

Abstract
This study compared the pharmacokinetic (PK) profile of a new liquid formulation of mepolizumab with the established lyophilized formulation. In this open‐label, parallel‐group, single‐dose study (NCT03014674; GSK ID: 204958), healthy participants were randomized (1:1:1) to receive a single mepolizumab dose (100 mg) administered subcutaneously as liquid in a single‐use prefilled syringe or single‐use prefilled autoinjector, or as a lyophilized formulation. Maximum plasma concentration, area under the plasma concentration–time curve from time zero (predose) to time of last quantifiable concentration (AUC0–t), and AUC from time zero to infinity (AUC0–∞) as well as additional PK parameters, safety assessments, and blood eosinophil count were evaluated. In total, 244 participants received study drug. All PK parameters were similar across the 3 groups; 90% confidence intervals for maximum plasma concentration, AUC0–t, and AUC0–∞ treatment ratios (liquid prefilled syringe or autoinjector vs lyophilized formulation) were within conventional bioequivalence bounds (0.80‐1.25), demonstrating statistical PK comparability. On‐treatment adverse event incidence was 29% to 38%. Mepolizumab liquid formulation administered via prefilled syringe or autoinjector had similar PK properties to the lyophilized formulation, with no safety concerns identified.
Keywords: asthma, biologics, clinical trials, pharmacokinetics and drug metabolism, pulmonary
Mepolizumab is a humanized, immunoglobulin G1 (IgG1), anti‐interleukin (IL)‐5 monoclonal antibody, which prevents IL‐5 from binding to the IL‐5 receptor complex expressed on eosinophils.1, 2 This results in the suppression of downstream IL‐5–mediated responses such as T‐helper cell type 2 immune responses.3, 4 In patients with severe eosinophilic asthma, mepolizumab treatment has been shown to reduce exacerbation frequency, decrease oral corticosteroid dependence, and improve health‐related quality of life and lung function, compared with placebo.5, 6, 7, 8 In addition, mepolizumab has been demonstrated to increase remission duratio, and reduce relapses compared with placebo in patients with eosinophilic granulomatosis with polyangiitis (EGPA).9
Mepolizumab exhibits dose‐proportional and time‐independent pharmacokinetics (PK) after both intravenous and subcutaneous (SC) administration, consistent with other IgG1 monoclonal antibodies that target soluble cytokines.10, 11, 12 After SC administration, mepolizumab is absorbed slowly, disperses into a central volume of distribution equal to plasma volume with a distribution half‐life of 1 to 2 days, and has a steady‐state volume of distribution of 1.5 to 2 times plasma volume. Mepolizumab is catabolized by ubiquitous proteolytic enzymes (not restricted to hepatic tissue) and does not undergo target‐mediated clearance.11 It is cleared slowly with a terminal phase elimination half‐life of approximately 20 days, irrespective of administration route, with mean systemic clearance of 0.21 L/day (for a 70‐kg individual).10, 11, 12 In a prior study in healthy participants, the absolute bioavailability of a single dose of SC mepolizumab ranged from 64% to 75%, across administration sites (abdomen, upper arm, and thigh).13
Mepolizumab is approved for use in severe eosinophilic asthma and EGPA,1, 2 and is also currently being explored for the treatment of patients with eosinophilic chronic obstructive pulmonary disease,14 nasal polyposis,15 and hypereosinophilic syndrome.16
The efficacy and safety of mepolizumab following both intravenous and SC administration have been demonstrated in a number of clinical studies across a range of eosinophilic diseases.5, 6, 7, 8, 9, 14, 15, 17, 18, 19, 20 The current formulation of mepolizumab in severe eosinophilic asthma and EGPA is a sterile, single‐use, preservative‐free, lyophilized drug product (referred to as reconstituted lyophilized drug product) for SC administration, which must be reconstituted with sterile water for injection using aseptic techniques.21 It is not uncommon to modify the formulation or presentation of a medicine to improve convenience of use.22 Mepolizumab as a liquid drug product in a ready‐to‐use prefilled syringe (PFS) or prefilled autoinjector (AI) would eliminate the need for reconstitution steps and may also allow administration by the patient or caregiver at home. This may help to improve patient autonomy and flexibility in physician visits, increase patient productivity, and reduce health care burden and costs compared with health care provider administration options.23, 24
The aim of this study was to compare the PK and safety profiles and to determine the relative bioavailability of the liquid drug product in a single‐use PFS or single‐use AI with the established reconstituted lyophilized drug product following administration of a single SC dose of mepolizumab 100 mg in healthy participants.
Methods
Study Design
The study protocol, amendments, and informed consent were reviewed and approved by national, regional, or investigational center ethics committees or institutional review boards (German Ethics Committee in Berlin: Landesamt fuer Gesundheit und Soziales, Ethik‐Kommission des Landes Berlin; UK Ethics Committee in Bristol: South Central–Berkshire B Research Ethics Committee; US Institutional Review Board in California: Aspire Institutional Review Board). Written informed consent was obtained from each participant before any procedures. The study was conducted at 3 centers: 1 in Germany (Parexel International, Berlin, Germany), 1 in the United Kingdom (Parexel International, Harrow, Middlesex, UK), and 1 in the United States (Parexel International, Baltimore, Maryland), and was conducted according to the ethical principles outlined in the current Declaration of Helsinki, International Conference for Harmonisation Good Clinical Practice, and the applicable country‐specific regulatory requirements.
This was a randomized, open‐label, 3‐arm, parallel‐group, single‐dose study in healthy participants that was conducted at clinical study centers in Germany, the United Kingdom, and the United States between January 6, 2017 and August 11, 2017 (ClinicalTrials.gov number: NCT03014674; GSK ID: 204958). Each participant received a single dose of mepolizumab 100 mg SC on day 1. Participants were monitored and had blood samples collected for at least 8 hours after dosing before being discharged from the study center. The overall follow‐up phase after drug administration was 85 days.
Participants
Healthy participants ≥18 years of age, with a body weight of ≥50 kg and a body mass index ≥19 to ≤30 kg/m2, with no clinically relevant abnormalities as determined from medical history, physical examination, vital signs, and laboratory tests were enrolled in this study. Eligible female participants were postmenopausal, premenopausal with nonreproductive potential, or premenopausal and not pregnant or lactating. Study exclusion criteria, typical for studies with healthy participants, were as follows: alanine aminotransferase level >1.5 × upper limit of normal, bilirubin level >1.5 × upper limit of normal, or QT interval corrected using Fridericia's formula >450 milliseconds; current or chronic history of liver disease or known hepatic or biliary abnormalities (with the exception of Gilbert's syndrome or asymptomatic gallstones); clinically relevant abnormality identified at the screening medical assessment (physical examination/medical history), clinical laboratory tests, or 12‐lead electrocardiogram (ECG); use of prescription or nonprescription drugs, including vitamins and herbal and dietary supplements (including St John's wort) within 7 days (or 14 days if the drug was a potential enzyme inducer) or 5 half‐lives (whichever was longer) before the first dose of study medication and until study completion, unless in the opinion of the investigator and the medical monitor the medication would not interfere with the study procedures or compromise participant safety; a history of regular alcohol consumption within 6 months of the study defined as an average weekly intake of >14 units for females and >21 units for males; urinary cotinine levels (>500 ng/mL) indicative of smoking or history or regular use of tobacco‐ or nicotine‐containing products within 6 months before screening; a history of sensitivity to any of the study medications or components thereof or a history of drug or other allergy that, in the opinion of the investigator or medical monitor, contraindicated their participation; any of the following: presence of hepatitis B surface antigen, positive hepatitis C antibody test result at screening or within 3 months before first dose of study treatment, a positive test for HIV antibody, or a known preexisting helminth infestation within 6 months before day 1; participation in a clinical trial and treatment with an investigational product within the following time period before the first dosing day in the current study: 30 days, 5 half‐lives, or twice the duration of the biological effect of the investigational product (whichever was longer); exposed to more than 4 new chemical entities within 12 months before the first dosing day; a positive prestudy drug/alcohol screen; or vulnerable, defined as individuals whose willingness to volunteer in a clinical trial may have been unduly influenced by the expectation, whether justified or not, of benefits associated with participation, or of a retaliatory response from senior members of a hierarchy in case of refusal to participate.
Interventions
Participants were randomized (1:1:1) to receive a single dose of mepolizumab 100 mg SC on day 1 of the study, administered either as a liquid drug product in a PFS (BD UltraSafe PLUS; Becton Dickinson, Franklin Lakes, New Jersey), a liquid drug product in an AI (YpsoMate; Ypsomed AG, Burgdorf, Switzerland), or as a reconstituted lyophilized drug product from a vial and administered with a standard syringe. The site of injection was also randomized (1:1:1) to the abdomen, upper arm, or thigh. Randomization was performed using an interactive response system. The randomization sequence was centrally computer generated using a permuted‐block design of block size 9, and was stratified by body weight (<70 kg, ≥70 to <80 kg, and ≥80 kg) measured at day −1 to ensure similar body weight distribution across the 3 treatment groups. Mepolizumab liquid drug product was provided as a fixed‐dose, fully disposable, prefilled glass syringe, which was assembled in either a PFS or an AI. Mepolizumab lyophilized drug product was provided in sterile vials and reconstituted with sterile water for injection immediately before use. All treatments were administered subcutaneously by a health care professional.
End Points and Assessments
The PK parameters for comparability assessment between the liquid (in PFS and AI) and the reconstituted lyophilized drug products were maximum plasma concentration (Cmax), area under the plasma concentration–time curve from time zero (predose) to time of last quantifiable concentration (AUC0–t), and AUC from time zero to infinity (AUC0–∞). Additional PK parameters, safety and tolerability, immunogenicity assessments, and blood eosinophil count were also evaluated. The additional PK parameters investigated were time to Cmax, apparent clearance following SC dosing, apparent volume of distribution following SC dosing, terminal phase elimination rate constant, terminal phase elimination half‐life, last time point where the concentration was above the limit of quantification, and the percentage of AUC obtained by extrapolation. Blood samples for PK assessments were taken on day 1 (before dosing and 2 hours and 8 hours after dosing) and days 2 to 10, 15, 22, 29, 43, 57, and 85. Safety and tolerability assessments included the incidence of adverse events (AEs), serious AEs (SAEs) including systemic reactions and injection‐site reactions, hematology and clinical chemistry (assessed at screening and days −1, 5, and 85), vital signs (assessed at screening and days −1 to 7, 43, and 85), and 12‐lead ECG (assessed at screening and days 1 and 85). AEs and SAEs were assessed from days −1 to 85, with SAEs additionally assessed at screening. Immunogenicity assessments included the frequency of positive antidrug antibodies (ADAs) and neutralizing antibodies, and occurred at screening and days 1 (before dosing), 29, 43, and 85. Pharmacodynamic effects of mepolizumab on blood eosinophil counts relative to baseline levels were assessed on days 1 (before dosing), 3, 5, 10, 29, 57, and 85. Mepolizumab PK and immunogenicity assays have been described previously.10, 12 For the PK assay, the limit of sensitivity was 50 ng/mL; the between‐run precision (% coefficient of variation) and between‐run accuracy (% bias) were ≤6.9% and ±3%, respectively. For the ADA assay, the limit of sensitivity was 0.4 ng/mL, and the between‐run precision (%coefficient of variation) was ≤10.9%.
Sample Size and Statistical Analysis
The sample size for this study was based on the number of participants needed to demonstrate a 2‐sided 90% confidence interval (CI) for µ(test)/µ(reference) within the bioequivalence range (0.80‐1.25) for Cmax, AUC0–t, and AUC(0–∞). Assuming standard deviations on the loge scale of 0.32 for Cmax and 0.27 for AUC0–t and AUC0–∞, a true difference between the formulations of 5% and within‐participant correlation between AUC and Cmax of 0.8, the joint power with 72 participants per treatment group was estimated to be 89%. To allow for a dropout rate of 10%, 243 participants were enrolled to ensure that 216 completed the study. A minimum of 27 participants were randomized within each of the 3 body weight categories to ensure that there were at least 3 participants within each body weight category, injection site, and mepolizumab treatment combination. Mepolizumab PK parameters were derived (as data permitted) by standard noncompartmental analysis using Phoenix WinNonlin Version 6.3 (Certara, L.P. [Pharsight], St Louis, Missouri). Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, North Carolina).
The statistical analysis was to compare the Cmax, AUC0–t, and AUC0–∞ for each test treatment (liquid drug product in PFS or AI) with the reconstituted lyophilized drug product. Separate models were used for each treatment comparison. The parameters Cmax, AUC0–t, and AUC0–∞ were loge transformed and analyzed separately using a fixed‐effects analysis of covariance model, including treatment group and injection site (abdomen, upper arm, or thigh) as categorical variables, and baseline body weight as a continuous covariate fitted on the loge scale. Estimates and 2‐sided 90%CIs for the ratio of each of the test treatments to the reference treatment were summarized and plotted. Each treatment comparison was considered separately, and no adjustment for multiplicity was made. A post hoc statistical analysis to assess the effect of injection site was conducted combining data from all 3 treatment groups, using a fixed‐effects analysis of covariance model adjusting for injection site, treatment group, and baseline body weight. AEs were coded using the Medical Dictionary for Regulatory Activities and summarized by preferred term. The ratio to baseline for blood eosinophils was loge transformed and compared between treatments using a mixed‐model repeated‐measures analysis, adjusting for baseline blood eosinophil count (loge scale) and baseline body weight (loge scale) as fixed effects. Treatment group, visit, and injection site were fitted as categorical fixed effects variables. Treatment‐by‐visit and baseline blood eosinophil count‐by‐visit interaction effects were included in the model as fixed effects. Estimates and 95%CIs for the ratio of each test treatment to the reference treatment were summarized at each visit.
Results
Patient Population
Of the 449 participants screened between January 6, 2017, and May 9, 2017, 246 participants were randomized, and 244 participants received the study drug (2 participants were randomized in error). A total of 80 participants received mepolizumab (100 mg SC) administered as liquid drug product in a PFS, 79 as liquid drug product in an AI, and 85 received the reconstituted lyophilized drug product (Figure 1). All participants, except one who withdrew owing to travel expenses, completed the trial (Figure 1). Participant demographics at baseline were similar between treatment groups (Table 1).
Figure 1.
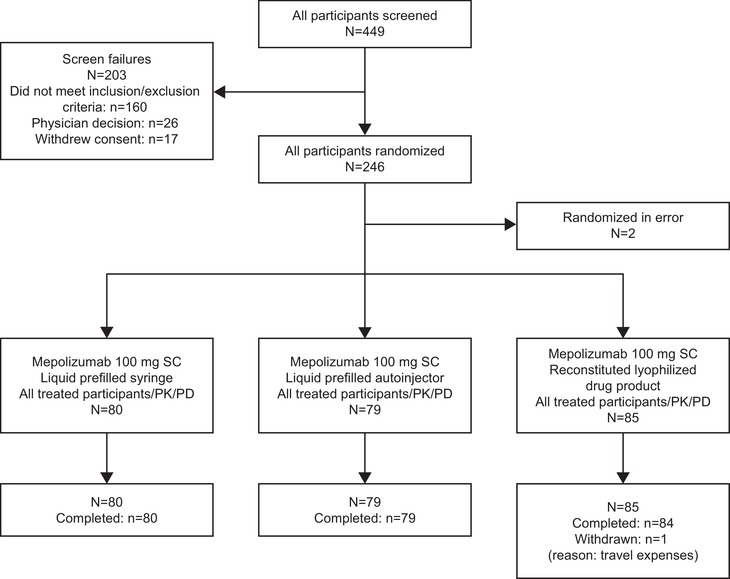
Participant disposition. PD, pharmacodynamic; PK, pharmacokinetic; SC, subcutaneous.
Table 1.
