

Abstract
Hereditary transthyretin‐mediated (hATTR) amyloidosis is a rare, inherited, progressively debilitating, and often fatal disease caused by deposition of mutated transthyretin (TTR) protein. Patisiran is an RNA interference therapeutic comprising a novel small interfering ribonucleic acid (ALN‐18328) formulated with 2 novel lipid excipients, DLin‐MC3‐DMA and PEG2000‐C‐DMG, in a lipid nanoparticle targeted to inhibit hepatic TTR synthesis. Here we report the pharmacokinetics (PK) of ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG from a phase 2 multiple‐ascending‐dose study and its open‐label extension (OLE) in patients with hATTR amyloidosis. Twenty‐nine patients received 2 intravenous infusions of patisiran of 0.01, 0.05, 0.15, or 0.3 mg/kg at 3‐ or 4‐week intervals; of these, 27 patients received 0.3 mg/kg once every 3 weeks over 24 months in the OLE study. Plasma PK profiles of ALN‐18328 and DLin‐MC3‐DMA exhibited 2 phases, the first characterized by a short distribution half‐life and the second by a minor peak and relatively long terminal elimination half‐life. PK exposures to 3 analytes increased proportionally across the dose range of 0.01 to 0.3 mg/kg. For ALN‐18328, mean terminal elimination half‐life was 3.2 days, mean total clearance was 3.0 mL/h/kg, and urinary excretion was negligible. All 3 analytes exhibited stable PK profiles with chronic dosing over 2 years. The 2‐ to 3‐fold plasma accumulation (AUCτ) of ALN‐18328 at steady state is attributable to the association of ALN‐18328 with the cationic lipid DLin‐MC3‐DMA. There was no appreciable accumulation of PEG2000‐C‐DMG.
Keywords: amyloidosis, lipid nanoparticle, patisiran, pharmacokinetics (PK), RNAi therapeutic, small interfering ribonucleic acid (siRNA)
Hereditary transthyretin (TTR)‐mediated (hATTR) amyloidosis is an inherited, rapidly progressive, and often fatal disease caused by mutations in the TTR gene.1, 2 TTR is primarily produced in the liver and forms a tetramer that transports vitamin A and the hormone thyroxine in association with retinol‐binding protein (RBP) in the plasma and cerebrospinal fluid.1, 2, 3 The pathogenic mutations in the TTR gene in patients with hATTR amyloidosis result in a misfolded TTR protein that accumulates as amyloid deposits at multiple sites including peripheral nerves, heart, kidney, and the gastrointestinal tract.1, 4 This gives rise to a heterogeneous clinical presentation, including neuropathy and/or cardiomyopathy, as well as other disease manifestations.1, 2, 5, 6, 7
hATTR amyloidosis affects approximately 50 000 people worldwide.1 V30M is the most common TTR mutation in Europe,8 with prevalence reaching up to 1 in 1000 in endemic areas in Portugal, Sweden, and Japan.1, 8 The most common TTR mutation in the United States is V122I, with a reported prevalence of approximately 4% in African Americans.9 hATTR amyloidosis can occur at any stage of adult life; penetrance varies widely, although it is typically highest in older patients.1, 10 hATTR amyloidosis has a rapid progression with a median survival of 4.7 years following diagnosis, reduced to 3.4 years for patients presenting with cardiomyopathy.11, 12, 13, 14
Treatment options for hATTR amyloidosis include TTR stabilizers (tafamidis and diflunisal), and TTR‐lowering therapies (orthotopic liver transplantation, inotersen, and patisiran).1, 15, 16, 17 The TTR‐lowering pharmacotherapies (inotersen and patisiran) inhibit production of the pathogenic protein, which is associated with clinical benefit in other forms of amyloidosis and represents a significant advance in the treatment of patients with hATTR amyloidosis.18 Patisiran is a first‐in‐class RNA interference therapeutic and was approved in the United States and Europe in August 2018 to treat the polyneuropathy caused by hATTR amyloidosis.19, 20 The recommended dosage is 0.3 mg/kg patisiran, administered as an intravenous infusion once every 3 weeks for patients weighing < 100 kg and 30 mg once every 3 weeks for patients weighing ≥ 100 kg.19, 20
The patisiran drug substance is a novel, synthetic, double‐stranded small interfering ribonucleic acid (siRNA) formed by 2 partially complementary single strands with 21 nucleotides per strand. Patisiran is formulated as a lipid nanoparticle (LNP) composed of the siRNA (ALN‐18328) and 4 lipid excipients, of which 2 are constituents of other approved drugs (DSPC [1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine] and cholesterol)21, 22 and 2 are novel excipients (DLin‐MC3‐DMA [(6Z,9Z,28Z,31Z)‐heptatriaconta‐6,9,28,31‐tetraen‐19‐yl‐4‐(dimethylamino)butanoate] and PEG2000‐C‐DMG [α‐(3′‐{[1,2‐di(myristyloxy)proponoxy]carbonylamino}propyl)‐ω‐methoxy, polyoxyethylene]).23 The siRNA is encapsulated in the LNP to protect it from degradation by endogenous nucleases and to facilitate its targeted delivery into hepatocytes, the primary site of TTR synthesis.23, 24, 25, 26, 27 The ratio of LNP components is optimized to ensure delivery to the liver, and the final LNP product composition has been described elsewhere.28, 29
DLin‐MC3‐DMA is important for particle formation, fusogenicity, cellular uptake, and endosomal release of the siRNA.23, 24 PEG2000‐C‐DMG aids LNP stability in the circulation and provides optimal circulation time, enabling uptake of patisiran into the liver.23, 27 DSPC and cholesterol provide physicochemical stability to the LNP.23 Following intravenous administration, the LNPs are opsonized by apolipoprotein E (ApoE) and then enter the liver, where they bind to ApoE receptors on the surface of hepatocytes.23, 30 Once ALN‐18328 enters the cytoplasm of hepatocytes, it controls TTR gene expression by binding to the RNA‐induced silencing complex (RISC), which, in turn, specifically cleaves wild‐type and mutant TTR messenger RNA, thereby reducing TTR synthesis.31, 32
In a phase 1 clinical study of patisiran in healthy subjects, single‐dose ALN‐18328 plasma exposures increased in an approximately dose‐proportional manner over the dose range of 0.01 to 0.5 mg/kg, yielding a rapid and dose‐dependent reduction of serum TTR.26 Maximum TTR reduction from baseline was generally reached around 10 days after single‐dose administration, with maximum mean TTR reduction of 87.6% and 93.8%, seen at 0.3 and 0.5 mg/kg doses, respectively.28 In the multiple‐ascending‐dose (MAD) phase 2 clinical study in patients with hATTR amyloidosis with polyneuropathy, maximum TTR reduction of 82.9% to 86.7% was observed at the highest dose of 0.3 mg/kg, with greater sustained TTR reduction from baseline achieved with patisiran 0.3 mg/kg administered intravenously once every 3 weeks compared with dosing once every 4 weeks.33 Consequently, the patisiran dose of 0.3 mg/kg once every 3 weeks was evaluated in the subsequent open‐label extension (OLE) study, in which it resulted in rapid and sustained mean serum TTR reduction of 82% over a period of 24 months.34 In the pivotal phase 3 trial in patients with hATTR amyloidosis with polyneuropathy (APOLLO), a median TTR reduction of 81% over a period of 18 months was observed following patisiran 0.3 mg/kg treatment.17 Consistent with the role of TTR as a major transporter of the RBP‐retinol complex in serum, reductions of vitamin A and RBP levels paralleled the reduction in TTR but to a lesser extent (62% and 45%, respectively).35 APOLLO data also showed that patisiran had an acceptable benefit:risk profile and significantly improved polyneuropathy, quality of life, motor strength, disability, gait speed, nutritional status, and autonomic symptoms compared with placebo.17
In the APOLLO trial, patisiran was generally well tolerated, with the majority of adverse events (AEs) being mild or moderate in severity.17 In addition, the proportion of patients who experienced AEs leading to discontinuation was lower in the patisiran group than in the placebo group in APOLLO.17 Patisiran treatment was also generally well tolerated in other clinical studies.26, 33, 36
Here we report the plasma and urine pharmacokinetic (PK) properties of the key LNP constituents ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG in the phase 2 MAD and OLE studies in patients with hATTR amyloidosis with polyneuropathy. Safety and pharmacodynamic (PD) data for both MAD and OLE studies have been reported elsewhere.33, 34
Methods
The PK analysis was based on data from the phase 2 MAD and phase 2 OLE clinical studies.33, 34
Study Designs
Both studies were conducted according to the guidelines of the International Council on Harmonization, the World Health Organization Declaration of Helsinki, and the Health Insurance Portability and Accountability Act of 1996. Written informed consent was obtained from all participants in the study. The study protocols were approved by local institutional review boards and ethics committees, and all subsequent protocol amendments underwent the same approval procedure.
