

Abstract
Tiara[5]arenes (T[5]s), a new class of five‐fold symmetric oligophenolic macrocycles that are not accessible from the addition of formaldehyde to phenol, were synthesized for the first time. These pillar[5]arene‐derived structures display both unique conformational freedom, differing from that of pillararenes, with a rich blend of solid‐state conformations and excellent host–guest interactions in solution. Finally we show how this novel macrocyclic scaffold can be functionalized in a variety of ways and used as functional crystalline materials to distinguish uniquely between benzene and cyclohexane.
Keywords: five-fold symmetry, host–guest chemistry, nonporous adaptive crystals, polymorphism, tiara[5]arene
I have a tiara! Tiara[5]arenes, a class of five‐fold symmetric oligophenol macrocycles, which is not accessible by direct phenol‐formaldehyde condensation, was synthesized through the metamorphosis of pillar[5]arenes.
Introduction
More than half a century since Pederson's seminal discovery of synthetic host molecules,1 the scope of macrocyclic chemistry is still expanding.2 Apart from well‐established classes such as cyclodextrins,3 cucurbiturils4 and crown ethers,5 a large number of synthetic macrocycles has emerged. Many of these are derived de facto from one set of reactions: the condensation of phenols with aldehydes.6 This gave rise (Figure 1) not only to bakelite,7 but also to cyclotriveratrylenes,8 resorcinarenes,9 calixarenes,10 and in recent times to pillararenes11 and other analogues.12 Pillararenes, named after their characteristic cylindrical pillar‐shaped cavities, have gained considerable popularity in the past decade on account of their facile syntheses, versatility towards derivatization, and rich host–guest chemistry. The synthesis of the pentameric homologue, pillar[5]arene13 (P[5]), is especially high‐yielding, allowing the construction of a wide range of supramolecular architectures based on symmetric P[5]s with identical functionalities on both rims. Furthermore, recent developments from our group and others have opened up rim‐specific functionalizations, leading to the so‐called rim‐differentiated14, 15, 16 P[5]s (RD‐P[5]s) with different top and bottom rims (R1≠R2; Figure 1) to be prepared and further functionalized in a tailor‐made fashion.
Figure 1.
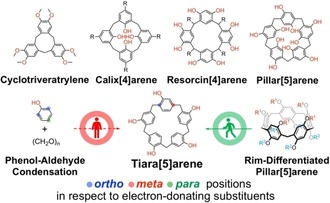
Tiara[5]arene, a para‐bridged oligophenolic macrocycle, which is not attainable by condensing phenol and formaldehyde, can be synthesized by taking a detour through rim‐differentiated pillar[5]arene.
No para‐bridged macrocycle arising from simply reacting phenol and formaldehyde is so far known, since one of the anchoring positions of the methylene bridges on the aromatic unit is not compatible with the well‐known regioselectivity of the electrophilic substitution of phenol. Noting, for example, the extensive chemistry developed for calixarenes, which could be regarded as meta‐bridged analogues, the conception of such unconventional macrocycles would open up as yet uncharted chemical space. Whereas the vast majority of current pillararene functionalizations are executed on the alkoxy moieties,17 we reasoned that the specific removal of the O atoms on one side of RD‐P[5]s to create a C−H functionalized rim would allow the construction of for example, C−C, C−N, and C−X bonds directly linked to the aromatic units, creating new opportunities for host–guest chemistry and novel scaffold construction approaches.
Herein we present the synthesis and solid‐state structures of RD‐P[5]‐derived macrocycles composed of five phenolic units para‐bridged covalently by methylenes, subsequently referred to as tiara[5]arenes (T[5]s) or tiararenes. Next, we discuss their host–guest chemistry, derivatization strategies, and remarkable potential as nonporous adaptive crystals (NACs),18 especially the ability to distinguish between rather similar guests, such as benzene and cyclohexane.
Results and Discussion
The key step in the synthesis of tiara[5]arenes is the initial formation of the RD‐P[5] precursor. Instead of a heads‐or‐tails statistical cyclization characterized by very low yields, our synthesis started with a monomer that aligns in a head‐to‐tail manner, resulting in a highly selective synthesis of RD‐P[5]s, that is, 55 % selectivity among all four P[5] consitutional isomers and 20 % yields of isolated product.14 In the beginning we set out to improve the yield of the RD‐P[5] synthesis, widening a bottleneck in our synthesis. One major limitation to be overcome in the oligocyclization is the retro‐Friedel–Crafts reaction; after formation of the aryl−CH2−aryl moiety, the reaction can reverse.19 The result is the generation of the other three undesired P[5] isomers, rather than an exclusive synthesis of RD‐P[5] that would have occurred had this retro Friedel–Crafts reaction not occurred (see the Supporting Information). Reduction of the water content by using CaH2‐dried 1,2‐dichloroethane as solvent suppressed this retro effect and routinely increased the yield of (OBn)5‐RD‐P[5] from 2015 to 25 %, leading to a gram‐scale production in one step (Scheme 1).
