

Abstract
Short half‐life proteins regulate many essential processes, including cell cycle, transcription, and apoptosis. However, few well‐characterized protein‐turnover pathways have been identified because traditional methods to measure protein half‐life are time and labor intensive. To overcome this barrier, we developed a protein stability probe and high‐content screening pipeline for novel regulators of short half‐life proteins using automated image analysis. Our pilot probe consists of the short half‐life protein c‐MYC (MYC) fused to Venus fluorescent protein (MYC‐Venus). This probe enables protein half‐life to be scored as a function of fluorescence intensity and distribution. Rapid turnover prevents maximal fluorescence of the probe due to the relatively longer maturation time of the fluorescent protein. Cells expressing the MYC‐Venus probe were analyzed using a pipeline in which automated confocal microscopy and image analyses were used to score MYC‐Venus stability by two strategies: assaying the percentage of cells with Venus fluorescence above background, and phenotypic comparative analysis. To evaluate this high‐content screening pipeline and our probe, a kinase inhibitor library was screened by confocal microscopy to identify known and novel kinases that regulate MYC stability. Compounds identified were shown to increase the half‐life of both MYC‐Venus and endogenous MYC, validating the probe and pipeline. Fusion of another short half‐life protein, myeloid cell leukemia 1 (MCL1), with Venus also demonstrated an increase in percent Venus‐positive cells after treatment with inhibitors known to stabilize MCL1. Together, the results validate the use of our automated microscopy and image analysis pipeline of stability probe‐expressing cells to rapidly and quantitatively identify regulators of short half‐life proteins. © 2019 The Authors. Cytometry Part A published by Wiley Periodicals, Inc. on behalf of International Society for Advancement of Cytometry.
Keywords: protein stability, high‐content screening, automated fluorescence microscopy, unsupervised data analysis
The median protein half‐life in cells is estimated to be between 31 and 46 h 1, 2, yet a unique subset of proteins possess distinctly shorter half‐lives, on the order of 30 min to 1 h. These fast‐turnover proteins are key regulators that control several essential cellular processes including cell cycle, transcription, and apoptosis 3, 4, 5, 6. Moreover, these normally highly regulated proteins often contribute to disease, due to mutations or alterations that confer increased stability and functionality 7, 8, 9, 10. A few known modes of regulation of these critical short half‐life proteins have taken decades to characterize, as traditional techniques to measure protein half‐life, such as pulse‐chase or protein synthesis inhibition, are both labor and time intensive 11. Progress in this field has been hampered by a lack of more efficient tools, suggesting that many regulatory mechanisms that control rapid protein turnover remain largely unknown.
To advance our understanding of protein turnover mechanisms in mammalian cells, a highly sensitive, specific, rapid, and informative assay is required, as traditional techniques are not amenable to automation or high‐throughput screening. Existing rapid fluorescence‐based assays to measure specific and/or global protein stability typically rely on the paired expression of two fluorescent proteins, one of which is tagged to the protein of interest 12, 13. By measuring changes in the ratio of fluorescence between the two fluorophores, it is possible to measure changes in protein abundance using fluorescence‐activated cell sorting (FACS). However, as a ratiometric approach, this type of assay relies heavily on equal translation of both proteins to avoid false positives and/or false negatives. Furthermore, ratiometric measures compound noise particularly when the denominator is small. Therefore, when expression levels vary between cells as is the case for cells in culture, the detection sensitivity is severely compromised. Finally, FACS‐based analysis is relatively low throughput, a large number of cells are required for processing, and valuable data on subcellular fluorescence distribution are lost.
To circumvent the drawbacks of existing protein turnover assays and to take advantage of the wealth of data afforded by high‐content imaging, we have developed a novel probe and high‐content screening pipeline to identify novel regulators of short half‐life proteins. The Venus fluorescent protein is bright, monomeric, and has a folding time of 40 min in vitro. Moreover, Venus fluorescence in a tissue section of mouse cerebellum was first observed at 4 h postexpression 14, 15. These data suggest that the Venus maturation time is similar to or longer than the half‐life of most short half‐life (~30 min) proteins in mammalian cells. Here we have exploited this difference, and the stochastic nature of protein folding, to build a protein stability probe. By constructing an in‐frame hybrid fusion protein consisting of a short half‐life protein fused to the amino terminus of Venus, the fluorescence features of the fusion protein function as a readout of protein half‐life. When the short half‐life protein is stabilized, more fusion proteins mature per cell, resulting in acquisition of increased cellular fluorescence.
Here, our probe and pipeline have been piloted with two short half‐life proteins that are essential to normal development and, when deregulated, directly contribute to cancer initiation and progression; c‐MYC (MYC) and myeloid cell leukemia 1 (MCL1). The transcription factor MYC regulates a diverse range of biological activities 3 and is deregulated in over 50% of cancers 7, 16, 17. Despite the important role of MYC in cancer, only one mechanism of MYC turnover has been well characterized; phosphorylation at threonine 58 (T58) by glycogen synthase kinase 3 (GSK3) 18 recruits the E3‐ubiquitin ligase complex comprised of SKP1, CUL1, and the F‐box protein FBXW7 (SCFFBXW7), which targets MYC for ubiquitin‐mediated proteasomal degradation (Fig. 1A). MYC half‐life is only 30 min in nontransformed cells, and loss of the GSK3‐SCFFBXW7 axis of MYC degradation results in only a twofold increase (60 min) 19, 20, suggesting additional pathways regulate MYC degradation. Like MYC, MCL1 also has a short half‐life (~40 min 21), is regulated by SCFFBXW7 22, and increased stability of MCL1 can directly promote tumorigenesis 8. A more complete picture of the pathways regulating these and other short half‐life proteins associated with disease may contribute to the discovery of mechanisms to control their activity and impact disease progression.
Figure 1.
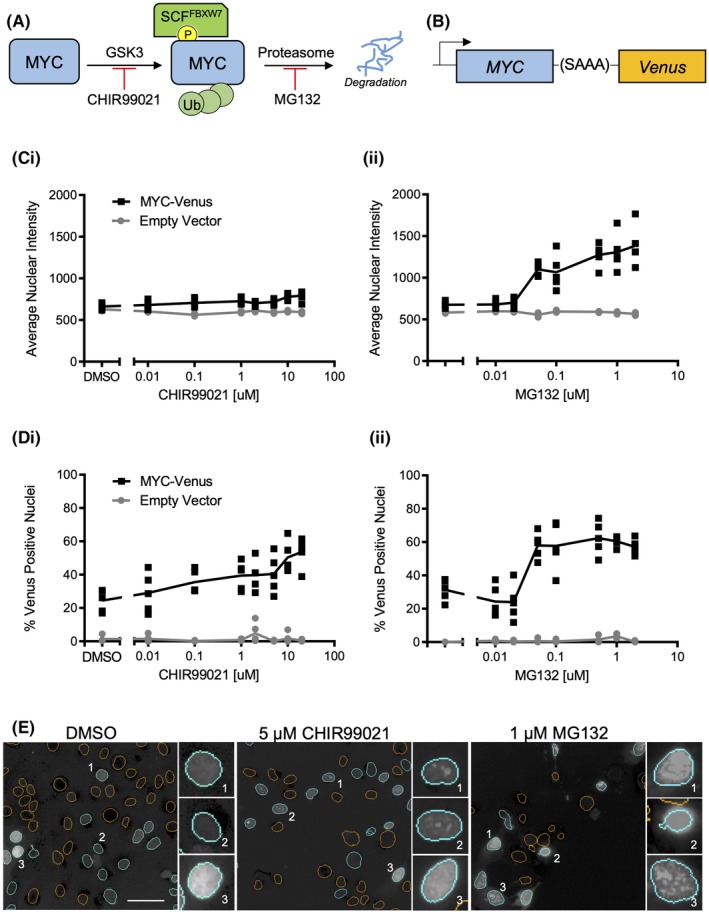
MYC‐Venus enables measurement of changes in MYC stability in live cells. (A) c‐MYC is regulated by GSK3 phosphorylation, which results in recognition by SCFFBXW7, leading to ubiquitylation and degradation. (B) Venus fluorescent protein is fused to the c‐terminus of MYC via a short, uncharged linker. (C) The mean and SD of average nuclear intensity for control and MYC‐Venus expressing MCF10A cells treated with the indicated concentrations of (i) GSK3 inhibitor CHIR99021 or (ii) proteasome inhibitor MG132 for 16 h; n = 3, representative well from one of three independent replicates is shown; each symbol represents one of five fields of view within a well (not all data points are visible due to overlap; total number of cells analyzed is reported in Table S3). (D) Percent Venus‐positive nuclei for control and MYC‐Venus expressing MCF10A cells treated with either the GSK3 inhibitor CHIR99021 (i) or the proteasome inhibitor MG132 (ii) at the indicated concentrations for 16 h; n = 3, a representative well from one of three independent replicates is shown; each symbol represents one of five fields of view within a well (not all data points are visible due to overlap; total number of cells analyzed is reported in Table S3). (E) Representative images of Venus fluorescence in cells treated with DMSO, 5 μM CHIR99021 or 1 μM MG132. Orange nuclear segmentation outlines indicate a nucleus scored as Venus negative; cyan outlines indicate a nucleus scored as Venus positive. Insets represent various patterns of fluorescence observed in these populations. Scale bar = 50 μm.
To examine stabilization of MYC, a protein stability probe was generated by fusing Venus in frame to the carboxyl terminus of MYC so that degradation of the two polypeptides would be coupled and retain the rapid turnover properties of MYC. Because protein folding is stochastic, coupling the two proteins most frequently results in degradation of MYC‐Venus prior to the final folding of Venus required for fluorescence. Stabilization of MYC‐Venus results in increased fluorescence as more fusion proteins mature and become fluorescent. This MYC‐Venus probe was used to screen a 320‐compound kinase inhibitor library. Unexpectedly, compounds that increase MYC stability could not be identified in a screen by measuring an increase in average nuclear fluorescence intensity across the population of cells due to variations in the fluorescence intensities between cells. Instead, the response to compounds was measured using a high‐content screening pipeline that assayed individual cells for both their fluorescence intensity and phenotype within a population. While enumerating Venus‐positive cells and phenotype is not necessarily directly linked to protein half‐life, we demonstrate that in concert with an appropriate data analysis pipeline these data can be used to identify known and novel regulators of the turnover of short half‐life proteins. Compounds that increase MYC stability were identified by an increase in the percentage of cells with Venus expression above a predetermined threshold (Venus positive) or by phenotypic similarity to cells treated with compounds that resulted in an increased percentage of Venus‐positive nuclei. As the canonical MYC stability pathway is regulated by kinases (Fig. 1A), identification of known kinase inhibitors in the library validated the probe and pipeline. Feasibility was further demonstrated using MCL1, as the MCL1‐Venus protein stability probe showed an increase in the percentage of Venus‐positive cells in response to inhibitors known to increase MCL1 stability. Thus, we show that production of an in‐frame fusion protein with a short half‐life protein at the amino terminus of the Venus fluorescence protein can serve as a protein stability probe that has utility in our high‐content screening pipeline to identify regulators of protein turnover.
Results
Detecting Regulators of a Short Half‐Life Protein by Fusion to the Venus Fluorescent Protein
To identify regulators of the short half‐life protein MYC, the Venus fluorescent protein was fused to the carboxyl terminus of MYC via a short, uncharged S‐A‐A‐A linker (Fig. 1B). This fusion protein was stably expressed in the nontransformed, immortal MCF10A epithelial breast cell line. The adherent MCF10A cell line is amenable to high‐content screening, provides a genomically stable background 23, and MYC degradation in this cell line is rapid. Inhibition of the canonical pathway of MYC degradation extends half‐life from ~30 to ~60 min 19, suggesting that MYC turnover is regulated in these cells by both canonical and novel pathways. The half‐life of the MYC‐Venus fusion protein (~30 min) is not statistically different from that of endogenous MYC, suggesting that degradation of the fusion protein is controlled by mechanisms regulating MYC turnover (Fig. S1A). Moreover, MYC‐Venus is localized to nuclei (Fig. S1B) and retains the ability to regulate expression of MYC target genes 49, 50 (Fig. S1C).
CHIR99021, an inhibitor of both GSK3α and β 24 previously shown to stabilize MYC in cells 25, was used as the primary positive control (Fig. 1A). The proteasome inhibitor MG132 serves as a second positive control, as inhibiting the proteasome prevents the degradation of many proteins, including MYC (Fig. 1A) 26. Following the treatment of MYC‐Venus expressing MCF10A cells with either CHIR99021 or MG132, cells were stained with Draq5 nuclear dye and imaged on the PerkinElmer Opera™ High‐Content confocal screening instrument. The images were analyzed using Columbus software, and nuclear segmentation was based on the Draq5 signal. Venus intensity was then measured within each of the identified nuclei.
Unexpectedly, while there was a measurable difference in Venus fluorescence intensity between dimethyl sulfoxide (DMSO) and MG132‐treated MYC‐Venus expressing cells, the dynamic range of the assay was negligible between DMSO and CHIR99021‐treated cells (Fig. 1Ci,ii).
To minimize the effect of cell‐to‐cell variation, individual cells within the population were instead scored only as either Venus positive or Venus negative. A cell was scored as positive when the nuclear Venus intensity was greater than or equal to 3 standard deviation (SDs) above the mean intensity in the Venus channel of nonfluorescent control cells. Using this method, both positive controls, GSK3 inhibitor CHIR99021 and MG312, resulted in concentration‐dependent increases in the percent positive nuclei compared to DMSO, suggesting that this approach provides a dynamic range suitable for screening (Fig. 1Di,ii). Similar trends for dynamic range between average nuclear intensity and percent Venus‐positive cells were observed for three independent replicates (Fig. S1D) demonstrating that the latter approach improves dynamic range. As expected, after 16 h of exposure to the negative control DMSO, approximately 33% of the MCF10A cells expressing MYC‐Venus had measurable nuclear fluorescence above background, and addition of the primary positive control CHIR99021 increased the percent Venus‐positive nuclei to approximately 50% at the highest dose of 20 μM (Fig. 1Di). Additionally, treatment with increasing doses of MG132 also increased the percent Venus‐positive nuclei (Fig. 1Dii).
Different phenotypes of fluorescence were also observed in cells treated with DMSO, CHIR99021 or MG132. For example, cells were observed with primarily low‐intensity diffuse fluorescence (DMSO Insets 1 and 2), while the positive controls exhibited punctate (CHIR99021 Insets 1 and 2; MG132 Inset 3), or high‐intensity diffuse fluorescence (DMSO Inset 3, CHIR99021 Inset 3, and MG132 Insets 1 and 2) (Fig. 1E). These observations suggest that cellular fluorescence phenotype can also be used to score compounds for MYC‐Venus stabilization in a high‐content screen.
Screening a Library of Small‐Molecule Inhibitors
To perform a high‐content screen to identify kinase inhibitors regulating MYC stability, MCF10A control cells and cells expressing MYC‐Venus were exposed to the control compounds used above, and 320 kinase inhibitors. Concentrations of 1 and 0.1 μM were selected based on the clinically achievable concentration (typically 1 μM 27), which is also the typical IC50 of kinase inhibitors. After 16 h incubation, MCF10A control cells and cells expressing MYC‐Venus were imaged by automated confocal microscopy. To eliminate false positives due to fluorescent compounds, kinase inhibitors for which the percentage of fluorescent cells in the control plates (cells not expressing MYC‐Venus) was at least 3 SDs above the DMSO controls were not advanced (eight compounds total) (see flowchart in Fig. S2 and red box in Fig. S3A). Based on the results of previous experiments (Fig. 1), the positive controls used for screening included the GSK3 inhibitor CHIR99021 at a concentration range of 1, 5, and 10 μM, and the proteasome inhibitor MG132 at 0.01, 0.1, and 1 μM. The strictly standardized mean difference (SSMD) β values for the 10 μM concentration of CHIR99021 (Fig. S3B) demonstrate for each replicate that the assay has sufficient dynamic range 28. There was also a high degree of concordance between the three independent replicates of the screen (Fig. S3C,D), enabling a pipeline to be established to identify hits (Fig. S2). The first criterion was based on an increase in the percent Venus‐positive nuclei. To establish an appropriate threshold, the mean (Fig. S3E) and SD of the percentage of Venus‐positive nuclei for the 312 nonfluorescent kinase inhibitors were used to calculate Z‐scores for individual kinase inhibitors on a per‐replicate basis. The ranked average Z‐scores across all three replicates were then calculated separately for the 0.1 and 1 μM screens (Fig. 2A, 1 μM screen shown).
Figure 2.
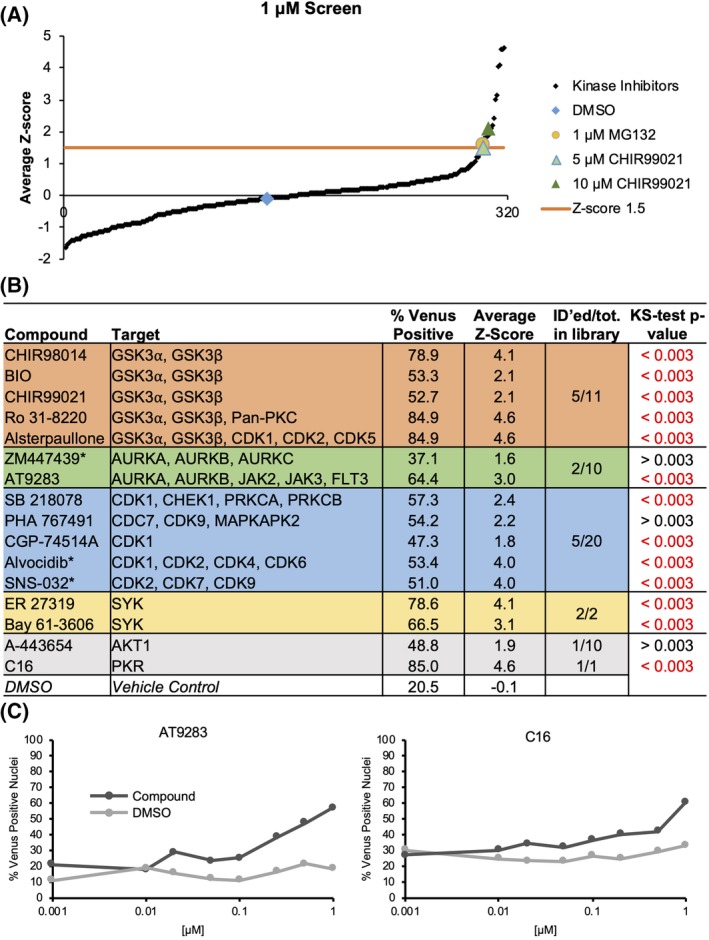
Screening of a kinase inhibitor library. (A) The ranked average Z‐score for each compound at 1 μM, calculated from percent Venus‐positive MCF10A cells expressing MYC‐Venus. Z‐score threshold of 1.5 SDs above the library mean is indicated by the orange line. Concentrations of control compounds are indicated in light green (CHIR99021, 5 μM), dark green (CHIR99021, 10 μM) or yellow (MG132, 1 μM) (n = 3, 16 h). (B) Inhibitors with Z‐score >1.5 in either 0.1 (indicated with an asterisk) or 1 μM concentration screens are grouped by target as reported in supplier catalogues (Selleck (Houston, TX, USA), Sigma (St. Louis, MO, USA), Tocris (Bristol, UK), AdooQ (Irvine, CA, USA), Enzo (Farmingdale, NY, USA), Cayman (Ann Arbor, MI, USA)). With GSK3 inhibitors in orange (4/11 in library), AURK inhibitors in green (2/10 in library), CDK inhibitors in blue (6/21 in library), and SYK inhibitors in yellow (2/2 in library). Inhibitors whose targets are unique in the library are shown in gray. Average percent Venus‐positive scores as well as Z‐scores are listed for each compound (n = 3). Inhibitors with Z‐scores above 1.5 were tested across eight concentrations of library‐sourced compound for 16 h to measure changes in percent Venus‐positive nuclei (n = 1). The KS statistical test (last column) was used to evaluate significant changes between DMSO and drug treatment. Based on the Bonferroni multiple‐testing correction, a false discovery rate was established to distinguish a significant P‐value cutoff of P = 0.003. P‐values in black are not significant. (C) The change in percentage of Venus‐positive nuclei in MCF10A cells expressing MYC‐Venus is shown across an eight‐point concentration curve using compounds within the library. Results for all compounds are shown in Figure S4. Shown are curves for AT9283 and C16, which are both significantly different from DMSO across the total of eight points (n = 1; total number of cells analyzed is reported in Table S4).
Based on the Z‐scores of the positive controls CHIR99021 (1.5 at 5 μM) and MG132 (1.6 at 1 μM), a percent Venus‐positive Z‐score >1.5 in either the 0.1‐ or the 1 μM screen was established as a threshold to identify compounds for further analysis (Fig. 2A). Using this threshold, 16 kinase inhibitors were selected for further examination (Figs. 2B and S2).
Validation and Prioritization of Compounds that Increased the Percentage of Venus‐Positive Nuclei
The reported targets of 14 of the 16 kinase inhibitors fall into four classes: GSK3, Aurora kinase (AURK), cyclin‐dependent kinase (CDK), and spleen tyrosine kinase (SYK) inhibitors (Fig. 2B). Two additional compounds that did not fall into these classes include A‐443654 and C16, which reportedly target AKT1 and protein kinase R (PKR), respectively. GSK3 inhibitors were an expected class, as they have been previously described to stabilize MYC 25, 29. Interestingly, the role of AURK inhibitors in destabilizing MYCN has been described 30, 31; however, the data on their regulation of c‐MYC are conflicting 30, 32. Five GSK3 inhibitors present in the library had Z‐scores ≥2.1 at 1 μM including the library sample of CHIR99021, which had a Z‐score >1.5 in all three replicates of the screen. In addition, five CDK inhibitors, two AURK inhibitors, and two SYK inhibitors in the library had Z‐scores >1.5 in either the 0.1 μM screen or the 1 μM screen. Some inhibitors within these classes in the library did not score as hits, likely due to the particular dose and time chosen for the assay. Two of the identified compounds, Alvocidib and SNS‐032, had Z‐scores >1.5 in the 0.1 μM screen with an average of 49.0% and 28.5% of cells remaining after treatment with Alvocidib and SNS‐032 as compared to DMSO, respectively, but at 1 μM both exhibited significant toxicity and Z‐scores below 1. This demonstrates the benefit of screening inhibitor libraries at two concentrations to accommodate potential variations in response between different inhibitors, thereby maximizing the number of evaluable compounds.
The 16 compounds with Z‐scores >1.5 were initially validated by assaying changes in the percent Venus‐positive nuclei over an eight‐point concentration curve at 16 h, using compounds from the library plates (Figs. 2C, S2, and S4). Changes in the percent Venus‐positive nuclei compared to DMSO were assessed using the Kolmogorov–Smirnov (KS) test to evaluate whether the concentration–response data represent different distributions (Figs. 2B,C and S4). While the KS test is sensitive to small deviations near the center of the distribution, this is a preliminary analysis and false positives are eliminated later in the pipeline. Most compounds showed a significant increase in the percentage of Venus‐positive nuclei over increasing doses of inhibitor as compared to DMSO and passed the KS test (Figs. 2B,C and S4). Three compounds (A‐443654, PHA 767491, and ZM447439) did not pass the KS test (Fig. 2B), and therefore were not advanced (Fig. S2). Furthermore, as GSK3 inhibitors are already known to be regulators of MYC stability, some compounds (CHIR98014, BIO) were not further characterized, whereas others were retained as additional positive controls (Ro 31‐8220, Alsterpaullone; Fig. S2). Similarly, the Aurora A inhibitor AT9283 was also retained as a positive control. Using this approach, a total of 10 compounds were advanced for additional validation analysis.
Phenotype‐Based Compound Identification
The GSK3 inhibitor CHIR99021 in the library resulted in an average Z‐score of 2.1 at 1 μM; however, CHIR99021, sourced from a different supplier and used as a positive control, had a Z‐score of 0.3 at this same concentration and therefore would have been missed by the original criteria of Z‐score >1.5. To capture other potential false negatives and increase the sensitivity of the assay further, we evaluated the rich phenotypic fluorescence data from MYC‐Venus expressing cells in response to each compound as captured by confocal microscopy. Based on the hypothesis that compounds that stabilize MYC by similar mechanisms result in similar spatial distributions of Venus fluorescence within the nucleus, we clustered the image data using unsupervised machine learning. For this analysis, we selected 83 compounds that had average Z‐scores in the 1 μM screen at least as high as the 1 μM CHIR99021 positive control (Z‐score >0.3) (Fig. S2). To mitigate the effect of the large number of potential false positives that could be generated by reducing the threshold to Z‐score 0.3, our pipeline employed a landmark‐based approach. For this analysis, the compounds that resulted in a Z‐score >1.5 served as landmarks for cluster selection. Clusters containing landmarks also contain compounds that generated MYC‐Venus phenotypes similar to those that scored >1.5 but that resulted in Z‐scores >0.3 and <1.5 (Fig. S2).
As shown in Figure 3A, treatment with different inhibitors yielded a range of phenotypes, both between and within treatments. For example, a treatment that results in a “Punctate” phenotype (Fig. 3A, left, Insets 1 and 2) also has some nuclei with dim diffuse fluorescence (Fig. 3A, left, Inset 3). A different treatment leads to cellular fluorescence described as “Bright Diffuse” nuclei (Fig. 3A, middle) comprised largely of nuclei that exhibit high Venus intensity (Fig. 3A, middle, Insets 1 and 2), with some nuclei being less intense (e.g. Fig. 3A, middle, Inset 3). Similarly, a treatment leading to an image described as “Dim Diffuse” (Fig. 3A, right) consists primarily of nuclei with low‐intensity, diffuse fluorescence across the entire nucleus (Fig. 3A, right, Insets 1 and 2) with some nuclei in the image being very bright (Fig. 3A, right, Inset 3).
Figure 3.
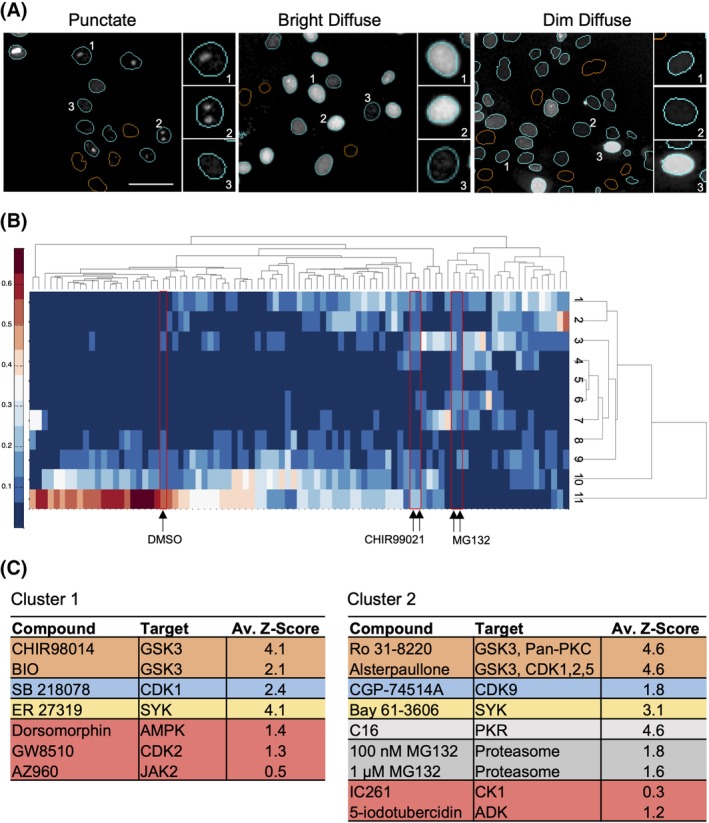
Identification and rescue of false‐negative inhibitors through phenotype analysis. (A) Representative images of different phenotypes of Venus fluorescence. Multiple phenotypes are observed for treated and untreated cells; therefore, the image is named by the most frequent phenotype observed. Left: Sample “Punctate” image of cells treated with 10 μM CHIR99021, Insets 1 and 2 indicate examples of punctate Venus fluorescence, Inset 3 indicates a dim diffuse phenotype of fluorescence. Middle: Sample “Bright Diffuse” image of cells treated with 1 μM C16, Insets 1 and 2 represent a bright but diffuse phenotype of fluorescence, Inset 3 represents a discrete pattern of fluorescence. Right: Sample “Dim Diffuse” image of cells treated with 1 μM ER 27319, Insets 1 and 2 represent very dim but diffuse patterns of fluorescence while Inset 3 represents a bright diffuse pattern of fluorescence. Scale bar = 50 μm. (B) A hierarchically clustered heat map depicts the proportion of cells (see color scale to left) for each of the 83 inhibitors (columns) that fell into each of 11 phenotype clusters (rows). Red boxes indicate the location of images of cells treated with DMSO, and the two highest concentrations for CHIR99021 and MG132. (C) Results from unsupervised data analysis of all three replicates were concatenated and clustered again to yield seven treatment clusters (see “Materials and Methods” section). The two clusters shown yielded the highest proportion of controls and compounds with Z‐score >1.5 in the intensity‐based analysis (colors as in Fig. 2 except MG132, dark gray). The two clusters included five additional compounds with Z‐scores >0.3 and <1.5 (red).
To capture this interimage and intraimage heterogeneity, 52 nuclear Venus channel intensity and texture features were calculated for the image of each Venus‐positive cell for the 83 kinase inhibitors with Z‐scores >0.3. In the first replicate, 11 unique cell phenotype clusters were identified. For each compound, the proportion of cells in each phenotype cluster was calculated and represented as a heat‐map organized by hierarchical clustering to group those inhibitors that resulted in similar distributions of cell phenotypes (Fig. 3B). As expected, the positive controls CHIR99021 and MG132 cluster near to each other, while DMSO has a very different distribution of cell phenotypes. The process was repeated separately for the other two replicates resulting in a total of 33 phenotypic clusters. To generate a phenotypic profile for each treatment, the proportions of cells in each phenotypic cluster across the three replicates were concatenated. These data were then used to cluster the data for the treatments (83 kinase inhibitors, and positive and negative control compounds) by Affinity Propagation 33. This approach clustered together those compounds that had the most similar distributions of cell phenotypes across all three replicates (Table S1). Clusters 1 and 2 of the seven treatment clusters were enriched for compounds with a Z‐score >1.5 and included five additional compounds with a Z‐score >0.3 and <1.5 (Fig. 3C, indicated in red). These clusters represent treatments that result in similar Venus‐positive cell phenotypes, regardless of the percent Venus‐positive nuclei. The five compounds identified by phenotypic analysis in Clusters 1 and 2 (Dorsomorphin, GW8510, AZ960, IC261, and 5‐iodotubercidin) were advanced for further characterization (Fig. S2). Both the positive control CHIR99021 and the library sample of CHIR99021 were assigned to Cluster 6, along with PHA767491 (CDK) and AT9283 (AURKA) (Table S1), suggesting that this cluster could also provide additional compounds to mine. Concatenating the data from the individual replicates provided an exhaustive set of potential hits; however, automated screens of large compound libraries do not typically have replicates. Therefore, the cell phenotypes for each individual replicate were clustered independently (Table S2). Each of these analyses captured four or five of the five compounds identified from the concatenated data set, suggesting that unsupervised clustering will have utility in large automated screens.
Validation with Independent Compound Stocks
To characterize compounds identified as putative inhibitors of MYC turnover, independent compound stocks were acquired for the 10 selected compounds with Z‐score >1.5, and the five compounds identified by phenotype clustering. Control cells or cells expressing MYC‐Venus were treated with eight concentrations of the compounds, and the concentration that resulted in the largest increase in percent Venus‐positive nuclei over DMSO, while minimizing toxicity and compound fluorescence, was then selected (indicated by the arrows; Figs. 4A,B and S5A,B). Where ambiguous, two concentrations were selected to cover the maximal percentage Venus‐positive signal under minimal antiproliferative responses (Figs. 4B and S5A,B). Cell number per well was used to identify either growing cells (>200 cells, blue), or toxicity (<200 cells, red). As expected the percent Venus‐positive nuclei generally increased with increasing concentrations of compound to a maximum between the concentrations of 1 and 10 μM (e.g., Fig. 4A, Ro 31‐8220, CGP 74514A). At elevated compound concentrations cellular toxicity was often evident, which in turn usually corresponded to a decrease in both total and Venus‐positive nuclei (e.g., Fig. 4A; ER 27319, GW8510). At the indicated doses (arrows), all of the increases in percent Venus‐positive nuclei were significant compared to the number of nuclei that scored positive at an equivalent concentration due to compound fluorescence. To preferentially follow‐through on potential novel regulators of MYC stability, we did not further characterize the GSK3 inhibitors (Alsterpaullone and Ro 31‐8220) or the identified AURK inhibitor (AT9283), as they have previously been shown to regulate stability of the MYC family of proteins 29, 31. SB 218078 was also excluded as there was no significant difference between MYC‐Venus fluorescence and compound fluorescence at the indicated dose (Fig. S5). Thus, 12 compounds were advanced in the pipeline for further analysis (Fig. S2).
Figure 4.