Participant Demographics
| Mepolizumab 100 mg SC | ||||
|---|---|---|---|---|
| Liquid PFS (N = 80) | Liquid AI (N = 79) | Reconstituted Lyophilized Drug Product (N = 85) | Total (N = 244) | |
| Age, years | ||||
| Mean (SD) | 47.5 (14.94) | 46.5 (15.00) | 46.1 (15.06) | 46.7 (14.95) |
| Range | 19‐76 | 22‐80 | 19‐75 | 19‐80 |
| ≥65, n (%) | 11 (14) | 12 (15) | 12 (14) | 35 (14) |
| Female, n (%) | 38 (48) | 36 (46) | 40 (47) | 114 (47) |
| Body weight, kg, mean (SD) | 74.68 (11.80) | 73.69 (10.29) | 73.57 (12.81) | 73.97 (11.67) |
| BMI, kg/m2, mean (SD) | 25.07 (2.77) | 24.91 (2.71) | 24.79 (2.77) | 24.92 (2.74) |
| Ethnicity | ||||
| Hispanic/Latino, n (%) | 2 (3) | 3 (4) | 3 (4) | 8 (3) |
| Race, n (%) | ||||
| White | 62 (78) | 61 (77) | 64 (75) | 187 (77) |
| Black | 18 (23) | 15 (19) | 18 (21) | 51 (21) |
| Asian | 0 | 1 (1) | 1 (1) | 2 (<1) |
| Multiple | 0 | 1 (1) | 1 (1) | 2 (<1) |
| American Indian/Alaska Native | 0 | 0 | 1 (1) | 1 (<1) |
| Native Hawaiian/Other Pacific Islander | 0 | 1 (1) | 0 | 1 (<1) |
AI, prefilled autoinjector; BMI, body mass index; PFS, prefilled syringe; SC, subcutaneous; SD, standard deviation.
PK End Points
Arithmetic mean plasma concentration–time profiles were similar following a single dose of mepolizumab (100 mg SC) of the liquid drug product delivered by a PFS or an AI, or of the reconstituted lyophilized drug product (Figure 2). PK parameters of Cmax, AUC0–t, and AUC0–∞ were similar across the 3 mepolizumab treatment groups (Table 2). The 90%CIs for the treatment ratios (liquid drug product in PFS vs reconstituted lyophilized drug product, and liquid drug product in AI vs reconstituted lyophilized drug product) for Cmax, AUC0–t, and AUC0–∞ were all contained within the conventional bioequivalence bounds of 0.80 to 1.25, demonstrating statistical PK comparability between the liquid and reconstituted lyophilized drug products (Table 3). The ratio for all estimates ranged from 1.02 (90%CI, 0.95‐1.09) to 1.08 (1.01‐1.15). Likewise, all the other PK parameters were similar across the 3 mepolizumab treatment groups (Table 2). In addition, mepolizumab adjusted geometric mean exposure observed for all 3 treatment groups combined across the 3 injection sites (abdomen, upper arm, or thigh) investigated did not markedly differ (Table 4).
Figure 2.
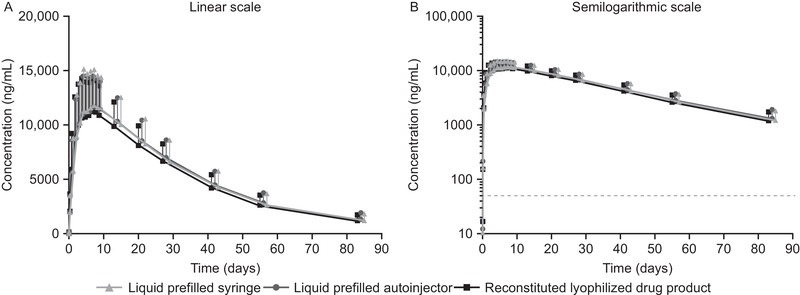
Arithmetic mean (+SD) plasma concentrations of mepolizumab over time by treatment group using (A) linear and (B) semilogarithmic scale. The dashed line indicates the lower limit of quantification: 50 ng/mL. SD, standard deviation.
Table 2.
Plasma Mepolizumab PK Parameters Following a Single SC 100 mg Dose by Treatment
| Liquid PFS (N = 80) | Liquid AI (N = 79) | Reconstituted Lyophilized Drug Product (N = 85) | ||||
|---|---|---|---|---|---|---|
| Arithmetic Mean (±SD) | Geometric Mean (±95%CI) | Arithmetic Mean (±SD) | Geometric Mean (±95%CI) | Arithmetic Mean (±SD) | Geometric Mean (±95%CI) | |
| Cmax (µg/mL) | 12.55 (3.427) | 12.07 (11.32‐12.87) | 12.40 (3.094) | 11.98 (11.27‐12.74) | 12.00 (3.291) | 11.57 (10.92‐12.27) |
| AUC0–t (µg • day/mL) | 432.45 (117.841) | 415.15 (388.36‐443.78) | 446.76 (101.053) | 434.49 (411.34‐458.94) | 420.29 (108.615) | 403.84 (377.83‐431.63) |
| AUC(0–∞) (µg • day /mL) | 475.47 (137.295) | 454.11 (423.03‐487.48) | 494.09 (122.357) | 478.06 (450.47‐507.34) | 466.22 (119.575)a | 450.83 (425.67‐477.47)a |
| tmax (days), median (range) | 7.06 (1.9‐14.0) | 7.05 (2.9‐21.0) | 7.04 (0.9‐14.1) | |||
| CL/F (L/day) | 0.233 (0.092) | 0.220 (0.205‐0.236) | 0.217 (0.062) | 0.209 (0.197‐0.222) | 0.230 (0.064)a | 0.222 (0.209‐0.235)a |
| Vz/F (L) | 7.22 (2.299) | 6.94 (6.53‐7.37) | 6.92 (1.823) | 6.74 (6.41‐7.08) | 7.20 (1.619)a | 7.02 (6.69‐7.37)a |
| t1/2, days | 22.40 (4.843) | 21.83 (20.72‐23.01) | 22.90 (4.896) | 22.34 (21.21‐23.53) | 22.36 (4.173)a | 21.95 (21.03‐22.92)a |
| tlast, days, median (range) | 83.99 (55.9‐87.9) | 83.98 (81.1‐87.1) | 83.97 (14.0‐87.0) | |||
| %AUCextrapolated | 8.49 (4.095) | 7.20 (6.19‐8.37) | 9.01 (4.264) | 7.64 (6.55‐8.90) | 8.52 (3.583)a | 7.67 (6.88‐8.54)a |
AI, prefilled autoinjector; AUC0–t, area under the plasma concentration–time curve from time zero (predose) to time of last quantifiable concentration; AUC0–∞, area under the plasma concentration–time curve from time zero to infinity; %AUCextrapolated, percentage of AUC(0–∞) obtained by extrapolation; CI, confidence interval; CL/F, apparent clearance following SC dosing; Cmax, maximum plasma concentration; PFS, prefilled syringe; PK, pharmacokinetic; SC, subcutaneous; SD, standard deviation; tlast, last time point where the concentration is above the limit of quantification; tmax, time to Cmax; t1/2, terminal phase elimination half‐life; Vz/F, apparent volume of distribution following SC dosing.