Scheme 1.
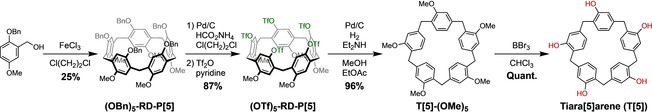
The metamorphosis of rim‐differentiated pillar[5]arenes (RD‐P[5]s) into tiara[5]arenes (T[5]s).
Subsequently, the benzyl groups were removed using ammonium formate as a hydrogen source in the presence of Pd/C, and the deprotected penta‐OH intermediate was converted directly to the air‐stable (OTf)5‐RD‐P[5] in 87 % yield in two steps. This penta‐triflate was hydrodeoxygenated by reduction (H2 over Pd/C) under slightly basic conditions to convert all five OTf groups into H atoms, providing access to the permethylated tiara[5]arene (T[5]‐(OMe)5) in 96 % yield. Near‐quantitative O‐demethylation with excess of BBr3 finally yields the parent tiara[5]arene (T[5]), a new macrocyclic scaffold consisting of phenol units linked by methylene bridges anchored on the ortho‐ and meta‐positions with respect to the OH groups. The total yield over five steps is 20 %, allowing us to synthesize 410 mg of T[5] from per gram of the (OBn)5‐RD‐P[5] precursor produced.
Tiara[5]arene and T[5]‐(OMe)5 show in solution (for example, observed in the NMR spectra) an averaged C 5 symmetry on account of their high conformational freedom similar to [15]paracyclophane.20, 21 The structural flexibility is also evidenced by the rich polymorphism of T[5]‐(OMe)5 displayed in the solid state, a selection of which are displayed in Figure 2 (see the Supporting Information for more polymorphs). A solvent‐free T[5]‐(OMe)5 structure22 (space group P , triclinic) obtained on crystallization by vapor diffusion of CH3OH into CHCl3 solution, features a C 1 conformation (Figure 2 a), with the cavity‐forming rings alternatingly up and down. With the odd number of aryl rings, this alternation leaves one remaining ring to be rotated nearly 90° with respect to the other four rings. While this rotation has also been observed for [15]paracyclophane and for the leaning pillar[6]arene,23 this rotational distortion is not intrinsic to these C−H functionalized para‐bridged macrocycles.
Figure 2.
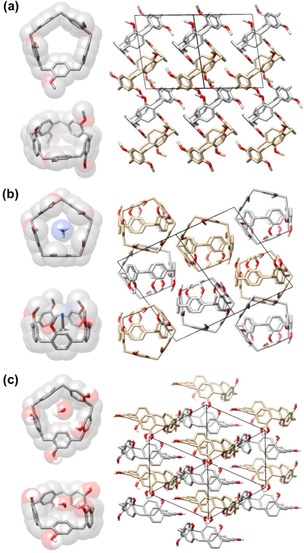
X‐ray crystal structures of a) T[5]‐(OMe)5, b) CH3CN⊂T[5]‐(OMe)5, and c) H2O⊂T[5], showing assorted solid‐state conformations of the tiara[5]arene scaffold. Packing modes in (a), (b), and (c) are viewed from [1 0], [110], and [111] directions, respectively. Most hydrogen atoms and solvent molecules are omitted for clarity. C silver/gold for different enantiomeric conformers, O red, N blue, H white.
C 1‐symmetrical conformations can also be found in single crystals obtained under different conditions, such as vapor diffusion of CH3OH into trans‐1,2‐dichloroethene or EtOAc solutions, although in both cases the T[5]‐(OMe)5 conformations deviate from the one shown in Figure 2 a: from 1,2‐dichloroethene, apart from 2 up, 2 down, 1 flat macrocycles, a 3 up, 1 down, 1 flat conformation can also be obtained. In the crystal structure obtained in EtOAc, another 3 up, 2 down geometry can be also observed. In other words, T[5]s by themselves do not typically display pillar‐like structures.
The inclusion of a CH3CN solvent molecule changes (Figure 2 b) this picture. Similar to many P[5] derivatives, in the CH3CN⊂T[5]‐(Me)5 crystal structure24 (space group P , triclinic) all five aromatic units are now aligned almost perpendicularly to the pentagonal plane formed by the carbon atoms on the methylene bridges, and result in a more or less five‐fold symmetric tiara‐like cavity of diameter ca. 8 Å. A CH3CN molecule is located inside. Both C−H⋅⋅⋅π (distances 2.95, 3.29, and 3.41 Å) and C−H⋅⋅⋅N interactions (distances 2.74, 2.77, 2.98, 3.04, and 3.14 Å) can be found between the T[5]‐(OMe)5 macrocycle and the CH3CN guest molecule. Interestingly, the CH3CN is oriented in only one of the two possible directions, indicating a strong orientational preference, presumably as a result of dipole stabilization. This complexation geometry is borne out by wB97XD/6–311+G(d,p) calculations which confirm a preference of 6.2 kcal mol−1 for the experimentally observed orientation of the CH3CN molecule in the cavity (see detailed computed structures in the Supporting Information).
The parent T[5] macrocycle can also be crystallized25 in the space group P (triclinic) by slow cooling in PhMe (Figure 2 c). A distorted conformation, with one flat and four more vertical phenolic units relative to the pentagonal scaffold, can be found in the structure, even although the orientations and dihedral angles involving the aromatic rings differ from those of the T[5]‐(OMe)5 polymorphs (Figure 2 a,b and the Supporting Information). Multiple hydrogen bonds are present between adjacent macrocycles, via interactions between the phenol groups (O−H⋅⋅⋅O distances 1.89, 1.69, and 2.00 Å). Apart from PhMe solvent molecules in the interstices between the T[5] macrocycles, an H2O molecule (most likely absorbed from the surroundings) is also found in the cavity. An H‐atom of this water is hydrogen‐bonded with a phenolic O‐atom of the adjacent T[5] (O−H⋅⋅⋅OH2, distance 1.92 Å).
The observation of these solvent‐inclusion complexes prompted us to study the host–guest chemistry of tiara[5]arene. Cationic guests, such as methyl viologen (MV2+), methylpyridinium (Py+), and a linear alkyl chain 1,6‐dicyanohexane guest were screened (Figure 3 and the Supporting Information) by NMR titration experiments. In a 1:1 mixture of the T[5] host (5 mm in MeOD‐d 4) and 5 mm Py⋅Cl, all 1H NMR signals of the cationic guest showed a pronounced upfield shift (Δδ of Hα, Hβ and Hγ=3.2, 0.4, and 0.6 ppm, respectively), as a result of the shielding effect commensurate with host–guest complexation. Similar upfield shifts (Δδ of Hα and Hβ=2.0 and 0.7 ppm, respectively) were observed for an analogous experiment with MV2+. Owing to the absence of one set of electron‐donating OH group in the aromatic unit, the electron density of the T[5] cavity is relatively poor compared to those of regular P[5]s. Furthermore, the high conformational freedom of the T[5] scaffold, evidenced by its very simple 1H NMR spectrum, would also lead to a comparatively increased entropy loss upon guest binding. Nonetheless, the T[5] host still exhibits good binding affinities to these cationic guests in CH3OH, with association constants K a of (1.2±0.2)×103 and (1.0±0.2)×103 m −1 for Py⋅Cl and MV⋅2Cl in a 1:1 binding mode, respectively. In contrast, 1,6‐dicyanohexane, which is commonly used in P[5] host–guest systems,26 shows a binding constant value of typically 1–2 orders of magnitude lower (see the Supporting Information), demonstrating the significant difference in binding preference towards guests between homologous T[5] and P[5] macrocycles.
Figure 3.
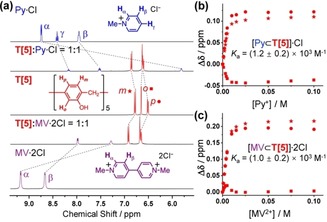
Host–guest interactions between tiara[5]arene and cationic species. a) Partial 1H NMR spectra (400 MHz, MeOD‐d 4, 298 K) of T[5], chloride salts of methylpyridinium and methyl viologen, and their 1:1 mixtures at 5 mm concentrations. b),c) The association constants of b) [Py⊂T[5]]⋅Cl and c) [MV⊂T[5]]⋅2Cl 1:1 complexes in MeOD‐d 4 are determined by NMR titration method.
Subsequent functionalization on either T[5] or T[5]‐(OMe)5 could be achieved (Scheme 2) in a variety of ways. First, for T[5] the OH groups can functionalized, in analogy to our previous work15 on rim‐differentiated P[5]s, by alkylation, esterification, or Suzuki–Miyaura coupling and SuFEx reactions, after being converted to triflates and sulfonyl fluorides, respectively. The corresponding penta‐triflate T[5]‐(OTf)5, for example, was synthesized in 80 % yield (see the Supporting Information).
Scheme 2.
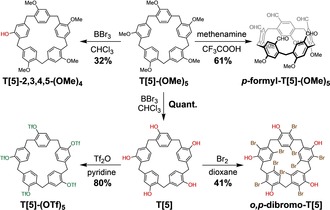
Versatile derivatizations of the tiara[5]arene scaffold at the ortho, para, and phenolic positions.