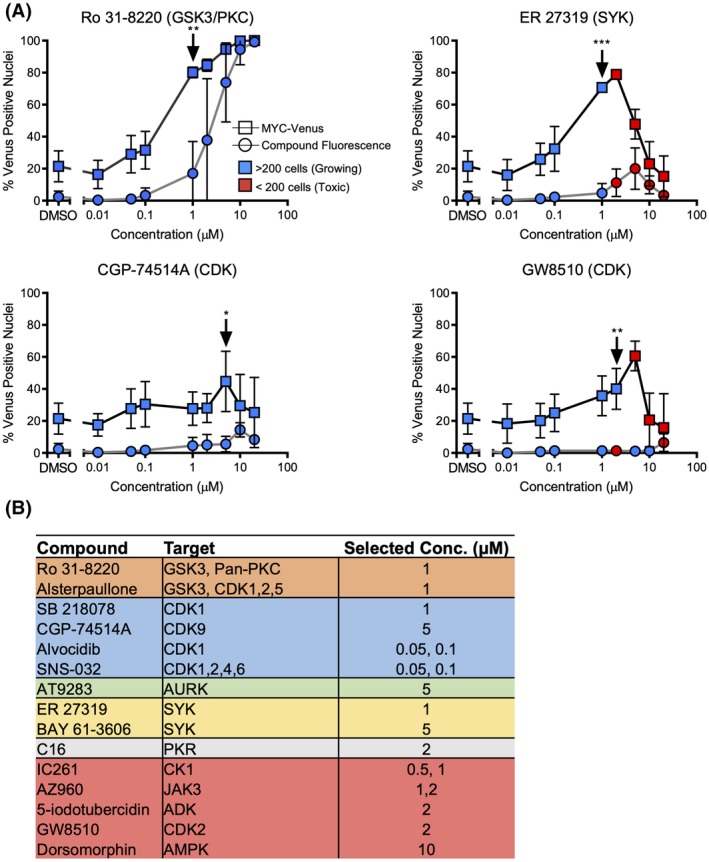
Validation of inhibitors that modulate MYC‐Venus levels. (A) The mean and SD in the percentage of Venus‐positive nuclei following 16 h of treatment with the eight indicated concentrations of independently sourced inhibitors for MCF10A cells expressing MYC‐Venus or empty vector (used here to infer compound fluorescence; n = 3). The color of each point (see inset) indicates the average number of cells per well at that concentration. The concentration–response curves for the indicated kinase inhibitors demonstrate variations in responses to the inhibitors. Arrows indicate concentrations selected for further analysis for each compound. *P < 0.05, **P < 0.01, ***P < 0.001. Student t‐test of % Venus‐positive nuclei for MYC‐Venus cells versus empty vector (compound fluorescence) cells, n = 3. (B) Table of compounds, targets and concentrations selected for each compound based on concentration response curves. Where the optimal concentration was unclear, two concentrations were selected to include both toxic and nontoxic responses.
Changes in MYC mRNA and Protein Stability
Changes in the percent Venus‐positive nuclei could result from changes in MYC‐Venus protein stability, increased expression due to induction of gene transcription, and/or stability of the MYC mRNA transcript. To determine whether the identified compounds significantly increased the mRNA abundance for either endogenous MYC or MYC‐Venus, RNA was harvested from compound‐treated MCF10A control and cells expressing MYC‐Venus, and mRNA levels for these and control genes were measured by quantitative RT‐PCR (qRT‐PCR) (Fig. S6A,B). While variation in RNA levels for both endogenous MYC and ectopic MYC‐Venus was observed, none were statistically significant.
Three of the major inhibitor classes discovered in this screen, SYK, CDK, and PKR, have not been previously reported to regulate MYC stability. Therefore, one representative compound from each class (ER 27319, GW8510, and C16, respectively), all the compounds identified by image‐based clustering (IC261, AZ960, 5‐iodotubercidin, GW8510, and Dorsomorphin), and the positive control GSK3 inhibitor (CHIR99021) were advanced for the evaluation of protein half‐life for both endogenous MYC and MYC‐Venus (Fig. S2). After treatment of cells with the protein synthesis inhibitor cycloheximide, cells were harvested at different time points and protein stability was assayed by immunoblotting (Figs. 5A–D and S7). Two of the seven compounds, C16, and GW8510 targeting PKR and CDK2, respectively, significantly increased the stability of both endogenous MYC and MYC‐Venus proteins, as did the positive control CHIR99021. C16 was originally identified by an increase in Venus‐positive nuclei, while GW8510 was identified by phenotypic clustering, demonstrating that our probe and pipeline enabled the identification of novel modulators of MYC stability. One compound ER 27319 significantly extended the half‐life of MYC‐Venus but not endogenous MYC (Fig. 5). A more detailed assessment of the published specificities of C16 and GW8510 kinase inhibitory activities in vitro (Fig. S7C) revealed that C16 effectively inhibits multiple kinases including GSK3, CDKs 1–3, and 5, and AURK, in addition to PKR 51. By contrast, GW8510 inhibits CDKs 2, 3, and 5 52. Multiple target specificity of these compounds is not surprising given the well‐known difficulty in identification of kinase inhibitors with exquisite specificity for a single kinase. As both compounds share activity against CDKs 2, 3, and 5 future experiments should address the possibility that one or more of these represent the most relevant target(s).
Figure 5.
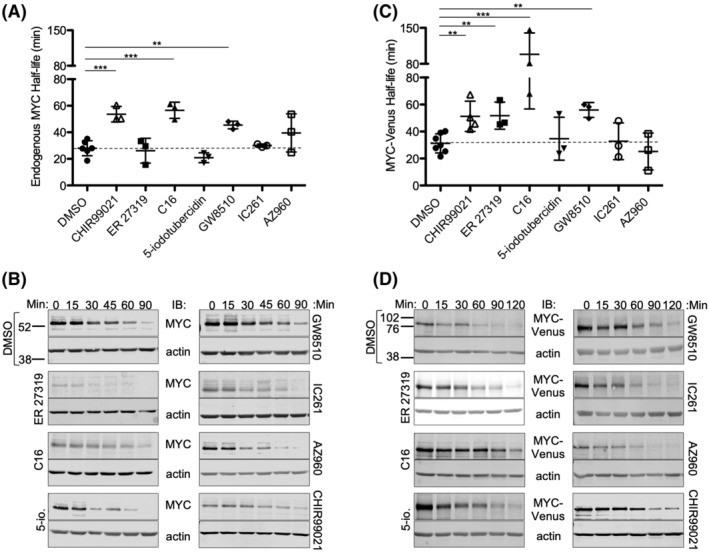
Validation of inhibitors that modulate the turnover of endogenous MYC and exogenous MYC‐Venus. (A,C) The mean and SD in the half‐lives of (A) endogenous MYC protein in MCF10A control cells, or (C) MYC‐Venus protein in MCF10A cells expressing exogenous MYC‐Venus after 16 h treatment with DMSO and CHIR99021 as negative and positive controls, or the indicated kinase inhibitors at concentrations identified in Figure 4, are plotted. ***P = <0.001, **P < 0.01. Student t‐test with Bonferroni multiple testing correction, n = 3–7. (B,D) Representative immunoblots for (B) control cells expressing endogenous MYC or (D) MCF10A cells expressing MYC‐Venus were treated with indicated drug for 16 h and then 10 μg/ml cycloheximide was added and cells were harvested and processed at the indicated time points. Migration positions of molecular weight markers (kDa) shown at the left for blots of extracts from cells treated with DMSO apply to all other blots shown.
MCL1‐Venus can be Used to Measure Changes in MCL1 Stability
To further test the utility of a Venus‐fusion protein as a probe to rapidly identify regulators of the turnover of the fusion partner, we expressed a MCL1‐Venus fusion protein in MCF10A cells. When cells expressing MCL1‐Venus were imaged, Venus fluorescence was detected primarily in the cytoplasm but also in the nucleus of cells treated with DMSO (Fig. 6A), consistent with reports of MCL1 localization to both compartments 34, 35. As MCL1 is also turned over by the GSK3 pathway, exposure of the cells to a GSK3 inhibitor or MG132 was expected to stabilize the MCL1‐Venus fusion protein, thereby increasing the cell fluorescence and therefore also the number of cells exhibiting more fluorescence than cells treated with DMSO. A panel of GSK3 inhibitors was tested with MCL1‐Venus, and CHIR98014 was found to give the greatest dynamic range as compared to DMSO (data not shown). Background for fluorescence for MCL1‐Venus was higher than for MYC‐Venus, likely due to a difference in half‐life, nevertheless, the principle of the assay remains the same. The difference in fluorescence between DMSO‐treated cells and those treated with either positive control suggests that in this case screening is feasible using either average cell intensities or number of cells with MCL1‐Venus intensity above an arbitrary threshold that resulted in 20% of the DMSO‐treated cells being scored Venus‐positive (Fig. 6Bi,ii). Nevertheless as seen above, the dynamic range of the assay was greater using number of positive cells compared to analyses based on the measurement of MCL1‐Venus intensities (Fig. 6).
Figure 6.
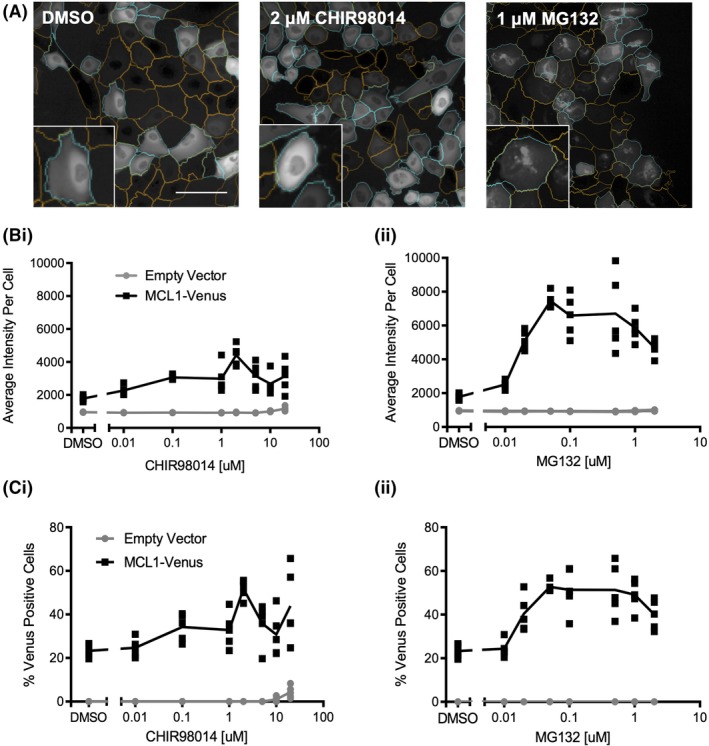
MCL1‐Venus enables measurement of changes in MCL1 stability in live cells. (A) Representative images of Venus fluorescence in cells treated with DMSO, 2 μM CHIR98014 or 1 μM MG132. Orange nuclear segmentation outlines indicate a cell scored as Venus negative; cyan outlines indicate a cell scored as Venus positive. Insets represent various patterns of fluorescence observed in these populations. Scale bar = 50 μm. (B) The mean and SD of average nuclear intensity for control and MCL1‐Venus expressing MCF10A cells treated with the indicated concentrations of (i) GSK3 inhibitor CHIR98014 or (ii) proteasome inhibitor MG132 for 16 h; n = 3, a representative well from one of three independent replicates is shown; each symbol represents one of five fields of view within a well (not all data points are visible due to overlap; total number of cells analyzed is reported in Table S5). (C) Percent Venus‐positive nuclei for control and MCL1‐Venus expressing MCF10A cells treated with either the GSK3 inhibitor CHIR98014 (i) or the proteasome inhibitor MG132 (ii) at the indicated concentrations for 16 h; n = 3, a representative well from one of three independent replicates is shown; each symbol represents one of five fields of view within a well (not all data points are visible due to overlap; total number of cells analyzed is reported in Table S5).
Together these data demonstrate that measuring the percentage of MCL1‐Venus‐positive cells provides a large enough dynamic range that imaging this fusion protein could be used to detect pathways that modulate MCL1 stability. Sample images of cells treated with CHIR98014 or MG132 (Fig. 6A) demonstrate that in addition to there being an increase in the number of cells with Venus fluorescence above that in cells treated with DMSO, there are also obvious differences in the staining pattern within cells treated with MG132 as compared to exposure to CHIR98014‐ and DMSO‐treated MCL1‐Venus‐expressing cells. This result suggests that phenotypic screening as performed here for MYC‐Venus may also be useful for identifying modulators of MCL1 stability.
Discussion
There is an unmet need for a robust, rapid, assay that can be used to measure the relative turnover rate of short half‐life proteins quantitatively. Many of these proteins regulate critical cell processes and are differentially regulated in disease states. The protein stability probe characterized here consists of a short half‐life protein (MYC and MCL1) fused in‐frame with a fluorescent protein (Venus). The probe, in combination with the high‐content image‐based analysis pipeline validated here, enables rapid and effective measurement of protein stability based on the fluorescent properties of cells expressing the probe.
When developing this probe and validation pipeline, we first assayed changes in the fluorescence intensity of the fusion protein but found that a population‐based intensity measure was not able to distinguish between treated and untreated cells due to variations in intensity across the populations. Instead, thresholding on individual cells successfully limited the impact of cell‐to‐cell variations in fluorescence intensity. We presume that these variations are at least in part a result of the stochastic nature of protein folding in cells. Importantly, we show that an exogenously expressed protein stability probe retains the protein turnover regulatory mechanisms of the endogenous short half‐life protein (Fig. S1A). As a result, the two kinase inhibitors identified, as well as the positive control CHIR99021, extend the half‐life of MYC‐Venus and endogenous MYC equivalently (Fig. 5). Nevertheless, one compound was identified (ER 27319) that stabilized MYC‐Venus but did not stabilize endogenous MYC for reasons that remain to be investigated. Another major advantage of our pipeline is the ability to identify kinase inhibitors that conferred stability based on the phenotypic distribution of Venus within cells following exposure to a given stimulus. Thus, combining a protein stability probe with the automated image analysis pipeline described here should enable rapid identification of compounds that modulate the stability of a given short half‐life protein.
The relationship between MYC stability and Venus fluorescence depends on the different conditions found in each cell of a population. Venus fluorescent protein folding is stochastic and influenced by pH and ion concentration 36, conditions that may differ across each cell in a population. When measuring the half‐life of MYC using an inhibitor of protein synthesis followed by immunoblotting, the readout of MYC degradation time is averaged across a whole population, obscuring the cell‐to‐cell variation inherently present in a population. In contrast, image‐based screening utilizing MYC‐Venus fluorescence permits the detection of variations in protein stability at the single‐cell level rather than averaging across the population. This may be particularly useful when the response of the population is not Gaussian and instead driven by a specific subpopulation of cells.
General Utility of the Probe and Pipeline
The single fluorescent protein reporter and pipeline described here can be employed with any confocal microscope and software‐controlled automated stage, in combination with commercial or open access high‐content screening software (e.g. CellProfiler), making this approach readily accessible to most researchers. The basis of the assay to measure changes in stability arises primarily from differences between protein turnover and Venus maturation time. As a consequence, it should be possible to adapt the assay to proteins with different half‐lives by selecting a fluorescence protein with the appropriate maturation time to maximize the difference between stabilized and destabilized proteins. As shown here, the maturation time of Venus is most appropriate for measuring increased stability of proteins with a half‐life of less than 1 h. In contrast, to use Venus to measure protein destabilization for the same proteins, we predict that a protein with a cellular half‐life of ~2 h would be optimal. This assay could potentially be extended to in vivo work with the use of a near‐infrared fluorescent protein such as mCardinal 37, which has a maturation half‐time of 27 min 38.
One potential drawback to the technique is that it is essential that the fluorescence protein is turned over with its fusion partner for single‐cell fluorescence intensity to correlate with protein half‐life. For example, we observed that when Venus‐MCL1 was used instead of MCL1‐Venus, the MCL1 protein was degraded and intact Venus accumulated in the cytoplasm (data not shown). For this reason, it may be necessary to test fluorescent proteins and fusion junctions for the protein of interest to determine a combination that yields sufficient dynamic range to conduct a screen. In addition, it is desirable that fusion of the fluorescence protein does not alter turnover or the function of the target protein. Here, we demonstrated that MYC‐Venus turns over with similar kinetics to endogenous MYC and that the fusion protein activates transcription of known MYC target genes (Fig. S1). We have also demonstrated that the MCL1‐Venus fusion protein inhibits apoptosis similarly to MCL1 (data not shown). Finally, in our experiments we identified one compound that stabilized MYC‐Venus but not endogenous MYC (Fig. 5), demonstrating the importance of evaluating both ectopic and endogenous protein half‐life to eliminate false‐positive compounds.
Previous efforts to develop a screen to assay protein stability focused on dual‐fluorescent reporter systems, where, for example, the expression of Discosoma sp. red fluorescent protein (dsRed), and enhanced green fluorescent protein (EGFP) tagged to the protein of interest, are expressed from the same construct via an internal ribosome entry site (IRES). Reporters of this type have been used for screening by FACS, where relative stability is inferred for individual cells from the ratio of dsRed to EGFP‐fusion protein fluorescence 13. However, IRES efficiency may not be uniform across assay conditions as IRES sequences tend to be less efficiently used in unstressed cells 39, while during stress, translation of IRES sequences occurs in an eIF2a‐independent manner 40. As the dual fluorescent protein assays rely entirely on changes in the ratio of the fluorescence intensities of the two proteins, differences in translation can result in false positives or negatives, thereby confounding assay results. In addition, the large size of the IRES sequence (often longer than 500 base pairs) can be problematic when working with viral vectors of limited capacity. Moreover, FACS‐based analysis is a measure of total intensity of each fluorescent protein per cell which as shown by our data is highly variable (Fig. 1) confounding the results for short half‐life proteins. As a result, it would not have been possible to identify any of the kinase inhibitors identified here, including the positive controls, using population intensity values such as those obtained by FACS. Moreover, FACS requires a comparatively larger number of cells and the information regarding cell phenotypes that we show here can be used to identify bona fide modulators of protein half‐life (Fig. 5) is lost compared to high‐content image‐based analyses 41, 42, 43.
Here, we have provided proof‐of‐concept evidence that by designing and implementing a high‐content screening pipeline, a Venus fluorescence protein‐based protein stability probe can be used to assay changes in the turnover time for short half‐life proteins in relatively high throughput. Using this research tool, we have identified six known and two novel regulators of the MYC short half‐life protein from a collection of 320 kinase inhibitors. Moreover, we have shown that this probe is not restricted to MYC but is also applicable to MCL1, another short half‐life protein. These results open the door to large‐scale screening for regulators of these, and other, short half‐life proteins.
Materials and Methods
Cell Culture
MCF10A cells were a kind gift from Dr. Senthil Muthuswamy and have been short tandem repeat (STR) profiled 44 and demonstrated free of mycoplasma using the MycoAlert™ Mycoplasma Detection Kit (Lonza, LT07‐318; Basel, Switzerland). Cells were cultured as described previously 45. MYC‐Venus and MCL1‐Venus cDNAs were cloned into the pQCXIP vector 46 and control cells were generated with an empty pQCXIP vector. Expression from the pQCXIP vector is driven by a cytomegalovirus (CMV) promoter, ensuring heterologous expression of the fusion proteins. The Phoenix‐AMPHO retroviral packaging cell line (a kind gift from Dr. Gary Nolan) was transfected with the pQCXIP constructs, and used to generate retroviral particles by calcium phosphate precipitation, and viral supernatant was harvested after 48 h and used to infect MCF10A cells.
Immunoblotting
Cells were seeded in six‐well plates at 100,000 cells per well, treated with the indicated drug 24 h after seeding, and 16 h following treatment the cells were lysed. Where indicated, cells were treated with the protein synthesis inhibitor cycloheximide (10 µg/ml) and then harvested at the indicated timepoints. Lysates were prepared from subconfluent cells by aspirating media, washing with phosphate‐buffered saline (PBS), and adding 100 μl hot sodium dodecyl sulfate (SDS) lysis buffer (1% SDS, 0.1 M Tris pH 6.8) to the plate to lyse cells, followed by manual scraping. The cell lysates were collected and boiled for 5 min. Loading dye was added and protein was quantified using the Pierce 660‐nm Protein Assay (Thermo Fisher Scientific, 22662; Waltham, MA, USA) in combination with the Ionic Detergent Compatibility Reagent (Thermo Fisher Scientific, 22663; Waltham, MA, USA). Between 20 and 30 μg of protein was loaded per lane of a 10% SDS‐polyacrylamide gel electrophoresis (PAGE) gel. The following antibodies were used for detection: MYC (mAb 9E10, commercially available from Thermo Fisher Scientific [13‐2500] (Waltham, MA, USA), purified in‐house from mouse ascites), Actin (Sigma, A2066; St. Louis, MO, USA), Lamin A and C (Abcam, ab8984; Cambridge, UK), and Tubulin (Calbiochem, CP06; San Diego, CA, USA). Secondary antibodies (LI‐COR, IRDye 800CW and 680RD; Lincoln, NE, USA) were used in conjunction with the LI‐COR Odyssey for imaging.
Protein Half‐Life Quantification
Images of immunoblots were imported into ImageJ (Bethesda, MD, USA), and the band densities corresponding to MYC and actin were quantified for each time point. The MYC band density for each lane was first normalized to the respective actin band density, and then the MYC/actin value for each time point was normalized to the MYC/actin value calculated for time zero. Normalized values were analyzed in GraphPad Prism (San Diego, CA, USA) using a one‐phase decay regression analysis to calculate the protein half‐life in response to each treatment.
Cytoplasmic Nuclear Fractionation
Cells were seeded in 10‐cm dishes at 2,000,000 cells per dish, and harvested 48 h later. Media was aspirated, and the plate was washed twice with cold PBS, and cells manually removed by scraping in PBS. The cell suspension was centrifuged at 425g for 2 min at 4°C. The supernatant was aspirated and the pellet was resuspended in 500 μl of cell lysis buffer (10 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) [pH 7.9], 10 mM KCl, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.1 mM ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid (EGTA), 1 mM dithiothreitol (DTT), 0.5% NP‐40, protease inhibitor cocktail [Sigma, P8340;St. Louis, MO, USA]). The resuspended pellet was disaggregated by frequent vortexing during a 15‐min incubation on ice and then centrifuged at 950g for 5 min at 4°C. The supernatants were isolated as the cytoplasmic fractions. The pellets were resuspended in 200 μl nuclear resuspension buffer (20 mM HEPES [pH 7.9], 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 μl TurboNuclease [BioVision, #9207; Milpitas, CA, USA], protease inhibitor cocktail [Sigma, P8340; St. Louis, MO, USA]), and incubated on ice for 15 min with frequent vortexing. The nuclear suspensions were centrifuged at 20,800g for 10 min at 4°C to remove chromatin, and the supernatants were isolated as the nuclear protein fractions. Loading dye was added to samples before boiling for 5 min and running on 10% SDS‐PAGE gel.
Confocal Imaging
Cells were seeded at 2,000 cells per well in 384‐well PerkinElmer (Waltham, MA, USA) Cell Carrier Ultra plates. Twenty‐four hours after seeding, the kinase inhibitors dissolved in DMSO were added to the cells at 0.1 and 1 μM final concentration. The plates were then incubated for 16 h at 37°C before staining with 5 μM final concentration of the nucleic acid dye Draq5 (Biostatus; Leicestershire, UK). The properties of Draq5 are such that it stains the nuclei strongly, and the cytoplasm weakly, facilitating detection of both compartments. Plates were imaged using automated spinning disc confocal high‐content screening microscopes (PerkinElmer, Waltham, MA, USA) with a 20× air long working distance lens on an Opera QEHS (MYC‐Venus), or a 20× water long working distance lens on an Opera Phenix (MCL1‐Venus). Five fields of view were captured per well. Laser intensities at 488 and 640 nm were adjusted such that a single‐exposure setting was used to simultaneously record the fluorescence of Venus and Draq5 using two cameras with 520/35 and 690/50 emission filters, respectively.
Z‐score Calculation
To prioritize compounds for further evaluation from across the three replicates at both concentrations, a Z‐score was calculated for each query based on the mean and SD of the percent Venus‐positive nuclei for all inhibitors in the library (Fig. S3E). The Z‐scores for each query were averaged across the three replicates for both the 0.1 and the 1 μM screens.
Image Analysis for Percent Venus‐Positive Nuclei
The Columbus software package (PerkinElmer; Waltham, MA, USA) was used to determine the percent Venus‐positive nuclei as follows. The only image preprocessing applied was basic flat‐field correction algorithm in Columbus. Identification of individual cell regions employed a Columbus segmentation algorithm based on the Draq5 channel signal. In this way, each cell was assigned nuclear and cytoplasmic areas from which the Venus intensity was measured. The intensity in the nuclear region was used to identify cells expressing MYC‐Venus, while the signal analyzed for MCL1‐Venus came from the cytoplasmic area. Venus‐positive nuclei were identified as being 3 or more SDs above the mean intensity in the Venus channel of nonfluorescent control cells treated with DMSO alone for MYC‐Venus. For MCL1‐Venus, the threshold was selected such that ~20% of the DMSO‐treated MCL1‐Venus cells were scored as Venus positive. This value was equivalent to 0.05 SDs above the mean cytoplasmic intensity in the Venus channel of MCL1‐Venus expressing cells treated with DMSO.
Unsupervised Data Analysis
The nuclear areas for each cell were identified using the Draq5 signal and a segmentation routine in the Opera image analysis software, Acapella (PerkinElmer; Waltham, MA, USA). Next, a custom Acapella script (available at http://www.andrewslab.ca) was used to calculate 52 Venus channel intensity and texture features for the nuclear region of the image of each cell scored as Venus positive. Intensity features included 10 features describing nuclear intensity, SD of intensity, and quantiles of intensity. Texture features included 42 features describing nuclear texture moments and radials, as well as threshold‐adjacency statistics as previously described 47, 48. For each screen replicate, the feature data for 200 randomly selected micrographs of individual cells for each kinase inhibitor treatment were clustered using the affinity propagation 33 algorithm implemented in MatLab (Natick, MA, USA). To determine the optimal number of cell phenotype clusters for each individual replicate, the affinity propagation routine was run iteratively over increasing preference values until the number of clusters increased exponentially. The selected preference value was the last point before the exponential increase.
To provide landmarks within the unsupervised image clusters, the feature data from micrographs of cells exposed to kinase inhibitors that resulted in a Z‐score >1.5 in the screen, DMSO (negative control); 1, 5, and 10 μM CHIR99021; and 0.01, 0.1, and 1 μM MG132 (positive controls) were also included in the analysis. The proportion of cells assigned to each of the clusters was then used as a multiparametric descriptor of the response of the cells to that drug. The multiparametric descriptors for each drug from the three biological replicates were analyzed separately and concatenated by hierarchical agglomeration clustering. Concatenation enabled clustering across the replicates while preserving the phenotypic relationships between the responses to the kinase inhibitors within each replicate.
Concentration Response Curves
Concentration response curves were generated for compounds with an average Z‐score >1.5 in the 1 μM screen or were identified by image‐based clustering. For each, eight concentrations between 0.01 and 20 μM were selected. For the two compounds, Alvocidib and SNS‐032, with observed toxicity in the 1 μM screen, the range of concentrations tested was from 0.001 to 2 μM. For all of the kinase inhibitors, the concentration(s) selected for use in subsequent experiments (indicated by arrows in Fig. S5) was manually selected from the concentration response curves in Figure S5. Our strategy was to attempt to maximize the percentage of Venus‐positive nuclei while minimizing observed toxicity. To characterize toxicity, cell number relative to initial seeding density and average doubling time was used to identify growing cells or pronounced cellular toxicity in response to the compound at each concentration (blue and red, respectively, in Fig. S5). In some instances, the peak of the Venus‐positive cell curve correlated with increased cellular toxicity and so a slightly lower concentration that still yielded an average of ~40% Venus‐positive nuclei (in comparison with an average of 15% Venus‐positive cells in the DMSO treatment) was selected. In instances where peaks in the percent Venus‐positive nuclei occurred only at concentrations where the cell count was in the toxic range, two concentrations were selected to account for toxic and nontoxic effects alongside changes in MYC‐Venus. For IC261, an additional concentration at 0.5 μM was selected alongside the identified concentration of 1 μM, as the eight‐point concentration curve did not yield a clear peak in the percent Venus‐positive nuclei.
Quantitative RT‐PCR
Cells were seeded at 100,000 cells per well in six‐well plates, and after 24 h incubation compounds were added and incubation continued for another 16 h. Total RNA was harvested from cells using TRIzol reagent according to the manufacturer's instructions (Invitrogen, 15596026; Carlsbad CA, USA). To quantify levels of specific mRNAs, 500 ng of total cellular RNA was used to synthesize cDNA with a SuperScript III Kit (Invitrogen, 18080093; Carlsbad CA, USA). Then specific cDNAs were quantified during qRT‐PCR using SYBR Green (Applied Biosystems, 4309155; Foster City, CA, USA) for detection and the following primers:
MYC: (F) AGGGTCAAGTTGGACAGTGTC, (R) TGGTGCATTTTCGGTTGTGG.
MYC‐Venus: (F) AGCTACGGAACTCTTGTGCT, (R) ATCAGCTTCAGGGTCAGCTT.
B2M: (F) GGCTATCCAGCGTACTCCAAA, (R) CGGCAGGCATACTCATCTTTTT.
NCL: (F) CTTCTGGACTCATCTGCGCC, (R) CCTTTGGAGGAGGAGCCATTT.
LDHA: (F) ACGTCAGCATAGCTGTTCCA, (R) AATGAGATCCGGAATCGGCG.
HK2: (F) GATGGGACAGAACACGGAGAG, (R) GTCCTCAGGGATGGCATAGA.
ABBREVIATIONS
- AURK
Aurora kinase
- CDK
cyclin‐dependent kinase
- CMV
cytomegalovirus
- DMSO
dimethyl sulfoxide
- DS
Sodium Dodecyl Sulfate
- DTT
Dithiothreitol
- EDTA
Ethylenediaminetetraacetic acid
- EGFP
enhanced Green Fluorescent Proteinds
- EGTA
ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid
- FACS
fluorescence‐activated cell sorting
- GSK3
glycogen synthase kinase 3
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
- IRES
Internal ribosome entry site
- KS
Kolmogorov–Smirnovq
- MCL1
myeloid cell leukemia 1
- MYC
c‐MYC
- PAGE
Polyacrylamide Gel Electrophoresis
- PBS
Phosphate buffered saline
- PKR
protein kinase R
- RED
Discosoma sp. red fluorescent protein
- RT‐PCR
quantitative real‐time polymerase chain reaction
- SCFFBXW7
SKP1, CUL1, and the F‐box protein FBXW7
- SD
Standard deviation
- SSMD
strictly standardized mean difference
- STR
short tandem repeat
- SYK
spleen tyrosine kinase
Conflict of Interest
The authors declare no conflicts of interest.
Author Contributions
KAH, LZP, and DWA designed the research and wrote the manuscript. KAH and JDM performed experiments and data analysis. SH performed data analysis. JY and LCL assisted in experimental planning. All authors contributed to manuscript editing and experimental discussion. LZP and DWA supervised the research.
Supporting information
Figure S1. (A) The mean and standard deviation in the half‐lives of endogenous MYC in MCF10A control cells (Empty Vector; 30.6 minutes) and MYC‐Venus (30.7 minutes) exogenously expressed in MCF10A cells are plotted. Student t‐test with Bonferroni multiple testing correction, n = 3‐5. (B) (i) Representative image of Venus fluorescence in growing, untreated MCF10A cells expressing MYC‐Venus. Orange nuclear segmentation outlines indicate a nucleus scored as Venus negative; cyan outlines indicate a nucleus scored as Venus positive. Venus fluorescence is observed in a subset of nuclei, indicative of MYC‐Venus localization to the nucleus. Scale bar = 50 μm. (ii) Immunoblot of MYC‐Venus in nuclear (Lamin A & C) and cytoplasmic (Tubulin) fractions of MCF10A cells expressing MYC‐Venus. (C) (i) The mean and standard deviation of mRNA levels normalized to housekeeping gene B2M (Beta 2 Microglobulin) for three MYC‐activated target genes49, 50 (NCL, Nucleolin; LDHA, Lactate Dehydrogenase A; HK2, Hexokinase 2) were measured in MCF10A control cells following serum starvation for 48 h to deplete the endogenous MYC protein, followed by treatment with ethanol (−) or with 100 nM 4‐hydroxytamoxifen (+) to induce MYC‐Venus expression for 24 hours prior to harvesting for RNA. Data is shown relative to vehicle‐treated control cells. One‐way ANOVA with Bonferroni post‐test, n = 3, *** p < 0.001, ** p < 0.01. (ii) Representative immunoblot of total protein harvested from cells treated as described in (Ci) was probed with anti‐MYC antibody to enable visualization of MYC‐Venus expression (expected molecular weight: 83 kDa). (D) The mean and standard deviation of average nuclear intensity for control and MYC‐Venus expressing MCF10A cells treated with the indicated concentrations of (i) GSK3 inhibitor CHIR99021 or (ii) proteasome inhibitor MG132 for 16 h. n = 3 (not all data points are visible due to overlap) (Total number of cells analyzed is reported in Table S6). (D) Percent Venus‐positive nuclei for control and MYC‐Venus expressing MCF10A cells treated with either the GSK3 inhibitor CHIR99021 (i) or the proteasome inhibitor MG132 (ii) at the indicated concentrations for 16 h. n = 3 (not all data points are visible due to overlap) (Total number of cells analyzed is reported in Table S6).
Figure S2. The screen conditions and steps of compound identification and validation are depicted in a flow chart. Compounds were advanced or not advanced as indicated at each step. Compounds previously known to regulate MYC stability and therefore excluded from further analysis, or not analyzed, are also indicated.
Figure S3. (A) The ranked percent Venus‐positive nuclei for each compound at 1 μM, from MCF10A control cells. Fluorescent inhibitors (red box) were identified as three standard deviations above the mean percent Venus‐positive nuclei in the library. (B) Summary of Strictly Standardized Mean Difference scores (SSMD β) calculated between DMSO and 10 μM CHIR99021 for each replicate. (C) Summary of R2 values between replicates 1 and 2, 1 and 3, 2 and 3 (D) The distribution of the percentage of Venus‐positive nuclei for each kinase inhibitor in replicates 1 and 3 is plotted, with all DMSO and 10 μM CHIR99021 wells shown in blue diamonds and green triangles, respectively. Reproducibility, quantified as R2, between replicates 1 and 3 was calculated from the linear fit of % Venus‐positive nuclei from each kinase inhibitor in each replicate. (E) The ranked percent Venus‐positive nuclei for all compounds in the kinase inhibitor library is plotted for MYC‐Venus expressing MCF10A cells (replicate 2 of 1 μM screen shown). The highest concentrations of CHIR99021 (10 μM) and MG132 (1 μM) are indicated by the green triangle and yellow circle, respectively. The Z‐scores for each compound were calculated from the mean and standard deviation of the library (blue line), and a Z‐score = 1.5 is shown as an orange line.
Figure S4. The percentage of Venus‐positive nuclei following 16 hours of treatment with the 8 indicated concentrations of library‐sourced inhibitors (dark grey) or DMSO (light grey) for MCF10A cells expressing MYC‐Venus, for 14 inhibitors with a Z‐score >1.5 (n = 1) (Total number of cells analyzed is reported in Table S4).
Figure S5. The mean and standard deviation in (A) the percentage of Venus‐positive nuclei or (B) number of nuclei, following 16 hours of treatment with the 8 indicated concentrations of independently sourced inhibitors for MCF10A cells expressing either MYC‐Venus or Empty Vector (used here to infer Compound Fluorescence) (n = 3). The color of each point (see inset) indicates the number of cells per well at that concentration. Arrows indicate concentrations selected for further analysis for each compound. Student t‐test of % Venus positive nuclei for MYC‐Venus cells versus Empty Vector (Compound Fluorescence) cells, n = 3.
Figure S6. The mean and standard deviation of (A) endogenous MYC mRNA from MCF10A control cells or (B) MYC‐Venus mRNA as compared to housekeeping gene B2M were measured following 16 h treatment with the indicated inhibitors at previously identified concentrations as shown in Figure 4B. (C) No significant changes (p < 0.003) in RNA following Wilcoxon Signed Rank test with Bonferroni multiple testing correction, n = 3–6.
Figure S7. (A) The mean and standard deviation in the half‐life of MYC‐Venus in MCF10A cells after 16 h treatment with DMSO and CHIR99021 as negative and positive controls, or Dorsomorphin at the previously identified concentration of 10 μM, is plotted. **p < 0.01. Student t‐test with Bonferroni multiple testing correction, n = 3. (B) Representative immunoblots for cells expressing MYC‐Venus treated with the indicated drug for 16 hours and then 10 μg/ml cycloheximide was added and cells were harvested and processed at the indicated timepoints. (C) The reported percentage of kinase activity remaining following treatment with the indicated concentration of either C1651 or GW851052 as compared to the vehicle control.
Table S1. Cluster assignment is shown for the 83 compounds with an average Z‐score >0.3, and for positive and negative control compounds (grey). Cell phenotypes were identified for each replicate based on Venus channel intensity and texture features. The proportion of cells in each phenotype cluster was calculated for each compound separately for each of 3 replicates and then concatenated. The concatenated cell phenotypes for each compound were then clustered by Affinity Propagation. This approach clustered together those compounds with the most similar distributions of cell phenotypes across all three replicates. Clusters 1 and 2 are enriched for compounds with an average Z‐score >1.5 (blue), and include five additional compounds with Z‐scores >0.3 and <1.5 (red).
Table S2. Cluster assignments for individual replicates for the 83 compounds with an average Z‐score >0.3, and for positive and negative control compounds (grey). Cell phenotypes were identified for each replicate based on Venus channel intensity and texture features. For each compound the proportion of cells of each phenotype was calculated and this data was clustered by Affinity Propagation. This approach clustered together those compounds that had the most similar distributions of cell phenotypes within each replicate. Compounds with an average Z‐score >1.5 are highlighted in blue. The five additional compounds with Z‐scores >0.3 and <1.5 identified in Supplemental Table 1 are highlighted in red.
Table S3. Total number of cells analyzed for MCF10A cells expressing empty vector or MYC‐Venus, treated with either CHIR99021 or MG132 for 16 h (n=1, 5 fields of view).
Table S4. Total number of cells analyzed for MCF10A cells expressing MYC‐Venus, treated with 8 concentrations of indicated compounds for 16 h (n=1).
Table S5. Total number of cells analyzed for MCF10A cells expressing MCL1‐Venus, treated with either CHIR988014 or MG132 for 16 h (n=1, 5 fields of view).
Table S6. Total number of cells analyzed for MCF10A cells expressing empty vector or MYC‐Venus, treated with either CHIR988014 or MG132 for 16 h (n=3).
Acknowledgments
LZP holds the Canada Research Chair (CRC) in Molecular Oncology, and DWA holds the CRC in Membrane Biogenesis. Funding in the LZP and DWA labs were provided in by a Canadian Institutes of Health Research (CIHR) project grant FRN156167 and Foundation grant FDN143312, respectively.
References
- 1. Sandoval PC, Slentz DH, Pisitkun T, Saeed F, Hoffert JD, Knepper MA. Proteome‐wide measurement of protein half‐lives and translation rates in vasopressin‐sensitive collecting duct cells. J Am Soc Nephrol 2013;24(11):1793–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature 2011;473(7347):337–342. [DOI] [PubMed] [Google Scholar]
- 3. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c‐Myc target gene network. Semin Cancer Biol 2006;16:253–264. [DOI] [PubMed] [Google Scholar]
- 4. Wisdom R, Johnson RS, Moore C. c‐Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J 1999;18:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti‐apoptotic MCL‐1 in the survival of hematopoietic stem cells. Science 2005;307(5712):1101–1104. [DOI] [PubMed] [Google Scholar]
- 6. Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA 1995;92(18):8493–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, Penn LZ. MYC deregulation in primary human cancers. Genes (Basel) 2017;8(151):1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okamoto T, Coultas L, Metcalf D, van Delft MF, Glaser SP, Takiguchi M, Strasser A, Bouillet P, Adams JM, Huang DCS. Enhanced stability of Mcl1, a prosurvival Bcl2 relative, blunts stress‐induced apoptosis, causes male sterility, and promotes tumorigenesis. Proc Natl Acad Sci USA 2014;111(1):261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kappelmann M, Bosserhoff A, Kuphal S. AP‐1/c‐Jun transcription factors: Regulation and function in malignant melanoma. Eur J Cell Biol 2014;93:76–81. [DOI] [PubMed] [Google Scholar]
- 10. Muller PAJ, Vousden KH, Norman JC. P53 and its mutants in tumor cell migration and invasion. J Cell Biol 2011. Jan 24;192(2):209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alvarez‐Castelao B, Ruiz‐Rivas C, Castaño JG. A critical appraisal of quantitative studies of protein degradation in the framework of cellular proteostasis. Biochem Res Int 2012;2012:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol 2012;30(7):708–714. [DOI] [PubMed] [Google Scholar]
- 13. Yen H‐CS, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science 2008;322(11):918–923. [DOI] [PubMed] [Google Scholar]
- 14. Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell‐biological applications. Nat Biotechnol 2002;20:1585–1588. [DOI] [PubMed] [Google Scholar]
- 15. Iizuka R, Yamagishi‐Shirasaki M, Funatsu T. Kinetic study of de novo chromophore maturation of fluorescent proteins. Anal Biochem 2011. Jul 15;414(2):173–178. [DOI] [PubMed] [Google Scholar]
- 16. Dang CV. MYC on the path to cancer. Cell 2012;149(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008;8(12):976–990. [DOI] [PubMed] [Google Scholar]
- 18. Welcker M, Orian A, Grim JA, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c‐Myc and cell size. Curr Biol 2004;14:1852–1857. [DOI] [PubMed] [Google Scholar]
- 19. Wasylishen AR, Stojanova A, Oliveri S, Rust AC, Schimmer AD, Penn LZ. New model systems provide insights into Myc‐induced transformation. Oncogene 2011;30(34):3727–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hann SR, Eisenman RN. Proteins encoded by the human c‐myc oncogene: Differential expression in neoplastic cells. Mol Cell Biol 1984;4(11):2486–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl‐1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 2003;17(12):1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011;471(7336):104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Privette LM, Weier JF, Nguyen HN, Yu X, Petty EM. Loss of CHFR in human mammary epithelial cells causes genomic instability by disrupting the mitotic spindle assembly checkpoint. Neoplasia 2008;10(7):643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, Ma ST, Reeder JW, Samuels I, Slabiak T, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003;52(3):588–595. [DOI] [PubMed] [Google Scholar]
- 25. Ye S, Tan L, Yang R, Fang B, Qu S, Schulze EN, Song H, Ying Q, Li P. Pleiotropy of glycogen synthase kinase‐3 inhibition by CHIR99021 promotes self‐renewal of embryonic stem cells from refractory mouse strains. PLoS One 2012;7(4):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gregory MA, Hann SR. c‐Myc proteolysis by the ubiquitin‐proteasome pathway: Stabilization of c‐Myc in Burkitt's lymphoma cells. Mol Cell Biol 2000;20(7):2423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oppermann S, Ylanko J, Shi Y, Hariharan S, Oakes CC, Brauer PM, Zúñiga‐Pflücker JC, Leber B, Spaner DE, Andrews DW. High‐content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood 2016;128(7):934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bray M‐A, Carpenter AE. Advanced assay development guidelines for image‐based high content screening and analysis Assay Guidance Manual Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2017; p. 1–31. [PubMed] [Google Scholar]
- 29. Mayes PA, Dolloff NG, Daniel CJ, Liu JJ, Hart LS, Kuribayashi K, Allen JE, Jee DIH, Dorsey JF, Liu YY, et al. Overcoming hypoxia‐induced apoptotic resistance through combinatorial inhibition of GSK‐3β and CDK1. Cancer Res 2011;71(15):5265–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otto T, Horn S, Brockmann M, Eilers U. Stabilization of N‐Myc is a critical function of Aurora A in human Neuroblastoma. Cancer Cell 2009;15:67–78. [DOI] [PubMed] [Google Scholar]
- 31. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, et al. Small molecule inhibitors of Aurora‐A induce proteasomal degradation of N‐Myc in childhood neuroblastoma. Cancer Cell 2013;24(1):75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee KB, Jin H, Ye S, Park BH, Kim SM. Recombinant human bone morphogenetic protein‐2 inhibits gastric cancer cell proliferation by inactivating Wnt signaling pathway via c‐Myc with aurora kinases. Oncotarget 2016;7(45):73473–73485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frey BJ, Dueck D. Clustering by passing messages between data points. Science 2007;315(5814):972–976. [DOI] [PubMed] [Google Scholar]
- 34. Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl‐1 overlap with, but are not identical to, those of Bcl‐2. J Cell Biol 1995;128(6):1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomas LW, Lam C, Edwards SW. Mcl‐1; the molecular regulation of protein function. FEBS Lett 2010;584:2981–2989. [DOI] [PubMed] [Google Scholar]
- 36. Hsu STD, Blaser G, Behrens C, Cabrita LD, Dobson CM, Jackson SE. Folding study of venus reveals a strong ion dependence of its yellow fluorescence under mildly acidic conditions. J Biol Chem 2010;285(7):4859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Canty L, Hariharan S, Liu Q, Haney SA, Andrews DW. Peak emission wavelength and fluorescence lifetime are coupled in far‐red, GFP‐like fluorescent proteins. PLoS One 2018;13(11):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chu J, Haynes RD, Corbel SY, Li P, González EG, Burg JS, Ataie NJ, Lam AJ, Cranfill PJ, Baird MA, et al. Non‐invasive intravital imaging of cellular differentiation with a bright red‐excitable fluorescent protein. Nat Methods 2014;11(5):572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chan HY, Sivakamasundari V, Xing X, Kraus P, Yap SP, Ng P, Lim SL, Lufkin T. Comparison of IRES and F2A‐based locus‐specific multicistronic expression in stable mouse lines. PLoS One 2011;6(12):e28885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thakor N, Holcik M. IRES‐mediated translation of cellular messenger RNA operates in eIF2a‐independent manner during stress. Nucleic Acids Res 2012;40(2):541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng Y, Mitchison TJ, Bender A, Young DW, Tallarico JA. Multi‐parameter phenotypic profiling: Using cellular effects to characterize small‐molecule compounds. Nat Rev Drug Discov 2009;8(7):567–578. [DOI] [PubMed] [Google Scholar]
- 42. Mitchison TJ. Small‐molecule screening and profiling by using automated microscopy. Chembiochem 2005;6(1):33–39. [DOI] [PubMed] [Google Scholar]
- 43. Jones TR, Carpenter AE, Lamprecht MR, Moffat J, Silver SJ, Grenier JK, Castoreno AB, Eggert US, Root DE, Golland P, et al. Scoring diverse cellular morphologies in image‐based screens with iterative feedback and machine learning. Proc Natl Acad Sci 2009;106(6):1826–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lourenco C, Kalkat M, Houlahan KE, De Melo J, Long J, Done SJ, Boutros PC, Penn LZ. Modeling the MYC‐driven normal‐to‐tumour switch in breast cancer. Dis Model Mech 2019;12(7):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF‐10A mammary epithelial acini grown in three‐dimensional basement membrane cultures. Methods 2003;30(3):256–268. [DOI] [PubMed] [Google Scholar]
- 46. Julius MA, Yan Q, Zheng Z, Kitajewski J. Q vectors, bicistronic retroviral vectors for gene transfer. Biotechniques 2000;28(4):702–708. [DOI] [PubMed] [Google Scholar]
- 47. Collins TJ, Ylanko J, Geng F, Andrews DW. A versatile cell death screening assay using dye‐stained cells and multivariate image analysis. Assay Drug Dev Technol 2015;13(9):547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hamilton NA, Pantelic RS, Hanson K, Teasdale RD. Fast automated cell phenotype image classification. BMC Bioinformatics 2007;8(110):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stojanova A, Tu WB, Ponzielli R, Kotlyar M, Chan PK, Boutros PC, Khosravi F, Jurisica I, Raught B, Penn LZ. MYC interaction with the tumor suppressive SWI/SNF complex member INI1 regulates transcription and cellular transformation. Cell Cycle 2016;15(13):1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim J, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. Evaluation of Myc E‐box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 2004;24(13):5923–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao Y, Davies SP, Augustin M, Woodward A, Patel UA, Kovelman R, Harvey KJ. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J 2013;451:313–328. [DOI] [PubMed] [Google Scholar]
- 52. Johnson K, Liu L, Majdzadeh N, Chavez C, Chin PC, Morrison B, Wang L, Park J, Chugh P, Chen HM, et al. Inhibition of neuronal apoptosis by the cyclin‐dependent kinase inhibitor GW8510: Identification of 3′ substituted indolones as a scaffold for the development of neuroprotective drugs. J Neurochem 2005;93(3):538–548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) The mean and standard deviation in the half‐lives of endogenous MYC in MCF10A control cells (Empty Vector; 30.6 minutes) and MYC‐Venus (30.7 minutes) exogenously expressed in MCF10A cells are plotted. Student t‐test with Bonferroni multiple testing correction, n = 3‐5. (B) (i) Representative image of Venus fluorescence in growing, untreated MCF10A cells expressing MYC‐Venus. Orange nuclear segmentation outlines indicate a nucleus scored as Venus negative; cyan outlines indicate a nucleus scored as Venus positive. Venus fluorescence is observed in a subset of nuclei, indicative of MYC‐Venus localization to the nucleus. Scale bar = 50 μm. (ii) Immunoblot of MYC‐Venus in nuclear (Lamin A & C) and cytoplasmic (Tubulin) fractions of MCF10A cells expressing MYC‐Venus. (C) (i) The mean and standard deviation of mRNA levels normalized to housekeeping gene B2M (Beta 2 Microglobulin) for three MYC‐activated target genes49, 50 (NCL, Nucleolin; LDHA, Lactate Dehydrogenase A; HK2, Hexokinase 2) were measured in MCF10A control cells following serum starvation for 48 h to deplete the endogenous MYC protein, followed by treatment with ethanol (−) or with 100 nM 4‐hydroxytamoxifen (+) to induce MYC‐Venus expression for 24 hours prior to harvesting for RNA. Data is shown relative to vehicle‐treated control cells. One‐way ANOVA with Bonferroni post‐test, n = 3, *** p < 0.001, ** p < 0.01. (ii) Representative immunoblot of total protein harvested from cells treated as described in (Ci) was probed with anti‐MYC antibody to enable visualization of MYC‐Venus expression (expected molecular weight: 83 kDa). (D) The mean and standard deviation of average nuclear intensity for control and MYC‐Venus expressing MCF10A cells treated with the indicated concentrations of (i) GSK3 inhibitor CHIR99021 or (ii) proteasome inhibitor MG132 for 16 h. n = 3 (not all data points are visible due to overlap) (Total number of cells analyzed is reported in Table S6). (D) Percent Venus‐positive nuclei for control and MYC‐Venus expressing MCF10A cells treated with either the GSK3 inhibitor CHIR99021 (i) or the proteasome inhibitor MG132 (ii) at the indicated concentrations for 16 h. n = 3 (not all data points are visible due to overlap) (Total number of cells analyzed is reported in Table S6).
Figure S2. The screen conditions and steps of compound identification and validation are depicted in a flow chart. Compounds were advanced or not advanced as indicated at each step. Compounds previously known to regulate MYC stability and therefore excluded from further analysis, or not analyzed, are also indicated.
Figure S3. (A) The ranked percent Venus‐positive nuclei for each compound at 1 μM, from MCF10A control cells. Fluorescent inhibitors (red box) were identified as three standard deviations above the mean percent Venus‐positive nuclei in the library. (B) Summary of Strictly Standardized Mean Difference scores (SSMD β) calculated between DMSO and 10 μM CHIR99021 for each replicate. (C) Summary of R2 values between replicates 1 and 2, 1 and 3, 2 and 3 (D) The distribution of the percentage of Venus‐positive nuclei for each kinase inhibitor in replicates 1 and 3 is plotted, with all DMSO and 10 μM CHIR99021 wells shown in blue diamonds and green triangles, respectively. Reproducibility, quantified as R2, between replicates 1 and 3 was calculated from the linear fit of % Venus‐positive nuclei from each kinase inhibitor in each replicate. (E) The ranked percent Venus‐positive nuclei for all compounds in the kinase inhibitor library is plotted for MYC‐Venus expressing MCF10A cells (replicate 2 of 1 μM screen shown). The highest concentrations of CHIR99021 (10 μM) and MG132 (1 μM) are indicated by the green triangle and yellow circle, respectively. The Z‐scores for each compound were calculated from the mean and standard deviation of the library (blue line), and a Z‐score = 1.5 is shown as an orange line.
Figure S4. The percentage of Venus‐positive nuclei following 16 hours of treatment with the 8 indicated concentrations of library‐sourced inhibitors (dark grey) or DMSO (light grey) for MCF10A cells expressing MYC‐Venus, for 14 inhibitors with a Z‐score >1.5 (n = 1) (Total number of cells analyzed is reported in Table S4).
Figure S5. The mean and standard deviation in (A) the percentage of Venus‐positive nuclei or (B) number of nuclei, following 16 hours of treatment with the 8 indicated concentrations of independently sourced inhibitors for MCF10A cells expressing either MYC‐Venus or Empty Vector (used here to infer Compound Fluorescence) (n = 3). The color of each point (see inset) indicates the number of cells per well at that concentration. Arrows indicate concentrations selected for further analysis for each compound. Student t‐test of % Venus positive nuclei for MYC‐Venus cells versus Empty Vector (Compound Fluorescence) cells, n = 3.
Figure S6. The mean and standard deviation of (A) endogenous MYC mRNA from MCF10A control cells or (B) MYC‐Venus mRNA as compared to housekeeping gene B2M were measured following 16 h treatment with the indicated inhibitors at previously identified concentrations as shown in Figure 4B. (C) No significant changes (p < 0.003) in RNA following Wilcoxon Signed Rank test with Bonferroni multiple testing correction, n = 3–6.
Figure S7. (A) The mean and standard deviation in the half‐life of MYC‐Venus in MCF10A cells after 16 h treatment with DMSO and CHIR99021 as negative and positive controls, or Dorsomorphin at the previously identified concentration of 10 μM, is plotted. **p < 0.01. Student t‐test with Bonferroni multiple testing correction, n = 3. (B) Representative immunoblots for cells expressing MYC‐Venus treated with the indicated drug for 16 hours and then 10 μg/ml cycloheximide was added and cells were harvested and processed at the indicated timepoints. (C) The reported percentage of kinase activity remaining following treatment with the indicated concentration of either C1651 or GW851052 as compared to the vehicle control.
Table S1. Cluster assignment is shown for the 83 compounds with an average Z‐score >0.3, and for positive and negative control compounds (grey). Cell phenotypes were identified for each replicate based on Venus channel intensity and texture features. The proportion of cells in each phenotype cluster was calculated for each compound separately for each of 3 replicates and then concatenated. The concatenated cell phenotypes for each compound were then clustered by Affinity Propagation. This approach clustered together those compounds with the most similar distributions of cell phenotypes across all three replicates. Clusters 1 and 2 are enriched for compounds with an average Z‐score >1.5 (blue), and include five additional compounds with Z‐scores >0.3 and <1.5 (red).
Table S2. Cluster assignments for individual replicates for the 83 compounds with an average Z‐score >0.3, and for positive and negative control compounds (grey). Cell phenotypes were identified for each replicate based on Venus channel intensity and texture features. For each compound the proportion of cells of each phenotype was calculated and this data was clustered by Affinity Propagation. This approach clustered together those compounds that had the most similar distributions of cell phenotypes within each replicate. Compounds with an average Z‐score >1.5 are highlighted in blue. The five additional compounds with Z‐scores >0.3 and <1.5 identified in Supplemental Table 1 are highlighted in red.
Table S3. Total number of cells analyzed for MCF10A cells expressing empty vector or MYC‐Venus, treated with either CHIR99021 or MG132 for 16 h (n=1, 5 fields of view).
Table S4. Total number of cells analyzed for MCF10A cells expressing MYC‐Venus, treated with 8 concentrations of indicated compounds for 16 h (n=1).
Table S5. Total number of cells analyzed for MCF10A cells expressing MCL1‐Venus, treated with either CHIR988014 or MG132 for 16 h (n=1, 5 fields of view).
Table S6. Total number of cells analyzed for MCF10A cells expressing empty vector or MYC‐Venus, treated with either CHIR988014 or MG132 for 16 h (n=3).