blasi A, Patel VC, Adelmeijer J, Azarian S, Aziz F, Fernández J, et al. Plasma levels of circulating DNA are associated with outcome, but not with activation of coagulation in decompensated cirrhosis and ACLF. JHEP Reports 2019. 10.1016/j.jhepr.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials