

Abstract
Astroglia or astrocytes, the most abundant cells in the brain, are interposed between neuronal synapses and microvasculature in the brain gray matter. They play a pivotal role in brain metabolism as well as in the regulation of cerebral blood flow, taking advantage of their unique anatomical location. In particular, the astroglial cellular metabolic compartment exerts supportive roles in dedicating neurons to the generation of action potentials and protects them against oxidative stress associated with their high energy consumption. An impairment of normal astroglial function, therefore, can lead to numerous neurological disorders including stroke, neurodegenerative diseases, and neuroimmunological diseases, in which metabolic derangements accelerate neuronal damage. The neurovascular unit (NVU), the major components of which include neurons, microvessels, and astroglia, is a conceptual framework that was originally used to better understand the pathophysiology of cerebral ischemia. At present, the NVU is a tool for understanding normal brain physiology as well as the pathophysiology of numerous neurological disorders. The metabolic responses of astroglia in the NVU can be either protective or deleterious. This review focuses on three major metabolic compartments: (i) glucose and lactate; (ii) fatty acid and ketone bodies; and (iii) D‐ and L‐serine. Both the beneficial and the detrimental roles of compartmentalization between neurons and astroglia will be discussed. A better understanding of the astroglial metabolic response in the NVU is expected to lead to the development of novel therapeutic strategies for diverse neurological diseases.
Keywords: astrocyte, D‐serine, ketone body, lactate, neurovascular unit (NVU)
INTRODUCTION
Astroglia are one of the three types of glial cells in the brain: astroglia (astrocytes), oligodendroglia (oligodendrocytes), and microglia.1, 2, 3 Astroglia are the most abundant cells in the human brain and outnumber neurons by a factor of 1.4 in the human cerebral cortex.4, 5 In addition, their unique anatomical location, which is interposed between neurons and cerebral microvessels and was depicted more than 100 years ago in a sketch by a legendary neuropathologist, Santiago Ramón y Cajal, has been attracting the attention of many neuroscientists.6 In fact, neurons do not have any direct contact with microvessels despite their strict dependence on a continuous supply of glucose and oxygen from outside the brain through the cerebral blood flow. In contrast, 99% of the surfaces of brain capillaries are covered by astroglial foot processes (end‐feet), indicating that all essential materials supplied from the cerebral circulation must interact with astroglia before reaching the neurons.7 The other side of the astroglial end‐feet envelopes synapses in the brain cortex. Thus, synapses composed of presynaptic and postsynaptic neurons as well as astroglial end‐feet are known as tripartite synapses.4, 5, 8, 9 Moreover, astroglial cells are connected to each other via gap junctions with connexin 43 channels, forming a functional syncytium overall.10, 11, 12
The cardinal roles of astroglia in tripartite synapses are the maintenance of normal synaptic function through metabolic support by taking advantage of their anatomical location, which is interposed between synapses and microvessels.13, 14 Accumulating evidence supports the notion that astroglia are also key players in the regulation of cerebral blood flow.15, 16, 17, 18, 19, 20 The neurovascular unit (NVU) is a conceptual framework that was originally used to better understand the pathophysiology of cerebral ischemia.21, 22, 23 Now, the NVU is a tool that can be used to understand normal brain physiology as well as the pathophysiology of numerous neurological disorders.24, 25, 26, 27, 28, 29 Conversely, the malfunction of astroglia in the NVU (i.e., “astrogliopathy”)30, 31, 32, 33, 34 induces neuronal dysfunction, leading to various neurological disorders including cerebrovascular disease (e.g., stroke and small vessel disease‐like Binswanger's disease and cerebral autosomal‐dominant arteriopathy with subcortical infarct and leukoencephalopathy [CADASIL]/35 cerebral autosomal recessive arteriopathy with subcortical infarct and leukoencephalopathy [CARASIL]),36, 37 neurodegenerative disease (e.g., Alzheimer's disease,38, 39 Parkinson's disease,40, 41 and amyotrophic lateral sclerosis [ALS]),42, 43, 44 and neuroimmunological disease (e.g., multiple sclerosis [MS],45, 46, 47, 48 and neuromyelitis optica spectrum disorder [NMOSD]).49, 50 This review will focus on the supportive roles of astroglia in the NVU from the perspective of three major metabolic compartments with neurons: (i) glucose and lactate; (ii) fatty acid and ketone bodies (KBs); and (iii) D‐ and L‐serine.
GLUCOSE AND LACATE
Oxidative metabolism of glucose is mandatory for generating action potentials
The adult human brain weighs approximately 2% of the total body weight and consumes 25% of the total body glucose consumption and 20% of the oxygen consumption. More precisely, the cerebral metabolic rate of glucose consumption (CMRglc) and oxygen (CMRO2) in the adult brain is 31 pmol/100 g/min and 156 pmol/100 g/min, respectively.51, 52 The ratio of CMRO2/CMRglc at resting state is approximately 5.5, which is close to 6.0, the theoretical stoichiometry for the complete oxidation of glucose with oxygen (C6H12O6 + 6O2 → 6CO2 + 6H2O). The measured data indicate that almost all glucose is metabolized oxidatively except for the consumption of some additional glucose for non‐oxidative metabolism. In fact, the brain is strictly dependent on glucose for its energy production.51, 52 High CMRO2 and CMRglc levels produce ATP efficiently with a ratio of 6.0, driving Na+,K+‐ATPase to maintain a steep ionic gradient across the cellular membrane. When an action potential is generated, a rapid influx of Na+ and a slow efflux of K+ follow. The functional activation of the brain is locally related to glucose consumption, indicating that glucose utilization in the whole brain reflects mainly the neuronal consumption of glucose.
Glucose utilization by astroglia
Using fluorescently labeled glucose analogs, the cellular uptake of glucose can be evaluated in the brain cortex in vivo.53, 54 Glucose uptake occurs in neurons and astroglia via different glucose transporters (GLUTs), that is, GLUT3 and GLUT1, respectively (Fig. 1).55, 56 Since GLUT1 is also expressed in endothelial cells in brain capillaries, glucose supplied by the cerebral circulation can cross the blood–brain barrier (BBB). The congenital deficiency of GLUT1 induces intractable seizures beginning in infancy as well as mental retardation because of the unavailability of glucose to neural cells arising from limited glucose transportation through the endothelium.57, 58 Surprisingly, the brain has almost no storage of glucose, and glucose must be supplied continuously via the blood circulation.53, 54 More precisely, astroglia contain small amounts of glucose in the form of glycogen granules in their cell bodies.59 Glycogen is degraded by a glycogen phosphorylase (glycogenolysis), which is the astroglia‐specific enzyme, forming glucose‐1‐phosphate (G1P).60 G1P then enters the glycolytic metabolic pathway in astroglia (Fig. 2). A total amount of glycogen content measured in the brain can maintain its function for only 3 min, based on the CMRglc.61 If glucose derived from astroglial glycogen was available for neurons, it would be of some help. Unfortunately, G1P cannot cross the cell membrane because of its low lipid solubility. Instead, lactate or pyruvate, the end‐products of glycolysis, can exit astroglia via monocarboxylate transporter 1 (MCT1) and MCT4; they can then re‐enter neurons via monocarboxylate transporter 2 (MCT2), which is utilized as an energy source for neuronal tricarboxylic acid (TCA) cycle substrates (Fig. 1).62, 63, 64 Not only glycogen‐derived lactate/pyruvate, but also lactate/pyruvate from blood‐supplied glucose can be transferred to the neurons (Fig. 2).65 If this intercellular compartmentalization between neurons and astroglia operates in the resting and/or activated brain, the measured CMRglc would reflect mainly astroglial glucose utilization, since neurons utilize lactate derived from astroglia. This hypothetical model, termed the “astrocyte‐neuron lactate shuttle hypothesis (ANLSH)”, was originally proposed by Pellerin and Magistretti in 1994 based on data obtained using cultured cells66 and was supported by our findings67, 68 but has remained controversial for more than a quarter of a century.69, 70
Figure 1.
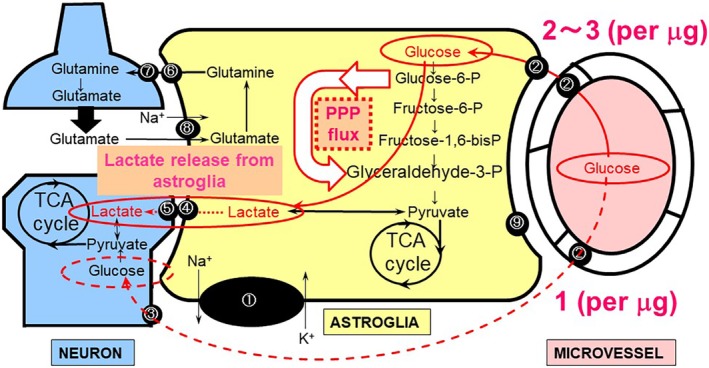
Metabolic compartment of glucose between astroglia and neurons in the neurovascular unit (NVU). Astroglia are interposed between microvessels and neuronal synapses, forming the NVU. Glucose supplied from outside the brain can be transported to and utilized by both astroglia and neurons (red lines). Glucose utilization by cultured astroglia (2–3 pmol/μg protein: red line) is two times higher than that in cultured neurons (1 pmol/μg protein: red broken line), and astroglia produce lactate even under normoxic conditions (aerobic glycolysis). Neuronal activation induces glutamate release from the pre‐synaptic nerve terminal; the glutamate is then taken up by astroglia, and this uptake, in turn, accelerates glucose consumption, leading to further lactate release. Lactate, then, serves as an energy substrate for neurons (astrocyte‐neuron lactate shuttle hypothesis, ANLSH). The basal pentose‐phosphate pathway (PPP) flux measured in cultured astroglia is approximately seven times higher than that in cultured neurons. Neuronal activation induces astroglial glycolysis, leading to increased flux to the PPP. Increases in NADPH in astroglia serve as a redox regulator that maintains the reduced form of glutathione. ① Na+,K+‐ATPase. ② Glucose transporter 1 (GLUT1). ③ Glucose transporter 3 (GLUT3). ④ Monocarboxylate transporter 1 (MCT1) and MCT4 (astrocytic form). ⑤ MCT2 (neuronal form). ⑥ System N transporter (astrocytic form). ⑦ System A transporter (neuronal form). ⑧ Na+‐dependent glutamate transporter‐1 (GLT‐1) and glutamate aspartate transporter (GLAST). ⑨ Fatty acid‐binding protein (FABP).
Figure 2.
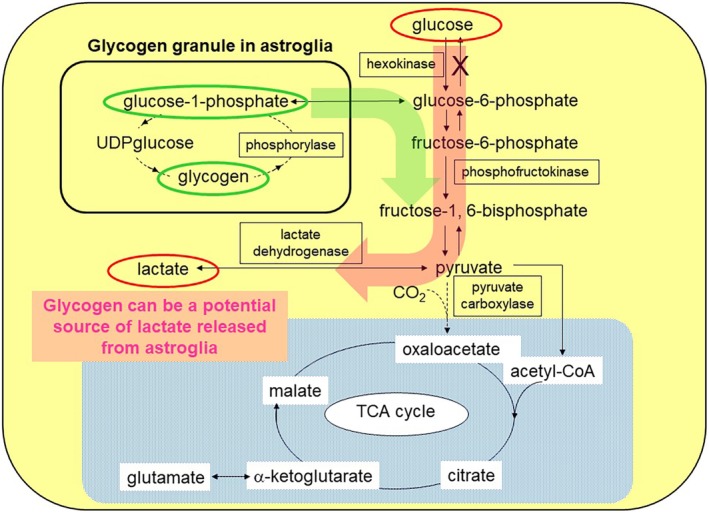
Glycogen deposits in astroglia are a potential source for lactate production. An astroglia‐specific enzyme, glycogen phosphorylase, degrades glycogen deposits in astroglia. In addition to glucose‐derived glucose‐6‐phosphate (pink arrow), glycogen‐derived glucose‐1‐phosphate (green arrow) is metabolized in a glycolytic pathway, producing lactate. When astroglia are cultured under high glucose conditions, the glycogen content and lactate production both increase.
Aerobic glycolysis in astroglia
Astroglia seem to be more strictly dependent on glucose, and their metabolism, at least in cultured astroglia in vitro, seems to be more glycolytic than that in cultured neurons.71 In fact, astroglia can survive if mitochondrial oxidative metabolism is inhibited, while neurons cannot.72 Glucose uptake or utilization (phosphorylation by hexokinase [HK]) can be quantified using cultured astroglia and neurons in vitro and in vivo using modern molecular techniques,73 while the quantitation of oxygen consumption is more difficult. However, importantly, the importance of the ANLSH is the capacity for lactate production by astroglia irrespective of an adequate supply of oxygen, that is, aerobic glycolysis. Although under cerebral ischemia, both astroglia and neurons produce large amounts of lactate that accumulate in tissues, experimental data show that astroglia in vitro produce a large amount of lactate under normal (21%) oxygen environments.74 In similar environments, cultured neurons produce less lactate from glucose. These phenomena have led to the hypothesis that glucose supplied from the cerebral circulation is taken up by mainly astroglia taking advantage of their anatomical location and is metabolized in astroglia to produce lactate, which is exported to neurons via MCT1 and 4. Neurons can technically utilize both glucose and lactate, as they express both GLUT3 and lactate transporter (MCT2). In contrast to GLUT1 deficiency, the congenital deficiency of neuron‐specific GLUT has not been reported. Only a mouse model of GLUT3‐knockout (KO) has been reported to exhibit autism‐like phenotypes, suggesting that neurons can survive without glucose under an environment where lactate is available.75 In fact, neuronal lactate dehydrogenase (LDH) isozyme favors the conversion of lactate to pyruvate, while astroglial LDH isozyme favors the formation of lactate from pyruvate.76 Supporting this notion, in vitro studies show that glucose consumption by neurons and that by astroglia are similar when lactate is not available. However, interestingly, when both glucose and lactate are available, cultured neurons preferentially utilize lactate as the TCA cycle substrate.77, 78, 79
The ANLSH emphasizes that astroglial lactate production is enhanced by glutamate stimulation in association with neuronal activation.66 Glutamate released from activated pre‐synaptic neurons is known to be rapidly taken up by astroglia that envelop the synapse.80, 81 The astroglial uptake of glutamate is dependent on the Na+‐dependent glutamate transporter‐1 (GLT‐1) and glutamate aspartate transporter (GLAST) (Fig. 1).80, 81 Co‐transported Na+ and glutamate increase the intracellular concentration of Na+ ([Na+]i), leading to the activation of Na+,K+‐ATPase; this, in turn, accelerates ATP production in astroglia. Whether ATP production in astroglia is dependent on glycolysis or the mitochondrial oxidative metabolism of glucose continues to be a matter of long‐lasting debate.69, 70, 82, 83 An in vitro experiment showed that glutamate stimulation does, indeed, increase glucose consumption and lactate production by astroglia (i.e., aerobic glycolysis).66, 67, 68, 74 The functional activation of in vivo brain also induces transient increases in lactate production locally in distinct lesions of an activated site where glucose utilization is eventually increased.84, 85 However, in vivo studies have not been able to determine the origin of the lactate because of a lack of cellular resolution. Even though astroglia produce lactate from glucose, astroglia are not necessarily devoid of mitochondrial function. Of note, glutamate that is taken up by astroglia can also serve as a substrate of the TCA cycle after its conversion to α‐ketoglutarate, implying that increased lactate production is not necessarily a reflection of solely glycolytic activation and that oxidative metabolism can also be activated using substrates other than glucose. Moreover, glutamate‐derived α‐ketoglutarate can also produce lactate during TCA cycle metabolism.86 Glutamate reportedly inhibits the neuronal utilization of glucose,87 implying a shift in the energy source from glucose to lactate (Fig. 3).
Figure 3.
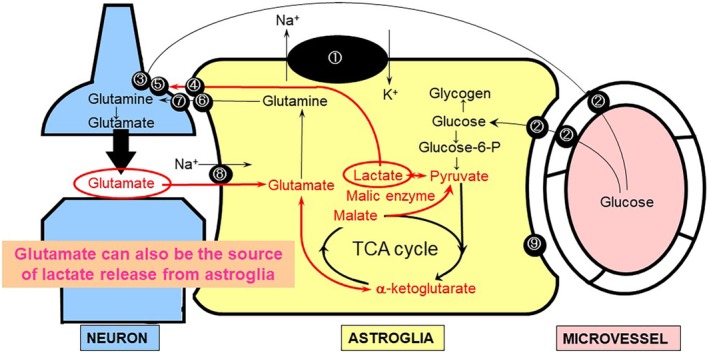
Glutamate taken up by astroglia serves as an astroglial tricarboxylic acid (TCA) cycle intermediate, leading to lactate production. Glutamate released from pre‐synaptic neurons is taken up by astroglia and recycled back to neurons (glutamate‐glutamine cycle). Some of the glutamate in astroglia is converted to α‐ketoglutarate, which enters the TCA cycle (red line) as an intermediate substrate, leading to astroglial lactate production by malic enzyme. ① Na+,K+‐ATPase. ② Glucose transporter 1 (GLUT1). ③ Glucose transporter 3 (GLUT3). ④ Monocarboxylate transporter 1 (MCT1) and MCT4 (astrocytic form). ⑤ MCT2 (neuronal form). ⑥ System N transporter (astrocytic form). ⑦ System A transporter (neuronal form). ⑧ Na+‐dependent glutamate transporter‐1 (GLT‐1) and glutamate aspartate transporter (GLAST). ⑨ Fatty acid‐binding protein (FABP)
Dual roles of lactate
Irrespective of accumulating evidence supporting the ANLSH, the fate of lactate, if it is really produced by astroglia via a glutamate signal from activated neurons, has remained a matter of controversy for more than 25 years.69, 70 The original ANLSH followed by numerous reports proposed that activated neurons consume lactate produced by astroglia.66, 88, 89, 90, 91, 92, 93, 94 A strong argument for this is based on a kinetic property of MCT2 expressed in neurons, which has a low kilometer value. The transportation of lactate into neurons becomes saturated at a low concentration of lactate, and neurons cannot utilize additional lactate even if it is produced by activated astroglia.95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105 Conversely, whether neuronal ATP production is exclusively dependent on the uptake of glucose by neurons remains uncertain. The in vivo truth remains to be clarified.
Another possible role of lactate is as a ligand of hydroxycarboxylic acid receptor 1 (HCAR1) expressed in neurons.106, 107 HCAR1, a G‐protein coupled receptor, suppresses neuronal excitability by increasing the intracellular cAMP concentration. Irrespective of the origin of lactate, direct evidence supports that extracellular lactate regulates synaptic activity (i.e., mainly inhibitory effects) and that the disinhibition of neuronal excitation can cause intractable epilepsy. Assuming that lactate is mainly produced by astroglia, the impairment of lactate production by astroglia can lead to neuronal damage through hyper‐excitation (i.e., excitotoxicity). In fact, neuronal degeneration in ALS, in which an unknown mechanism induces motor neuron death, has been hypothesized to be induced by the impairment of astroglial glutamate uptake as well as the hyper‐excitation of spinal motor neurons.108
Astroglial lactate production, especially that derived from glycogen (i.e., glycogenolysis), has been implicated in the formation of long‐term memory.109, 110 The exact mechanism by which lactate consolidates memories has not been elucidated and remains somewhat controversial.103, 111, 112 Several experiments support the idea that lactate produced in astroglia may serve as an energy source for neuronal synapse remodeling and gene expression, but not for the TCA cycle.113 The role of lactate as a ligand for HCAR1 remains to be elucidated.
Neuroprotective roles of astroglial glycolysis and PPP
Of note, the functional activation of neurons increases glucose utilization locally, and both neurons and astroglia contribute to glucose consumption. We have focused on the roles of glucose metabolism, especially glycolysis in astroglia, from the perspective of their supportive aspects in the protection of neurons against oxidative stress.30, 31, 32, 33, 34 Neuronal energy production is dependent on mitochondrial oxidative metabolism, which produces small amounts of reactive oxygen species (ROS). Oxidative stress has been implicated in the pathogenesis of numerous neurological disorders (i.e., stroke, ALS, Parkinson's disease) as well as normal aging of the brain.30, 31, 32, 33, 34 The glutathione system acts as an intrinsic protective mechanism. Glutathione peroxidase reduces ROS by converting it to H2O2 in concert with the conversion of a reduced form of glutathione (GSH) to an oxidized form (GSSG).30, 114, 115 The maintenance of the GSH concentration is dependent on nicotinamide adenine dinucleotide phosphate (NADPH), a product of the pentose‐phosphate pathway (PPP), which is a shunt pathway of glycolysis (Fig. 1).30, 51, 52 Therefore, the influx to the PPP in glycolysis is an index of PPP activity and reflects the anti‐oxidative function of many kinds of cells. PPP flux is regulated by glucose‐6‐phosphate dehydrogenase (G6PDH), which is a rate‐limiting enzyme of this shunt pathway.30, 51, 52 The basal influx to the PPP in astroglia is approximately seven times as high as that in neurons, suggesting that glycolysis has anti‐oxidative roles in astroglia.30, 31
Increased glucose phosphorylation by HK leads to PPP flux through the allosteric regulation of G6PDH. Thus, a high glucose environment can activate PPP flux in astroglia.30, 31 In diabetic patients, a high plasma glucose concentration is associated with a high glucose content in the brain because of the facilitated diffusion of glucose by GLUT1. Whether a high glucose level in the brain readily increases glucose phosphorylation is debatable.30, 31 Because the Km (Michaelis constant) of HK is low, in clear contrast to glucokinase (GK) in the liver, glucose phosphorylation becomes saturated at a low glucose concentration.51, 52 The existence of high‐Km HK in the brain has been postulated. Our study showed that PPP flux increases according to the glucose concentration in an assay solution.30, 31 These observations might reflect the presence of high‐Km GK, like HK, in the brain. If this is true for in vivo brain, neuronal activation would induce astroglial glucose utilization via glutamate, leading to astroglial PPP activation. Importantly, the synthetic activity of glutathione, which consists of three amino acids, glutamine, cysteine, and alanine, is higher in astroglia than in neurons.116, 117 As mentioned above, the glycolytic activity as well as the PPP flux are dominant in astroglia, leading to the anti‐oxidative mechanism is astroglia.30, 31 When glutathione is synthesized, it must be transferred in a reduced form.116, 117 In addition, the reduced form of GSH itself can exert an anti‐oxidative role (see dopamine‐induced neurotoxicity).
Transcriptional regulation of PPP flux by the Keap1/Nrf2 system
Another mechanism of PPP flux regulation is the transcriptional control of a key PPP enzyme. The rate‐limiting enzyme of PPP flux is G6PDH, and its transcription is under the control of the transcriptional factor nuclear factor‐erythroid‐2‐related factor 2 (Nrf2).118, 119 Nrf2 is anchored by an adaptor protein of Kelchlike ECH‐associated protein 1 (Keap1) in the cytosol. As a complex, Keap1/Nrf2 is degraded constantly by the proteasome system, and Nrf2 is not in an active state in terms of transcriptional control. When cells are exposed to various stress, a conformational modification of Keap1 or Nrf2 occurs and results in dissociated Nrf2 being released and binding to the antioxidant response element (ARE), where it initiates the transcription of anti‐stress response proteins including G6PDH.118, 119, 120, 121 Importantly, glutathione synthesis enzymes are also under the regulation of the Keap1/Nrf2 system. Astroglial glycolysis and its shunt pathway, the PPP, seem to play an important role in protecting neurons against oxidative stress through the Keap1/Nrf2 system.30, 31
Dopamine released from dopaminergic neurons is known to be auto‐oxidized to form dopamine quinone and ROS, which, in turn, damage neurons. Astroglia releases GSH and reduces dopamine‐induced ROS toxicity in Nrf2‐depenent manners. We have found that astroglia protect neurons against dopamine exposure by reducing ROS, while astroglia prepared from Nrf2‐KO mice are incapable of such activity.34 An in vivo model supports the notion that Nrf2‐KO mice are susceptible to a Parkinson's disease‐like phenotype and that an Nrf2 activator can modify the progression of Parkinson's disease.34
Neuro‐inflammation plays a cardinal role in the pathogenesis of stroke.33, 122, 123, 124 During the early phase of stroke, ischemic cell damage makes neurons release various kinds of molecules, such as damage‐associated molecular pattern, which, in turn, act as Toll‐like receptor 4 (TLR4) ligands. Microglia in which TLRs are expressed abundantly produce various pro‐inflammatory cytokines, accelerating neuronal damage. Nitric oxide (NO) is thought to be one of the molecules that plays a pro‐inflammatory role.122, 123, 124 Such actions of microglia switch astroglia from acting as neuroprotectors to acting as neurodamagers.125 However, we found that upon stimulation with lipopolysaccharide (LPS), a classical TLR ligand, microglia produce NO that diffuses into astroglia, activating the PPP through the S‐nitrosylation of Keap1, which facilitates Nrf2 translocation to the nucleus and triggers G6PDH transcriptional activation.33
FATTY ACID AND KB
KBs produced from fatty acids serve as energy substrates for neurons
Fatty acids and KBs form the second metabolic compartment between neurons and astroglia (Fig. 4).126, 127, 128, 129, 130, 131 KBs, consisting of acetoacetate (AA), acetone, and β‐hydroxybutyrate (BHB), are alternative substrates for glucose. KBs, rather than glucose, are the main energy substrates in the brains of infants. KBs and lactate share the same MCTs. In adults, KBs are produced in the liver under reduced glucose availability, such as starvation and insulin resistance. KBs produced in the liver are transported into the brain where they are utilized by neurons as well as glial cells as substrates for the TCA cycle. After being taken up by neural cells via MCT1 or MCT2, KBs are converted to acetyl‐CoA; a further metabolic process subsequently occurs in the TCA cycle (Fig. 5). In contrast to lactate or pyruvate, which must also be converted to acetyl‐CoA by pyruvate dehydrogenase complex (PDHC) before entering the TCA cycle, KBs do not require PDHC activity. PDHC is a key enzyme in glucose metabolism that links glycolysis and the TCA cycle. Despite being of vital importance, PDHC is susceptible to oxidative stress (Fig. 5).32, 132, 133 Therefore, neurons are not capable of utilizing lactate, which accumulates during cerebral ischemia, after reperfusion (re‐oxygenation) because of PDHC impairment, with ROS being produced in association with the resupply of O2 to damaged tissue.
Figure 4.
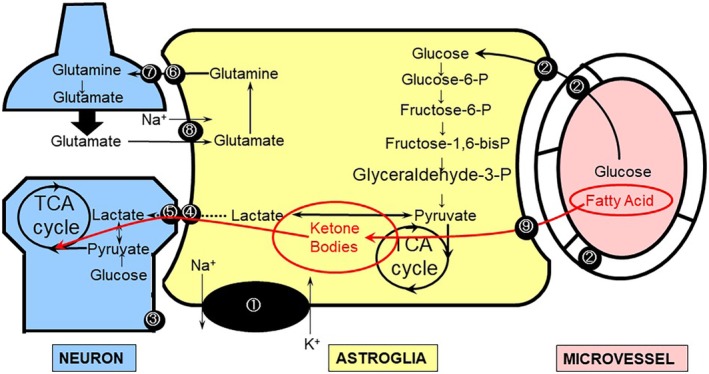
Ketone bodies (KBs) produced by astroglia serve as energy substrates for neurons. Fatty acids supplied from the blood are transported to astroglia in the brain, generating KBs; these KBs can fuel neurons as a tricarboxylic acid (TCA) cycle substrate (red line). ① Na+,K+‐ATPase. ② Glucose transporter 1 (GLUT1). ③ Glucose transporter 3 (GLUT3). ④ Monocarboxylate transporters 1 (MCT1) and MCT4 (astrocytic form). ⑤ MCT2 (neuronal form). ⑥ System N transporter (astrocytic form). ⑦ System A transporter (neuronal form). ⑧ Na+‐dependent glutamate transporter‐1 (GLT‐1) and glutamate aspartate transporter (GLAST). ⑨ Fatty acid‐binding protein (FABP).
Figure 5.
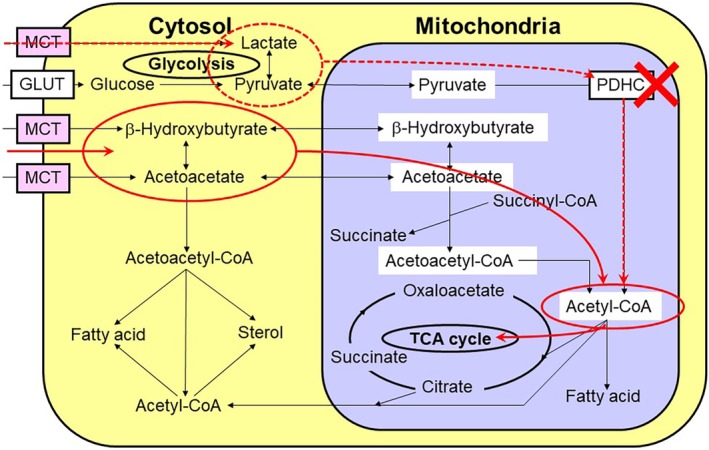
Ketone bodies (KBs) are a better energy source for neurons after ischemia/reperfusion. During ischemia/hypoxia, both lactate and KBs are generated in astroglia. The malfunction of the pyruvate dehydrogenase complex (PDHC) as a result of reperfusion injury enables neurons to utilize KBs, rather than lactate, as a more efficient tricarboxylic acid (TCA) cycle substrate.
In contrast, KBs, for which PDHC is not required to enter the TCA cycle, can serve as energy substrates for the mitochondrial TCA cycle in neurons after re‐oxygenation. In addition, KBs play neuroprotective roles in several different ways.32, 126, 127, 128 More importantly, KBs can be supplied from inside the brain: astroglia are capable of generating KBs and together form a metabolic compartment with neurons (Fig. 4).32, 126, 127, 128 KBs are produced from fatty acids as well as an amino acid (leucine).134 The main sources of KB production are long‐chain fatty acids. We have evaluated KB production from palmitic acid in astroglia, compared with that in neurons, and our results suggested that KB produced in astroglia can serve as an alternative energy substrate for the TCA cycle in neurons.32
Astroglia produce KBs that act on neurons through transporters and receptors
Fatty acids are bound to albumin in the blood, and only free fatty acids can cross the BBB.135, 136 Although the brain levels of soluble fatty acids have not been reported, fatty acids have long been known to be BBB permeable. Neural cells also reportedly express fatty acid‐binding proteins (FABPs) that take up free fatty acids.137, 138, 139, 140 Long‐chain fatty acids (> 12 carbons) such as palmitic acid (PAL, 16 carbons), a predominant fatty acid in the body, are then metabolized to produce long‐chain fatty acid acyl‐CoA. Fatty acid acyl‐CoA is then transported into mitochondria by carnitine palmitoyltransferase I (CPT‐I), which exists in the outer membrane of mitochondria and undergoes β‐oxidation. Because astrocytes envelop microvessels in the brain, they are likely to be the main site of fatty acid metabolism in the brain (Fig. 6).7
Figure 6.

Ketone body (KB) production by astroglia. A long‐chain fatty acid (palmitic acid) is transported into hepatocytes in the liver, generating acetyl‐CoA through β‐oxidation; acetyl‐CoA, in turn, serves as a tricarboxylic acid (TCA) cycle substrate. Astroglia are equipped with a similar metabolic activity and are capable of generating KBs. As KBs are not utilized in the TCA cycle of astroglia, they are transported out through monocarboxylate transporters (MCTs).
The initial step is β‐oxidation, and we measured KB production using 14C‐labeled palmitic acid. Long‐chain fatty acids (e.g., PAL) are an important source for KB production.132, 133 Although FABP is reportedly expressed in the endothelium and helps with the transportation of fatty acids into the brain, the kinetic properties of the transportation of long‐chain fatty acids into the brain remains a topic of debate. Another source of KB production in the brain is amino acids. Leucine can be converted to KB.134
KB production by astroglia is regulated by AMP‐dependent kinase (AMPK), an energy sensor in cells (Fig. 7). When astroglia are exposed to hypoxia and/or hypoglycemia, KB production by astroglia is stimulated by metformin, an oral diabetic drug. Guzmán and Blázquez126 reported that AMPK regulates astroglial ketogenesis by phosphorylating acetyl‐CoA carboxylase (ACC), thereby inhibiting ACC activity and reducing cytosolic malonyl‐CoA—a major physiological inhibitor of CPT‐I (a rate‐limiting enzyme of fatty acid metabolism). In fact, 5‐amino‐1‐β‐D‐ribofuranosylimidazole‐4‐carboxamide (AICAR), a cell‐permeable analog of AMP that activates AMPK,141 enhances ketogenesis in astroglia. Because AMPK is a sensor of AMP/ATP and acts as an indicator of the energy reserve in cells, Guzmán and Blázquez126 speculated that hypoxia/ischemia may stimulate astroglial ketogenesis; they proved that chemical hypoxia induced by 1 mmol/L of NaN3, which inhibits cytochrome oxidase and thus the mitochondrial respiratory chain,142 for 1 h did indeed enhance ketogenesis in cultured astroglia.143
Figure 7.
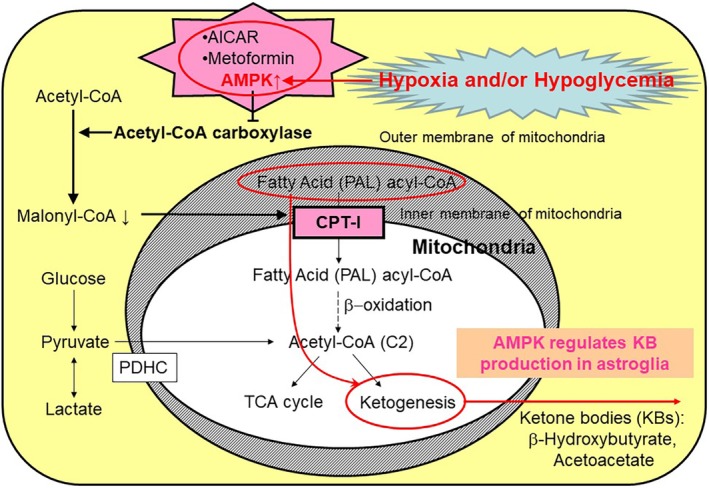
Regulation of ketone body (KB) production by astroglia. Hypoxia and/or hypoglycemia activates adenosine monophosphate‐dependent kinase (AMPK), which phosphorylates and inhibits acetyl‐CoA carboxylase. Decreases in malonyl‐CoA disinhibit carnitine palmitoyltransferase I (CPT‐I), leading to increased KB production. Both 5‐amino‐1‐β‐D‐ribofuranosylimidazole‐4‐carboxamide (AICAR) and metformin activate AMPK and induce increases in KB production in astroglia.
We previously reported that hypoxia and/or hypoglycemia enhances astroglial KB production in vitro and that metformin, an AMPK activator, also induces KB production in astroglia (Fig. 7).32 Moreover, neurons that had been exposed to hypoxic conditions exhibited reduced oxidative capacities for lactate and pyruvate, while the oxidation of BHB was preserved, suggesting astroglial metabolic support through KB production under ischemia/reperfusion.32 Even without the presence of long‐chain fatty acids, astroglia are capable of producing KBs from leucine, although the physiological relevance of this mechanism remains to be determined.
BHB as a ligand for HCAR2
The recent discovery that BHB acts as an endogenous ligand for hydroxycarboxylic acid receptor 2 (HCAR2) has expanded the role of BHB beyond that of a mere energy substrate.144, 145, 146 An experimental ischemic model showed that the activation of HCAR2 reduced the infarct volume, and this beneficial effect was lost in animals with the genetic deletion of HCAR2.145 Interestingly, HCAR2 was expressed mostly in peripheral blood cells. Astroglia‐derived BHB can activate HCAR2 in brain cells more efficiently, and accumulating evidence suggests that microglia in the brain also express HCAR2.147
AMINO ACID AND D‐/L‐SERINE
Glutamate and NMDA receptor co‐agonists
Glutamate plays a cardinal role in synaptic function through glutamate receptors in postsynaptic neurons. Especially, the N‐methyl‐D‐aspartate (NMDA) receptor can act as a double‐edged sword. The NMDA receptor is necessary for normal memory function, and NMDA deficits can induce psychiatric disorders. In contrast, overstimulation of the NMDA receptor induces Ca2+ influx and leads to neuronal death, that is, excitotoxicity. These two faces of the glutamate‐NMDA receptor's nature make it difficult to use NMDA antagonists as neuroprotective agents against stroke and neurodegenerative diseases, even though NMDA blockers do exert strong neuroprotection.148, 149 Another aspect of the NMDA receptor is the discovery of co‐agonists that work in concert with glutamate.150, 151, 152, 153, 154, 155 At present, two different co‐agonists have been identified: D‐serine and glycine. The former acts as a co‐agonist of glutamate in synaptic NMDA receptors, and the latter acts in extra‐synaptic NMDA receptors.154
D‐serine is converted from L‐serine in pre‐synaptic neurons
D‐serine is an enantiomer of L‐serine and is thought to be converted from L‐serine by serine racemase (SRR).156, 157, 158, 159 SRR is almost exclusively expressed in pre‐synaptic neurons, indicating that pre‐synaptic neurons release both glutamate and D‐serine into the synaptic cleft and modulate postsynaptic function (Fig. 8).155, 156 Of note, L‐serine is more abundant in astroglia than in neurons. The de novo synthesis of L‐serine occurs exclusively in astroglia, and its synthetic pathway branches at glycolysis, implying the presence of another metabolic compartment between neurons and astroglia (Fig. 8).157, 158, 159
Figure 8.

D‐ and L‐serine form amino acid compartmentalization between neurons and astroglia. The de novo synthesis of L‐serine occurs in the astroglial glycolytic pathway. L‐serine is transported to neurons, where it is converted to D‐serine by serine racemase (SRR) and acts as a co‐agonist of N‐methyl‐D‐aspartate (NMDA) receptors (red line). ① Na+,K+‐ATPase. ② Glucose transporter 1 (GLUT1). ③ Glucose transporter 3 (GLUT3). ④ Monocarboxylate transporter 1 (MCT1) and MCT4 (astrocytic form). ⑤ MCT2 (neuronal form). ⑥ Astrocytic Na+‐independent asc‐type amino acid transporter‐1 (ASCT‐1). ⑦ ASCT‐2.
Following the first report of the effect of SRR deletion on ischemic brain damage,160 we have evaluated the neuroprotective effect of the elimination of D‐serine in a mouse ischemic stroke model.161 SRR‐KO mice exhibited smaller infarct volumes and better functional recovery, compared with control mice, after experimental middle cerebral artery occlusion and reperfusion, indicating that D‐serine deletion can, at least in stroke, be used as a neuroprotective strategy.161 However, L‐serine deletion does not necessarily enable neuroprotection.
De novo synthesis of L‐serine in astroglia and possible neuroprotection
L‐serine is produced in astroglia from the glycolytic pathway via enzyme 3‐phosphoglycerate dehydrogenase (3PGDH).157, 158, 159 The astrocyte‐specific knockout of this enzyme dramatically reduces L‐serine in the brain and probably also reduces D‐serine in neurons. Using the middle cerebral artery occlusion (MCAO) model, the effect of reducing L‐serine on the stroke volume was evaluated. Surprisingly, the infarct volume was not significantly reduced, suggesting an opposite function, that is, neuroprotection, of L‐serine (unpublished data). In fact, Wang et al.162 found that L‐serine infusion reduced the infarct volume in a mouse MCAO model, and they speculated that this action of L‐serine may be dependent on a vasodilatory effect, since L‐serine increases cerebral blood flow.163 Furthermore, a neuro‐restorative role of L‐serine, in addition to its neuroprotective role, has been postulated.164
SUMMARY AND UNSOLVED ISSUES
Metabolic interaction between astroglia and neurons
Accumulating evidence indicates astroglial metabolic supports through at least three compartments under both normal physiological as well as pathophysiological conditions. The cardinal metabolic compartments are as follows: (i) glucose and lactate; (ii) fatty acids and KBs; and (iii) D‐ and L‐serine. Of note, most available evidence is based on in vitro studies using rodent neural cell cultures. A major criticism is that rodent neurons and astroglia might not be an appropriate model for human brain cells.165 In particular, the high glycolytic metabolic activity with lactate production might be a characteristic of rodent cell cultures only. Recent technology utilizing induced‐pluripotent stem cells (iPSCs) has enabled us to evaluate human neurons and astroglia in vitro. Thus far, only a limited number of studies have focused on the metabolic compartments between these types of cells.166 We recently induced cortical astroglia and spinal motor neurons from iPSCs prepared from normal healthy adults and measured the glycolytic activities of both cell types. So far, the astroglial glycolytic capacity seems to be higher than the neuronal capacity (unpublished data). Further confirmation of these findings using in vivo human brain studies is warranted.
Compartmentalization between oligodendroglia and astroglia
In the white matter of the brain, the axons of neurons are myelinated by oligodendrocytes, which enable saltatory conduction. Similar to the gray matter of the brain, small vessels are completely covered by astroglial end‐feet.7 Thus, substrates for energy production as well as structure construction must be supplied through the astroglia to the oligodendroglia. More precisely, at Ranvier nodes, where the ionic flux is most active, the astroglial end‐feet are in direct contact with axons.167, 168 Assuming that axons utilize lactate preferentially as an energy substrate for the TCA cycle, how lactate is supplied to the axons is an interesting and unsolved issue: is lactate supplied by oligodendroglia or astroglia? Astroglia can supply lactate directly at Ranvier nodes, similar to their actions at tripartite synapses. However, a recent report has elucidated that oligodendroglia metabolize glucose glycolytically to produce lactate.169, 170 Glucose supplied from microvessels can be used as a direct substrate for oligodendroglia to support neuronal energy metabolism.9, 171, 172 The CMRglc in the white matter is much less than that in the gray matter, and the exact contributions of astroglia, oligodendroglia, and neurons (axons) to CMRglc and the pathways of glucose transport to oligodendroglia and neurons remain to be solved.
The fate of KBs produced in astroglia in the white matter should be evaluated from the perspective of myelin formation. KBs can be utilized in lipid synthesis for the cell membrane.173, 174, 175 Membrane formation is dependent on cholesterol, which can be supplied directly from astroglia as KBs. A dysfunction in cholesterol trafficking has been implicated in memory impairment.175 How astroglial metabolic support is needed for myelination by oligodendroglia remains to be elucidated.176
Energy metabolism in microglia
Microglia are the last type of glial cells in the brain. Although the origin of this type of glial cells seems to differ from that of other glial cells, microglia reside in the brain during the early developmental stage and play a pivotal role in the immunological response of the brain by regulating synaptic pruning and scavenging damaged neurons.177, 178 As mentioned in previous chapters, microglia have a strong influence on astroglial responses of either a harmful or beneficial nature.33, 125 We have examined the interaction between astroglia and microglia via NO. Upon stimulation by LPS, a classical ligand of TLR4, microglia are activated to produce NO which, in turn, induces neuroprotective astroglia through the Keap1/Nrf2 system.33 Moreover, LPS stimulation activates NADPH oxidase (NOX), which is highly expressed in microglia. In fact, microglia have a strong capacity to produce ROS and, therefore, the anti‐oxidative system should also be equipped to protect itself. A limited number of studies have revealed that the microglial PPP is active probably to keep the glutathione system active, as in astroglia. Our observations suggest that the microglial PPP flux is as high as the astroglial one (unpublished data), suggesting a high glycolytic metabolism in microglia.
Another issue to be solved is how microglial energy production is regulated in the light of physiological and pathophysiological aspects.179, 180 Namely, microglia can be ameboid in shape and can travel upon various stimulations. The physical movement of cells requires more energy than that of cells in a quiescent state. Whether microglia utilize glucose in the extracellular space or lactate or KBs is an interesting question that remains to be answered. Fatty acids could be another candidate, as cardiac and skeletal muscles preferentially utilize fatty acids over glucose. The alteration of MCT expression in microglia has been reported, indicating that lactate or KBs may also be energy substrates for microglial energy metabolism.181
DISCLOSURE
The author declares no conflict of interest.
ACKNOWLEDGMENT
The author thanks the Departments of Neurology and Physiology, Keio University School of Medicine, for their support. This work was supported in part by JSPS KAKENHI Grant Number 19K08002 and 17K09762.
REFERENCES
- 1. Somjen GG. Nervenkitt: Notes on the history of the concept of neuroglia. Glia 1988; 1: 2–9. [DOI] [PubMed] [Google Scholar]
- 2. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci 2003; 26: 523–530. [DOI] [PubMed] [Google Scholar]
- 3. Allen NJ, Barres BA. Neuroscience: Glia ‐ more than just brain glue. Nature 2009; 457: 675–677. [DOI] [PubMed] [Google Scholar]
- 4. Peters A, Palay SL, Webster HD. The Fine Structure of the Nervous System: Neurons and their Supporting Cells, 3rd edn. New York: Oxford University Press, 1991. [Google Scholar]
- 5. Pope A. Neuroglia: quantitative aspects In: Schoffeniels E, Franck G, Hertz L, Tower DB, (eds). Dynamic Properties of Glia Cells. Pergamon: Oxford, 1978. [Google Scholar]
- 6. García‐Marín V, García‐López P, Freire M. Cajal's contributions to glia research. Trends Neurosci 2007; 30: 479–487. [DOI] [PubMed] [Google Scholar]
- 7. Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010; 58: 1094–1103. [DOI] [PubMed] [Google Scholar]
- 8. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci 1999; 22: 208–215. [DOI] [PubMed] [Google Scholar]
- 9. Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature 2010; 468: 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dermietzel R, Hertberg EL, Kessler JA, Spray DC. Gap junctions between cultured astrocytes: Immunocytochemical, molecular, and electrophysiological analysis. J Neurosci 1991; 11: 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enkvist MO, McCarthy KD. Astroglial gap junction communication is increased by treatment with either glutamate or high K+ concentration. J Neurochem 1994; 62: 489–495. [DOI] [PubMed] [Google Scholar]
- 12. Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 2010; 11: 87–99. [DOI] [PubMed] [Google Scholar]
- 13. Escartin C, Rouach N. Astroglial networking contributes to neurometabolic coupling. Front Neuroenergetics 2013; 5: 4 10.3389/fnene.2013.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Charvériat M, Naus CC, Leybaert L, Sáez JC, Giaume C. Connexin‐dependent Neuroglial networking as a new therapeutic target. Front Cell Neurosci 2017; 11: 174 10.3389/fncel.2017.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science 1987; 234: 896–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harder DR, Alkayed NJ, Lange AR, Gebremedhin D, Roman RJ. Functional hyperemia in the brain: Hypothesis for astrocyte‐derived vasodilator metabolites. Stroke 1998; 29: 229–234. [DOI] [PubMed] [Google Scholar]
- 17. Zonta M, Angulo MC, Gobbo S et al Neuron‐to‐astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 2003; 6: 43–50. [DOI] [PubMed] [Google Scholar]
- 18. Jakovcevic D, Harder DR. Role of astrocytes in matching blood flow to neuronal activity. Curr Top Dev Biol 2007; 79: 75–97. [DOI] [PubMed] [Google Scholar]
- 19. Filosa JA, Morrison HW, Iddings JA, Du W, Kim KJ. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience 2016; 323: 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cauli B, Hamel E. Brain perfusion and astrocytes. Trends Neurosci 2018; 41: 409–413. [DOI] [PubMed] [Google Scholar]
- 21. Report of the Stroke Progressive Review Group . National Institute of Neurological Disorders and Stroke. Department of Health and Human Services, National Institutes of Health, Maryland, NIH Publication Number 02‐5117, 2002; 1–116.
- 22. del Zoppo GJ. Stroke and neurovascular protection. N Engl J Med 2006; 354: 553–555. [DOI] [PubMed] [Google Scholar]
- 23. Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron 2010; 67: 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalaria RN, Hase Y. Neurovascular ageing and age‐related diseases. Subcell Biochem 2019; 91: 477–499. [DOI] [PubMed] [Google Scholar]
- 25. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011; 12: 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 2018; 21: 1318–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sweeney MD, Montagne A, Sagare AP et al Vascular dysfunction‐the disregarded partner of Alzheimer's disease. Alzheimers Dement 2019; 15: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Presta I, Vismara M, Novellino F et al Innate immunity cells and the neurovascular unit. Int J Mol Sci 2018; 19: pii: E3856 10.3390/ijms19123856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thurgur H, Pinteaux E. Microglia in the neurovascular unit: Blood‐brain barrier‐microglia interactions after central nervous system disorders. Neuroscience 2019; 405: 55–67. [DOI] [PubMed] [Google Scholar]
- 30. Takahashi S, Izawa Y, Suzuki N. Astrogliopathy as a loss of astroglial protective function against glycoxidative stress under hyperglycemia. Rinsho Shinkeigaku 2012; 52: 41–51. [DOI] [PubMed] [Google Scholar]
- 31. Takahashi S, Izawa Y, Suzuki N. Astroglial pentose phosphate pathway rates in response to high‐glucose environments. ASN Neuro 2012; 4: pii: e00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi S, Iizumi T, Mashima K, Abe T, Suzuki N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 2014; 6: pii: 1759091414550997 10.1177/1759091414550997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iizumi T, Takahashi S, Mashima K et al A possible role of microglia‐derived nitric oxide by lipopolysaccharide in activation of astroglial pentose‐phosphate pathway via the Keap1/Nrf2 system. J Neuroinflammation 2016; 13: 99 10.1186/s12974-016-0564-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mashima K, Takahashi S, Minami K et al Neuroprotective role of Astroglia in Parkinson disease by reducing oxidative stress through dopamine‐induced activation of pentose‐phosphate pathway. ASN Neuro 2018; 10: 1759091418775562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hase Y, Chen A, Bates LL et al Severe white matter astrocytopathy in CADASIL. Brain Pathol 2018; 28: 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hara K, Shiga A, Fukutake T et al Association of HTRA1 mutations and familial ischemic cerebral small‐vessel disease. N Engl J Med 2009; 360: 1729–1739. [DOI] [PubMed] [Google Scholar]
- 37. Chen J, Van Gulden S, McGuire TL et al BMP‐responsive protease HtrA1 is differentially expressed in astrocytes and regulates Astrocytic development and injury response. J Neurosci 2018; 38: 3840–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodríguez‐Arellano JJ, Parpura V, Zorec R, Verkhratsky A. Astrocytes in physiological aging and Alzheimer's disease. Neuroscience 2016; 323: 170–182. [DOI] [PubMed] [Google Scholar]
- 39. Verkhratsky A, Parpura V, Rodriguez‐Arellano JJ, Zorec R. Astroglia in Alzheimer's disease. Adv Exp Med Biol 2019; 1175: 273–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teismann P, Tieu K, Cohen O et al Pathogenic role of glial cells in Parkinson's disease. Mov Disord 2003; 18: 121–129. [DOI] [PubMed] [Google Scholar]
- 41. Halliday GM, Stevens CH. Glia: Initiators and progressors of pathology in Parkinson's disease. Mov Disord 2011; 26: 6–17. [DOI] [PubMed] [Google Scholar]
- 42. Yamanaka K, Komine O. The multi‐dimensional roles of astrocytes in ALS. Neurosci Res 2018; 126: 31–38. [DOI] [PubMed] [Google Scholar]
- 43. Valori CF, Guidotti G, Brambilla L, Rossi D. Astrocytes in motor neuron diseases. Adv Exp Med Biol 2019; 1175: 227–272. [DOI] [PubMed] [Google Scholar]
- 44. Liddelow SA, Sofroniew MV. Astrocytes usurp neurons as a disease focus. Nat Neurosci 2019; 22: 512–513. [DOI] [PubMed] [Google Scholar]
- 45. Kira J. Autoimmunity in neuromyelitis optica and opticospinal multiple sclerosis: Astrocytopathy as a common denominator in demyelinating disorders. J Neurol Sci 2011; 311: 69–77. [DOI] [PubMed] [Google Scholar]
- 46. Ludwin SK, VTs R, Moore CS, Antel JP. Astrocytes in multiple sclerosis. Mult Scler 2016; 22: 1114–1124. [DOI] [PubMed] [Google Scholar]
- 47. Ponath G, Park C, Pitt D. The role of astrocytes in multiple sclerosis. Front Immunol 2018; 9: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nellessen A, Nyamoya S, Zendedel A et al Nrf2 deficiency increases oligodendrocyte loss, demyelination, neuroinflammation and axonal damage in an MS animal model. Metab Brain Dis 2019; 1–10. [Epub ahead of print]. 10.1007/s11011-019-00488-z [DOI] [PubMed] [Google Scholar]
- 49. Masaki K, Suzuki SO, Matsushita T et al Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS One. 2013; 8: e72919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: Lessons learned from neuromyelitis optica. Brain Pathol 2014; 24: 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Clarke DD, Sokoloff L. Circulation and energy metabolism of the brain In: Siegel G, Agranoff B, Albers RW, Fisher S, (eds). Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 6th edn. Philadelphia, PA: Lippincott‐Raven, 1999; 637–669. [Google Scholar]
- 52. Dienel GA. Energy metabolism in the brain In: Byrne JH, Roberts JL, (eds). From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience, 2nd edn. Academic Press: London, 2009; 49–110. [Google Scholar]
- 53. Itoh Y, Abe T, Takaoka R, Tanahashi N. Fluorometric determination of glucose utilization in neurons in vitro and in vivo. J Cereb Blood Flow Metab 2004; 24: 993–1003. [DOI] [PubMed] [Google Scholar]
- 54. Lundgaard I, Li B, Xie L et al Direct neuronal glucose uptake heralds activity‐dependent increases in cerebral metabolism. Nat Commun 2015; 6: 6807 10.1038/ncomms7807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia 1997; 21: 2–21. [DOI] [PubMed] [Google Scholar]
- 56. Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J Cereb Blood Flow Metab 2007; 27: 1766–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Klepper J, Voit T. Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: Impaired glucose transport into brain: A review. Eur J Pediatr 2002; 161: 295–304. [DOI] [PubMed] [Google Scholar]
- 58. Brockmann K. The expanding phenotype of GLUT1‐deficiency syndrome. Brain Dev 2009; 31: 545–552. [DOI] [PubMed] [Google Scholar]
- 59. Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose‐6‐phosphatase activity under normal and experimental conditions: 1. Neurons and glia. J Electron Microsc Tech 1986; 3: 413–437. [DOI] [PubMed] [Google Scholar]
- 60. Pfeiffer B, Elmer K, Roggendorf W, Reinhart PH, Hamprecht B. Immunohistpchmical demonstration of glycogen phosphorylase in rat brain slices. Histochemistry 1990; 94: 73–80. [DOI] [PubMed] [Google Scholar]
- 61. Sokoloff L. Energy metabolism and effects of energy depletion or exposure to glutamate. Can J Physiol Pharmacol 1992; 70: S107–S112. [DOI] [PubMed] [Google Scholar]
- 62. Pierre K, Magistretti PJ, Pellerin L. MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J Cereb Blood Flow Metab 2002; 22: 586–595. [DOI] [PubMed] [Google Scholar]
- 63. Pellerin L, Bergersen LH, Halestrap AP, Pierre K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res 2005; 79: 55–64. [DOI] [PubMed] [Google Scholar]
- 64. Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J Neurochem 2005; 94: 1–14. [DOI] [PubMed] [Google Scholar]
- 65. Bergersen LH. Lactate transport and signaling in the brain: Potential therapeutic targets and roles in body‐brain interaction. J Cereb Blood Flow Metab 2015; 35: 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 1994; 91: 10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takahashi S, Driscoll BF, Law MJ, Sokoloff L. Role of sodium and potassium ions in regulation of glucose metabolism in cultured astroglia. Proc Natl Acad Sci U S A 1995; 92: 4616–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sokoloff L, Takahashi S, Gotoh J, Driscoll BF, Law MJ. Contribution of astroglia to functionally activated energy metabolism. Dev Neurosci 1996; 18: 344–352. [PubMed] [Google Scholar]
- 69. Souza DG, Almeida RF, Souza DO, Zimmer ER. The astrocyte biochemistry. Semin Cell Dev Biol 2019; 95: 142–150. [DOI] [PubMed] [Google Scholar]
- 70. Bordone MP, Salman MM, Titus HE et al The energetic brain ‐ A review from students to students. J Neurochem 2019; 151: 139–165. 10.1111/jnc.14829 [DOI] [PubMed] [Google Scholar]
- 71. Jakoby P, Schmidt E, Ruminot I, Gutiérrez R, Barros LF, Deitmer JW. Higher transport and metabolism of glucose in astrocytes compared with neurons: A multiphoton study of hippocampal and cerebellar tissue slices. Cereb Cortex 2014; 24: 222–231. [DOI] [PubMed] [Google Scholar]
- 72. Supplie LM, Düking T, Campbell G et al Respiration‐deficient astrocytes survive as glycolytic cells in vivo. J Neurosci 2017; 37: 4231–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Barros LF, Bolanos JP, Bonvento G et al Current technical approaches to brain energy metabolism. Glia 2018; 66: 1138–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abe T, Takahashi S, Suzuki N. Oxidative metabolism in cultured rat astroglia: Effects of reducing the glucose concentration in the culture medium and of D‐aspartate or potassium stimulation. J Cereb Blood Flow Metab 2006; 26: 153–160. [DOI] [PubMed] [Google Scholar]
- 75. Zhao Y, Fung C, Shin D et al Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism spectrum disorders. Mol Psychiatry 2010; 15: 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bittar PG, Charnay Y, Pellerin L, Bouras C, Magistretti PJ. Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J Cereb Blood Flow Metab 1996; 16: 1079–1089. [DOI] [PubMed] [Google Scholar]
- 77. Itoh Y, Esaki T, Shimoji K et al Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci U S A 2003; 100: 4879–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bouzier‐Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab 2003; 23: 1298–1306. [DOI] [PubMed] [Google Scholar]
- 79. Bouzier‐Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: A comparative NMR study. Eur J Neurosci 2006; 24: 1687–1694. [DOI] [PubMed] [Google Scholar]
- 80. Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65: 1–105. [DOI] [PubMed] [Google Scholar]
- 81. Bröer S, Brookes N. Transfer of glutamine between astrocytes and neurons. J Neurochem 2001; 77: 705–719. [DOI] [PubMed] [Google Scholar]
- 82. Fox PT, Raichle ME. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci U S A 1986; 83: 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science 1988; 241: 462–464. [DOI] [PubMed] [Google Scholar]
- 84. Prichard J1, Rothman D, Novotny E et al Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci U S A 1991; 88: 5829–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Madsen PL, Cruz NF, Sokoloff L, Dienel GA. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: Excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J Cereb Blood Flow Metab 1999; 19: 393–400. [DOI] [PubMed] [Google Scholar]
- 86. Sonnewald U. Glutamate synthesis has to be matched by its degradation ‐ where do all the carbons go? J Neurochem 2014; 131: 399–406. [DOI] [PubMed] [Google Scholar]
- 87. Porras OH, Loaiza A, Barros LF. Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J Neurosci 2004; 24: 9669–9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Aubert A, Costalat R, Magistretti PJ, Pellerin L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc Natl Acad Sci U S A 2005; 102: 16448–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Laughton JD, Bittar P, Charnay Y et al Metabolic compartmentalization in the human cortex and hippocampus: Evidence for a cell‐ and region‐specific localization of lactate dehydrogenase 5 and pyruvate dehydrogenase. BMC Neurosci 2007; 8: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pellerin L, Bouzier‐Sore AK, Aubert A et al Activity‐dependent regulation of energy metabolism by astrocytes: An update. Glia 2007; 55: 1251–1262. [DOI] [PubMed] [Google Scholar]
- 91. Jolivet R, Allaman I, Pellerin L, Magistretti PJ, Weber B. Comment on recent modeling studies of astrocyte‐neuron metabolic interactions. J Cereb Blood Flow Metab 2010; 30: 1982–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab 2012; 32: 1152–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mazuel L, Blanc J, Repond C et al A neuronal MCT2 knockdown in the rat somatosensory cortex reduces both the NMR lactate signal and the BOLD response during whisker stimulation. PLoS One 2017; 12: e0174990 10.1371/journal.pone.0174990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pellerin L. Neuroenergetics: Astrocytes have a sweet spot for glucose. Curr Biol 2018; 28: R1258–R1260. [DOI] [PubMed] [Google Scholar]
- 95. Dienel GA, Hertz L. Glucose and lactate metabolism during brain activation. J Neurosci Res 2001; 66: 824–838. [DOI] [PubMed] [Google Scholar]
- 96. Hertz L, Dienel GA. Lactate transport and transporters: General principles and functional roles in brain cells. J Neurosci Res 2005; 79: 11–18. [DOI] [PubMed] [Google Scholar]
- 97. Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 2007; 27: 219–249. [DOI] [PubMed] [Google Scholar]
- 98. Dienel GA, Ball KK, Cruz NF. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: Implications for glycogen turnover. J Neurochem 2007; 102: 466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dienel GA. Astrocytes are 'good scouts': Being prepared also helps neighboring neurons. J Cereb Blood Flow Metab 2010; 30: 1893–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dienel GA. Brain lactate metabolism: The discoveries and the controversies. J Cereb Blood Flow Metab 2012; 32: 1107–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci 2013; 36: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Dienel GA, Cruz NF. Contributions of glycogen to astrocytic energetics during brain activation. Metab Brain Dis 2015; 30: 281–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dienel GA. The metabolic trinity, glucose‐glycogen‐lactate, links astrocytes and neurons in brain energetics, signaling, memory, and gene expression. Neurosci Lett 2017; 637: 18–25. [DOI] [PubMed] [Google Scholar]
- 104. Dienel GA. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte‐neuron lactate shuttle in brain. J Neurosci Res 2017; 95: 2103–2125. [DOI] [PubMed] [Google Scholar]
- 105. Dienel GA. Brain glucose metabolism: Integration of energetics with function. Physiol Rev 2019; 99: 949–1045. [DOI] [PubMed] [Google Scholar]
- 106. Morland C, Lauritzen KH, Puchades M et al The lactate receptor, G‐protein‐coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J Neurosci Res 2015; 93: 1045–1055. [DOI] [PubMed] [Google Scholar]
- 107. de Castro AH, Briquet M, Schmuziger C et al The lactate receptor HCAR1 modulates neuronal network activity through the activation of Gα and Gβγ subunits. J Neurosci 2019; 39: 4422–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Noto Y, Shibuya K, Vucic S, Kiernan MC. Novel therapies in development that inhibit motor neuron hyperexcitability in amyotrophic lateral sclerosis. Expert Rev Neurother 2016; 16: 1147–1154. [DOI] [PubMed] [Google Scholar]
- 109. Suzuki A, Stern SA, Bozdagi O et al Astrocyte‐neuron lactate transport is required for long‐term memory formation. Cell 2011; 144: 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hertz L, Chen Y. Glycogenolysis, an astrocyte‐specific reaction, is essential for both Astrocytic and neuronal activities involved in learning. Neuroscience 2018; 370: 27–36. [DOI] [PubMed] [Google Scholar]
- 111. Alberini CM, Cruz E, Descalzi G, Bessières B, Gao V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018; 66: 1244–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dienel GA. Does shuttling of glycogen‐derived lactate from astrocytes to neurons take place during neurotransmission and memory consolidation? J Neurosci Res 2019; 97: 863–882. [DOI] [PubMed] [Google Scholar]
- 113. Descalzi G, Gao V, Steinman MQ, Suzuki A, Alberini CM. Lactate from astrocytes fuels learning‐induced mRNA translation in excitatory and inhibitory neurons. Commun Biol 2019; 2: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med 2009; 30: 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lu SC. Glutathione synthesis. Biochim Biophys Acta 1830; 2013: 3143–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem 2000; 74: 1434–1442. [DOI] [PubMed] [Google Scholar]
- 117. Bolaños JP. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J Neurochem 2016; 139 (Suppl 2): 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2‐regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002; 62: 5196–5203. [PubMed] [Google Scholar]
- 119. Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF‐E2‐related factor‐2‐dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 2003; 278: 12029–12038. [DOI] [PubMed] [Google Scholar]
- 120. Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med 2008; 74: 1526–1539. [DOI] [PubMed] [Google Scholar]
- 121. Vargas MR, Johnson JA. The Nrf2‐ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med 2009; 11: e17 10.1017/S1462399409001094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med 2011; 17: 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Carpentier PA, Duncan DS, Miller SD. Glial toll‐like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun 2008; 22: 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gorina R, Font‐Nieves M, Márquez‐Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88‐dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011; 59: 242–255. [DOI] [PubMed] [Google Scholar]
- 125. Liddelow SA, Guttenplan KA, Clarke LE et al Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017; 541: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Guzmán M, Blázquez C. Is there an astrocyte‐neuron ketone body shuttle? Trends Endocrinol Metab 2001; 12: 169–173. [DOI] [PubMed] [Google Scholar]
- 127. Guzmán M, Blázquez C. Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins Leukot Essent Fatty Acids 2004; 70: 287–292. [DOI] [PubMed] [Google Scholar]
- 128. Achanta LB, Rae CD. β‐Hydroxybutyrate in the brain: One molecule, multiple mechanisms. Neurochem Res 2017; 42: 35–49. [DOI] [PubMed] [Google Scholar]
- 129. Mukherjee S, Suresh SN. Neuron‐astrocyte liaison to maintain lipid metabolism of brain. Trends Endocrinol Metab 2019; 30: 573–575. [DOI] [PubMed] [Google Scholar]
- 130. Ioannou MS, Jackson J, Sheu SH et al Neuron‐astrocyte metabolic coupling protects against activity‐induced fatty acid toxicity. Cell 2019; 177: 1522–1535. [DOI] [PubMed] [Google Scholar]
- 131. Barber CN, Raben DM. Lipid metabolism crosstalk in the brain: Glia and neurons. Front Cell Neurosci 2019; 13: 212 10.3389/fncel.2019.00212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cardell M, Koide T, Wieloch T. Pyruvate dehydrogenase activity in the rat cerebral cortex following cerebral ischemia. J Cereb Blood Flow Metab 1989; 9: 350–357. [DOI] [PubMed] [Google Scholar]
- 133. Fukuchi T, Katayama Y, Kamiya T, McKee A, Kashiwagi F, Terashi A. The effect of duration of cerebral ischemia on brain pyruvate dehydrogenase activity, energy metabolites, and blood flow during reperfusion in gerbil brain. Brain Res 1998; 792: 59–65. [DOI] [PubMed] [Google Scholar]
- 134. Bixel MG, Hamprecht B. Generation of ketone bodies from leucine by cultured astroglial cells. J Neurochem 1995; 65: 2450–2461. [DOI] [PubMed] [Google Scholar]
- 135. Dhopeshwarkar GA, Mead JF. Uptake and transport of fatty acids into the brain and the role of the blood‐brain barrier system. Adv Lipid Res 1973; 11: 109–142. [DOI] [PubMed] [Google Scholar]
- 136. Smith QR, Nagura H. Fatty acid uptake and incorporation in brain: Studies with the perfusion model. J Mol Neurosci 2001; 16: 167–172. [DOI] [PubMed] [Google Scholar]
- 137. Mitchell RW, On NH, Del Bigio MR, Miller DW, Hatch GM. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem 2011; 117: 735–746. [DOI] [PubMed] [Google Scholar]
- 138. Hamilton JA, Brunaldi K. A model for fatty acid transport into the brain. J Mol Neurosci 2007; 33: 12–17. [DOI] [PubMed] [Google Scholar]
- 139. Katz R, Hamilton JA, Pownall HJ et al Brain uptake and utilization of fatty acids, lipids & lipoproteins: Recommendations for future research. J Mol Neurosci 2007; 33: 146–150. [DOI] [PubMed] [Google Scholar]
- 140. Pan Y, Scanlon MJ, Owada Y, Yamamoto Y, Porter CJ, Nicolazzo JA. Fatty acid‐binding protein 5 facilitates the blood‐brain barrier transport of Docosahexaenoic acid. Mol Pharm 2015; 12: 4375–4385. [DOI] [PubMed] [Google Scholar]
- 141. Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5‐aminoimidazole‐4‐carboxamide ribonucleoside. A specific method for activating AMP‐activated protein kinase in intact cells? Eur J Biochem 1995; 229: 558–565. [DOI] [PubMed] [Google Scholar]
- 142. Leary SC, Hill BC, Lyons CN et al Chronic treatment with azide in situ leads to an irreversible loss of cytochrome c oxidase activity via holoenzyme dissociation. J Biol Chem 2002; 277: 11321–11328. [DOI] [PubMed] [Google Scholar]
- 143. Blázquez C, Woods A, de Ceballos ML, Carling D, Guzmán M. The AMP‐activated protein kinase is involved in the regulation of ketone body production by astrocytes. J Neurochem 1999; 73: 1674–1682. [DOI] [PubMed] [Google Scholar]
- 144. Graff EC, Fang H, Wanders D, Judd RL. Anti‐inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 2016; 65: 102–113. [DOI] [PubMed] [Google Scholar]
- 145. Rahman M, Muhammad S, Khan MA et al The β‐hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun 2014; 5: 3944 10.1038/ncomms4944 [DOI] [PubMed] [Google Scholar]
- 146. Offermanns S, Schwaninger M. Nutritional or pharmacological activation of HCA(2) ameliorates neuroinflammation. Trends Mol Med 2015; 21: 245–255. [DOI] [PubMed] [Google Scholar]
- 147. Parodi B, Rossi S, Morando S et al Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol 2015; 130: 279–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog Neurobiol 2014; 115: 157–188. [DOI] [PubMed] [Google Scholar]
- 149. Wu QJ, Tymianski M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol Brain 2018; 11: 15 10.1186/s13041-018-0357-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schell MJ, Molliver ME, Snyder SH. D‐serine, an endogenous synaptic modulator: Localization to astrocytes and glutamate‐stimulated release. Proc Natl Acad Sci U S A 1995; 92: 3948–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fuchs SA, Berger R, Klomp LW, de Koning TJ. D‐amino acids in the central nervous system in health and disease. Mol Genet Metab 2005; 85: 168–180. [DOI] [PubMed] [Google Scholar]
- 152. Wolosker H, Dumin E, Balan L, Foltyn VN. D‐amino acids in the brain: D‐serine in neurotransmission and neurodegeneration. FEBS J 2008; 275: 3514–3526. [DOI] [PubMed] [Google Scholar]
- 153. Mothet JP, Parent AT, Wolosker H et al D‐serine is an endogenous ligand for the glycine site of the N‐methyl‐D‐aspartate receptor. Proc Natl Acad Sci U S A 2000; 97: 4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Papouin T, Ladépêche L, Ruel J et al Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012; 150: 633–646. [DOI] [PubMed] [Google Scholar]
- 155. Sasabe J, Suzuki M. Distinctive roles of D‐amino acids in the Homochiral world: Chirality of amino acids modulates mammalian physiology and pathology. Keio J Med 2019; 68: 1–16. [DOI] [PubMed] [Google Scholar]
- 156. Wolosker H, Sheth KN, Takahashi M et al Purification of serine racemase: Biosynthesis of the neuromodulator D‐serine. Proc Natl Acad Sci U S A 1999; 96: 721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Yamasaki M, Yamada K, Furuya S, Mitoma J, Hirabayashi Y, Watanabe M. 3‐Phosphoglycerate dehydrogenase, a key enzyme for l‐serine biosynthesis, is preferentially expressed in the radial glia/astrocyte lineage and olfactory ensheathing glia in the mouse brain. J Neurosci 2001; 21: 7691–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Furuya S. An essential role for de novo biosynthesis of L‐serine in CNS development. Asia Pac J Clin Nutr 2008; 17(Suppl 1): 312–315. [PubMed] [Google Scholar]
- 159. Wolosker H, Balu DT, Coyle JT. The rise and fall of the d‐serine‐mediated Gliotransmission hypothesis. Trends Neurosci 2016; 39: 712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Mustafa AK, Ahmad AS, Zeynalov E et al Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J Neurosci 2010; 30: 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Abe T, Suzuki M, Sasabe J et al Cellular origin and regulation of D‐ and L‐serine in in vitro and in vivo models of cerebral ischemia. J Cereb Blood Flow Metab 2014; 34: 1928–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wang GH, Jiang ZL, Chen ZQ, Li X, Peng LL. Neuroprotective effect of L‐serine against temporary cerebral ischemia in rats. J Neurosci Res 2010; 88: 2035–2045. [DOI] [PubMed] [Google Scholar]
- 163. Ren TJ, Qiang R, Jiang ZL et al Improvement in regional CBF by L‐serine contributes to its neuroprotective effect in rats after focal cerebral ischemia. PLoS One. 2013; 8: e67044 10.1371/journal.pone.0067044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sun L, Qiang R, Yang Y et al L‐serine treatment may improve neurorestoration of rats after permanent focal cerebral ischemia potentially through improvement of neurorepair. PLoS One. 2014; 9: e93405 10.1371/journal.pone.0093405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Bedner P, Jabs R, Steinhäuser C. Properties of human astrocytes and NG2 glia. Glia 2019. 10.1002/glia.23725 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 166. Thevenet J, De Marchi U, Domingo JS et al Medium‐chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte‐neuron lactate and ketone body shuttle systems. FASEB J 2016; 30: 1913–1926. [DOI] [PubMed] [Google Scholar]
- 167. Black JA, Waxman SG. The perinodal astrocyte. Glia 1988; 1: 169–183. [DOI] [PubMed] [Google Scholar]
- 168. Butt AM, Duncan A, Berry M. Astrocyte associations with nodes of Ranvier: Ultrastructural analysis of HRP‐filled astrocytes in the mouse optic nerve. J Neurocytol 1994; 23: 486–499. [DOI] [PubMed] [Google Scholar]
- 169. Fünfschilling U, Supplie LM, Mahad D et al Glycolytic oligodendrocytes maintain myelin and long‐term axonal integrity. Nature 2012; 485: 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Rosko L, Smith VN, Yamazaki R, Huang JK. Oligodendrocyte bioenergetics in health and disease. Neuroscientist 2019; 25: 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Cambron M, D'Haeseleer M, Laureys G, Clinckers R, Debruyne J, De Keyser J. White‐matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. J Cereb Blood Flow Metab 2012; 32: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zeis T, Allaman I, Gentner M et al Metabolic gene expression changes in astrocytes in multiple sclerosis cerebral cortex are indicative of immune‐mediated signaling. Brain Behav Immun 2015; 48: 313–325. [DOI] [PubMed] [Google Scholar]
- 173. Koper JW, Lopes‐Cardozo M, Van Golde LM. Preferential utilization of ketone bodies for the synthesis of myelin cholesterol in vivo. Biochim Biophys Acta 1981; 666: 411–417. [DOI] [PubMed] [Google Scholar]
- 174. Poduslo SE, Miller K. Ketone bodies as precursors for lipid synthesis in neurons, astrocytes, and oligodendroglia (myelin) in hyperthyroidism, hyperketonemia and hypoketonemia. Neurochem Int 1991; 18: 85–88. [DOI] [PubMed] [Google Scholar]
- 175. Kadish I, Thibault O, Blalock EM et al Hippocampal and cognitive aging across the lifespan: A bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci 2009; 29: 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kıray H, Lindsay SL, Hosseinzadeh S, Barnett SC. The multifaceted role of astrocytes in regulating myelination. Exp Neurol 2016; 283: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Ginhoux F, Prinz M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb Perspect Biol 2015. Jul 1; 7: a020537 10.1101/cshperspect.a020537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Tay TL, Carrier M, Tremblay MÈ. Physiology of microglia. Adv Exp Med Biol 2019; 1175: 129–148. [DOI] [PubMed] [Google Scholar]
- 179. Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia 2018; 66: 1200–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Aldana BI. Microglia‐specific metabolic changes in Neurodegeneration. J Mol Biol 2019; 431: 1830–1842. [DOI] [PubMed] [Google Scholar]
- 181. Moreira TJ, Pierre K, Maekawa F et al Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab 2009; 29: 1273–1283. [DOI] [PubMed] [Google Scholar]