Values are arithmetic mean (±SD) and geometric mean (95% I) except for tmax and tlast, for which the median and range are presented.
n = 84.
Table 3.
Adjusted Geometric Means and Treatment Ratios (90%CI) for the Derived Plasma Mepolizumab PK Parameters Cmax, AUC0–t, and AUC0–∞ by Treatment
| Adjusted Geometric Mean (SE Log) | ||||
|---|---|---|---|---|
| Parameter | Test Treatment (PFS or AI) | Test Treatment (PFS or AI) | Reconstituted Lyophilized Drug Product | Adjusted Treatment Ratio (90%CI) (vs Reconstituted Lyophilized Drug Product) |
| Cmax | Liquid PFS | 12.14 (0.027) | 11.51 (0.027) | 1.06 (0.99‐1.12) |
| (µg/mL) | Liquid AI | 12.01 (0.028) | 11.55 (0.027) | 1.04 (0.98‐1.11) |
| AUC0–t | Liquid PFS | 417.93 (0.031) | 401.30 (0.030) | 1.04 (0.97‐1.12) |
| (µg • day /mL) | Liquid AI | 435.46 (0.029) | 403.00 (0.028) | 1.08 (1.01‐1.15) |
| AUC0–∞ | Liquid PFS | 457.03 (0.029) | 447.92 (0.028) | 1.02 (0.95‐1.09) |
| (µg • day /mL) | Liquid AI | 478.94 (0.026) | 449.70 (0.026) | 1.07 (1.00‐1.13) |
AI, prefilled autoinjector; AUC0–t, area under the plasma concentration–time curve from time zero (predose) to time of last quantifiable concentration; AUC0–∞, area under the plasma concentration–time curve from time zero to infinity; CI, confidence interval; Cmax, maximum plasma concentration; PFS, prefilled syringe; PK, pharmacokinetic; SE, standard error.
Estimates are adjusted for injection site (abdomen, upper arm, thigh) and baseline body weight (loge scale).
Table 4.
Adjusted Geometric Means and Treatment Ratios (90%CI) for the Plasma Mepolizumab PK Parameters Cmax, AUC(0–t), and AUC(0–∞) by Injection Site (All Treatments Combined)
| Injection Site | Adjusted Geometric Mean (SE Log) | Comparison | Adjusted Ratio (±90%CI) | |
|---|---|---|---|---|
| Cmax (µg/mL) | Abdomen | 11.40 (0.028) | Abdomen vs upper arm | 1.04 (0.97‐1.11) |
| Upper arm | 11.00 (0.028) | Abdomen vs thigh | 0.86 (0.81‐0.92) | |
| Thigh | 13.25 (0.027) | Upper arm vs thigh | 0.83 (0.78‐0.88) | |
| AUC0–t (µg • day /mL) | Abdomen | 392.07 (0.029) | Abdomen vs upper arm | 0.96 (0.89‐1.02) |
| Upper arm | 410.26 (0.029) | Abdomen vs thigh | 0.87 (0.82‐0.93) | |
| Thigh | 450.00 (0.028) | Upper arm vs thigh | 0.91 (0.85‐0.97) | |
| AUC0–∞ (µg • day /mL) | Abdomen | 436.91 (0.028) | Abdomen vs upper arm | 0.97 (0.91‐1.04) |
| Upper arm | 449.40 (0.028) | Abdomen vs thigh | 0.88 (0.83‐0.94) | |
| Thigh | 495.40 (0.027) | Upper arm vs thigh | 0.91 (0.85‐0.97) |
AUC0–t, area under the plasma concentration‐time curve from time zero (predose) to time of last quantifiable concentration; AUC0–∞, area under the plasma concentration–time curve from time zero to infinity; CI, confidence interval; Cmax, maximum plasma concentration; SE, standard error.
Estimates are adjusted for treatment (reconstituted lyophilized drug product, liquid PFS, liquid AI) and baseline body weight (loge scale).
Safety
The overall incidence of any on‐treatment AEs was 38% (30 of 80) in the liquid drug product PFS group, 34% (27 of 79) in the liquid drug product AI group, and 29% (25 of 85) in the reconstituted lyophilized drug product group. AEs considered related to study treatment by the investigator were reported in 25% (20 of 80) of participants in the liquid drug product PFS group, 22% (17 of 79) in the liquid drug product AI group, and 20% (17 of 85) in the reconstituted lyophilized drug product group. The most commonly reported on‐treatment AEs in all treatment groups were headache and viral upper respiratory tract infections, with an incidence of 7% to 11% and 2% to 8%, respectively. The most frequently reported AEs considered by the investigator to be drug related were headache and fatigue, with an incidence of 5% to 9% and 3% to 6% across treatment groups, respectively. No on‐treatment SAEs or deaths were reported (Table 5). There were no reports of anaphylaxis or on‐treatment systemic allergic (type 1 hypersensitivity) reactions. The incidence of injection‐site reactions was similar (1%–3%; edema, pain, pruritus, and warmth) across all treatment groups. There were no apparent treatment‐related changes in clinical laboratory parameters, vital signs, and 12‐lead ECG.
Table 5.
Summary of AEs
| n (%) | Liquid PFS (N = 80) | Liquid AI (N = 79) | Total Liquid (N = 159) | Reconstituted Lyophilized Drug Product (N = 85) | Total (N = 244) |
|---|---|---|---|---|---|
| Any on‐treatment AE | 30 (38) | 27 (34) | 57 (36) | 25 (29) | 82 (34) |
| AE related to treatment | 20 (25) | 17 (22) | 37 (23) | 17 (20) | 54 (22) |
| AE leading to treatment discontinuation/study withdrawal | 0 | 0 | 0 | 0 | 0 |
| Any on‐treatment SAE | 0 | 0 | 0 | 0 | 0 |
| SAE related to treatment | 0 | 0 | 0 | 0 | 0 |
| Fatal SAEs | 0 | 0 | 0 | 0 | 0 |
| Any posttreatment SAE | 0 | 0 | 0 | 1 (1) | 1 (<1) |
| Common on‐treatment AEs (≥3%) | |||||
| Headache | 8 (10) | 9 (11) | 17 (11) | 6 (7) | 23 (9) |
| Viral URTI | 6 (8) | 3 (4) | 9 (6) | 2 (2) | 11 (5) |
| Fatigue | 1 (1) | 2 (3) | 3 (2) | 5 (6) | 8 (3) |
| On‐treatment AEs of special interest | |||||
| Anaphylaxisa | 0 | 0 | 0 | 0 | 0 |
| Allergic (type I hypersensitivity) reactions | 0 | 0 | 0 | 0 | 0 |
| Other systemic reactions | 3 (4) | 4 (5) | 7 (4) | 4 (5) | 11 (5) |
| Local injection‐site reactionsb | 2 (3) | 1 (1) | 3 (2) | 1 (1) | 4 (2) |
| Serious cardiac, vascular, and thromboembolic events | 0 | 0 | 0 | 0 | 0 |
| Serious ischemic events | 0 | 0 | 0 | 0 | 0 |
| Malignancies | 0 | 0 | 0 | 0 | 0 |
| All infectionsc | 8 (10) | 4 (5) | 12 (8) | 6 (7) | 18 (7) |
| Opportunistic infections | 1 (1) | 0 | 1 (<1) | 0 | 1 (<1) |
AE, adverse event; AI, prefilled autoinjector; PFS, prefilled syringe; SAE, serious adverse event; URTI, upper respiratory tract infection.
Considered by the investigator to represent systemic reactions meeting the Sampson's criteria for anaphylaxis.
As identified by the investigator in electronic case report form designed for collecting data on local injection‐site reactions.
cNo serious infections were reported.
Immunogenicity
Eleven (5%) participants tested positive for ADAs for at least 1 visit after baseline, and the number of ADA‐positive participants was similar across the 3 treatment groups (4%, 6%, and 4% for the liquid drug product PFS, liquid drug product AI, and reconstituted lyophilized drug product groups, respectively). None of the participants who tested positive for binding ADA tested positive for neutralizing antibodies.
Blood Eosinophil Count
At baseline, geometric mean blood eosinophil counts were similar across the 3 treatment groups (119 cells/µL, 106 cells/µL, and 102 cells/µL in the liquid drug product PFS, liquid drug product AI, and reconstituted lyophilized drug product groups, respectively). In all 3 treatment groups, geometric mean blood eosinophil counts were reduced from baseline at day 3 (48 hours; first measurement) following a single dose of mepolizumab 100 mg SC and remained below baseline levels at day 85 (62 cells/µL, 61 cells/µL, and 54 cells/µL in the liquid drug product PFS, liquid drug product AI, and reconstituted lyophilized drug product groups, respectively). Adjusted geometric mean ratios to baseline blood eosinophil count over time were comparable among the 3 treatment groups, with reductions from baseline at day 29 of 69%, 66%, and 67% in the liquid drug product PFS, liquid drug product AI, and reconstituted lyophilized drug product groups, respectively (Figure 3). The geometric mean ratios of blood eosinophil count for both test‐reference comparisons (liquid drug product in PFS vs reconstituted lyophilized drug product, and liquid drug product in AI vs reconstituted lyophilized drug product) at each visit were approximately 1. Moreover, the liquid drug product in both PFS and AI had statistically similar effects to the reconstituted lyophilized drug product on blood eosinophil counts based on the 0.70‐1.43 (±30%) bioequivalence bounds that are typical for more variable pharmacodynamic measures.
Figure 3.
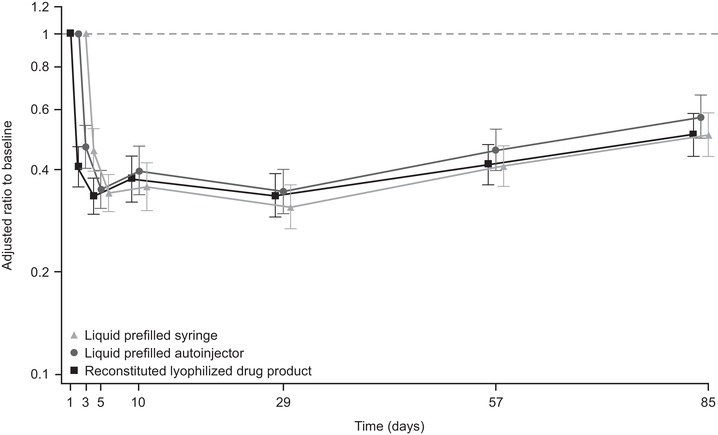
Adjusted geometric meana (95%CI) ratio to baseline blood eosinophil count (109/L) by visit. aThe estimates of the geometric mean were adjusted for baseline blood eosinophil count (loge scale), injection site (abdomen, upper arm, or thigh), baseline body weight (loge scale), and the interaction between baseline blood eosinophil count and visit. CI, confidence interval.
Discussion
This study assessed the clinical systemic exposure comparability in healthy participants of a single dose of mepolizumab 100 mg SC of a new liquid drug product delivered via 2 different devices, a PFS and an AI, with the current reconstituted lyophilized drug product. Our results demonstrated statistical PK comparability between the liquid and reconstituted lyophilized drug products, irrespective of the device used, with PK parameter estimates consistent with a previous study conducted in healthy participants with the reconstituted lyophilized drug product of mepolizumab.11 In addition, mepolizumab exposure observed across the 3 injection sites investigated (abdomen, upper arm, or thigh) did not differ markedly within the 3 treatment groups, suggesting that the site of administration does not affect exposure, reconfirming previous observations with the reconstituted lyophilized drug product.11
We found that mepolizumab liquid drug product delivered via either device was well tolerated and safety outcomes were consistent with the known safety profile of the reconstituted lyophilized drug product.5, 6, 7, 8, 9, 14, 15, 17, 19, 20 The AE profile was similar among the liquid drug product delivered by PFS and AI and the reconstituted lyophilized drug product groups. No SAEs, deaths, or cases of anaphylaxis or systemic allergic (type 1 hypersensitivity) reactions were reported. In addition, the incidence of injection‐site reactions and immunogenicity were low, 1% to 3% and 4% to 6% across the 3 treatment groups, respectively, consistent with the findings from previous studies of mepolizumab.6, 7, 14, 17
Moreover, statistically comparable reductions from baseline in blood eosinophil counts were also seen with the liquid and reconstituted lyophilized drug products, further reinforcing the comparability between the 2 drug products. The decreases from baseline were observed at 48 hours (day 3), which was the first time point measured after mepolizumab administration. This is consistent with previous observations in a clinical pharmacology study conducted with the reconstituted lyophilized drug product in patients with moderate/severe asthma and blood eosinophils >300 cells/µL at screening.10 Although the percentage reductions observed in our study were lower than those observed in other clinical studies of mepolizumab in patients, this may be due to the fact that healthy participants were enrolled in this study with geometric mean blood eosinophil counts at baseline ranging from 102 to 119 cells/µL across treatment groups. These levels are below the threshold of 150 cells/µL for mepolizumab treatment eligibility in patients with severe eosinophilic asthma,25 who are on standard‐of‐care therapy with high‐dose inhaled corticosteroids with or without oral corticosteroids.
The availability of a liquid formulation of mepolizumab that can be delivered via easy‐to‐use devices is likely to simplify treatment administration and provide convenience for patient self‐administration. Self‐administration of treatment is considered a key goal in the management of many diseases, as it can provide a range of benefits including increased patient autonomy, flexibility in physician visits, and increased patient productivity.23, 24 As a result of these potential advantages, self‐administration can provide significant economic benefits for patients, employers, and health care systems worldwide, as demonstrated in a review of studies assessing the impact of self‐administration on 26 chronic conditions.23 It is important to note that incorrect dosing is a potential risk with self‐administration.24 However, use of devices such as PFSs and AIs, together with appropriate patient training, should help to minimize the likelihood of this occurring. In addition, it is worth noting that the AI and PFS assessed in this study are intended for injections of a single fixed dose and have been tested in the real world by patients for self‐administration in the clinic and at home, suggesting that both devices may be ideal platforms for the delivery of mepolizumab.21, 26, 27
Conclusions
In conclusion, the PK of mepolizumab liquid drug product administered either via a PFS or AI is statistically comparable to the reconstituted lyophilized drug product, with similar effects on blood eosinophil counts and no safety concerns identified. Overall, based on these results and on those from other studies performed in patients with asthma,21, 26 this suggests that the liquid drug product of mepolizumab, administered either via a PFS or AI, is a viable alternative to the current reconstituted lyophilized drug product for patients with severe eosinophilic asthma and EGPA.
Declaration of Conflicting Interest
S.S., I.P., J.H.B., E.S.B., and M.C.K. are employees of GlaxoSmithKline (GSK) and hold stocks/shares. M.A. is an employee of PAREXEL International, which received funds from GSK for this study.
Funding
This study was funded by GSK (ClinicalTrials.gov number: NCT03014674; GSK ID: 204958).
Author Contributions
S.S., I.P., J.H.B., E.S.B., and M.C.K. contributed to the conception and design of the study. M.A. was involved in the acquisition of the data during the study. All authors contributed to the analysis and interpretation of the data and development of the manuscript and provided approval of the final draft to be published.
Protocol
The full study protocol can be accessed at https://clinicaltrials.gov/ProvidedDocs/74/NCT03014674/Prot_000.pdf.
Data Sharing
Anonymized individual participant data and study documents can be requested for further research from http://www.clinicalstudydatarequest.com.
Acknowledgments
This study was funded by GSK (NCT03014674; GSK ID: 204958). Editorial support (in the form of writing assistance, including development of the initial draft from the study report, assembling tables and figures, collating authors’ comments, grammatical editing, and referencing) was provided by Roisin McCorkell, MSc, at Fishawack Indicia Ltd, United Kingdom, and was funded by GSK.