Given the presence of OH or OMe activating groups, tiara[5]arenes can be easily functionalized by regioselective electrophilic substitutions. Under Duff formylation conditions, T[5]‐(OMe)5 was transformed successfully into penta‐para‐substituted p‐formyl‐T[5]‐(OMe)5 in 61 % yield.27 Various bromination conditions were also tested, with the relative mild water–bromine system providing the highest yield (41 %) of an isomerically pure decabromide derivative o,p‐dibromo‐T[5]. Interestingly, it was found that bromination is a fast reaction, that is, completed in minutes, and not only on the five para‐ but also at all five ortho‐positions. Mass spectrometry analysis shows good agreement in isotopic distributions with the decabromide product (see the Supporting Information).
Apart from highly symmetric penta‐ and deca‐functionalizations, we have also focused on strategies to address a single aromatic ring in T[5]s selectively. Such mono‐functionalized28 macrocycles are highly desirable building blocks for developing various supramolecular architectures and functional systems. Whereas cleavage of the ether bonds on perfunctionalized pillar[5]arenes results in numerous isomeric products,29 partial demethylation of penta‐methylate T[5]‐(OMe)5 by 1 equiv of BBr3 led to a much less complicated reaction product mixture.30 As a result, mono‐deprotected T[5]‐2,3,4,5‐(OMe)4 could be isolated in a reasonable yield of 32 % by column chromatography.31 We believe the field of mono‐functionalized tiararenes may be open to a wide range of applications not easily available to other macrocycles.
Finally, attention was given to the possibility of using tiararene‐based materials for separation purposes. Many oligophenol macrocycles, especially calix[4]arenes, have been found32 to exhibit single‐crystal‐to‐single‐crystal transformations in the solid state upon guest inclusion and/or gas absorption. More recently, pillararene‐based crystalline organic materials have also been shown33 to be effective for the separation of linear/branched alkanes, xylene isomers, styrene/ethylbenzene, toluene/methyl‐cyclohexane, and dihalobenzene isomers. Analogously, the triangular biphen[3]arene macrocycle was used12g to discriminate cis‐ over trans‐dichloroethene during preferential solid‐vapor uptake, and dual‐selective fractionations of chlorobenzene/chlorocyclohexane by two polymorphs of geminiarene featuring different conformations were also reported.12h These findings all intrigued us to investigate the solid‐state host–guest chemistry of tiara[5]arene‐based crystalline materials.
Permethylated T[5]‐(OMe)5 was chosen for the ease of straightforward crystallization by vapor diffusion of CH3OH into a CH3CN solution. The resulting CH3CN⊂T[5]‐(OMe)5 crystals were then subjected to vacuum at elevated temperature to remove solvent guest molecules from the cavity. Powder X‐ray diffraction (PXRD) characterization of T[5]‐(OMe)5 materials before and after activation confirms (Figure 4 a,b) their crystalline nature. Preliminary solid‐vapor sorption experiments were carried out to test the uptake capacity and selectivity of the activated T[5]‐(OMe)5 crystalline material. It was found that T[5]‐(OMe)5 strongly favors aromatic benzene derivatives over their aliphatic counterparts.
Figure 4.

Outline of the benzene/cyclohexane fractionation process involving tiara[5]arene‐based nonporous adaptive crystals. a) First, crystals of T[5]‐(OMe)5 obtained from CH3CN were subjected to heat/vacuum for solvent removal. b) The activated T[5]‐(OMe)5 crystalline material was exposed to a 1:1 benzene/cyclohexane mixture vapor (50:50 v/v) under ambient conditions. c) Subsequent PXRD and X‐ray crystallography analyses confirmed the phase change of T[5]‐(OMe)5 after vapor adsorption, while d) 1H‐NMR time‐dependent vapor‐solid sorption plot and gas chromatography analysis revealed the preferential uptake of benzene over cyclohexane.
The separation of benzene and cyclohexane is an important industrial process particularly in view of their interdependence, for example, cyclohexane is obtained from the catalytic hydrogenation of benzene.34 The minute 0.7 K difference between the boiling points of benzene (353.2 K) and cyclohexane (353.9 K) imposes a great challenge for various distillation and purification processes. To test its capability for benzene/cyclohexane fractionation, time‐dependent sorption experiments were conducted on activated T[5]‐(OMe)5 samples over a mixture of benzene/cyclohexane (50:50 v/v). 1H NMR analysis showed that benzene uptake increased rapidly within the first hour and eventually reached about 0.8 equiv per tiara[5]arene ring after saturation, whereas relatively small amount of cyclohexane is also present in the crystals. Gas chromatography confirmed (Figure 4 d) that benzene accounts for 92.3 % of total guest uptake in T[5]‐(OMe)5. Upon heating, the benzene molecules adsorbed are released and the T[5]‐(OMe)5 can be reactivated for repeated fractionation cycles (see the Supporting Information).
The PXRD patterns of activated T[5]‐(OMe)5 before and after benzene uptake are significantly different, indicating (Figure 4 c, bottom) the formation of a new crystalline phase upon the adsorption of benzene. To elucidate the structural transformations of the T[5]‐(OMe)5 crystalline materials resulting from the adsorption process, T[5]‐(OMe)5 was crystallized from benzene after slow evaporation and analyzed by X‐ray crystallography. The C6H6@T[5]‐(OMe)5 superstructure35 features (Figure 4 c, top) a distorted conformer which stacks along the crystallographic c‐axis. Instead of forming inclusion complexes with T[5]‐(OMe)5, the benzene molecules are found in the interstices between stacks of macrocycles. A simulated PXRD pattern of the C6H6@T[5]‐(OMe)5 crystal structure, bears resemblance to the experimental PXRD pattern of T[5]‐(OMe)5 after benzene exposure, indicating that the permethylated tiara[5]arene selectively recrystallized with benzene upon solid‐vapor contact.
Conclusion
In summary, we have synthesized an oligophenol‐based macrocycle, tiara[5]arene, which is not obtainable by the direct, century‐old condensation of phenol with formaldehyde. The synthesis detour took advantage of the efficient formation of rim‐differentiated pillar[5]arenes, followed by the selective hydrodeoxygenation of one rim. X‐ray crystallography studies revealed multifaceted conformations of tiara[5]arenes in the solid state. We demonstrated a range of functionalization routes specific for tiararenes, further expanding this class of compounds. In addition to the ability of binding cationic pyridinium and bipyridinium guests, tiara[5]arene‐based crystalline materials showed highly selective fractionations of aromatic/aliphatic compounds during solid‐vapor adsorption. We expect that these new macrocycles will find widespread applications in supramolecular chemistry, both in solution and in the solid state, and will serve as unique five‐fold symmetric building blocks for constructing complex architectures.36
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the 973 National Basic Research Program of China (2015CB856500), National Science Foundation of China (H.Z., 21871208), Tianjin City Thousand Talents Program (H.Z. and A.C.‐H.S.), Chinese National Thousand Talents Young Investigator Program (A.C.‐H.S.), SAFEA foreign young talent program (K.S.), Wageningen University, and Tianjin University. We thank Prof. Xiangyang Zhang and all staff of Instrumental Analysis Center, School of Pharmaceutical Science and Technology, Tianjin University for assistance in various characterizations, and Jia‐Rui Wu, Prof. Ying‐Wei Yang, Kaidi Xu, and Prof. Chunju Li for their help with the NACs experiments.
W. Yang, K. Samanta, X. Wan, T. U. Thikekar, Y. Chao, S. Li, K. Du, J. Xu, Y. Gao, H. Zuilhof, A. C.-H. Sue, Angew. Chem. Int. Ed. 2020, 59, 3994.
Contributor Information
Prof. Han Zuilhof, Email: han.zuilhof@wur.nl.
Prof. Andrew C.‐H. Sue, Email: andrew.sue@tju.edu.cn.
References
- 1.
- 1a. Pedersen C. J., J. Am. Chem. Soc. 1967, 89, 2495–2496; [Google Scholar]
- 1b. Pedersen C. J., J. Am. Chem. Soc. 1967, 89, 7017–7036. [Google Scholar]
- 2. Steed J. W., Atwood J. L., Supramolecular Chemistry, Wiley, Chichester, 2013. [Google Scholar]
- 3.
- 3a. Villiers A., C. R. Hebd. Seances Acad. Sci. 1891, 112, 536–538; [Google Scholar]
- 3b. D'Souza V. T., Lipkowitz K. B., Chem. Rev. 1998, 98, 1741–1742; [DOI] [PubMed] [Google Scholar]
- 3c. Crini G., Chem. Rev. 2014, 114, 10940–10975. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Behrend R., Meyer E., Rusche F., Justus Liebigs Ann. Chem. 1905, 339, 1–37; [Google Scholar]
- 4b. Freeman W. A., Mock W. L., Shih N. Y., J. Am. Chem. Soc. 1981, 103, 7367–7368; [Google Scholar]
- 4c. Kim J., Jung I.-S., Kim S.-Y., Lee E., Kang J.-K., Sakamoto S., Yamaguchi K., Kim K., J. Am. Chem. Soc. 2000, 122, 540–541. [Google Scholar]
- 5.
- 5a. Hiraoka M., Crown Ethers and Analogous Compounds, Elsevier, Amsterdam, 2016; [Google Scholar]
- 5b. Pedersen C. J., Science 1988, 241, 536–540. [DOI] [PubMed] [Google Scholar]
- 6. Baeyer A., Ber. Dtsch. Chem. Ges. 1872, 5, 280–282. [Google Scholar]
- 7.