The phase 2 MAD study was a multicenter, international, open‐label trial of patisiran in patients with hATTR amyloidosis with polyneuropathy.33 Eligible patients were adults (≥18 years) with biopsy‐proven ATTR amyloidosis and mild to moderate neuropathy. Patients were excluded if they had received a liver transplant. Nine cohorts comprising 3 or 4 patients received 2 doses of patisiran, administered as an intravenous infusion over 60 or 70 minutes. Patients in cohorts 1‐3 received 0.01, 0.05, and 0.15 mg/kg patisiran, respectively, once every 4 weeks, patients in cohorts 4 and 5 received 0.3 mg/kg patisiran once every 4 weeks, and patients in cohorts 6‐9 received 0.3 mg/kg patisiran once every 3 weeks. After the dose was determined using cohorts 1‐4, cohorts 5‐9 were used to confirm findings in prior cohorts, to assess alternative dosing frequency, and to evaluate the use of a reduced premedication regimen. Details of the original and reduced premedication regimens have been previously reported in the supplementary materials of Suhr et al.33
The phase 2 OLE study was a multicenter international trial of patisiran in patients with hATTR amyloidosis with polyneuropathy who were originally dosed in the phase 2 MAD study and were eligible to continue treatment.34 Patients received intravenous infusions of patisiran 0.3 mg/kg once every 3 weeks over approximately 70 minutes for approximately 2 years. Patients received an oral daily supplemental dose of the recommended daily allowance of vitamin A.
Evaluation of patisiran PK was included in objectives of both studies.
PK Assessments
In both the phase 2 MAD study and the phase 2 OLE study, serial blood samples were collected to analyze the plasma concentrations of ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG. Separate plasma samples were collected to analyze the plasma concentrations of free and encapsulated ALN‐18328. In addition, urine samples were collected in both studies for analysis of the urine concentrations of ALN‐18328 and 4‐dimethylaminobutyric acid (DMBA), a metabolite of DLin‐MC3‐DMA.
In the phase 2 MAD study, blood samples for assessment of PK parameters associated with the first dose were collected predose, at the end of infusion (EOI), and 5, 10, and 30 minutes and 1, 2, 4, 6, 24, 48, 168 (day 7), 336 (day 14), and 504 (day 21) hours postinfusion, with an additional sample collected at 672 hours (day 28) for the once‐every‐4‐week regimens. For the second dose, a similar sampling schedule was used, but with additional samples taken on days 35, 91, and 187 (once every 3 weeks) or days 84 and 180 (once every 4 weeks) to follow the terminal elimination phase. For patients in cohorts 3‐9, a separate plasma sample was collected on day 0 at EOI and 2 hours postinfusion, for analysis of free and encapsulated ALN‐18328 siRNA. For each patisiran administration, urine PK samples were collected predose (within 1 hour of the planned dosing start) and from 0 to 6 hours postinfusion.
In the OLE study, PK samples were collected after the first dose in week 1, after 12 doses in week 34, and after 36 doses in week 106. On each occasion, blood samples were collected predose, at EOI, and 1, 2, 4, 6, and 24 hours postdose and 3, 7, 17, and 21 days post‐EOI. In addition, sparse PK samples were taken at EOI (maximum concentration at EOI [Cmax]) and at the end of the dosing interval (Cmin) in weeks 4, 7, 10, 13, 25, 27, 40, 52, 54, 67, 79, 81, 94, and 106. In weeks 1, 34, and 106, spot urine samples were collected predose (within 1 hour of dosing start), at EOI, and 2, 6, and 24 hours post‐EOI and 3, 7, and 17 days postinfusion.
Antidrug Antibody Assessment
Serum samples for the assessment of antidrug antibodies (ADAs) were taken predose on day 0 and on days 7, 21 (once every 3 weeks), 28 (once every 4 weeks), 56, and 208 in the MAD study and predose in week 1 and weeks 4, 13, 25, 28, 31, 40, 52, 79, and 106 in the OLE study.
Determination of Plasma Concentrations
Plasma concentrations of encapsulated and free ALN‐18328 were measured with a validated ATTO‐Probe using a high‐performance liquid chromatography (HPLC)/fluorescence detection method.35 The lower limit of quantification (LLOQ) was 1 ng/mL. For encapsulated ALN‐18328, interassay precision ranged from 1.7% to 8.0% (percent coefficient of variation [CV%]), and the interassay accuracy ranged from ‐5.0% to 4.0% (percent difference between found and nominal concentrations). For free ALN‐18328, interassay precision ranged from 0.0% to 5.7% (CV%), and interassay accuracy ranged from ‐6.7% to 5.0%.
Plasma concentrations of PEG2000‐C‐DMG were determined using a validated liquid chromatography with tandem mass spectrometry (LC‐MS/MS) method. The LLOQ of the assay was 5 ng/mL. Interassay precision ranged from 2.2% to 8.3% (CV%), and interassay accuracy ranged from ‐2.7% to 3.0%. Plasma concentrations of DLin‐MC3‐DMA were determined with an LC‐MS/MS method. Two methods were developed and validated: low range (0.500‐100 ng/mL) and high range (50.0‐50 000 ng/mL). For the high‐range assay, interassay precision ranged from 0.0% to 4.5% (CV%), and interassay accuracy from ‐4.5% to 2.8%. For the low‐range assay, interassay precision ranged from 2.8% to 4.8% (CV%), and interassay accuracy from ‐2.7% to 0.8%.
All 3 analytes in human plasma were stable during storage (‐70°C) over a period of approximately 15‐66 months and during processing and sample analysis.
Determination of Urine Concentrations
Urine concentrations of ALN‐18328 were measured using a validated ATTO‐Probe with an HPLC/fluorescence detection method.35 The LLOQ of the assay was 1 ng/mL; interassay precision ranged from 0.0% to 8.9% (CV%), and interassay accuracy ranged from ‐0.7% to 6.0%.
Urine concentrations of DMBA in human urine treated with 0.1% to 1.0% CHAPS (3‐[(3‐cholamidopropyl)dimethylammonio]‐1‐propanesulfonate) were measured using a validated LC‐MS/MS method. The method used protein precipitation extraction followed by monitoring the MS/MS transition. The validation was conducted using the stable labeled isotope 4‐di‐(trideuteromethyl)‐aminobutanoic (2D2, 3D2, 4D2) acid in all standards and quality control samples, as measurable levels of endogenous DMBA are expected to be present in human urine. The assay had an LLOQ of 1 ng/mL. Interassay precision ranged from 0.8% to 2.0% (CV%), and interassay accuracy from ‐5.0% to ‐1.8%. Low levels of DMBA were present in the predose urine samples; therefore, all postdose values were corrected by subtracting the predose concentrations.
Both ALN‐18328 and DMBA in treated human urine were stable during storage (‐70°C) over a period of approximately 55‐70 months and during processing and sample analysis.
Determination of Serum ADAs
ADAs to PEG2000‐C‐DMG were detected using a previously described validated enzyme‐linked immunosorbent assay.35
PK and Statistical Analyses
The plasma and urine concentrations of ALN‐18328, DLin‐MC3‐DMA, PEG2000‐C‐DMG, and DMBA were analyzed using noncompartmental analysis employing Phoenix WinNonlin (version 6.3; Pharsight), Microsoft Excel, and R (version 3.1). All PK parameters were estimated using actual PK sampling times and actual doses for each component of patisiran.
Phase 2 MAD Study PK Parameters
For the phase 2 MAD study, the following PK parameters were estimated following the first and second infusions of patisiran: the Cmax; maximum concentration at the secondary peak (Cmax2); trough concentration at the end of the dosing interval (Cmin); the time to Cmax (tmax); area under the curve (AUC) from the start of infusion to the last quantifiable time point (AUC0‐last); AUC from the start of infusion to the lowest concentration in the first phase of the PK profile following dose administration (AUCp1); AUC from the end of the first PK phase to the last quantifiable concentration of the second PK phase (AUCp2); concentration at the end of the first PK phase (Cp1,min); half‐life for the first phase of the PK profile (t½α); terminal elimination half‐life for the second PK phase (t½β), calculated following administration of the second dose, as the PK sampling duration was sufficient to estimate t½β; systemic clearance (CL); and volume of distribution at steady state (Vss). The ratio of free to encapsulated ALN‐18328 plasma concentrations was computed at each sampling point. The amount of analyte excreted in urine (Ae) was calculated as the product of concentration and volume over the 6‐hour collection interval postdose. Renal clearance (CLR) was calculated as CLR = Ae/AUC0‐6, where AUC0‐6 is the plasma AUC from the start of infusion to 6 hours following dose administration. Analyte recovered in urine as a percentage of dose (fe) over 6 hours postdose was calculated as Ae/dose × 100%. Calculation of the DLin‐MC3‐DMA dose excreted in urine was based on the amount of DMBA measured in urine multiplied by the following molecular‐weight correction: molecular weight of DLin‐MC3‐DMA (642.093 g/mol)/molecular weight of DMBA (131.17 g/mol).
Dose proportionality of plasma PK parameters of ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG was assessed using a power model.37, 38 A statistical linear relationship between ln‐transformed PK parameters (AUC0‐last and Cmax) and ln‐transformed dose was fitted using the following equation, where Y represents the PK parameter of interest, β represents the slope, and ε represents the error:
The relationship between PK parameter and the ln‐transformed dose was statistically verified as being linear if the quadratic term LnDose2 was not statistically significant (P > .05). For ln‐transformed PK parameters (ie, AUC0‐last and Cmax) with statistically significant linear relationships, 95% confidence intervals (CIs) were derived for the β estimate. Dose proportionality was evaluated based on the establishment of a statistical linear relationship and the derived 95%CI for the β estimate being approximately within the United States Food and Drug Administration guidance for bioequivalence (0.80‐1.25).37, 38
Phase 2 OLE Study PK Parameters
In the phase 2 OLE study, the PK parameters were estimated in a manner similar to those described for the phase 2 MAD study. As the OLE was a multiple‐dose study, AUC was calculated during dosing interval τ (AUCτ). Steady‐state (ss) PK parameters were determined from values obtained in week 106. The accumulation ratio (Rac) was calculated as the ratio of PK parameters obtained in week 106 to those obtained after the first dose.
Time to reach steady state was evaluated by plotting mean Cmax and Cmin over 24 months of treatment. Data were excluded if there appeared to be sample handling errors such that PK values were inconsistent and were several orders of magnitude different from those of other subjects at the same visit. The following values were excluded: for ALN‐18328 and DLin‐MC3‐DMA, 2 Cmin values (weeks 13 and 40) and 1 Cmax value (week 13); for PEG2000‐C‐DMG, 3 Cmin values (weeks 13, 40, and 52) and 1 Cmax value (week 13).
Results
Patient Baseline Demographics
In total, 29 patients with hATTR amyloidosis enrolled in the MAD study were included in the PK analysis. All patients were white, and 69% of patients were male. The mean age ± standard deviation (SD) was 55.6 ± 15.6 years, and the mean body weight ± SD was 70.0 ± 15.6 kg. Twenty‐two patients (75.9%) had the V30M mutation, and 7 (24.1%) had other TTR mutations. The OLE study enrolled 27 patients from the MAD study, all of whom were included in the PK analysis. The gap between the last patisiran dose in the MAD study and the first dose in the OLE study ranged from 169 to 512 days. Patient disposition and demographic characteristics in the OLE study34 were similar to those of the MAD study.33
Free Versus Encapsulated ALN‐18328
In the MAD study, the majority of ALN‐18328 in circulation in the plasma was encapsulated in the LNP or associated with LNP constituents. One hour after EOI of the first and second doses, free ALN‐18328 plasma concentrations were less than 2% and 5%, respectively, of the encapsulated ALN‐18328 concentrations. Three hours after EOI, free ALN‐18328 was less than 1% of encapsulated levels after both the first and second doses.
Plasma PK of ALN‐18328
After the first dose in the MAD study (Figure 1A), peak concentrations of ALN‐18328 were observed at EOI and increased with increasing doses, ranging from 0.145 to 6.05 μg/mL (Supplemental Table S1). Subsequently, plasma concentrations of ALN‐18328 decreased rapidly during the first phase, with a t½α value ranging from 0.947 to 1.26 hours across the dose range. Plasma concentrations during the first phase reached a minimum at around 12 hours (range across doses, 0.00790 to 0.166 μg/mL). A second phase was observed thereafter, with a minor secondary peak (Cmax2) ranging from 0.0220 to 0.369 μg/mL across the doses seen within 24 hours after EOI. The ratio of Cmax2/Cmax1 was 5% to 15% and was similar across the different doses. Concentrations of ALN‐18328 declined more slowly in the second phase (t½β, 1.65 to 2.48 days) than in the first phase. The percentage of AUC comprising the second PK phase (77.5% to 90.7%) was similar across doses (Supplemental Table S1). The mean PK profiles and PK parameters from the second dose were similar to the first dose and are presented in Figure 1B and Supplemental Table S1.
Figure 1.
