

ABBREVIATIONS
- ABOi
ABO‐incompatible
- AML
acute myeloid leukemia
- CGIs
CpG islands
- ChIP
chromatin immunoprecipitation
- DHSs
DNase I–hypersensitive sites
- EMSAs
electrophoretic mobility shift assays
- HGVS
Human Genome Variation Society
- MDS
myelodysplastic syndrome
- SSP
single specific primer
- TFs
transcription factors
- vWF
von Willebrand factor
The ABO blood group system, discovered by Karl Landsteiner in 1900,1 is fundamental to the safety of blood transfusion, which requires identification of weak phenotypes or subgroups. The system is composed of two carbohydrate antigens, A and B, and their antibodies. The International Society of Blood Transfusion designates four antigens—A; B; A,B; and A1—based on the specificity of the antibodies. Biochemical and molecular genetic studies have clarified the molecular basis of the histo‐blood group ABO system.2, 3, 4 The functional A and B alleles at the ABO genetic locus encode two glycosyltransferases, α‐1‐3‐N‐acetylgalactosaminyltransferase (A‐transferase) and α‐1‐3‐galactosyltransferase (B‐transferase), respectively. Initial molecular genetic studies demonstrated that ABO is composed of seven exons spanning approximately 19.5 kb of genomic DNA (Fig. 1A)5 and that two critical single‐base substitutions in the last coding exon result in amino acid substitutions responsible for the different donor nucleotide‐sugar substrate specificity between the A‐ and B‐transferases.6, 7, 8 A single‐base deletion in Exon 6 is considered to shift the reading frame of codons and to abolish the transferase activity of A‐transferase in most O alleles.6
Figure 1.
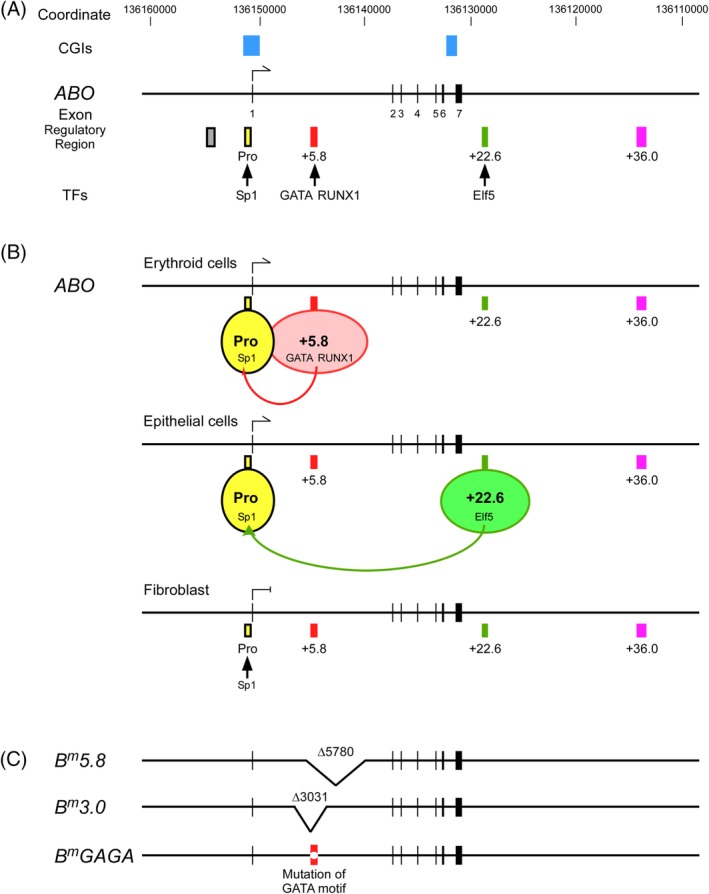
Schematics of transcriptional regulation of ABO expression. (A) A map of the 50‐kb region of genomic DNA from the upstream to downstream region of the human ABO gene. The diagram shows ABO gene Exons 1‐7 represented as vertical lines with coordinates in hg19 and CGIs. Below the diagram, regulatory regions including the proximal promoter, the erythroid cell‐specific regulatory element or the +5.8‐kb site, the epithelial cell‐specific regulatory element or the +22.6‐kb site, and the DHS region +36.0 are shown. Pro, promoter: +5.8, the +5.8‐kb site: +22.6, the +22.6‐kb site: +36.0, region +36.0. The colored boxes are represented as follows: yellow, the proximal promoter; light red, the +5.8‐kb site; dark green, the +22.6‐kb site; purple, region +36.0. Also, the CCAAT‐binding factor/NF‐Y enhancer region is shown as a gray box. Binding of TFs to individual regions is also indicated. (B) Schematic diagram of cell‐specific regulation of ABO expression. The top diagram indicates putative interaction of the proximal promoter with the +5.8‐kb site for ABO expression in erythroid cells. The second diagram from the top indicates putative interaction of the proximal promoter with the +22.6‐kb site for the ABO expression in epithelial cells. The third diagram from the top indicates the proximal promoter not interacting with either the +5.8‐kb site or the +22.6‐kb site without ABO expression in fibroblasts, in which neither GATA‐1,2 nor Elf5 is expressed. C. Schema of B m 5.8, B m 3.0, and B m GAGA. The deletions in B m 5.8 and B m 3.0 are each represented by a V‐shaped segment. Variant of the GATA motif in the +5.8‐kb site of B m GAGA is denoted as a clear circle in a red box. [Color figure can be viewed at http://wileyonlinelibrary.com]
On the other hand, some aspects remain to be explored.9, 10 The ABO antigens are expressed in a cell type–specific manner9; the isoantigens A, B, and H of blood groups A, B, and O are not confined to red blood cells (RBCs) but are also found in most secretions and on some epithelial cells. However, they are absent in connective tissue, muscle, and the central nervous system. Moreover, ABH antigens are known to undergo drastic changes during the development, differentiation, and maturation of cells in the epithelial and erythroid lineages.10 For example, studies of A‐antigen expression during the maturation of erythroid progenitors in a two‐phase liquid culture system showed that A‐positive cells gradually increased during erythroid maturation.11, 12, 13 Fluorescence‐activated cell sorting analysis with monoclonal antibodies has demonstrated the expression of A‐antigens on colony cells derived from blast‐forming units–erythroid and colony‐forming units–erythroid.14 In addition to these physiological processes, profound changes have also been documented in pathological conditions such as tumorigenesis. Reduction or complete deletion of A‐ or B‐antigen expression in bladder and oral cancers has been documented.15, 16, 17 Moreover, the loss of ABH antigens has been correlated with the progression of various cancers, including lung and bladder carcinomas.18 Finally, since Yamamoto and colleagues6 clarified the molecular genetic basis of the ABO system, a number of weak phenotypes have been found to be attributable to single nucleotide polymorphisms in the coding exons and splicing sites and hybrid formation between common alleles,19, 20 although other weak phenotypes for which no variant apparently exists in the coding exons and splicing sites have also been reported.21, 22, 23 Therefore, to understand the molecular mechanism responsible for the control of ABO gene expression in a cell type–specific manner, during normal cell differentiation, in cancer cells lacking A or B antigens, and in weak phenotypes, it is essential to grasp how ABO gene transcription is regulated.