- 7a. Baekeland L. H., US Patent 942 699, 1907;
- 7b. Crespy D., Bozonnet M., Meier M., Angew. Chem. Int. Ed. 2008, 47, 3322–3328; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3368–3374. [Google Scholar]
- 8. Hardie M. J., Chem. Soc. Rev. 2010, 39, 516–527. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Timmerman P., Verboom W., Reinhoudt D. N., Tetrahedron 1996, 52, 2663–2704; [Google Scholar]
- 9b. Biros S. M., J. Rebek, Jr. , Chem. Soc. Rev. 2007, 36, 93–104; [DOI] [PubMed] [Google Scholar]
- 9c. Pirondini L., Dalcanale E., Chem. Soc. Rev. 2007, 36, 695–706. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Böhmer V., Angew. Chem. Int. Ed. Engl. 1995, 34, 713–745; [Google Scholar]; Angew. Chem. 1995, 107, 785–818; [Google Scholar]
- 10b. Gutsche C. D., Calixarenes Revisited, Royal Society of Chemistry, Cambridge: 1998; [Google Scholar]
- 10c. Gutsche C. D., Calixarenes, An Introduction, Royal Society of Chemistry, Cambridge, 2008. [Google Scholar]
- 11.
- 11a. Ogoshi T., Yamagishi T.-a., Nakamoto Y., Chem. Rev. 2016, 116, 7937–8002; [DOI] [PubMed] [Google Scholar]
- 11b. Ogoshi T., Pillararenes, Royal Society of Chemistry, Cambridge, 2016. [Google Scholar]
- 12.
- 12a. Schneebeli S. T., Cheng C., Hartlieb K. J., Strutt N. L., Sarjeant A. A., Stern C. L., Stoddart J. F., Chem. Eur. J. 2013, 19, 3860–3868; [DOI] [PubMed] [Google Scholar]
- 12b. Boinski T., Cieszkowski A., Rosa B., Szumna A., J. Org. Chem. 2015, 80, 3488–3495; [DOI] [PubMed] [Google Scholar]
- 12c. Chen H., Fan J., Hu X., Ma J., Wang S., Li J., Yu Y., Jia X., Li C., Chem. Sci. 2015, 6, 197–202; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Zhang G.-W., Li P.-F., -Meng Z., Wang H.-X., Han Yi., Chen C.-F., Angew. Chem. Int. Ed. 2016, 55, 5304–5308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5390–5394; [Google Scholar]
- 12e. Dai L., Ding Z.-J., Cui L., Li J., Jia X., Li C., Chem. Commun. 2017, 53, 12096–12099; [DOI] [PubMed] [Google Scholar]
- 12f. Li B., Wang B., Huang X., Dai L., Cui L., Li J., Jia X., Li C., Angew. Chem. Int. Ed. 2019, 58, 3885–3889; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3925–3929; [Google Scholar]
- 12g. Wang Y., Xu K., Li B., Cui L., Li J., Jia X., Li C., Angew. Chem. Int. Ed. 2019, 58, 10281–10284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10387–10390; [Google Scholar]
- 12h. Wu J., Yang Y., J. Am. Chem. Soc. 2019, 141, 12280–12287. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Ogoshi T., Kanai S., Fujinami S., Yamagishi T.-a., Nakamoto Y., J. Am. Chem. Soc. 2008, 130, 5022–5023; [DOI] [PubMed] [Google Scholar]
- 13b. Ogoshi T., Aoki T., Kitajima K., Fujinami S., Yamagishi T.-a., Nakamoto Y., J. Org. Chem. 2011, 76, 328–331. [DOI] [PubMed] [Google Scholar]
- 14. Guo M., Wang X., Zhan C., Demay-Drouhard P., Li W., Du K., Olson M. A., Zuilhof H., Sue A. C.-H., J. Am. Chem. Soc. 2018, 140, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Demay-Drouhard P., Du K., Samanta K., Wan X., Yang W., Srinivasan R., Sue A. C.-H., Zuilhof H., Org. Lett. 2019, 21, 3976–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Kou Y., Tao H., Cao D., Fu Z., Schollmeyer D., Meier H., Eur. J. Org. Chem. 2010, 6464–6470; [Google Scholar]
- 16b. Zhang Z., Luo Y., Xia B., Han C., Yu Y., Chen X., Huang F., Chem. Commun. 2011, 47, 2417–2419; [DOI] [PubMed] [Google Scholar]
- 16c. Al-Azemi T. F., Vinodh M., Alipour F. H., Mohamod A. A., J. Org. Chem. 2017, 82, 10945–10952; [DOI] [PubMed] [Google Scholar]
- 16d. Ding J., Chen J., Mao W., Huang J., Ma D., Org. Biomol. Chem. 2017, 15, 7894–7897. [DOI] [PubMed] [Google Scholar]
- 17. Strutt N. L., Zhang H., Schneebeli S. T., Stoddart J. F., Acc. Chem. Res. 2014, 47, 2631–2642. [DOI] [PubMed] [Google Scholar]