References
- 1. Menzella F, Lusuardi M, Galeone C, Taddei S, Zucchi L. Profile of anti‐IL‐5 mAb mepolizumab in the treatment of severe refractory asthma and hypereosinophilic diseases. J Asthma Allergy. 2015;8:105‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ennis D, Lee JK, Pagnoux C. Mepolizumab for the treatment of eosinophilic granulomatosis with polyangiitis [published online ahead of print May 31, 2019]. Expert Opin Biol Ther. 10.1080/14712598.2019.1623875. [DOI] [PubMed] [Google Scholar]
- 3. Foster PS, Maltby S, Rosenberg HF, et al. Modeling TH 2 responses and airway inflammation to understand fundamental mechanisms regulating the pathogenesis of asthma. Immunol Rev. 2017;278(1):20‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farne HA, Wilson A, Powell C, Bax L, Milan SJ. Anti‐IL5 therapies for asthma. Cochrane Database Syst Rev. 2017;9:CD010834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid‐sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371(13):1189‐1197. [DOI] [PubMed] [Google Scholar]
- 6. Chupp GL, Bradford ES, Albers FC, et al. Efficacy of mepolizumab add‐on therapy on health‐related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): a randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre, phase 3b trial. Lancet Respir Med. 2017;5(5):390‐400. [DOI] [PubMed] [Google Scholar]
- 7. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198‐1207. [DOI] [PubMed] [Google Scholar]
- 8. Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double‐blind, placebo‐controlled trial. Lancet. 2012;380(9842):651‐659. [DOI] [PubMed] [Google Scholar]
- 9. Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med. 2017;376(20):1921‐1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pouliquen IJ, Kornmann O, Barton SV, Price JA, Ortega HG. Characterization of the relationship between dose and blood eosinophil response following subcutaneous administration of mepolizumab. Int J Clin Pharmacol Ther. 2015;53(12):1015‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith DA, Minthorn EA, Beerahee M. Pharmacokinetics and pharmacodynamics of mepolizumab, an anti‐interleukin‐5 monoclonal antibody. Clin Pharmacokinet. 2011;50(4):215‐227. [DOI] [PubMed] [Google Scholar]
- 12. Tsukamoto N, Takahashi N, Itoh H, Pouliquen I. Pharmacokinetics and pharmacodynamics of mepolizumab, an anti‐interleukin 5 monoclonal antibody, in healthy Japanese male subjects. Clin Pharmacol Drug Dev. 2016;5(2):102‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ortega H, Yancey S, Cozens S. Pharmacokinetics and absolute bioavailability of mepolizumab following administration at subcutaneous and intramuscular sites. Clin Pharmacol Drug Dev. 2014;3(1):57‐62. [DOI] [PubMed] [Google Scholar]
- 14. Pavord ID, Chanez P, Criner GJ, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med. 2017;377(17):1613‐1629. [DOI] [PubMed] [Google Scholar]
- 15. Bachert C, Sousa AR, Lund VJ, et al. Reduced need for surgery in severe nasal polyposis with mepolizumab: randomized trial. J Allergy Clin Immunol. 2017;140(4):1024‐1031.e1014. [DOI] [PubMed] [Google Scholar]
- 16. Kuang FL, Fay MP, Ware J, et al. Long‐term clinical outcomes of high‐dose mepolizumab treatment for hypereosinophilic syndrome. J Allergy Clin Immunol Pract. 2018;6(5):1518‐1527.e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lugogo N, Domingo C, Chanez P, et al. Long‐term efficacy and safety of mepolizumab in patients with severe eosinophilic asthma: a multi‐center, open‐label, phase IIIb study. Clin Ther. 2016;38(9):2058‐2070.e2051. [DOI] [PubMed] [Google Scholar]
- 18. Rothenberg ME, Klion AD, Roufosse FE, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med. 2008;358(12):1215‐1228. [DOI] [PubMed] [Google Scholar]
- 19. Straumann A, Conus S, Grzonka P, et al. Anti‐interleukin‐5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo‐controlled, double‐blind trial. Gut. 2010;59(1):21‐30. [DOI] [PubMed] [Google Scholar]
- 20. Assa'ad AH, Gupta SK, Collins MH, et al. An antibody against IL‐5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology. 2011;141(5):1593‐1604. [DOI] [PubMed] [Google Scholar]
- 21. Bel EH, Bernstein DI, Bjermer L, et al. Usability of mepolizumab single‐use prefilled syringe for patient self‐administration [published online ahead of print April 24, 2019]. J Asthma. 10.1080/02770903.2019.1604745. [DOI] [PubMed] [Google Scholar]
- 22. Modi NB. Application of pharmacokinetics and pharmacodynamics in product life cycle management. A case study with a carbidopa‐levodopa extended‐release formulation. AAPS J. 2017;19(3):607‐618. [DOI] [PubMed] [Google Scholar]
- 23. Noone J, Blanchette CM. The value of self‐medication: summary of existing evidence. J Med Econ. 2018;21(2):201‐211. [DOI] [PubMed] [Google Scholar]
- 24. World Health Organization . Guidelines for regulatory assessment of medicinal products for use in self‐medication. http://apps.who.int/medicinedocs/pdf/s2218e/s2218e.pdf. Published 2000. Accessed December 21, 2018.
- 25. Ortega H, Menzies‐Gow A, Llanos JP, et al. Rapid and consistent improvements in morning PEF in patients with severe eosinophilic asthma treated with mepolizumab. Adv Ther. 2018;35(7):1059‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bernstein D, Pavord ID, Chapman KR, et al. Usability of mepolizumab single‐use prefilled autoinjector for patient self‐administration [published online ahead of print June 28, 2019]. J Asthma. 10.1080/02770903.2019.1630641. [DOI] [PubMed] [Google Scholar]
- 27. Lange J, Richard P, Bradley N. Usability of a new disposable autoinjector platform device: results of a formative study conducted with a broad user population. Med Devices (Auckl). 2015;8:255‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]