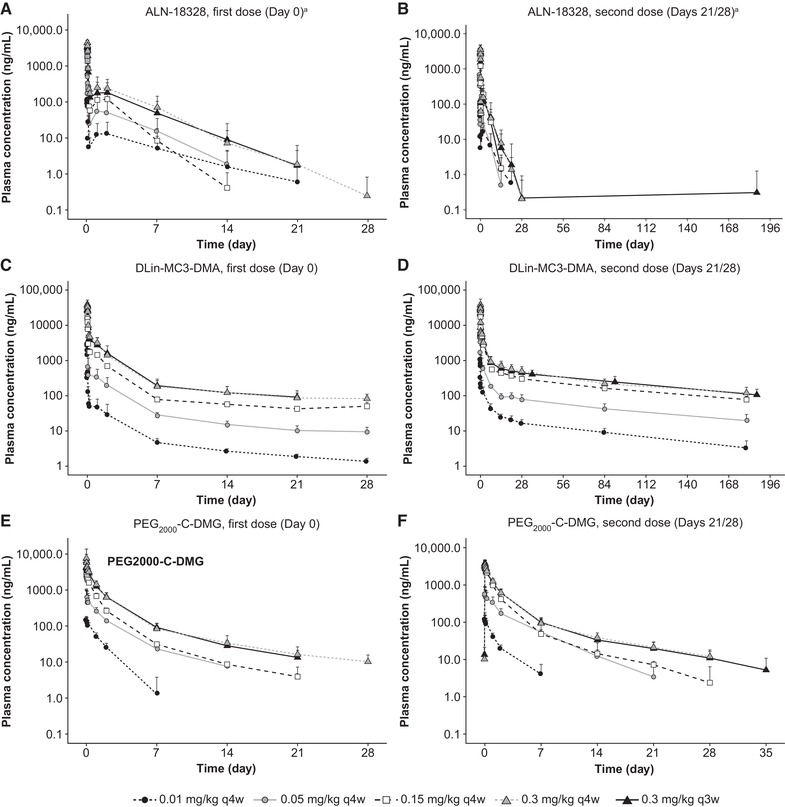
Mean plasma concentration‐time profiles of ALN‐18328 (A, B), DLin‐MC3‐DMA (C, D), and PEG2000‐C‐DMG (E, F) after the first dose (left) and second dose (right) of patisiran in patients with hATTR amyloidosis in the phase 2 MAD study. Error bar represents standard deviation. aContains data from Suhr et al (2015).33 ALN‐18328, patisiran drug substance (small interfering ribonucleic acid); DLin‐MC3‐DMA, (6Z,9Z,28Z,31Z)‐heptatriaconta‐6,9,28,31‐tetraen‐19‐yl‐4‐(dimethylamino)butanoate; hATTR, hereditary transthyretin‐mediated amyloidosis; MAD, multiple ascending dose; PEG2000‐C‐DMG, α‐(3′‐{[1,2‐di(myristyloxy)proponoxy]carbonylamino}propyl)‐ω‐methoxy, polyoxyethylene; q3w, once every 3 weeks; q4w, once every 4 weeks.
Similarly, in the OLE study, ALN‐18328 PK profiles in weeks 1, 34, and 106 exhibited 2 distinct PK phases (Figure 2A). Following repeat 0.3 mg/kg once‐every‐3‐week dosing, mean ALN‐18328 Cmax and Cmin increased over time (Figure 2B), reached steady state by week 24, and stabilized thereafter: mean Cmax,ss was 7.15 μg/mL (Rac, 1.75) and Cmin,ss was 0.0210 μg/mL (Rac, 3.23) in week 106. The Rac for ALN‐18328 AUCτ (week 106) was 3.20 (Table 1). Steady‐state (week 106) t½α,ss was 0.789 hours, t½β,ss was 3.16 days, and the percentage of AUC comprising the second PK phase was 83.3% (Table 1).
Figure 2.
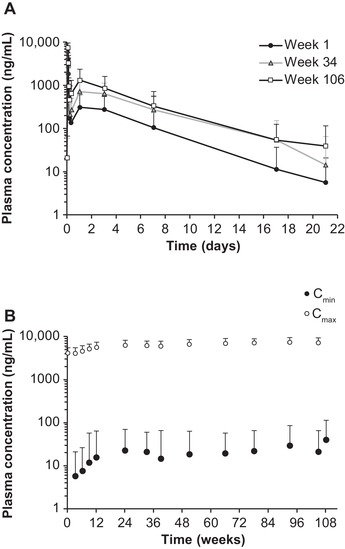
Plasma PK profiles for ALN‐18328 following intravenous administration of patisiran 0.3 mg/kg once every 3 weeks for 24 months of treatment in the OLE study. (A) Mean ALN‐18328 plasma concentration‐time profiles in weeks 1, 34, and 106. (B) Mean ALN‐18328 plasma Cmin and Cmax over time. Error bar represents standard deviation. ALN‐18328, patisiran drug substance (small interfering ribonucleic acid); Cmax, maximum concentration at EOI; Cmin, concentration at the end of the dosing interval; EOI, end of infusion; OLE, open‐label extension; PK, pharmacokinetic.
Table 1.
PK Parameter Estimates Following Single‐ and Multiple‐Dose Administration of Patisiran at 0.3 mg/kg Once Every 3 Weeks in the Phase 2 Open‐Label Extension Study
| ALN‐18328 | DLin‐MC3‐DMA | PEG2000‐C‐DMG | |
|---|---|---|---|
| PK Parameter | n = 27 | n = 27 | n = 27 |
| Week 1 | |||
| Cmax, μg/mL | 4.10 ± 1.28 | 42.5 ± 13.1 | 5.07 ± 1.31 |
| tmax, hour | 1.25 (1.03‐2.67) | 1.25 (1.03‐2.67) | 1.27 (1.03‐3.42) |
| AUCτ, μg·h/mL | 57.5 ± 57.4 | 796 ± 292 | 179 ± 168 |
| AUCp2, % | 81.7 | 71.0 | NA |
| Cmax2, μg/mL | 0.367 ± 0.30 | 7.94 ± 2.30 | NA |
| Cmin, μg/mL | 0.0065 ± 0.015 | 0.628 ± 1.10 | 0.0179 ± 0.007 |
| Week 34 | |||
| Cmax, μg/mL | 6.12 ± 1.69 | 41.1 ± 9.25 | 4.84 ± 1.42 |
| AUCp2, % | 91.5 | 82.2 | NA |
| Cmax2, μg/mL | 0.781 ± 0.492 | 9.65 ± 2.49 | NA |
| Cmin, μg/mL | 0.0211 ± 0.0377 | 1.50 ± 0.367 | 0.0296 ± 0.0085 |
| Week 106 | |||
| Cmax,ss, μg/mL | 7.15 ± 2.14 | 40.2 ± 11.5 | 4.22 ± 1.22 |
| tmax, hour | 1.30 (1.17‐2.10) | 1.30 (1.17‐2.10) | 1.30 (1.17‐3.10) |
| AUCτ, μg·h/mL | 184 ± 159 | 1403 ± 514 | 145 ± 65 |
| AUCp2,ss, % | 83.3 | 84.4 | NA |
| Cmax2,ss μg/mL | 1.57 ± 2.04 | 9.90 ± 4.10 | NA |
| Cmin,ss, μg/mL | 0.0210 ± 0.0442 | 1.75 ± 0.698 | 0.0236 ± 0.00930 |
| t½α,ss, hour | 0.789 ± 0.170 | 0.875 ± 0.112 | 17.1 ± 5.35 |
| t½β,ss, day | 3.16 ± 1.75 | 10.9 ± 7.9 | 3.76 ± 1.05 |
| Vss a, L/kg | 0.255 ± 0.198 | 0.470 ± 0.238 | 0.130 ± 0.050 |
| CLss a, mL/kg/h | 3.03 ± 2.50 | 2.08 ± 0.771 | 2.08 ± 0.586 |
| Rac b for Cmin | 3.23 | 2.79 | 1.32 |
| Rac b for Cmax | 1.75 | 0.95 | 0.833 |
| Rac b for AUCτ | 3.20 | 1.76 | 0.812 |
ALN‐18328, patisiran drug substance (small interfering ribonucleic acid); AUC, area under the concentration‐time curve; AUCp2, AUC in second PK phase and percent of second PK phase = AUCp2/AUCτ; AUCτ, AUC during the dosing interval; CLss, total clearance at steady state; Cmax, maximum concentration at EOI; Cmax2, the maximum concentration at the secondary peak; Cmin, concentration at the end of the dosing interval; DLin‐MC3‐DMA, (6Z,9Z,28Z,31Z)‐heptatriaconta‐6,9,28,31‐tetraen‐19‐yl‐4‐(dimethylamino)butanoate; EOI, end of infusion; NA, not applicable; PEG2000‐C‐DMG, α‐(3′‐{[1,2‐di(myristyloxy)proponoxy]carbonylamino}propyl)‐ω‐methoxy, polyoxyethylene; PK, pharmacokinetic; Rac, accumulation ratio; SD, standard deviation; t½α, half‐life of the first phase of the PK profile; t½β, terminal half‐life (half‐life of the second phase of the PK profile); tmax, time to reach Cmax; Vss, apparent volume of distribution at steady state; ss, steady state.
All PK parameters are expressed as mean ± SD, except for tmax, which is presented as median (range).
tmax occurred approximately at the EOI.
Urine PK of ALN‐18328
After the first and second doses in the MAD study, the majority of patients (61% and 71%, respectively) had no detectable ALN‐18328 in urine samples from the 6‐hour collection period. The fraction of the dose excreted in urine over the dose range of 0.01 to 0.3 mg/kg was approximately 0.1% after the first dose and 0.2% after the second dose (Supplemental Table S1). In the OLE study, at all 3 times examined (weeks 1, 34, and 106), ALN‐18328 was measurable in urine up to 6 hours postinfusion but was not detected from 24 hours through to the day 17 postdose.
Plasma PK of DLin‐MC3‐DMA
The plasma PK profile of DLin‐MC3‐DMA was similar to that of ALN‐18328, exhibiting a rapid decline followed by a minor secondary peak and a long terminal phase (Figure 1C). However, the distinction between the 2 phases was not as marked as that seen with ALN‐18328, and the minor second peak was not as prominent. The half‐life of the first phase (t½α) ranged from 0.86 to 1.9 hours across the different doses (Supplemental Table S2) and was similar to that estimated for ALN‐18328. Also similar to ALN‐18328, the ratio of Cmax2/Cmax1 was 16% to 21% across the different doses. The terminal‐phase half‐life of DLin‐MC3‐DMA was much longer (t½β, 14.6 to 28.7 days) than that of ALN‐18328. The percentage of AUC comprising the second PK phase was 85% to 92% across the different doses (Supplemental Table S2). The mean PK profiles and PK parameters from the first and second doses in the MAD study were similar (Figure 1C,D and Supplemental Table S2).
Similarly, in the OLE study, DLin‐MC3‐DMA PK profiles in weeks 1, 34, and 106 exhibited 2 distinct PK phases (Figure 3A). Following repeat dosing once every 3 weeks, mean DLin‐MC3‐DMA Cmax and Cmin both increased over time (Figure 3B), reached steady state by week 24, and stabilized thereafter: mean Cmax,ss was 40.2 μg/mL (Rac, 0.95) and Cmin,ss was 1.75 μg/mL (Rac, 2.79) in week 106. The Rac for DLin‐MC3‐DMA AUCτ (week 106) was 1.76 (Table 1). The t½α,ss (0.875 hours) and t½β,ss (10.9 days) and the contribution of the second PK phase to the overall AUC (84.4%) after achieving steady state (week 106) were similar to the values in the MAD study and after the first dose in OLE study. The mean CLss and Vss values were 2.08 mL/kg/h and 0.470 L/kg, respectively.
Figure 3.
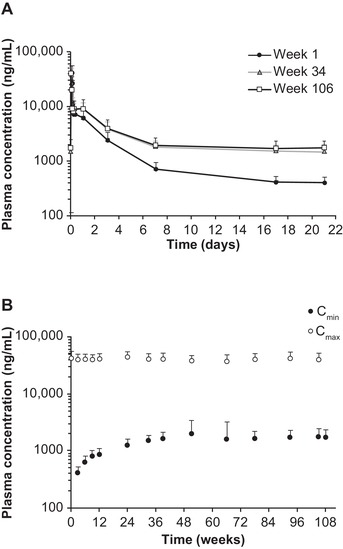
Plasma PK profiles for DLin‐MC3‐DMA following intravenous administration of patisiran 0.3 mg/kg once every 3 weeks for 24 months of treatment in the OLE study. (A) Mean DLin‐MC3‐DMA plasma concentration‐time profiles in weeks 1, 34, and 106. (B) Mean DLin‐MC3‐DMA plasma Cmin and Cmax over time. Error bar represents standard deviation. Cmax, maximum concentration at EOI; Cmin, concentration at the end of the dosing interval; DLin‐MC3‐DMA, (6Z,9Z,28Z,31Z)‐heptatriaconta‐6,9,28,31‐tetraen‐19‐yl‐4‐(dimethylamino)butanoate; EOI, end of infusion; OLE, open‐label extension; PK, pharmacokinetic.
Urine PK of DLin‐MC3‐DMA
All subjects in the MAD study had low but measurable urine concentrations of DMBA in the 0‐ to 6‐hour interval after the first and second doses, with calculated fe values of 0.193% to 1.88% (Supplemental Table S2). In the OLE study, urine concentrations of DMBA reached maximum 24 hours after EOI and generally approached predose values before the next infusion.
Plasma PK of PEG2000‐C‐DMG
After the first dose in the MAD study (Figure 1E), plasma concentrations of PEG2000‐C‐DMG peaked at EOI and decreased in a multiexponential manner. Mean Cmax of PEG2000‐C‐DMG increased with increasing doses, ranging from 0.169 to 7.29 μg/mL (Supplemental Table S3). Unlike ALN‐18328 and DLin‐MC3‐DMA, a minor peak was not observed in the PEG2000‐C‐DMG plasma PK profile. Mean t½β was consistent in the dose range from 0.15 to 0.3 mg/kg and ranged from 4.67 to 8.46 days. The mean PK profiles and PK parameters from the second dose in the MAD study were similar to the first dose and are presented in Figure 1F and Supplemental Table S3.
In the OLE study, PEG2000‐C‐DMG PK profiles were very similar at the 3 times examined (weeks 1, 34, and 106; Figure 4A), indicating no accumulation. Mean PEG2000‐C‐DMG Cmax was stable over time, and mean Cmin increased only slightly up to week 24 following 0.3 mg/kg once‐every‐3‐week chronic dosing (Figure 4B): mean Cmax,ss was 4.22 μg/mL (Rac, 0.833) and Cmin,ss was 0.0236 μg/mL (Rac, 1.32) in week 106. The Rac for PEG2000‐C‐DMG AUCτ (week 106) was 0.812 (Table 1). The mean CLss and Vss values were 2.08 mL/kg/h and 0.130 L/kg, respectively.
Figure 4.
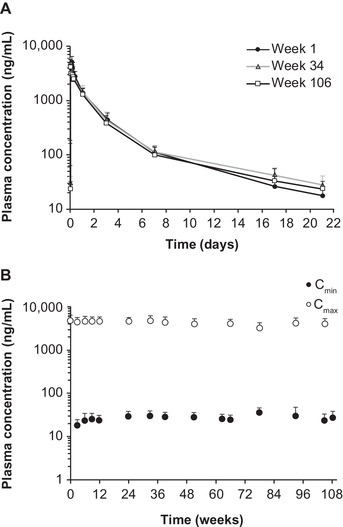
Plasma PK profiles for PEG2000‐C‐DMG following intravenous administration of patisiran 0.3 mg/kg once every 3 weeks for 24 months of treatment in the OLE study. (A) Mean PEG2000‐C‐DMG plasma concentration‐time profiles in weeks 1, 34, and 106. (B) Mean PEG2000‐C‐DMG plasma Cmin and Cmax over time. Error bar represents standard deviation. Cmax, maximum concentration at EOI; Cmin, concentration at the end of the dosing interval; EOI, end of infusion; OLE, open‐label extension; PEG2000‐C‐DMG, α‐(3′‐{[1,2‐di(myristyloxy)proponoxy]carbonylamino}propyl)‐ω‐methoxy, polyoxyethylene; PK, pharmacokinetic.
Patisiran Dose Proportionality
The dose‐proportionality evaluation in the phase 2 MAD study indicated that PK parameters for all 3 analytes had a linear relationship with dose (Supplemental Table S4). Pearson correlation coefficient for PK exposure‐dose plots ranged from 0.558 to 0.972. The 95%CI for β estimates for the linear term included 1, and the coefficient for the quadratic term LnDose2 was not found to be statistically significant (P > .05) in the model. These results indicate that PK exposures to ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG increased in an approximately dose‐proportional manner across the patisiran dose range of 0.01 to 0.3 mg/kg.
ADA Assessment
In the MAD study, 2 patients in the patisiran 0.3 mg/kg once‐every‐3‐week dose group had treatment‐emergent ADAs. One patient was confirmed ADA positive on day 56 (titer, 40) and tested negative for ADAs at all other times. The second patient was confirmed ADA‐positive on days 7 (titer, 80), 56 (titer, 320), and 208 (titer, 320) and tested ADA negative at all other points. There was no impact of ADAs on the PK of ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG (data not shown). Nadir TTR reduction in these 2 patients (75% and 95.4%) were similar to the previously reported maximum mean TTR reduction for all patients (82.9% to 86.7%).33
In the OLE study, 1 patient who had developed ADAs in the prior MAD study tested ADA positive, with a low titer (80) at screening and on days 21 and 84. There was no impact of ADAs on the PK of ALN‐18328 or TTR reduction in this patient (data not shown).
Discussion
In this analysis, we report the first full characterization of the PK of 3 patisiran LNP components — the siRNA, ALN‐18328, and the novel lipid excipients DLin‐MC3‐DMA and PEG2000‐C‐DMG — from 2 clinical studies in which frequent PK samples were collected. In the OLE study, sparse PK samples were collected over 2 years of treatment. The PK profiles of the 3 analytes were multiphasic, and the PK exposure to each analyte increased dose proportionally over a wide dose range (0.01 to 0.3 mg/kg). For ALN‐18328 and DLin‐MC3‐DMA, plasma concentrations accumulated over time and reached steady state after 24 weeks of dosing. In contrast, there was no noticeable accumulation of PEG2000‐C‐DMG over time. All 3 analytes displayed stable PK with chronic dosing of patisiran 0.3 mg/kg once every 3 weeks. Transient ADA positivity in 2 patients did not affect PK exposure to the 3 analytes or TTR reductions from baseline.
We also compared PK profiles of the 3 analytes from the MAD and OLE studies with PK profiles obtained from the previously reported phase 1 study (data on file).26 PK profiles of all 3 analytes were generally consistent across studies and between patients with hATTR amyloidosis and healthy volunteers.
Further understanding of analyte PK was provided by population PK and PK/PD modeling analyses performed on pooled data from 5 clinical studies in healthy volunteers and patients with hATTR amyloidosis with polyneuropathy.39, 40 The PK of ALN‐18328 including the secondary PK peak were described using a semimechanistic model with linear elimination from the liver. In contrast, the PK of the 2 lipids was described by 3‐compartment mammillary models with linear elimination.39 Serum TTR reduction was best described by an indirect response model linking ALN‐18328 plasma concentration to inhibition of the synthesis rate of TTR.40
Taken together, the PK data from the MAD and OLE studies provide insights into the mechanism of hepatocellular uptake of the LNP, release of ALN‐18328 into the cytoplasm, and LNP release into systemic circulation. The proposed mechanism is illustrated in Figure 5 and explained in the following paragraphs in the context of the PK behavior of the siRNA and the lipids. In the first 4 hours after dosing, the plasma concentration‐time profiles of ALN‐18328 and DLin‐MC3‐DMA showed a similar PK distribution phase, in which decline was more rapid than that of PEG2000‐C‐DMG (data on file). As ALN‐18328 and DLin‐MC3‐DMA are associated with LNPs, the rate of decline in their concentrations can be explained by the rapid uptake of LNPs by the liver (Figure 5). Conversely, following intravenous dosing, PEG lipids have been shown to desorb rapidly from the LNP in the circulation and to transfer to circulating endogenous lipoproteins and blood cells.27 This situation would explain the shallower distribution profile of PEG2000‐C‐DMG in plasma.
Figure 5.
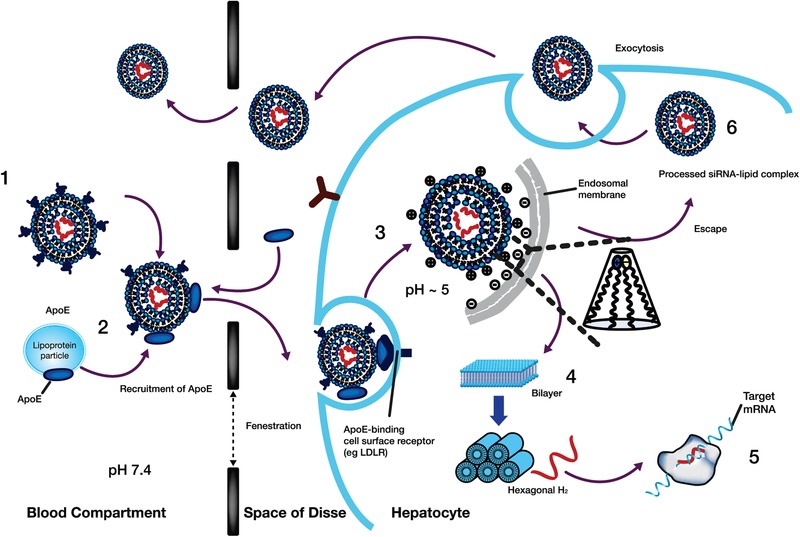
Proposed mechanism of liver uptake of patisiran LNP and release from the liver following intravenous administration. (1) After intravenous administration of patisiran, PEG2000‐C‐DMG dissociates from the LNP. (2) Removal of the PEG coating allows endogenous ApoE to associate with the LNP, facilitating uptake into hepatocytes via an ApoE‐dependent process. (3) On internalization via endocytosis, the ionizable DLin‐MC3‐DMA lipid is protonated (positively charged), as the pH decreases in the endosome. (4) The positively charged DLin‐MC3‐DMA lipid interacts with the negatively charged endosomal lipid, resulting in disintegration of the LNP, destabilization of the endosome membrane, and release of ALN‐18328 into the cytoplasm. (5) ALN‐18328 binds to RISC, leading to the targeted degradation of TTR mRNA and subsequent reductions in the target protein levels. (6) A proportion of internalized LNPs undergo exocytosis egress from late endosomes/lysosomes back into the circulation. ALN‐18328, patisiran drug substance (small interfering ribonucleic acid); ApoE, apolipoprotein E; IV, intravenous; LDLR, low‐density lipoprotein receptor; LNP, lipid nanoparticle; mRNA, messenger RNA; PEG2000‐C‐DMG, α‐(3′‐{[1,2 di(myristyloxy)proponoxy]carbonylamino}propyl)‐ω‐methoxy, polyoxyethylene; PEG, PEG2000‐C‐DMG; DLin‐MC3‐DMA, (6Z,9Z,28Z,31Z)‐heptatriaconta‐6,9,28,31‐tetraen‐19‐yl‐4‐(dimethylamino)butanoate; RISC, RNA‐induced silencing complex; siRNA, small interfering ribonucleic acid; TTR, transthyretin.
The plasma PK profiles of ALN‐18328 and DLin‐MC3‐DMA exhibited 2 PK phases with similar PK properties, including the similarity of t½α, the extent of the contribution of AUCp2 to overall AUC, and the ratio of Cmax2/Cmax in both MAD and OLE studies. Consistent with these observations is the finding that <5% of ALN‐18328 was free in the circulation. To explain these data, we propose that the 2 PK phases of ALN‐18328 and DLin‐MC3‐DMA correspond to the PK of ALN‐18328 and DLin‐MC3‐DMA in 2 different states. The first phase represents the PK of ALN‐18328 and DLin‐MC3‐DMA encapsulated within LNPs after PEG2000‐C‐DMG shedding, which are actively taken up by hepatocytes via the ApoE‐dependent low‐density lipoprotein (ApoE‐dependent LDL) receptor pathway (Figure 5).23, 30 The multiphasic nature of the analyte PK profiles was supported by the respective population PK models.39 It was previously reported that on internalization into hepatocytes via endocytosis, the ionizable DLin‐MC3‐DMA lipid is protonated (positively charged) as the endosomal pH is acidic.41, 42 The positively charged DLin‐MC3‐DMA lipid can then interact with negatively charged endosomal lipid, resulting in disintegration of the LNP, destabilization of the endosome membrane, and release of ALN‐18328 into the cytoplasm of the hepatocyte.26 After delivery into the cytoplasm, ALN‐18328 binds to RISC, leading to the targeted degradation of TTR mRNA and subsequent reductions in TTR protein levels.
Consistent with rapid ingress of ALN‐18328 into the cytosol of the hepatocyte, a 20% to 30% reduction in TTR level was observed within 24 hours after the first dose. TTR levels continued to decline and reached a nadir between 10 and 14 days postdose. This nadir was stably maintained with repeat administration of patisiran once every 3 weeks, as seen in the 2‐year OLE study and 18‐month APOLLO study.17, 26, 33
Imaging studies have shown that only a small fraction of the siRNA in the LNP is released into the cytosol and that about 70% of the siRNA that enters hepatocytes undergoes exocytosis through egress of LNPs from late endosomes/lysosomes back into the circulation.25 We propose that the second phase of the PK profiles for plasma ALN‐18328 and DLin‐MC3‐DMA corresponds to the exocytosis of the siRNA‐lipid complex from hepatocytes back into systemic circulation (Figure 5) but not as intact LNPs. This proposal is consistent with the prolonged half‐life of ALN‐18238 in plasma in the terminal phase compared with the short half‐life in the first phase during the first few hours after administration. The finding also suggests that the exocytosed siRNA‐lipid complex is not actively re‐endocytosed by the ApoE‐dependent LDL receptor pathway. In a Quantitative Whole‐Body Autoradiography study conducted in rats, 90% of the radiolabeled patisiran was distributed to the liver 4 hours postdose.28 In this rat study, the radiolabeled patisiran in the liver declined to 34.5%, 18%, and 0.45% after 24, 168, and 1344 hours postdose, respectively (data on file). Thus, we propose that the egressed LNP is redistributed and eliminated by an alternative pathway in the liver. The estimated t½β values for DLin‐MC3‐DMA after the second dose in the MAD study (65.2 to 80.8 days) and during a 21‐day dosing interval in the OLE study (t½β,ss, 10.9 days) are very different. To reconcile the difference, we evaluated the PK profile for DLin‐MC3‐DMA using a population PK modeling approach by pooling data from 5 patisiran clinical studies.39 The DLin‐MC3‐DMA PK profile was described using a 3‐compartment mammillary model. The elimination half‐life for each of the 3 phases is determined by the α, β, and γ phases, respectively. The observed t½β,ss of 10.9 days during a dosing interval of 21 days in the OLE study is reflective of the β phase and the initial part of the γ phase of the 3‐compartment model, whereas the observed t½β of 65.2 to 80.8 days after the second dose in the MAD study is in agreement with the γ‐phase half‐life (∼ 60 days). Based on the equation for calculating the effective half‐life by dosing interval and observed accumulation ratio,43 the derived effective half‐life for DLin‐MC3‐DMA was 17 days, indicating that the terminal phase of the DLin‐MC3‐DMA profile is a minor contributor to the overall AUC, as reflected by the accumulation in Cmin alone and not Cmax.
The estimated t½β of ALN‐18328 was only 2‐4 days in both studies. Based on t½β values, 97% of the ALN‐18328 administered would be expected to be eliminated by approximately 12‐20 days postdose, leading to no accumulation with once‐every‐3‐week dosing. However, following multiple dosing of patisiran 0.3 mg/kg once every 3 weeks in the OLE study, the ALN‐18328 plasma concentration reached steady state at 24 weeks, with an accumulation ratio of 3.23‐fold for AUCτ. This level of accumulation of ALN‐18328 was unexpected, given its t½β of only 2‐4 days. The disparity between the t½β and the observed accumulation after multiple once‐every‐3‐week dosing can be explained by the finding that >95% of ALN‐18328 is associated with LNP constituents, likely to be DLin‐MC3‐DMA in circulation, such that the PK of ALN‐18328 is dependent on the PK of DLin‐MC3‐DMA.
Following multiple‐dose intravenous administration of patisiran at 0.3 mg/kg once every 3 weeks, the Vss for all 3 analytes was higher than the blood volume in a typical 70‐kg human, indicating that all 3 analytes distributed outside the vasculature, although this was not as pronounced for PEG2000‐C‐DMG. Rat autoradiography studies with radiolabeled patisiran demonstrated that most of the radioactivity (>90%) was rapidly distributed to the liver.28
The majority of patients in the phase 2 MAD study did not have measurable ALN‐18328 in urine, suggesting that the renal route of elimination is a negligible elimination pathway for ALN‐18328. This finding is consistent with an in vitro metabolism study that suggested that ALN‐18328 is metabolized by nucleases.19, 20 In vitro human metabolic profiling of ALN‐18328 in the serum and liver S9 fractions revealed degradation of both sense and antisense strands by exonuclease cleavage to nucleotides of various lengths (data on file). In contrast, DLin‐MC3‐DMA is primarily metabolized to DMBA by hydrolysis,20 and consistent with this result, a small proportion of DMBA was found to be excreted in the urine up to 6 hours postdose. This finding indicates that urinary excretion of DMBA is a pathway for DLin‐MC3‐DMA elimination. No data on renal elimination of PEG2000‐C‐DMG in humans are available, but PEG2000‐C‐DMG was not detected in the urine in rats or monkeys. In monkeys, approximately 44% of PEG2000‐C‐DMG was excreted in the feces.28 Nonclinical data suggest that PEG2000‐C‐DMG is not extensively metabolized and is eliminated unchanged through the hepatobiliary tract to the feces.28 Although PEG2000‐C‐DMG coated the surface of the LNP, ADAs against PEG were infrequent with repeat dosing over 2 years and did not affect the PK of the analytes.
Conclusion
The patisiran analytes ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG displayed multiphasic PK profiles and exhibited dose proportionality over the evaluated dose range of 0.01 to 0.3 mg/kg. All 3 analytes exhibited time‐independent PK with chronic dosing of 0.3 mg/kg once every 3 weeks over 2 years. Half‐life and accumulation data suggested that the PK of ALN‐18328 was associated with LNP lipids, likely to be DLin‐MC3‐DMA, and that a secondary peak in siRNA concentration may be driven by exocytosis of siRNA‐lipid complexes from the liver. Renal elimination was negligible for ALN‐18328, whereas urinary excretion is an elimination pathway for DLin‐MC3‐DMA. The incidence of ADAs was low and had no impact on PK exposure or the TTR reduction effect.
Conflicts of Interest
The data in this article were obtained from the phase 2 multiple‐ascending‐dose study of patisiran and its open‐label extension, both funded by Alnylam Pharmaceuticals. Authors are full‐time employees of Alnylam Pharmaceuticals.
Funding
This study was funded by Alnylam Pharmaceuticals.
Data‐Sharing Statement
The data that support the findings of this study are available from coauthor Gabriel J. Robbie (grobbie@alnylam.com) on reasonable request.
Supporting information
Supplemental Table S1. Mean (CV%) Plasma and Urine PK Parameters of ALN‐18328 in the MAD Study
Supplemental Table S2. Mean (CV%) PK Parameters of DLin‐MC3‐DMA and Urine Excretion of DMBA in the MAD Study
Supplemental Table S3. Mean (CV%) Plasma PK Parameters of PEG2000‐C‐DMG in the MAD Study
Supplemental Table S4. Dose Proportionality of ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG Across Patisiran Dose Range of 0.01 to 0.3 mg/kg in the MAD Study
Acknowledgments
The authors are grateful to the dedicated patients who participated in the study, investigators, coordinators, study monitors, and study team members who supported trial conduct and completion. The authors thank Ms. Emilie Simard, Dr. Martin Béliveau, and Dr. J.F. Marier at Certara Strategic Consulting for the data analysis and Dr. Valerie Clausen and Yuanxin Xu for management of bioanalytical analysis. Editorial assistance was provided by Adelphi Communications Ltd., funded by Alnylam Pharmaceuticals.