- 18. Jie K., Zhou Y., Li E., Huang F., Acc. Chem. Res. 2018, 51, 2064–2072. [DOI] [PubMed] [Google Scholar]
- 19. Holler M., Allenbach N., Sonet J., Nierengarten J.-F., Chem. Commun. 2012, 48, 2576–2578. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Gribble G. W., Nutaitis C. F., Tetrahedron Lett. 1985, 26, 6023–6026; [Google Scholar]
- 20b. Liu P., Li Q., Zeng H., Shi B., Liu J., Huang F., Org. Chem. Front. 2019, 6, 309–312. [Google Scholar]
- 21.The rotational freedom of pillar[5]arenes has been investigated in detail, both experimentally and theoretically; see:
- 21a. Ogoshi T., Kitajima K., Aoki T., Fujinami S., Yamagishi T.-a., Nakamoto Y., J. Org. Chem. 2010, 75, 3268–3273; [DOI] [PubMed] [Google Scholar]
- 21b. Strutt N. L., Schneebeli S. T., Stoddart J. F., Supramol. Chem. 2013, 25, 596–608; [Google Scholar]
- 21c. Wang X., Chen R.-x., Sue A. C.-H., Zuilhof H., Aquino A. J. A., Lischka H., Comput. Theor. Chem. 2019, 1161, 1–9. [Google Scholar]
- 22.Crystallographic data for T[5]-(OMe)5: C40H40O5; colorless block, 0.2×0.2×0.2 mm3; triclinic, space group P ; a=11.30470(10), b=12.60500(10); c=12.78350(10) Å; α=76.1580(10), β=88.8410(10), γ=66.9490(10)°; V=1621.97(3) Å3; Z=2; ρ calc=1.230 g cm−3; 2θ max=149.378; T=160(10) K; 45 791 reflections collected, 6363 independent, 411 parameters; μ=0.634 mm−1; R1=0.0513 [I>2σ(I)], wR2=0.1472 (all data).[37]
- 23. Wu J.-R., Mu A. U., Li B., Wang C.-Y., Fang L., Yang Y.-W., Angew. Chem. Int. Ed. 2018, 57, 9853–9858; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10001–10006. [Google Scholar]
- 24.Crystallographic data for CH3CN⊂T[5]-(OMe)5: C42H43NO5; colorless block, 0.2×0.2×0.2 mm3; triclinic, space group P ; a=12.0935(2), b=17.8479(3); c=17.9763(2) Å; α=95.1170(10), β=106.5750(10), γ=105.9900(10)°; V=3515.19(9) Å3; Z=4; ρ calc=1.213 g cm−3; 2θ max=149.272; T=159.99(10) K; 34 158 reflections collected, 12 350 independent, 1069 parameters; μ=0.626 mm−1; R1=0.0701 [I>2σ(I)], wR2=0.16 (all data).[37]
- 25.Crystallographic data for H2O⊂T[5]: C42H40O6; yellow block, 0.2×0.2×0.2 mm3; triclinic, space group P ; a=10.1845(2), b=12.1980(2); c=15.5764(2) Å; α=98.7530(10), β=102.834(2), γ=107.544(2)°; V=1748.27(6) Å3; Z=2; ρ calc=1.217 g cm−3; 2θ max=149.204; T=310.60(10) K; 33 007 reflections collected, 6919 independent, 604 parameters; μ=0.644 mm−1; R1=0.0532 [I>2σ(I)], wR2=0.1600 (all data).[37]
- 26.
- 26a. Shu X., Chen S., Li J., Chen Z., Weng L., Jia X., Li C., Chem. Commun. 2012, 48, 2967–2969; [DOI] [PubMed] [Google Scholar]
- 26b. Wang Y., Ping G., Li C., Chem. Commun. 2016, 52, 9858–9872. [DOI] [PubMed] [Google Scholar]
- 27.The formylation turned out to be rather specific in relation to reaction conditions, as under for example, Vilsmeier-Haak (POCl3/DMF) or Rieche formylation (Cl2CHOCH3/TiCl4), neither p- nor o-formyl-functionalized tiara[5]arenes could be obtained.
- 28. Lavendomme R., Leroy A., Luhmer M., Jabin I., J. Org. Chem. 2014, 79, 6563–6570. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Ogoshi T., Demachi K., Kitajima K., Yamagishi T.-a., Chem. Commun. 2011, 47, 7164–7166; [DOI] [PubMed] [Google Scholar]
- 29b. Strutt N. L., Forgan R. S., Spruell J. M., Botros Y. Y., Stoddart J. F., J. Am. Chem. Soc. 2011, 133, 5668–5671; [DOI] [PubMed] [Google Scholar]
- 29c. Chen Y., He M., Li B., Wang L., Meier H., Cao D., RSC Adv. 2013, 3, 21405–21408; [Google Scholar]
- 29d. Al-Azemi T. F., Mohamod A. A., Vinodha M., Alipoura F. H., Org. Chem. Front. 2018, 5, 10–18. [Google Scholar]
- 30.Partial demethylation of T[5]-(OMe)5 results in six different partially deprotected products in total.