The DNA sequences in and around specific genes provide the code that dictates when, where, and at what level specific genes are transcribed.24, 25 This code comprises three parts: the core promoter where RNA production initiates and is directed toward Exon 2, the region proximal to the core promoter, and the more distant regulatory sequences that enhance RNA production. It has become obvious that enhancers usually work in groups (i.e., the locus control region and super‐enhancers), each being bound by several transcription factors (TFs), forming a so‐called enhanceosome. These enhanceosomes are nucleated by pioneer TFs early during differentiation, and these TFs are subsequently replaced by other TFs that trigger recruitment of the preinitiation complex, involving RNA polymerase II, to the promoter. Enhancers also interact with each other through a multilooped structure. Thus, for elucidation of ABO regulation, it is important to reveal regulatory regions such as the core promoter, the region proximal to it, and the more distant enhancer, as well as the TFs that bind to those regions.
Although A‐ or B‐antigen expression is dependent upon many steps including the structure of ABO, transcriptional regulation of ABO, translational regulation, modification or localization of A‐ or B‐transferase, and H antigen expression, in this review we focus on the transcriptional regulation of the ABO gene through regulatory regions and TFs, and outline the molecular basis for weak phenotypes with variants in those regions.
REGULATORY REGIONS FOR ABO EXPRESSION
ABO gene regulatory regions have been identified by in vitro studies and genetic studies of weak phenotypes. The variants are described according to the HGVS nomenclature using the nucleotide sequences of accession numbers NG_006669.1 and NM_020469.1 as a reference in the genetic study sections. The relationship between the descriptions of variants in Intron 1 according to the Human Genome Variation Society (HGVS) nomenclature and those in the original reports is shown in Table 1. However, the positions that were used in the in vitro experiments described in the original papers remain unchanged. The positions reported in the original papers have been described according to the nucleotide sequences of accession number NT_035014.4 as a reference.
Table 1.
Relationship between the descriptions of variants in Intron 1 according to the HGVS nomenclature and those in the original reports*
| Variants according to HGVS nomenclature Reference sequence: NG_006669.1 (NM_020469.2) | Variants in the original reports Reference sequence: NT_035014.4†, NG_006669.1‡ or KC841929§ | Phenotype | Reference |
|---|---|---|---|
| c.28 + 4077_7107del | +4105_ + 7136del† | Bm | 26 |
| c.28 + 5110_10889del | +5137_ + 10914del† | Bm | 27 |
| c.28 + 5443_11354del | c.28 + 5443_29–1655del‡ | Ax | 28 |
| c.28 + 5859G > C | c.28 + 5830G > C§ | Am | 29 |
| c.28 + 5861 T > G | +5890 T > G† | Ael, Bel, Bm | 30, 31 |
| c.28 + 5864G > A | +5893G > A† | A3 | 32 |
| c.28 + 5865_5887del | +5892_ + 5914del† | Am | 33 |
| c.28 + 5875C > T | +5904C > T† | B3 or Bw | 31 |
| c.28 + 5880A > G | +5909A > G† | A3 | 32 |
| c.28 + 5885C > T | c.28 + 5885C > T‡ | B3 | 34 |
The original reports are listed in the Reference section. The specific reference numbers are shown in the Reference column.
Variants were described according to the reference sequence of NT_035014.4.
Variant was described according to the reference sequence of NG_006669.1. It is likely that c.28 + 5443_11354del is easier to indicate the deletion size than c.28 + 5443_29–1655del.
Variant was described according to the reference sequence of KC841929.
The proximal promoter of ABO
In vitro studies
Initially, Yamamoto et al.5 demonstrated two transcription initiation sites upstream of the ATG translation start site in ABO using human pancreatic cDNA. Consistently, similar transcription initiation sites were found upstream of the translation start site using erythroid cells differentiated in vitro from AC133−CD34+ cells and K562 cells by us and others.35, 36
For demonstrating the proximal promoter of ABO, we carried out transfection experiments into gastric cancer KATOIII cells and erythroleukemia HEL cells using luciferase reporter plasmids prepared from a genomic clone of human ABO. Those experiments defined the proximal ABO promoter between −150 and −2 relative to the translation start site in those cells (Figs. 1A and 2), and the promoter showed constitutive activity regardless of the cell types examined.37, 38 Electrophoretic mobility shift assays (EMSAs) demonstrated that the GC box at −56 to −44 in the promoter bound a ubiquitous TF Sp1 or Sp1‐like protein(s) (Fig. 1A), whereas mutations of the recognition motif that abrogated binding of those factors reduced the promoter activity in both cell types.38 Thus, Sp1 or Sp1‐like protein(s) seemed to be important for proximal promoter activity. The nucleotide sequence in ABO reveals two CpG islands (CGIs) (Fig. 1A), one extending from the immediate 5′ flanking region through the first exon and into Intron 1, and the other extending from Intron 6 to Exon 7. Thus, the ABO proximal promoter is located within a CGI. Because a promoter within a CGI would include a few transcription initiation sites, the presence of several transcription initiation sites in ABO is relevant.
Figure 2.
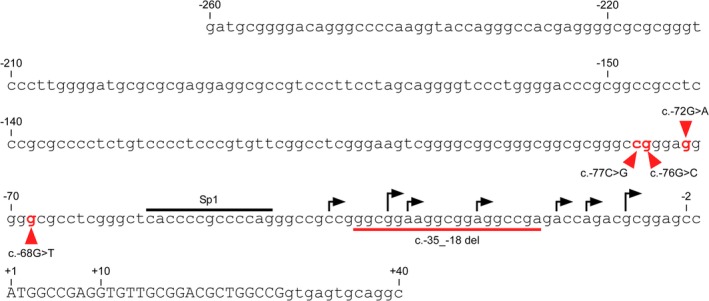
Nucleotide sequence of the 5′ ‐flanking region in ABO. The sequence is given in full, from position −260 to +40, relative to the translation start ATG site of ABO. The upper case letters denote the coding sequence of Exon 1, and the lower case letters the noncoding genomic sequence. High arrows above the sequence indicate the transcription initiation sites that were determined by 5′ ‐RACE using human pancreas cDNA by Yamamoto et al.,5 and low arrows denote the transcription initiation sites that were determined by in vitro erythroid culture of AC133−CD34+ cells.35 The proximal ABO promoter is located between −150 and −2 relative to the ATG translation start site.37, 38 The recognition motif for TF Sp1 is indicated by an overbar. Nucleotide substitutions at −77, −76, −72, and –68 are indicated in red, and the deletion between −35 and −18 is denoted by an underbar. [Color figure can be viewed at http://wileyonlinelibrary.com]
Genetic studies
Cai et al.39 reported a nucleotide deletion between −35 and −18, i.e., c.−35_−18del, in the proximal ABO promoter in the B3 phenotype (Fig. 2), which reduced the promoter activity in a plasmid‐based reporter assay. Similarly, three single‐nucleotide substitutions at −77, −76, or −68 in the ABO promoter, i.e., c.−77C > G, c.−76G > C, or c.−68G > T, respectively, have been reported in A3 and B3, each substitution reducing the promoter activity in luciferase reporter assays.32, 40 Recently, another single‐nucleotide substitution at −72 in the ABO promoter, i.e., c.−72G > A, was reported in B3.41 These genetic variants confirm the functional significance of the proximal ABO promoter in vivo.
Conclusion
ABO transcription is likely regulated by the proximal promoter and cell‐specific regulatory regions (Fig. 1B), because the proximal promoter shows constitutive activity regardless of cell type.35
Regulatory region for erythroid cell–specific expression of ABO
For delineation of distal regulatory regions involved in ABO regulation in erythroid cells, we used both in vitro experiments and genetic approaches.
In vitro studies
First, because DNase I–hypersensitive sites (DHSs) are associated with transcriptional regulatory regions including the promoter and distal enhancer, we prepared luciferase reporter plasmids on the basis of DHSs within a 15‐kb region of genomic DNA in and around ABO in erythroleukemia K562 cells with publicly available data from DNase‐Seq and FAIRE‐seq on the University of California, Santa Cruz (UCSC) Genome Browser. Subsequent plasmid‐based reporter assays demonstrated a distal regulatory region between +5653 and +6154 relative to the translation start site, named the +5.8‐kb site (Fig. 1A), in K562 cells, and the regulatory activity of this region was specific to erythroid cells.27 According to the HGVS nomenclature, the +5.8‐kb site corresponds to c.28 + 5624_6125. The same site was referred to the GATA binding site by the others.41 EMSAs and chromatin immunoprecipitation (ChIP) assays demonstrated that the region bound hematopoietic TF GATA‐1 or ‐2, and RUNX1 (Fig. 1A), whereas mutations of the recognition motifs that abrogated binding of those factors reduced the regulatory activities of the +5.8‐kb site in K562 cells.27, 33, 42 Thus, binding of GATA‐1 or ‐2, and RUNX1 to the +5.8‐kb site seemed to be crucial for the erythroid cell–specific activity of the region (Fig. 1B).
Genetic and serologic studies
Comparison of the genomic DNA in the ABO gene between humans and these primate species demonstrated high conservation between the ATG translation start codon and the stop codon of the ABO gene, except for a few regions. The +5.8‐kb site is conserved among humans, chimpanzees, and orangutans, showing similar expression of the A and B antigens on RBCs. However, it is intriguing to note that the site is not conserved in rhesus monkeys and marmosets, in which the A and B antigens are expressed only slightly on RBCs. Therefore, a comparative approach could indicate involvement of the site in ABO expression in human erythroid lineage cells.
Genetic studies demonstrated a 5.8‐kb deletion of c.28 + 5110_10889del in Intron 1 of ABO and a 3.0‐kb deletion of c.28 + 4077_7107del in individuals with the Bm phenotype, termed B m 5.8 and B m 3.0, respectively (Fig. 1C).26, 27 These deletions involved the +5.8‐kb site. The Bm phenotype is characterized by the discrepancy of B antigen expression between RBCs and secretions43: Bm RBCs are not agglutinated by anti‐B or anti‐A,B antibody, whereas the saliva of Bm secretors contains about as much B substance as that of a normal B secretor. However, the B antigens on RBCs can only be detected by sensitive techniques such as adsorption and elution of anti‐B. Bm erythrocytes contain abundant H sites, which can be converted into B sites by in vitro treatment with B‐transferase derived from normal B individuals. B‐transferase activity was detected in serum of Bm individuals, although the activity was distinctly reduced in all cases. The Bm trait is inherited as a rare allele at the ABO locus, although a few nonhereditary cases have also been reported.44, 45 Thus, deletion of the erythroid cell–specific regulatory region or the +5.8‐kb site on the B m allele could explain the discrepancy of B‐antigen expression between RBCs and secretions in Bm. Further genetic studies found variants in the GATA motif of c.28 + 5861 T > G or c.28 + 5859G > C in Bm or Am, respectively (Fig. 3).29, 30 The former was termed B m GAGA. Am is analogous to Bm in blood group A. Moreover, deletion of the RUNX1 binding motif of c.28 + 5865_5887del was revealed in Am (Fig. 3).33 A similar discrepancy of blood antigen expression between RBCs and secretions is also observed in A3 and B3 phenotypes, where single nucleotide substitutions of c.28 + 5864G > A and c.28 + 5880A > G were found around the GATA and RUNX1 motifs in the +5.8‐kb site (Fig. 3).32 Similarly, single‐nucleotide substitution of c.28 + 5885C > T was reported around the RUNX1 motif in an individual with B3.34 These genetic studies confirmed the regulatory significance of the +5.8‐kb site for erythroid cell–specific expression of ABO in vivo.
Figure 3.

Alignment of variants within the +5.8‐kb site found in weak phenotypes. The wild‐type sequence between c.28 + 5852 and c.28 + 5891 in Intron 1 of ABO is shown at the top. The motifs for TFs GATA and RUNX1 are indicated by overbars. In alignment with the wild‐type sequence, the variants found within the +5.8‐kb site in weak phenotypes are shown. However, two reports have described variants in the coding exon of Am,46, 47 while a number of variants of exons and splicing sites are reportedly associated with A3 and B3.20 Those variants are described according to HGVS nomenclature using the nucleotide sequences of accession numbers NG_006669.1 and NM_020469.1 as a reference. Relationship between the variant descriptions according to the HGVS nomenclature and those in the original reports is shown in Table 1. Positions c.28 + 5852 and c.28 + 5891 correspond to +5881 and +5920 relative to the ABO translation start site, respectively, according to the nucleotide sequences of accession number NT_035014.4 as a reference, which was used in the original reports.30, 32, 33 [Color figure can be viewed at http://wileyonlinelibrary.com]
Conclusion
It appears that the proximal promoter and the +5.8‐kb site are required for ABO expression in an erythroid cell–specific manner (Fig. 1B), although it remains to be explored whether regions other than the +5.8‐kb site might affect ABO expression in erythroid cells.
Regulatory region for epithelial cell–specific expression of ABO
In vitro studies
For delineation of ABO regulation in epithelial cells, we prepared luciferase reporter plasmids on the estimation of enhancers within a 50‐kb region of genomic DNA in and around ABO in epithelial cells by publicly available data from DNase‐Seq and chromatin state segmentation (ChromHMM) on the UCSC Genome Browser. Subsequent plasmid‐based reporter assays indicated a distal regulatory region between +22563 and +22781 relative to the translation start site of ABO, termed the +22.6‐kb site, in KATOIII cells (Fig. 1A), and the regulatory activity of the region was specific to epithelial cells.48 Subsequently, we validated the significance of the +22.6‐kb site with use of KATOIII cells with homozygote deletion of the site constructed by the CRISPR/Cas9 system. EMSAs and ChIP assays demonstrated that the region bound an epithelial cell–specific TF, Elf5 (Fig. 1A), whereas variant of the recognition motif that abrogated binding of the factor reduced the regulatory activity of the site in KATOIII cells. Thus, binding of Elf5 seemed to be crucial for the epithelial cell–specific activity of the region.
Conclusion
It is likely that the proximal promoter and the +22.6‐kb site are required for ABO expression in epithelial cells (Fig. 1B), although regions other than the +22.6‐kb site might also have some influence.
Other regulatory regions for ABO expression
CCAAT‐binding factor/NF‐Y enhancer region
The initial luciferase reporter assays with KATOIII cells demonstrated that a positive element was located between −3931 and −3650 from the translation start site where four tandem copies of 43‐bp repeat units bound a positive TF, CCAAT‐binding factor/NF‐Y, through the CCAAT motif (Fig. 1A).37 However, similar regulatory activity was not observed in K562 cells and HEL cells of erythroid origin.18, 23 Thus, it was likely that the minisatellite was not involved in transcriptional regulation of ABO in erythroid cells. Genetic population studies revealed that both the B and O alleles are linked via four tandem copies of a 43‐bp repeat unit, and that the A1 allele is linked in the absence of this tandemly repetitive element.49, 50, 51 Seltsam et al.52 observed unexpected variations in the CCAAT‐binding factor/NF‐Y enhancer region including the repeat units in four individuals with weak B phenotypes, suggesting that those weak phenotypes might be caused by sequence variations in the enhancer region. On the other hand, Thuresson et al.53 reported a hybrid allele between O 2 and B which lacked three repeat units, although the B transcript level was similar to that in fresh peripheral blood samples from normal controls. Thus, it remains controversial whether ABO transcription is influenced by the CCAAT‐binding factor/NF‐Y enhancer region in erythroid cells.36, 52, 53
Distal promoter of ABO
At the 5′ end of the CGI involving the ABO proximal promoter in cultured cells expressing ABO, an alternative starting Exon 1a comprising 27 base pairs was found by 5′‐RACE (Fig. 1A). The level of transcription from Exon 1a was much lower than that from Exon 1.35 The luciferase reporter assays demonstrated that the sequence located between −864 and −699 was responsible for transcription from Exon 1a in both erythroid and epithelial cell lineages. However, significance of Exon 1a remained elusive.
The region proximal to the proximal ABO promoter
The plasmid‐based reporter assays demonstrated a negative regulatory element just upstream from the proximal ABO promoter in KATOIII cells and HEL cells,54 suggesting that ABO transcription is regulated by negative elements in the −307 to −150 region from the translation start site. EMSAs indicated that this region bound to a nuclear factor from KATOIII cells. However, we have not identified this factor.
INFERENCES REGARDING ABO REGULATION
Cell type–specific expression
ABO transcription is regulated by a constitutive proximal promoter and a cell‐specific regulatory region such as the +5.8‐kb site or the +22.6‐kb site (Fig. 1B). Luciferase reporter assays showed that the erythroid cell‐specific regulatory activity of the +5.8‐kb site was dependent upon binding of the erythroid cell–specific TF GATA‐1 or ‐2.27 In addition, variants in the GATA motif were found in Bm and Am,29, 30 in which B‐ or A‐antigen expression is reduced on RBCs, while a large amount of B or A substance is present in the saliva of secretors. Similarly, plasmid‐based reporter assays demonstrated that the epithelial cell–specific regulatory activity of the +22.6‐kb site was dependent upon binding of the epithelial cell–specific TF Elf5.48 In fibroblasts not expressing GATA‐1 or ‐2, or Elf5, it is plausible that abrogation of the siteʼs cell‐specific regulatory activity contributes to lack of ABO expression.48 Therefore, it is likely that the cell type–specific expression of ABO is dependent upon expression of cell‐specific TFs binding to those cell‐specific regulatory elements.
Cell differentiation–specific regulation
In vitro erythroid differentiation of CD34+ cells and AC133−CD34+ cells from peripheral blood mononuclear cells indicated that ABO was expressed at an early stage and disappeared later,35, 42 and that the period when ABO was expressed at a higher level preceded that of FUT1 expression (Fig. 4). Expression of RUNX1 and GATA‐2 characteristically decreases during erythroid differentiation of CD34+ cells.42 Thus, it seems likely that down regulation of ABO expression might be ascribed to reduction of RUNX1 in the later phase of erythroid differentiation. However, the mechanism of ABO expression at an early stage of erythroid differentiation remains to be explored.
Figure 4.
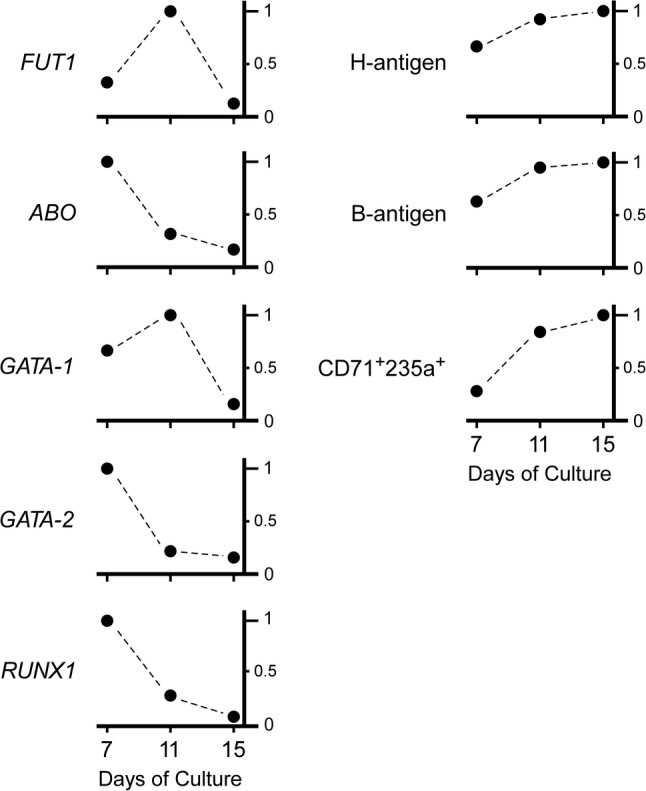
Schematic illustration of the expression of cell surface antigens and genes during the course of in vitro erythroid cell differentiation. The diagrams were constructed with the expression of genes such as FUT1, ABO, GATA‐1, GATA‐2, and RUNX1 as well as the cell surface expression of B antigen and H antigen 7 days, 11 days, and 15 days after in vitro erythroid cell differentiation from CD34+ cells reported previously.42 The time course of the relative expression of individual genes or antigens is indicated. The maximum gene expression or cell population showing antigen expression during erythroid differentiation was expressed as 1.0 on the vertical axis, whereas the relative expression ratios at other time points were calculated for individual genes and antigens. Also, changes in the proportions of cells expressing CD71 antigen and CD235 antigen that were not included in that report were added to the diagrams.
Loss of ABO antigens in malignancy
ABH antigens are often absent from glycoproteins and glycolipids of malignant tissue in the gastrointestinal tract, oral cavity, uterine cervix, lung, prostate, breast, and bladder.19, 43 ABO antigen expression decreases as a result of down regulated transcription of ABO, which is ascribed to at least two different mechanisms: allelic loss and hypermethylation of the ABO promoter region in a CGI.55, 56, 57, 58
There is abundant evidence that methylated CGIs at transcription start sites are associated with some silent genes. We and others have also demonstrated that hypermethylation of the ABO promoter could be responsible for absence of the ABO transcript and A‐antigen in gastric and colon cancer cell lines.56, 57 Using clinical samples of oral squamous cell cancer, Gao et al.58 showed that loss of A/B antigens was responsible for molecular events such as loss of the A/B allele or ABO promoter hypermethylation in two‐thirds of tissue samples they examined. Thus, an additional mechanism for loss of A/B antigens other than allelic loss or promoter hypermethylation remained to be clarified in one‐third of such cases. The origin of DNA hypermethylation is currently being clarified, and mutations in such genes as Ten‐eleven‐translocation 2, DNA methylatransferase 3A, and isocitrate dehydrogenase 1 and 2 encoding epigenetic regulators are known to be involved in DNA methylation.59 Besides mutated epigenetic regulators, mutations in splicing factors may also dysregulate epigenetics through splicing of epigenetic enzymes, and down regulation of transcription is associated with DNA hypermethylation of the promoter. Therefore, the causes and consequences of DNA hypermethylation need to be carefully distinguished.59
The association of weak A expression with acute myeloid leukemia (AML) is well documented.19, 43 Bianco‐Miotto et al.60, 61 have reported that loss of ABH antigens from RBCs of patients with myeloid malignancies is a frequent phenomenon and that DNA methylation of the ABO promoter underlies the loss of ABO allelic expression in a significant proportion of leukemia patients. Such myeloid malignancies include AML, myelodysplastic syndrome (MDS) and myeloproliferative disorders including chronic myeloid leukemia. Recently, we have reported a patient with MDS in whom blood typing demonstrated mixed‐field agglutination, prompting us to investigate the mechanism underlying blood group A‐antigen reduction on RBCs.62 Screening of somatic mutations using bone marrow cells demonstrated mutations in ASXL1, EZH2, RUNX1, and WT1. Experiments involving transient transfection into K562 cells showed that the expression of ABO was decreased by expression of the mutated RUNX1. Since the frame‐shift mutation of RUNX1 could encode an abnormally elongated protein without a transactivation domain that might act as a dominant negative inhibitor, it was plausible that this RUNX1 mutation might be a genetic factor contributing to A‐antigen loss on RBCs. This is another example of a factor other than DNA methylation that could be responsible for ABO antigen reduction in patients with leukemia. Therefore, there is a need for further investigation of patients with leukemia and ABO antigen reduction on RBCs to clarify which events are attributable to ABO‐antigen loss on RBCs in leukemia.
The weak phenotype Bm with deletion or mutation of the +5.8‐kb site
The prevalence of ABO subgroups was 0.048% among transfusion donors in Tokyo, Japan, in 2010‐2011.26 The total occurrence of the Bm and A1Bm subgroups was 0.024% at the Kanto‐Koshinetsu Blood Center, indicating that both are the most frequent ABO blood group variants in the Japanese population. A recent study demonstrated B m 5.8 in 1300 individuals, B m GAGA in two, and B m 3.0 in one among 1303 Japanese individuals with Bm and A1Bm with sequence‐specific polymerase chain reaction (SSP) targeting B m 5.8 (Fig. 1A).63 In practice, Bm is determined by serologic procedures and SSP targeting B m 5.8 at blood centers in Japan.26 Genetic diagnosis of B m 5.8 is beneficial because Bm accounts for almost one‐half of all weak phenotypes in the Japanese population. Based on the above observations, it seems likely that B m 5.8 might have been inherited over a long time period and spread throughout the Japanese population. Since the variant has not been reported in Korea and China, from where ancient people migrated to Japan, B m 5.8 could be specific to the Japanese population.64
Recently, the mutated GATA site in B m GAGA has been reported in individuals with Ael and Bel.31 Moreover, a 5.9‐kb deletion of Intron 1 including the +5.8‐kb site has been found in an individual with Ax.28 It seems intriguing to explore the reason that the same mutation or a similar deletion of the +5.8‐kb site could result in different phenotypes.
PROSPECT
As shown above, ABO transcription is regulated by the proximal promoter and cell‐specific regulatory regions. However, ABO transcription was not completely lost in the cells with biallelic deletions of the +22.6‐kb site.48 Also, reporter assays demonstrated transcriptional activity in the DHS region +36.0 (Fig. 1A),48 which was suggested to interact with the ABO transcription start site on the basis of publicly available data from GeneHancer Regulatory Elements and Gene Interactions on the UCSC Genome Browser. In addition, recent studies have suggested coordinated activity of multiple enhancers to control a single gene, regulation of multiple genes by the same enhancer, and competition or coordination between neighboring promoters.65, 66 Therefore, it would be informative to investigate elements other than the promoter and cell‐specific regulatory regions to clarify the regulatory mechanism of ABO transcription, and delineation of the regulatory mechanism of ABO transcription regulation would yield new insight into the regulatory network involving ABO and the genes associated with the ABO regulatory regions.
Histone deacetylase inhibitors are reported to reduce ABO expression in cultured cells, suggesting that ABO transcription may be regulated epigenetically.67 This also appears to be an intriguing phenomenon in the context of cardiovascular disorders because it has been reported that the levels of von Willebrand factor (vWF) are approximately 25% higher in individuals with blood group types other than O, and this seems to be the main reason for the higher risk of venous thromboembolism and coronary heart disease in non–group O individuals.68 Because it has been argued that addition of A/B‐antigens to vWF in endothelial cells might influence circulating levels of vWF,68 ABO suppression in cells might reduce the risk of developing these diseases. A similar argument could apply to organ transplantation. In cases of ABO‐incompatible (ABOi) liver transplantation, acute humoral rejection directed against donor‐oriented A/B antigens on endothelial cells of liver arteries or bile ducts is the most serious form of rejection, leading to graft loss.69, 70 Thus, a decrease in the amount of antigen on endothelial cells might ameliorate any adverse effects resulting from ABOi liver transplantation. Further studies may shed further light on ABO transcriptional regulation and provide clues for clinical applications.
CONCLUSION
Clarification of the mechanism of ABO transcriptional regulation has contributed to practical transfusion medicine, and delineation of the regulatory mechanism of ABO transcription regulation would yield new insight into the regulatory network involving ABO and the genes associated with the ABO regulatory regions on Chromosome 9.
CONFLICT OF INTEREST
The authors have disclosed no conflicts of interest.
REFERENCES
- 1. Landsteiner K. Zur Kenntnis der antifermentativen lytischen and agglutinierenden Wirkung des Blutserums and der lymph. Zentralbl Bakteriol 1900;27:357‐63. [Google Scholar]
- 2. Hakomori S. Antigen structure and genetic basis of histo‐blood groups A, B and O. their changes associated with human cancer. Biochim Biophys Acta 1999;1473:247‐66. [DOI] [PubMed] [Google Scholar]
- 3. Yamamoto F. Molecular genetics of ABO. Vox Sang 2000;78:91‐103. [PubMed] [Google Scholar]
- 4. Yamamoto F. Cloning and regulation of the ABO genes. Transfus Med 2001;11:281‐94. [DOI] [PubMed] [Google Scholar]
- 5. Yamamoto F, McNeill PD, Hakomori S. Genomic organization of human histo‐blood group ABO genes. Glycobiology 1995;5:51‐8. [DOI] [PubMed] [Google Scholar]
- 6. Yamamoto F, Clausen H, White T, et al. Molecular genetic basis of the histo‐blood group ABO system. Nature 1990;345:229‐33. [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto F, Hakomori S. Sugar‐nucleotide donor specificity of histo‐blood group A and B transferases is based on amino acid substitutions. J Biol Chem 1990;265:19257‐62. [PubMed] [Google Scholar]
- 8. Yamamoto F, McNeill PD. Amino acid residue at codon 268 determines both activity and nucleotide‐sugar donor substrate specificity of human histo‐blood group A and B transferases. in vitro mutagenesis study. J Biol Chem 1996;271:10515‐20. [DOI] [PubMed] [Google Scholar]
- 9. Clausen H, Hakomori S. ABH and related histo‐blood group antigens; immunochemical differences in carrier isotypes and their distribution. Vox Sang 1989;56:1‐20. [DOI] [PubMed] [Google Scholar]
- 10. Dabelsteen E, Buschard K, Hakomori S, et al. Pattern of distribution of blood group antigens on human epidermal cells during maturation. J Invest Dermatol 1984;82:13‐7. [DOI] [PubMed] [Google Scholar]
- 11. Wada H, Suda T, Miura Y, et al. Expression of major blood group antigens on human erythroid cells in a two phase liquid culture systems. Blood 1990;75:505‐11. [PubMed] [Google Scholar]
- 12. Bony V, Gane P, Bailly P, et al. Time‐course expression of polypeptides carrying blood group antigens during human erythroid differentiation. Br J Haematol 1999;107:263‐74. [DOI] [PubMed] [Google Scholar]
- 13. Edvardsson L, Dykes J, Olsson ML, et al. Clonogenicity, gene expression and phenotype during neutrophil versus erythroid differentiation of cytokine‐stimulated CD34+ human marrow cells in vitro . Br J Haematol 2004;127:451‐63. [DOI] [PubMed] [Google Scholar]
- 14. Sieff C, Bicknell D, Caine G, et al. Changes in cell surface antigen expression during hemopoietic differentiation. Blood 1982;60:703‐13. [PubMed] [Google Scholar]
- 15. Cordón‐Cardó C, Lloyd KO, Finstad CL, et al. Immunoanatomic distribution of blood group antigens in the human urinary tract. Influence of secretor status. Lab Invest 1986;55:444‐54. [PubMed] [Google Scholar]
- 16. Ørntoft TF. Expression and biosynthesis of ABH‐related carbohydrate antigens in normal and pathologic human urothelium. APMIS Suppl 1990;17:1‐34. [PubMed] [Google Scholar]
- 17. David L, Leitao D, Sobrinho‐Simoes M, et al. Biosynthetic basis of incompatible histo‐blood group A antigen expression: anti‐A transferase antibodies reactive with gastric cancer tissue of type O individuals. Cancer Res 1993;53:5494‐500. [PubMed] [Google Scholar]
- 18. Lee JS, Ro JY, Sahin AA, et al. Expression of blood‐group antigen A‐ a favorable prognostic factor in non‐small‐cell lung cancer. N Engl J Med 1991;324:1084‐90. [DOI] [PubMed] [Google Scholar]
- 19. Daniels G. Human Blood Groups. 3rd ed. West Sussex, UK: Wiley‐Blackwell; 2013. [Google Scholar]
- 20.[accessed 2020 Jan 19] Available from: https://www.isbtweb.org/workingparties/red-cell-immunogenetics-and-blood-group-terminology/.
- 21. Ogasawara K, Yabe R, Uchikawa M, et al. Molecular genetic analysis of variant phenotypes of the ABO blood group system. Blood 1996;88:2732‐7. [PubMed] [Google Scholar]
- 22. Hansen T, Namork E, Olsson ML, et al. Different genotypes causing indiscernible patterns of A expression on Ael red blood cells as visualized by scanning immunogold electron microscopy. Vox Sang 1998;75:47‐51. [PubMed] [Google Scholar]
- 23. Iwasaki M, Kobayashi K, Suzuki H, et al. Genetic analyses for the ABO blood group subtypes in Japanese. Jpn J Transfus Med 2000;46:532‐9. [Google Scholar]
- 24. Vernimmen D. Uncovering enhancer functions using the α–globin locus. PLoS Genet 2014;10:e1004668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell 2014;157:13‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sano R, Kuboya E, Nakajima T, et al. A 3.0‐kb deletion including an erythroid cell‐specific regulatory element in intron 1 of the ABO blood group gene in an individual with the Bm phenotype. Vox Sang 2015;108:310‐3. [DOI] [PubMed] [Google Scholar]
- 27. Sano R, Nakajima T, Takahashi K, et al. Expression of ABO blood‐group genes is dependent upon an erythroid cell‐specific regulatory element that is deleted in persons with the Bm phenotype. Blood 2012;119:5301‐10. [DOI] [PubMed] [Google Scholar]
- 28. Wu PC, Lin YH, Tsai LF, et al. ABO genotyping with next‐generation sequencing to resolve heterogeneity in donors with serology discrepancies. Transfusion 2018;58:2232‐42. [DOI] [PubMed] [Google Scholar]
- 29. Oda A, Isa K, Ogasawara K, et al. A novel mutation of the GATA site in the erythroid cell‐specific regulatory element of the ABO gene in a blood donor with the AmB phenotype. Vox Sang 2015;108:425‐7. [DOI] [PubMed] [Google Scholar]
- 30. Nakajima T, Sano R, Takahashi Y, et al. Mutation of the GATA site in the erythroid cell‐specific regulatory element of the ABO gene in a Bm subgroup individual. Transfusion 2013;53:2917‐27. [DOI] [PubMed] [Google Scholar]
- 31. Ying Y, Hong X, Xu X, et al. A novel mutation +5904 C>T of RUNX1 site in the erythroid cell‐specific regulatory element decreases the ABO antigen expression in Chinese population. Vox Sang 2018;113:594‐600. [DOI] [PubMed] [Google Scholar]
- 32. Takahashi Y, Isa K, Sano R, et al. Presence of nucleotide substitutions in transcriptional regulatory elements such as the erythroid cell‐specific enhancer‐like element and the ABO promoter in individuals with phenotypes A3 and B3, respectively. Vox Sang 2014;107:171‐80. [DOI] [PubMed] [Google Scholar]
- 33. Takahashi Y, Isa K, Sano R, et al. Deletion of the RUNX1 binding site in the erythroid cell‐specific regulatory element of the ABO gene in two individuals with the Am phenotype. Vox Sang 2014;106:167‐75. [DOI] [PubMed] [Google Scholar]
- 34. Tao C, Xiao J, Hu Y, et al. A novel B allele with c.28+5885C>T substitution in the erythroid cell‐specific regulatory element identified in an individual with phenotype B3 . Transfusion 2017;57:1318‐9. [DOI] [PubMed] [Google Scholar]
- 35. Kominato Y, Hata Y, Takizawa H, et al. Alternative promoter identified between a hypermethylated upstream region of repetitive elements and a CpG Island in human ABO histo‐blood group genes. J Biol Chem 2002;277:37936‐48. [DOI] [PubMed] [Google Scholar]
- 36. Thuresson B, Chester MA, Storry JR, et al. ABO transcript levels in peripheral blood and erythropoietic culture show different allele‐related patterns independent of the CBF/NF‐Y enhancer motif and multiple novel allele‐specific variations in the 5′‐ and 3′‐noncoding regions. Transfusion 2008;48:493‐504. [DOI] [PubMed] [Google Scholar]
- 37. Kominato Y, Tsuchiya T, Hata N, et al. Transcription of human ABO histo‐blood group genes is dependent upon binding of transcription factor CBF/NF‐Y to minisatellite sequence. J Biol Chem 1997;272:25890‐8. [DOI] [PubMed] [Google Scholar]
- 38. Hata Y, Kominato Y, Yamamoto F, et al. Characterization of the human ABO gene promoter in erythroid cell lineage. Vox Sang 2002;82:39‐46. [DOI] [PubMed] [Google Scholar]
- 39. Cai X, Jin S, Liu X, et al. Molecular genetic analysis of ABO blood group variations reveals 29 novel ABO subgroup alleles. Transfusion 2013;53:2910‐6. [DOI] [PubMed] [Google Scholar]
- 40. Isa K, Yamamuro Y, Ogasawara K, et al. Presence of nucleotide substitutions in the ABO promoter in individuals with phenotypes A3 and B3 . Vox Sang 2016;110:285‐7. [DOI] [PubMed] [Google Scholar]
- 41. Hellberg Å, Hult AK, Moser I, et al. A novel single‐nucleotide substitution in the proximal ABO promoter gives rise to the B3 phenotype. Transfusion 2019;59:E1‐3. [DOI] [PubMed] [Google Scholar]
- 42. Sano R, Nogawa M, Nakajima T, et al. Blood group B gene is barely expressed in in votro erythroid culture of Bm‐derived CD34+ cells without an erythroid cell‐specific regulatory element. Vox Sang 2015;108:302‐9. [DOI] [PubMed] [Google Scholar]
- 43. Schenkel‐Brunner H. Human blood groups. 2nd ed. Wien: Springer; 2000. p. 54‐150. [Google Scholar]
- 44. Kogure T, Iseki S. A family of Bm, due to a modifying gene. Proc Jpn Acad 1970;46:728‐32. [Google Scholar]
- 45. Marsh ML, Ferrari M, Nichols E, et al. Bm H: a weak B antigen variant. Vox Sang 1973;25:341‐6. [DOI] [PubMed] [Google Scholar]
- 46. Asamura H, Ota M, Takayanagi K, et al. Molecular genetic analysis of the Am phenotype of the ABO blood group system. Vox Sang 2002;83:263‐7. [DOI] [PubMed] [Google Scholar]
- 47. Lin M, Hou MJ, Twu YC, et al. A novel A allele with 664G>A mutation identified in a family with the Am phenotype. Transfusion 2005;45:63‐9. [DOI] [PubMed] [Google Scholar]
- 48. Sano R, Nakajima T, Takahashi Y, et al. Epithelial expression of human ABO blood‐group genes is dependent upon a downstream regulatory element functioning through an epithelial cell‐specific transcription factor, Elf5. J Biol Chem 2016;291:22594‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu LC, Chang CY, Twu YC, et al. Human histo‐blood group ABO glycosyltransferase genes: different enhancer structures with different transcriptional activities. Biochem Biophys Res Commun 2000;273:459‐66. [DOI] [PubMed] [Google Scholar]
- 50. Shimada I, Kominato Y, Hata N, et al. DNA polymorphisms in the 5′‐ flanking sequence of human ABO blood group genes and their association with the alleles for the common ABO phenotypes. Leg Med 1999;1:217‐25. [DOI] [PubMed] [Google Scholar]
- 51. Irshaid NM, Chester MA, Olsson ML. Allele‐related variation in minisatellite repeats involved in the transcription of the blood group ABO gene. Transfus Med 1999;9:219‐26. [DOI] [PubMed] [Google Scholar]
- 52. Seltsam A, Wagner FF, Grüger D, et al. Weak blood group B phenotypes may be caused by variations in the CCAAT‐binding factor/NF‐Y enhancer region of the ABO gene. Transfusion 2007;47:2330‐5. [DOI] [PubMed] [Google Scholar]
- 53. Thuresson B, Hosseini‐Maaf B, Hult AK, et al. A novel B weak hybrid allele lacks three enhancer repeats but generates normal ABO transcript levels. Vox Sang 2012;102:55‐64. [DOI] [PubMed] [Google Scholar]
- 54. Kominato Y, Hata Y, Matsui K, et al. Transcriptional regulation of the human ABO histo‐blood group genes is dependent on the N box upstream of the proximal promoter. Transfusion 2004;44:70‐8. [DOI] [PubMed] [Google Scholar]
- 55. Orlow I, Lacombe L, Pellicer I, et al. Genotypic and phenotypic characterization of the histoblood group ABO(H) in primary bladder tumors. Int J Cancer 1998;75:819‐24. [DOI] [PubMed] [Google Scholar]
- 56. Kominato Y, Hata Y, Takizawa T, et al. Expression of human histo‐blood group ABO genes is dependent upon DNA methylation of the promoter region. J Biol Chem 1999;274:37240‐50. [DOI] [PubMed] [Google Scholar]
- 57. Iwamoto S, Withers DA, Handa K, et al. Deletion of A‐antigen in a human cancer cell line is associated with reduced promoter activity of CBF/NF‐Y binding region, and possibly with enhanced DNA methylation of A transferase promoter. Glycoconj J 1999;16:659‐66. [DOI] [PubMed] [Google Scholar]
- 58. Gao S, Worm J, Guldberg P, et al. Genetic and epigenetic alterations of the blood group ABO gene in oral squamous cell carcinoma. Int J Cancer 2004;109:230‐7. [DOI] [PubMed] [Google Scholar]
- 59. Heuser M, Yun H, Thol F. Epigenetics in myelodysplastic syndromes. Semin Cancer Biol 2018;51:170‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bianco‐Miotto T, Farmer BJ, Sage RE, et al. Loss of red cells A, B, and H antigens is frequent in myeloid malignancies. Blood 2001;97:3633‐9. [DOI] [PubMed] [Google Scholar]
- 61. Bianco‐Miotto T, Hussey DJ, Day TK, et al. DNA methylation of the ABO promoter underlies loss of ABO allelic expression in a significant proportion of leukemic patients. PLoS One 2009;4:e4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hayakawa A, Sano R, Takahashi Y, et al. RUNX1 mutation in a patient with myelodysplastic syndrome and decreased erythrocyte expression of blood group A antigen. Transfusion 2020;60:184‐96. [DOI] [PubMed] [Google Scholar]
- 63. Ogasawara K, Miyazaki T, Ito S, et al. The B allele with a 5·8 kb deletion in intron 1 of the ABO gene is the major allele in Japanese individuals with Bm and A1Bm phenotypes. Vox Sang 2018;113:393‐6. [DOI] [PubMed] [Google Scholar]
- 64. Kominato Y, Ogasawara K. Is B m 5.8 specific to the Japanese population? Vox Sang 2019;114:185. [DOI] [PubMed] [Google Scholar]
- 65. Fulco CP, Munschauer M, Anyoha R, et al. Systematic mapping of functional enhancer‐promoter connections with CRISPR interference. Science 2016;354:769‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Engreitz JM, Haines JE, Perez EM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016;539:452‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takahashi Y, Kubo R, Sano R, et al. Histone deacetylase inhibitors suppress ABO transcription in vitro, leading to reduced expression of the antigens. Transfusion 2017;57:95‐103. [DOI] [PubMed] [Google Scholar]
- 68. Liumbruno GM, Franchini M. Beyond immunohaematology: the role of the ABO blood group in human diseases. Blood Transfus 2013;11:491‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haga H, Egawa H, Fujimoto Y, et al. Acute humoral rejection and C4d immunostaining in ABO blood type‐incompatible liver transplantation. Liver Transpl 2006;12:457‐64. [DOI] [PubMed] [Google Scholar]
- 70. Sakashita H, Haga H, Ashihara E, et al. Significance of C4d staining in ABO‐identical/compatible liver transplantation. Mod Pathol 2007;20:676‐84. [DOI] [PubMed] [Google Scholar]