Clinicaltrials.gov identifiers: NCT01617967 and NCT01961921
References
- 1. Hawkins PN, Ando Y, Dispenzeri A, Gonzalez‐Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50‐57. [DOI] [PubMed] [Google Scholar]
- 3. Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol‐binding protein. Science. 1995;268(5213):1039‐1041. [DOI] [PubMed] [Google Scholar]
- 4. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286‐1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damy T, Judge DP, Kristen AV, Berthet K, Li H, Aarts J. Cardiac findings and events observed in an open‐label clinical trial of tafamidis in patients with non‐Val30Met and non‐Val122Ile hereditary transthyretin amyloidosis. J Cardiovasc Transl Res. 2015;8(2):117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conceição I, Gonzalez‐Duarte A, Obici L, et al. “Red‐flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shin SC, Robinson‐Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79(6):733‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR‐FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR‐FAP. Curr Opin Neurol. 2016;29(Suppl 1):S3‐S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60(4):765‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(31):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR‐cardiac amyloidosis: emerging disease‐modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swiecicki PL, Zhen DB, Mauermann ML, et al. Hereditary ATTR amyloidosis: a single‐institution experience with 266 patients. Amyloid. 2015;22(2):123‐131. [DOI] [PubMed] [Google Scholar]
- 13. Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120‐1127. [DOI] [PubMed] [Google Scholar]
- 14. Gertz MA, Kyle RA, Thibodeau SN. Familial amyloidosis: a study of 52 North American‐born patients examined during a 30‐year period. Mayo Clin Proc. 1992;67(5):428‐440. [DOI] [PubMed] [Google Scholar]
- 15. Benson M, Waddington‐Cruz M, Wang A, et al. Safety and efficacy of inotersen in patients with hereditary transthyretin amyloidosis with polyneuropathy (hATTR‐PN). Orphanet J Rare Dis. 2017;12(Suppl 1):Abstract O8. [Google Scholar]
- 16. Benson MD, Waddington‐Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22‐31. [DOI] [PubMed] [Google Scholar]
- 17. Adams D, Gonzalez‐Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11‐21. [DOI] [PubMed] [Google Scholar]
- 18. Gertz MA, Mauermann ML, Grogan M, Coelho T. Advances in the treatment of hereditary transthyretin amyloidosis: a review. Brain Behav. 2019;9(9):e01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alnylam Pharmaceuticals Inc . US prescribing information: ONPATTRO (patisiran) lipid complex injection, for intravenous use : Food and Drug Administration; 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210922s000lbl.pdf. Accessed November 6, 2019.
- 20. European Medicines Agency . Summary of product characteristics: Onpattro 2 mg/mL concentrate for solution for infusion 2018. https://www.ema.europa.eu/documents/product-information/onpattro-epar-product-information_en.pdf. Accessed November 6, 2019.
- 21. Food and Drug Administration . Inactive ingredient search for approved drug products: DSPC (1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine). 2019. https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm?event=BasicSearch.page. Accessed November 6, 2019.
- 22. Food and Drug Administration . Inactive ingredient search for approved drug products: cholesterol. 2019. https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm?event=BasicSearch.page. Accessed November 6, 2019.
- 23. Cullis PR, Hope MJ. Lipid nanoparticle systems for enabling gene therapies. Mol Ther. 2017;25(7):1467‐1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leung AK, Hafez IM, Baoukina S, et al. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron‐dense nanostructured Core. J Phys Chem C Nanomater Interfaces. 2012;116(34):18440‐18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sahay G, Querbes W, Alabi C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31(7):653‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819‐829. [DOI] [PubMed] [Google Scholar]
- 27. Mui BL, Tam YK, Jayaraman M, et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol Ther Nucleic Acids. 2013;2:e139 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3894582/pdf/mtna201366a.pdf. Accessed November 6, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. European Medicines Agency . Assessment report: onpattro (patisiran) 2018. https://www.ema.europa.eu/documents/assessment-report/onpattro-epar-public-assessment-report_.pdf. Accessed November 6, 2019.
- 29. Center for Drug Evaluation and Research (CDER) Food and Drug Administration . Multi‐discipline review: patisiran. Population PK and/or PD analyses 2018. NDA210992. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000MultiR.pdf. Accessed November 6, 2019.
- 30. Akinc A, Querbes W, De S, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand‐based mechanisms. Mol Ther. 2010;18(7):1357‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans . Nature. 1998;391(6669):806‐811. [DOI] [PubMed] [Google Scholar]
- 32. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494‐498. [DOI] [PubMed] [Google Scholar]
- 33. Suhr OB, Coelho T, Buades J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi‐dose study. Orphanet J Rare Dis. 2015;10(109):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams D, Coelho T, Conceição I, et al. Phase 2 open‐label extension (OLE) study of patisiran, an investigational RNAi therapeutic for the treatment of polyneuropathy due to hereditary ATTR (hATTR) amyloidosis: Final 24‐month data. Presented at: American Academy of Neurology (AAN); April 22‐28, 2017; Boston, MA.
- 35. Zhang X, Goel V, Attarwala H, Sweetser MT, Clausen VA, Robbie GJ. Patisiran pharmacokinetics, pharmacodynamics, and exposure‐response analyses in the phase 3 APOLLO trial in patients with hereditary transthyretin‐mediated (hATTR) amyloidosis. J Clin Pharmacol. 2020;60(1):37‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams D, Coelho T, Conceição I, et al. Phase 2 open‐label extension (OLE) study of patisiran with or without a TTR stabilizer for the treatment of hereditary ATTR (hATTR) amyloidosis with polyneuropathy. Eur J Neurol. 2017;24(Suppl 1):31‐32 Abstract O1121. [Google Scholar]
- 37. Hummel J, McKendrick S, Brindley C, French R. Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharm Stat. 2009;8(1):38‐49. [DOI] [PubMed] [Google Scholar]
- 38. Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17(10):1278‐1283. [DOI] [PubMed] [Google Scholar]
- 39. Goel V, Jomphe C, Gosselin N, et al. Population pharmacokinetics (PK) of investigational patisiran in healthy volunteers and in patients. 15th International Congress on Neuromuscular Diseases (ICNMD) 2018:Poster.
- 40. Goel V, Gosselin N, Jomphe C, et al. Population pharmacokinetic (PK)/pharmacodynamic (PD) model of serum transthyretin (TTR) following patisiran‐LNP administration in healthy volunteers and patients with hereditary TTR mediated (hATTR) amyloidosis with polyneuropathy. Presented at: European Academy of Neurology (EAN); June 16‐19, 2018; Lisbon, Portugal.
- 41. Semple SC, Akinc A, Chen J, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28(2):172‐176. [DOI] [PubMed] [Google Scholar]
- 42. Jayaraman M, Ansell SM, Mui BL, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed Engl. 2012;51(34):8529‐8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rowland M, Tozer TN. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications. 4th ed. Baltimore, MD: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Mean (CV%) Plasma and Urine PK Parameters of ALN‐18328 in the MAD Study
Supplemental Table S2. Mean (CV%) PK Parameters of DLin‐MC3‐DMA and Urine Excretion of DMBA in the MAD Study
Supplemental Table S3. Mean (CV%) Plasma PK Parameters of PEG2000‐C‐DMG in the MAD Study
Supplemental Table S4. Dose Proportionality of ALN‐18328, DLin‐MC3‐DMA, and PEG2000‐C‐DMG Across Patisiran Dose Range of 0.01 to 0.3 mg/kg in the MAD Study