- 31.The side product mixtures, containing partially-deprotected T[5]-(OMe)5, were collected and recycled for complete demethylation to T[5] in 57 % yield (see the Supporting Information).
- 32.
- 32a. Atwood J. L., Barbour L. J., Jerga A., Science 2002, 296, 2367–2369; [DOI] [PubMed] [Google Scholar]
- 32b. Atwood J. L., Barbour L. J., Jerga A., Schottel B. L., Science 2002, 298, 1000–1002; [DOI] [PubMed] [Google Scholar]
- 32c. Atwood J. L., Barbour L. J., Jerga A., Angew. Chem. Int. Ed. 2004, 43, 2948–2950; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3008–3010; for a more recent comprehensive review, see [Google Scholar]
- 32d. Zhou Y., Jie K., Zhao R., Huang F., Adv. Mater. 2019, 1904824. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Ogoshi T., Sueto R., Yoshikoshi K., Sakata Y., Akine S., Yamagishi T.-a., Angew. Chem. Int. Ed. 2015, 54, 9849–9852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9987–9990; [Google Scholar]
- 33b. Jie K., Liu M., Zhou Y., Little M. A., Bonakala S., Chong S. Y., Stephenson A., Chen L., Huang F., Cooper A. I., J. Am. Chem. Soc. 2017, 139, 2908–2911; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33c. Jie K., Zhou Y., Li E., Zhao R., Liu M., Huang F., J. Am. Chem. Soc. 2018, 140, 3190–3193; [DOI] [PubMed] [Google Scholar]
- 33d. Jie K., Liu M., Zhou Y., Little M. A., Pulido A., Chong S. Y., Stephenson A., Hughes A. R., Sakakibara F., Ogoshi T., Blanc F., Day G. M., Huang F., Cooper A. I., J. Am. Chem. Soc. 2018, 140, 6921–6930; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33e. Jie K., Zhou Y., Li E., Zhao R., Huang F., Angew. Chem. Int. Ed. 2018, 57, 12845–12849; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13027–13031; [Google Scholar]
- 33f. Li E., Zhou Y., Zhao R., Jie K., Huang F., Angew. Chem. Int. Ed. 2019, 58, 3981–3985; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4021–4025. [Google Scholar]
- 34. Garcia Villaluenga J. P., Tabe-Mohammadi A., J. Membr. Sci. 2000, 169, 159–174. [Google Scholar]
- 35.Crystallographic data for C6H6@T[5]-(OMe)5: C52H50.77O5; colorless block, 0.15×0.15×0.1 mm3; monoclinic, space group P21/c; a=13.07000(10), b=25.2860(2); c=13.24560(10) Å; α=γ=90, β=106.9020(10); V=4188.42(6) Å3; Z=4; ρ calc=1.198 g cm−3; 2θ max=149.61; T=160(10) K; 78 807 reflections collected, 8439 independent, 550 parameters; μ=0.595 mm−1; R1=0.0409 [I>2σ(I)], wR2=0.1006 (all data).[37]
- 36.
- 36a. Olenyuk B., Levin M. D., Whiteford J. A., Shield J. E., Stang P. J., J. Am. Chem. Soc. 1999, 121, 10434–10435; [Google Scholar]
- 36b. Bacsa J., Less R. J., Skelton H. E., Soracevic Z., Steiner A., Wilson T. C., Wood P. T., Wright D. S., Angew. Chem. Int. Ed. 2011, 50, 8279–8282; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8429–8432; [Google Scholar]
- 36c. Ren C., Zhou F., Qin B., Ye R., Shen S., Su H., Zeng H., Angew. Chem. Int. Ed. 2011, 50, 10612–10615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10800–10803; [Google Scholar]
- 36d. Pasquale S., Sattin S., Escudero-Adán E. C., Martínez-Belmonte M., de Mendoza J., Nat. Commun. 2012, 3, 785; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36e. Fujita D., Ueda Y., Sato S., Yokoyama H., Mizuno N., Kumasaka T., Fujita M., Chem 2016, 1, 91–101; [Google Scholar]
- 36f. Chen Y.-S., Solel E., Huang Y.-F., Wang C.-L., Tu T.-H., Keinan E., Chan Y.-T., Nat. Commun. 2019, 10, 3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201913055 (T[5]-(OMe)5, CH3CN⊂T[5]-(OMe)5, H2O⊂T[5], and C6H6@T[5]-(OMe)5) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary