

Abstract
The frequent deregulation of MYC and its elevated expression via multiple mechanisms drives cells to a tumorigenic state. Indeed, MYC is overexpressed in up to ~50% of human cancers and is considered a highly validated anticancer target. Recently, we discovered that WDR5 binds to MYC and is a critical cofactor required for the recruitment of MYC to its target genes and have reported the first small molecule inhibitors of the WDR5-MYC interaction using structure-based design. These compounds display high binding affinity, but have poor physicochemical properties and are hence not suitable for in vivo studies. Herein, we conducted an NMR-based fragment screening to identify additional chemical matter and, using a structure-based approach, we merged a fragment hit with the previously reported sulfonamide series. Compounds in this series can disrupt the WDR5-MYC interaction in cells and as a consequence, we observed a reduction of MYC localization to chromatin.
Keywords: WDR5, MYC, structure-based design, NMR-based fragment screening, PPI
Graphical Abstract
INTRODUCTION
The MYC family of transcription factors regulate a vast array of cellular processes, including control of the cell cycle, cell growth, metabolism, differentiation, apoptosis, transformation, and angiogenesis.1 Dysregulation of MYC proteins causes broad transcriptional reprogramming to drive increased cell proliferation, oftentimes leading to tumor initiation and progression.2,3,4 Indeed, MYC is overexpressed in up to ~50% of human cancers, including lymphoma, melanoma, multiple myeloma, neuroblastoma, lung, colon, and breast cancer.5–7 Based on these data, MYC is considered to be an attractive target for cancer drug discovery. However, MYC is a challenging drug target since it is intrinsically disordered, without well-defined pockets.8 Alternative strategies for MYC inhibition are continuously sought.6,9,10 Recently, we reported that WDR5 is a critical cofactor required for the recruitment of MYC to a subset of its genomic targets.11 WDR5 is a scaffolding protein containing seven WD40 repeats arranged in a β-propeller shape, a motif that is commonly found within proteins that mediate protein-protein interactions.12 WDR5 has more than two dozen primary direct interaction partners whose binding has been mapped to one of two sites: the “WDR5-interacting” site (WIN site) or the “WDR5-binding motif” (WBM site).13 Co-immunoprecipitation (Co-IP) studies identified a specific, highly conserved region of MYC (MYC box IIIb or MbIIIb) that binds at the WBM site of WDR5 (Figure 1).11 Chromatin immunoprecipitation sequencing (ChIP-seq) and mutational experiments showed that MYC requires WDR5 to effectively recognize target genes on chromatin.11 The biological implication of disrupting the WDR5-MYC interaction was tested by CRISPR engineering the endogenous c-myc locus in a Burkitt’s Lymphoma cell line to carry a switchable c-myc allele that is defective for interaction with WDR5.14 When injected into the flanks of nude mice, the mutant cells displayed delayed tumor growth compared to cells expressing wild type MYC. Switching MYC to the WDR5 defective mutant in preformed tumors caused rapid and complete regression within a week.14 These mice survived the entire 60 day duration of the experiment, whereas control mice were all sacrificed by day 17 due to heavy tumor burden, effectively demonstrating that MYC can be therapeutically targeted through WDR5 to reverse malignancy.14 Microarray data from patient-derived pancreatic ductal adenocarcinoma (PDAC) xenografts revealed that WDR5 is overexpressed and required for tumor maintenance. Consequently, silencing WDR5 in pancreatic ductal adenocarcinomal (PDAC) cells showed a reduction of chromatin-bound MYC.15 In addition, inhibition of the WDR5-MYC interaction by mutation of key residues in MbIIIb caused accumulation of DNA damage, a similar effect to that observed when WDR5 was knocked down. Together, these studies suggest that the disruption of the WDR5-MYC interaction with a small molecule may have utility as a cancer therapy.13
Figure 1.
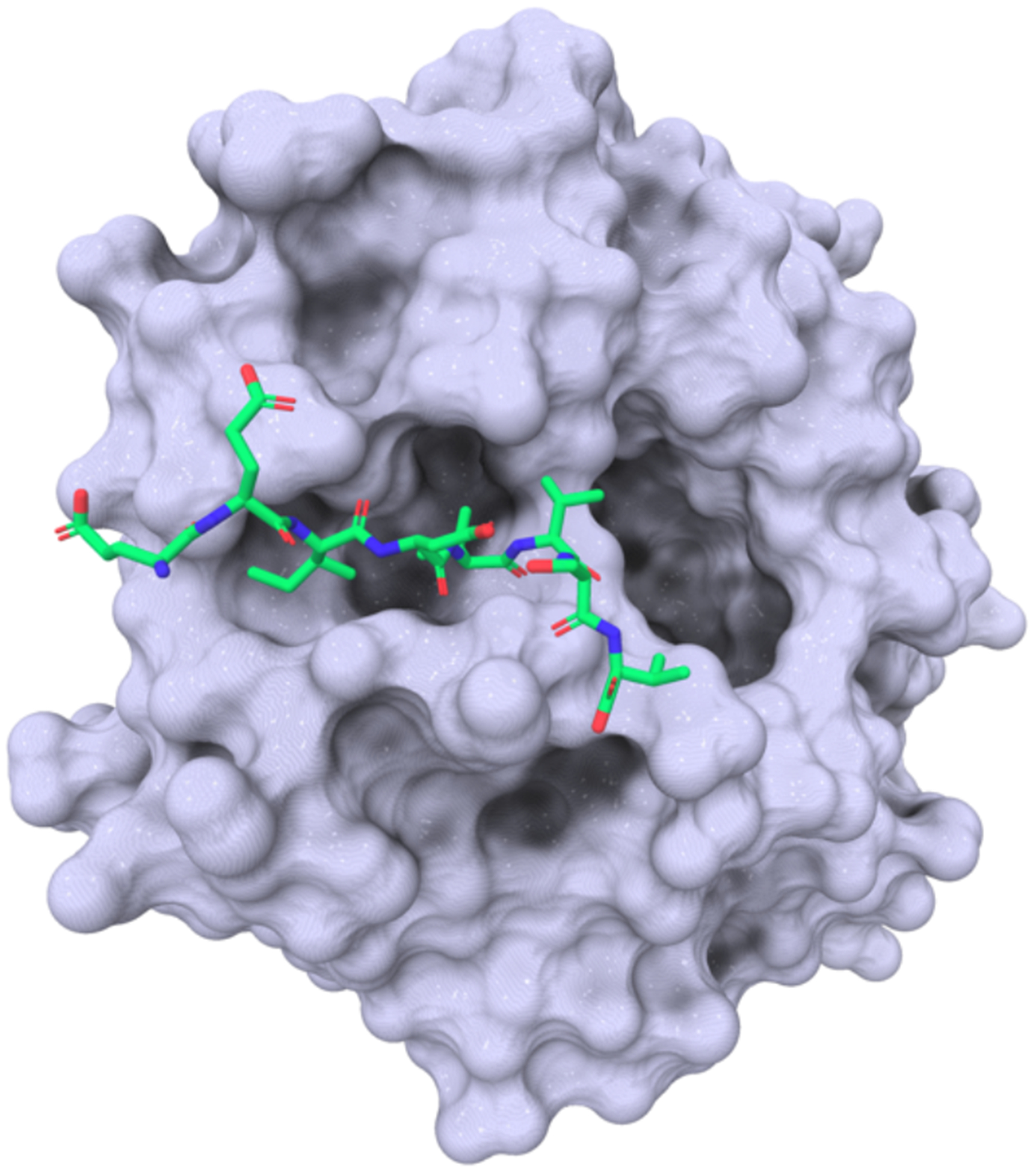
Crystal structure of MbIIIb MYC peptide bound to WDR5 at the WBM (PDB: 4Y7R).
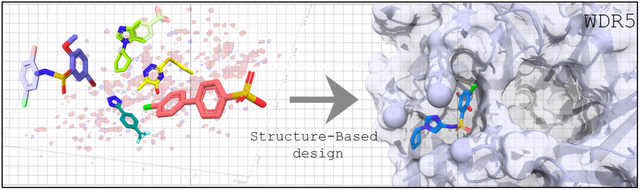
We have previously reported the first small molecules that bind to the WBM site of WDR5. These salicylic acid-based compounds, discovered from structure-based optimization of a high-throughput screening hit (1), are capable of demonstrating low nanomolar affinity for WDR5 and potent inhibition of histone methyltransferase activity. In addition to inhibiting MYC binding to WDR5 in the biochemical assays, these compounds can inhibit the WDR5-MYC interaction in cellular lysates.16 However, these bi-aryl sulfonamide series have challenging physicochemical profiles. Multiple subseries of compounds (including acid, amide, and sulfone variants) exhibit very low Fu, and many of the most potent examples contain multiple phenols that may be prone to glucuronidation or other metabolism.17,18 To identify additional chemical matter that may aid the discovery of compounds with improved properties, we conducted an NMR-based fragment screening campaign.19,20 By merging a fragment hit with the compounds previously reported using structure-guided-design, we have developed a new subseries of compounds that display high nanomolar binding affinity to WDR5. Overall, the compounds in this series showcase improved drug-like properties, and several of them are capable of disrupting the WDR5-MYC interaction in cell lysates. The best-in-class compound disrupts the WDR5-MYC interaction in whole cells and decreases the amount of MYC at genes requiring WDR5 while leaving MYC levels close to normal at genes where recruitment of MYC is independent of WDR5. Thus, the best-in-class compound can be used as a chemical probe to study the implications of disrupting the interaction between WDR5 and MYC in cells.
RESULTS AND DISCUSSION
Hit Identification from NMR-based fragment screening.
The HMQC spectrum of uniformly 15N-labeled WDR5 in complex with unlabeled MYC ‘MbIIIb’ peptide was obtained, showing peak shifts in specific regions (Figure 2A). Our ~14,000 compound fragment library was screened against WDR5 (aa. 23–334) using SOFAST 1H−15N HMQC, collecting the HMQC spectra of 15N-labeled WDR5 protein in the presence of mixtures of 12 fragments. Fragment mixtures that caused similar peak shifts as the unlabeled MYC peptide were identified as WBM site hits. Deconvolution of the mixtures containing such hits was accomplished by individual assessment of the compounds from each hit pool (Figure 2B); thus, identifying 43 hits (0.32% hit rate). Several of the hits that induced large chemical shift perturbations were selected for affinity determination by NMR titration. However, they all showed relatively weak binding, and did not achieve saturable binding at concentrations up to 2 mM, preventing the determination of an accurate Kd. A survey of the chemical structures of the fragment hits reveals some structural diversity; representative fragment hits F1–8 are shown in Figure 3. The 10-mer MYC peptide contains hydrophobic residues flanking multiple acidic amino acids, with Lys and Arg residues around this hydrophobic cleft. The known native substrate(s) of this site (C-MYC, RBBP5, KANSL2) have previously been shown to have a structurally similar motif (IDVV, VDVT, LDVV respectively).11 Likewise, we observed that the vast majority of the fragment hits contain an acidic functionality coupled to a hydrophobic motif.
Figure 2.
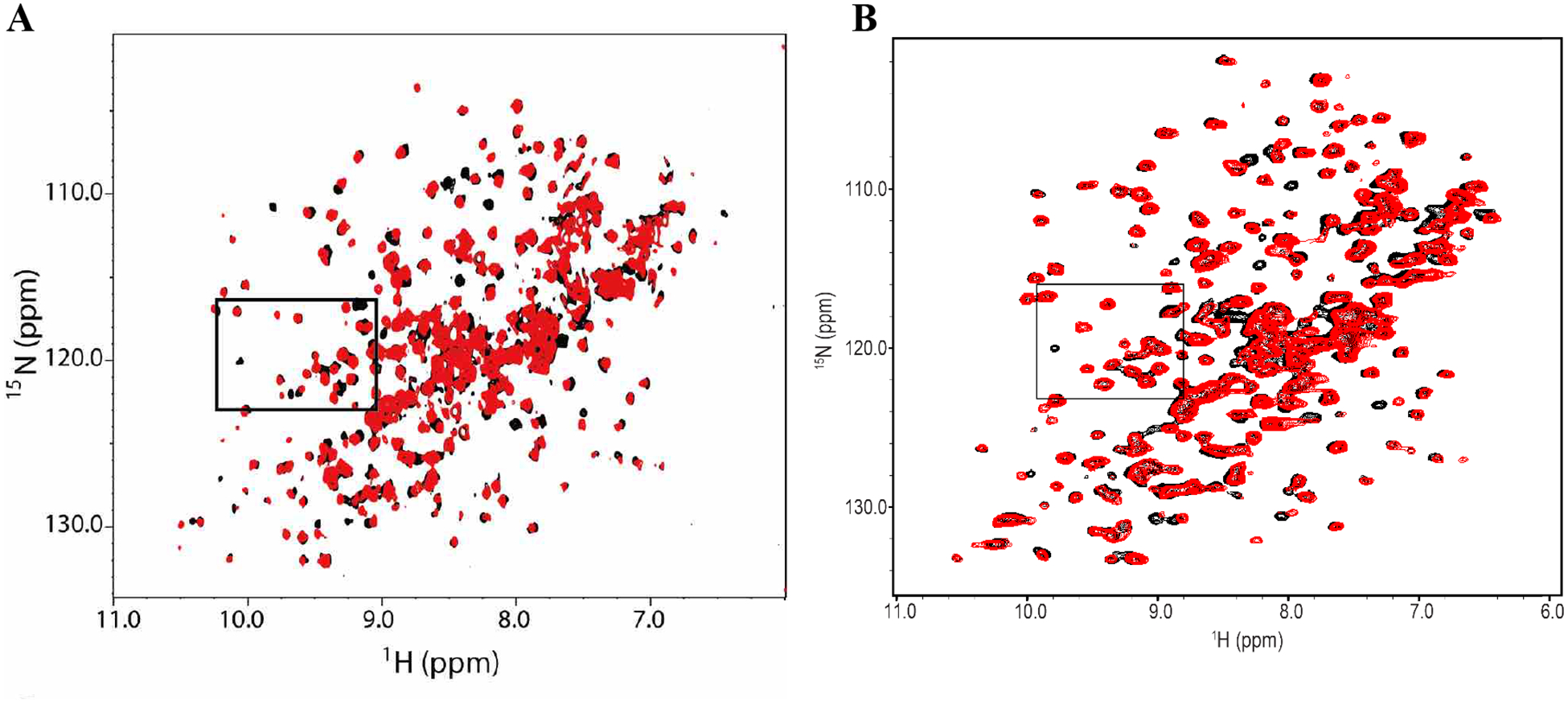
A) Overlay of the HMQC spectra of apo-WDR5 (black) and WDR5 with unlabeled MYC peptide (red). B) Overlay of HMQC spectra of apo-WDR5 (black) and WDR5 with MYC site fragment hit F2 (red). The shifts upon ligand (peptide or hit) binding are indicated.
Figure 3.
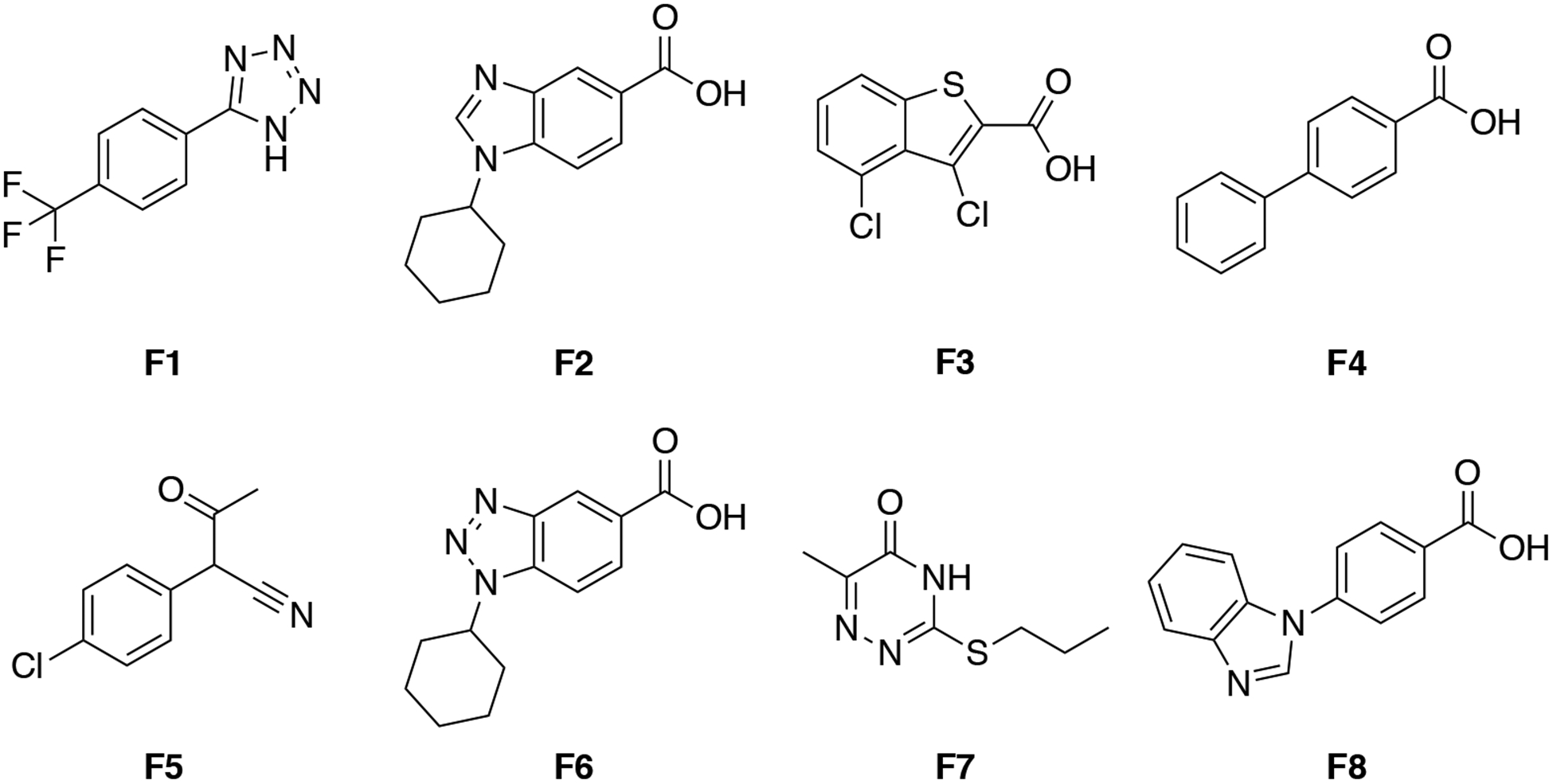
Representative MYC site fragment hits.
Structure Determination of Fragment Complexes.
Five X-ray co-crystal structures of fragments bound to WDR5 (F1, 2, 5, 6, and 7) were determined using co-crystallization and soaking methods (Figure 4). These co-crystal structures confirm the HMQC-indicated binding to the WBM site. However, the fragments are observed to occupy different parts of this cleft. F1, F2, and F6 bind to a pocket corresponding to that occupied by the Ile residue (herein designated as S1) of the conserved ‘IDVV’ WBM binding sequence (Figure 4A) and engage the amide group of N225 through a hydrogen bond. The cyclohexyl group of F2 and F6 fits into this hydrophobic groove very well, and establishes hydrophobic contacts with a Leu residue of WDR5. The trifluoromethyl group of F1 occupies the same region (Figure 4B, C, and E). In contrast, F5 and F7 bind to a different part of the binding cleft. While F7 retains the hydrogen bond with the amide backbone of N225, both F5 and F7 make hydrophobic contacts with the Val-binding shelf of the pocket (designated as S4), mimicking the IDVV conserved sequence. These two fragments make contacts with the loop region of WDR5, directed towards the central core of the protein that has not been demonstrated to be involved in MYC binding (Figure 4D and F).
Figure 4.
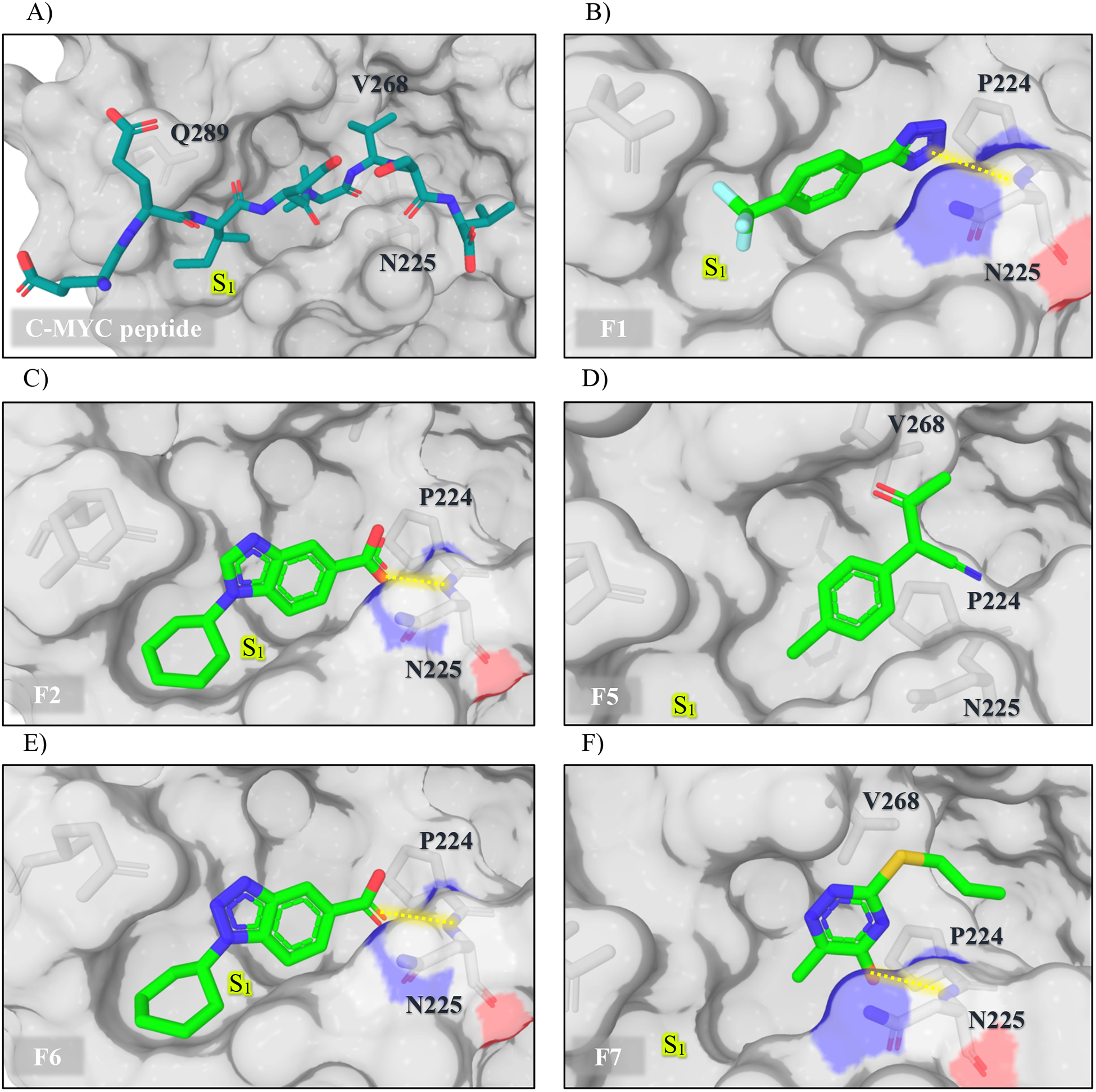
X-ray structures of WDR5 in complex with the C-MYC peptide and fragment hits. (A) C-MYC peptide, PDB: 4Y7R; (B) F1 (PDB:6UJJ); (C) F2 (PDB: 6UHY); (D) F5 (PDB: 6UJH); (E) F6 (PDB: 6UHZ); (F) F7 (PDB: 6UJL). WDR5 is displayed as the surface model, with the WBM site centered.
Compound Design.
Linking together two fragments bound in close proximity to one another in a protein is a proven strategy that can dramatically improve binding affinity.21,22 However, analysis of the X-ray co-crystal structures of the different fragment hits did not directly reveal an obvious opportunity to merge or link multiple fragments. Conversely, the overlay of the co-crystal structures of F2 with WDR5 and the previously reported biarylsulfonamide, 1, which was obtained from our HTS campaign16 (Figure 5A and 5B) showed that merging this chemical matter may represent a viable path forward (Figure 5C). The phenyl carboxylic acid of F2 interacts with Asn225 in a manner highly similar to the sulfonamide of 1, and provides a conformation able to access the S4 pocket of the WBM binding site. Retention of the bromide-containing aryl group of 1 was desired, as it should allow a halogen bond interaction between the bromine and the carbonyl group from Trp273. This was demonstrated to be a key binding interaction in the previously reported series.16 For the synthesis of these merged compounds, we decided to incorporate the phenol and the chlorine sulfonyl ring elements found on the best-in-class compounds in previous work.16 An additional consideration when designing the merged compound was the location of the N-substituent of the imidazole ring. The overlapped atoms (Figure 5C) show that the amine could be placed at the position corresponding to either C-4 or C-5 of the imidazole. We designed the compounds with the amine at C-4, due to anticipation of a better angle to fill the S1 pocket and an easier synthesis.
Figure 5.
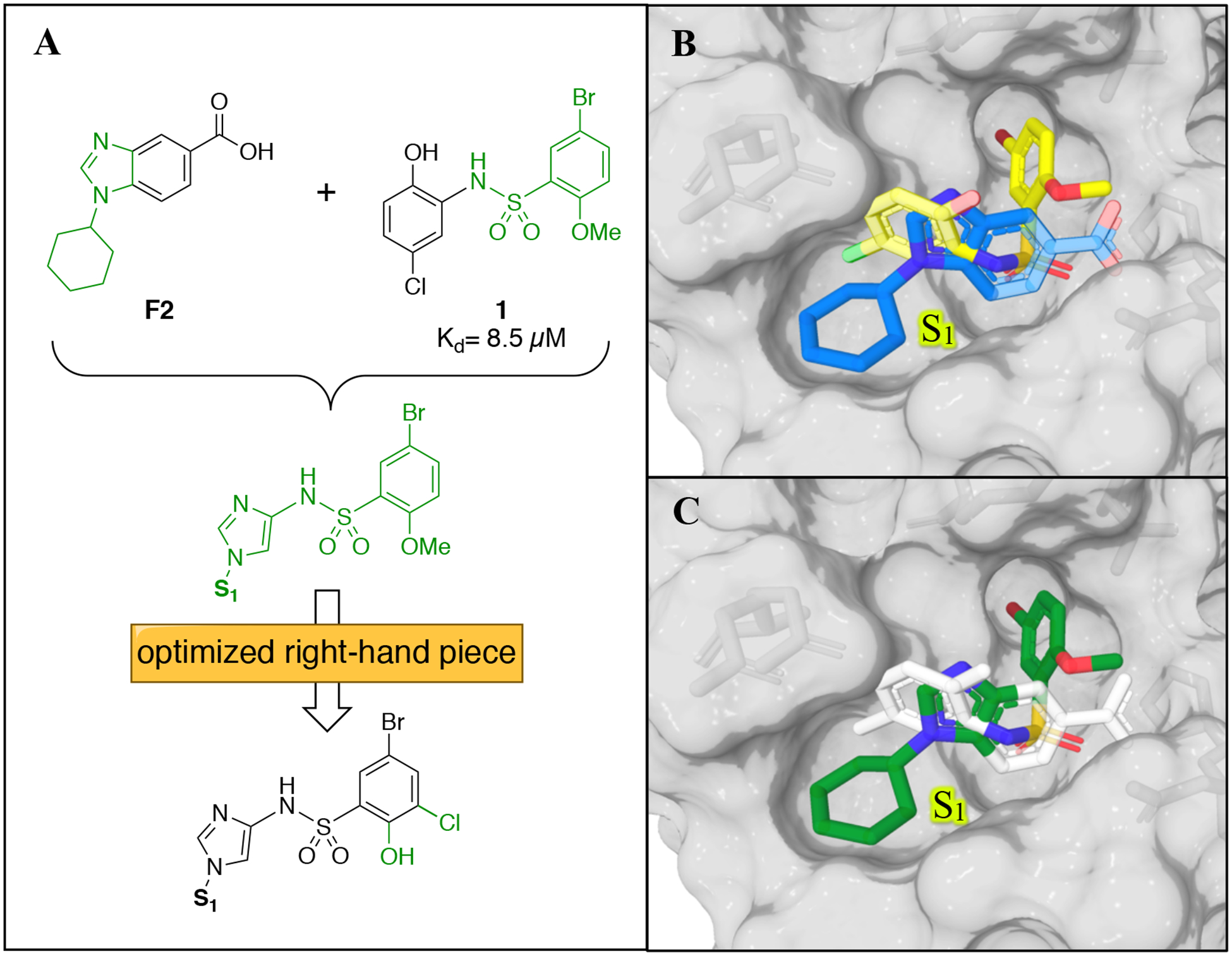
Merging strategy. (A) Merging the previously reported HTS hit 1 with F2 results in a new class of compounds. Incorporation of the previously reported best-in-class sulfonyl piece was expected to result in improved compounds. (B) X-ray co-crystal structure of F2 bound to WDR5 (blue) and overlaid with 1 (yellow). (C) Atoms from F2 and 1 to be merged are colored in green.
N-substituted imidazolyl analogues.
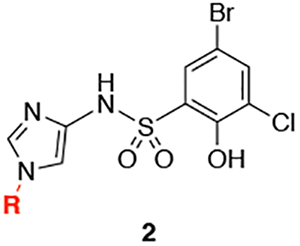
A competition fluorescence polarization-based assay (FPA) was used to evaluate the ability of all analogs to disrupt the complex between WDR5 and a FITC-labeled small molecule.16 From this, the binding affinity of the small molecule can be calculated. The synthesis of the initial merged compound 2a (Table 1) used sulfonyl chloride 17 (Scheme 1), which represents the best-in-class piece reported in our prior work. Evaluation of 2a in the FPA showed a binding affinity of 1.00 μM; representing a several hundred-fold improvement from the fragment F2 (from which we were unable to ascertain an accurate Kd via this FPA protocol) and a 8-fold improvement from 1 (Kd = 8.5 μM). We were highly encouraged by this initial success of the fragment-merging design strategy. To optimize the binding affinity of the merged compound, a survey of hydrophobic substituents, including different sizes of carbocycles, as well as various alkyl and acyl groups, were installed at the N-1 of the imidazole ring (Table 1). Inspired by other fragment hits (SF-9, see Supplemental Table 1), a phenyl ring was installed (2b), which could potentially generate multiple new vectors for optimization of the compounds. However, 2b showed reduced affinity, perhaps due to a planar relationship with the imidazole ring. An ortho-substituted aryl ring (2c) was also explored to induce a twist in the angle between the rings and to better occupy the pocket. Unfortunately, this was also detrimental to the binding affinity. Previous success was observed with small, cyclic alkyl groups in S1 .16 Indeed, flexible, linear chains are not well tolerated in this series (2d), and larger groups like dicyclopropylmethane (2e) also proved to be deleterious for binding affinity. The better fit of the cyclohexyl group in the S1 pocket encouraged the study of other acyclic and saturated groups. Smaller substituents, like cyclopropyl (2g) and isopropyl (2f), appear to not efficiently fill the pocket, as evidenced by the reduced binding affinity to WDR5. Cycloalkanes somewhat smaller than cyclohexane, such as cyclobutane (2h) and cyclopentane (2i), did show a 2.5-fold improvement in the binding affinity compared to 2a. We also sought to introduce solvent-facing polarity into the compounds by replacing the lipophilic cyclopentyl ring. However, the pyrrolidine of 2j was not tolerated. Likewise, the introduction of a methylene spacer between the imidazole and the cycloalkane (2k-2n) was detrimental for binding affinity. We also introduced acyl groups near the S1 pocket to possible engage Q289 with a hydrogen bond; however, 2o and 2p were very weak binders. Interestingly, modification of the phenol to a methoxy led to significant drops in affinity, allowing for the identification of structurally analogous negative control compounds (e.g., N.C., Supplemental Figure 1).
Table 1.
SAR of substituents at N-1 of the imidazole ring.
 |
|||||
|---|---|---|---|---|---|
| Compound | R | FPA Kd(μM)* | Compound | R | FPA Kd (μM)* |
| 2a | 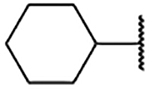 |
1.0 ±0.3 | 2i | 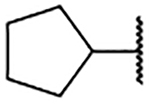 |
0.4 ±0.1 |
| 2b |  |
3.2 ±0.5 | 2j | 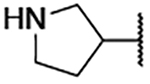 |
11 ±3 |
| 2c | 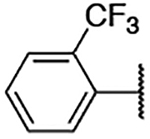 |
4.0 ±0.7 | 2k | 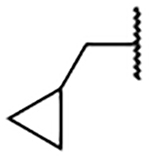 |
2.1 ±0.7 |
| 2d | 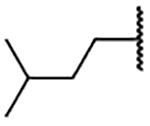 |
5.2 ±0.9 | 2l |  |
4.5 ±0.8 |
| 2e | 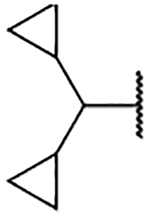 |
1.2 ±0.2 | 2m | 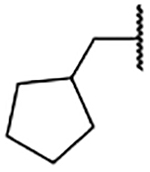 |
3.4 ±0.7 |
| 2f | 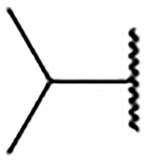 |
5.5 ±0.7 | 2n | 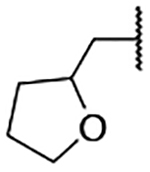 |
8 ± 2 |
| 2g | 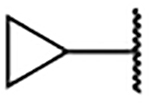 |
4.0 ±0.6 | 2o | 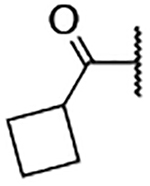 |
38 ±3 |
| 2h | 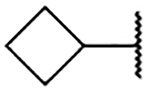 |
0.4 ±0.1 | 2p |  |
>40 |
Data displayed is the average of at least 3 independent replicates ± standard deviation.
Scheme 1.

Synthesis of Compounds 2a-p. Reagents and conditions: (a) Bromoalkane (any a, d, f, h-n), KI, Cs2CO3, MeCN, MW, 135 °C, 45 min, 10–100%; (b) HOAc, HNO3, Ac2O, 0 °C - r.t., 77%; (c) alkyl amine or aniline (c, e, or g), MeOH:H2O (1:1, v/v), 0 °C - r.t, 12–72%; (d) Acyl chloride (o or p), Et3N, MeCN, 75 °C, 3–19%; (e) ArI (b), CuI, L-proline, K2CO3, DMSO, 85 °C, 19%; (f) 10% Pd/C, EtOH, H2, 3 M HCl; (g) Pyridine, DCM, 0 °C - r.t, 1–48%.
We were able to obtain an X-ray structure of compound 2a bound to WDR5 (Figure 6). As anticipated, the compound forms a hydrogen bond to the NH of Asn225 and a halogen bond to the carbonyl of Trp273. Notably, the X-ray crystal structure also shows a putative interaction between the imidazole nitrogen and the side chain amine of Lys272. The cyclopentyl group sits in the hydrophobic pocket designated as S1 (Figure 6), which is partly formed from the hydrophobic side chains of Leu240, Leu249, and Leu288.
Figure 6.
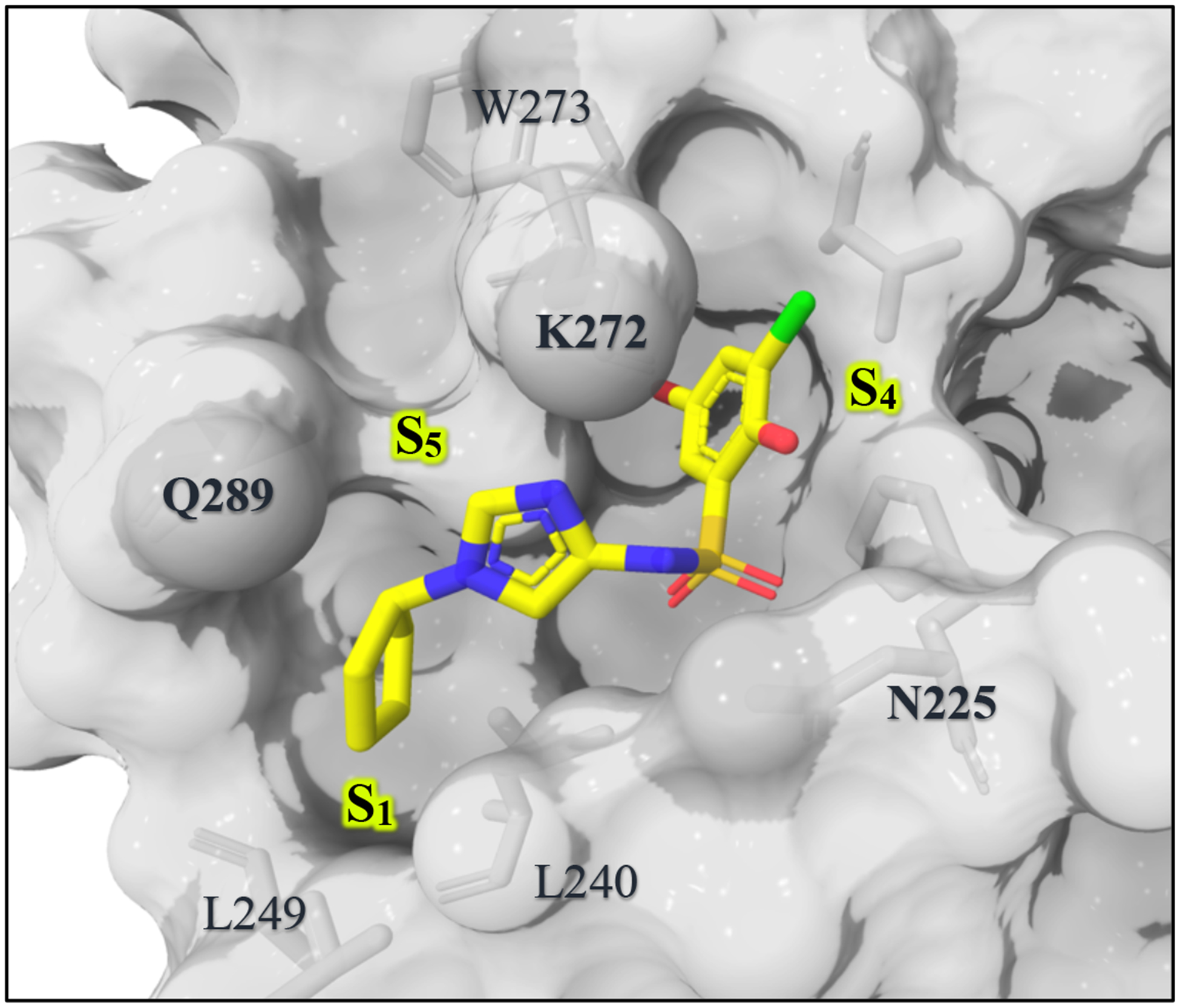
X-ray co-crystal structure of compound 2i bound to WDR5. PDB: 6UJL
Analogs with alternative heterocycles.
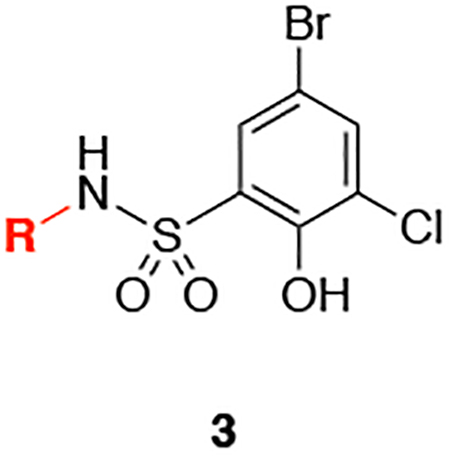
Having established preferences for the hydrophobic group at S1, we transitioned to examining various heterocyclic alternatives to the imidazole ring (Table 2). A 1,2,4-triazole ring (3a) demonstrated that a third nitrogen atom was disadvantageous in this pocket, as shown by a ~16-fold decrease in binding affinity relative to 2i. Based on the X-ray crystal structure of 2i, one of the nitrogen atoms of 3a is most likely pointing towards the hydrophobic region, explaining this significant loss of affinity. Incorporation of pyrazole rings allowed us to walk the nitrogen atoms around the ring (3bd). The regioisomers 3b and 3c did not improve the binding affinity, indicating that the angles and/or the nitrogen distribution were not favorable. On the other hand, analog 3d showed affinity comparable to 2i. Likewise, the thiazole 3e was tolerated, with only a 1.6-fold decrease in binding affinity relative to 2i. Finally, the larger and more lipophilic benzothiazole 3f (AlogP = 4.1, compared to AlogP = 3.5 for 2i, Supplemental Table 3) was not tolerated. Based on this SAR, we decided to retain the imidazole to continue the SAR study.
Table 2.
SAR of heterocycles.
 | ||
|---|---|---|
| Compound | R | FPA Kd (μM)* |
| 2i |  |
0.4 ±0.1 |
| 3a | 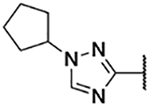 |
6.5 ±0.8 |
| 3b | 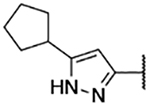 |
10 ± 1 |
| 3c | 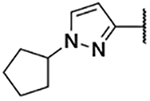 |
2.8 ±0.4 |
| 3d | 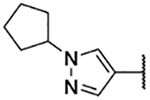 |
0.5 ±0.1 |
| 3e | 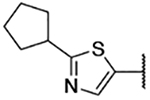 |
0.6 ±0.1 |
| 3f | 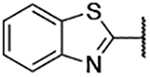 |
32 ± 7 |
Data displayed is the average of at least 3 independent replicates ± standard deviation.
Growing towards the S5 pocket.
Examination of the X-ray co-crystal structure of 2i bound to WDR5 reveals an additional opening in close proximity to the compound, caused by the side-chain rotation of K272 and Q289 (S5 in Figure 6). We hypothesized that occupation of that region may lead to further increases in affinity, and C-2 of the imidazole ring appears to offer a vector to explore this area. First, small groups like methyl (4) and ethyl (5) were incorporated onto C-2 of the imidazole (Table 3). Assessment by FPA assay showed that these substitutions are well tolerated. To further guide compound optimization, 4 was co-crystalized with WDR5 and the X-ray structure (Figure 7a) confirmed that the methyl group points towards the S5 region. Therefore, we proposed that larger and extended linear groups may also be tolerated. An overlay of the crystal structures of 4 and 30 (Supplemental Figure 1)16 further suggests that incorporation of an amide could allow an additional option to reach to the S5 region (Figure 7b). Compounds 6, 7, and 8 were synthesized to test this idea. Interestingly, the ester and the amide bound better than the acid; this is the opposite observation compared to the bi-aryl sulfonamide series. We were encouraged to design and synthesize three amide analogs (9, 10, and 11) with the intention to occupy the S 5 region. Unfortunately, the FPA data for these three analogs showed a considerable reduction of the binding affinity. Analog 8 was co-crystalized with WDR5 to help understand the cause of potency loss upon introduction of larger groups (Figure 7c). The crystal structure revealed that the access to S5 was blocked by the Q289 side chain, which rotates to engage in a hydrogen bond interaction with the carbonyl oxygen of the methyl amide of 8, in a similar manner to our previously published compounds. As a consequence of establishing this hydrogen bond, the methyl amide is forced to point towards the solvent rather than along the targeted shelf of the protein. The lack of improvement in binding affinity in 9, 10, and 11 can be explained by a blocked access to the S5 region, and the loss in binding affinity might be related to secondary amines lacking a hydrogen that could form an intramolecular H-bond with the imidazole nitrogen.
Table 3.
C-2 substitution.
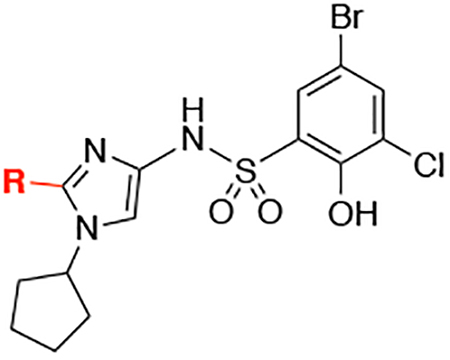 | ||
|---|---|---|
| Compound | R | FPA Kd (μM)* |
| 4 | 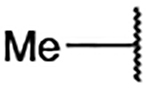 |
0.17 ±0.04 |
| 5 | 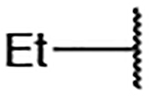 |
0.22 ±0.08 |
| 6 | 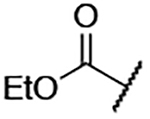 |
0.11 ±0.03 |
| 7 | 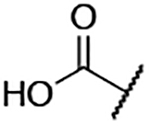 |
0.7 ±0.2 |
| 8 | 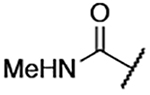 |
0.17 ±0.03 |
| 9 | 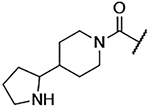 |
5 ± 1 |
| 10 | 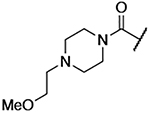 |
1.1 ±0.1 |
| 11 | 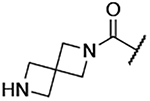 |
2.3 ±0.5 |
| 12 | 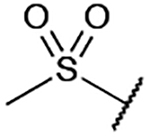 |
0.10 ±0.01 |
Data displayed is the average of at least 3 independent replicates ± standard deviation.
Figure 7.
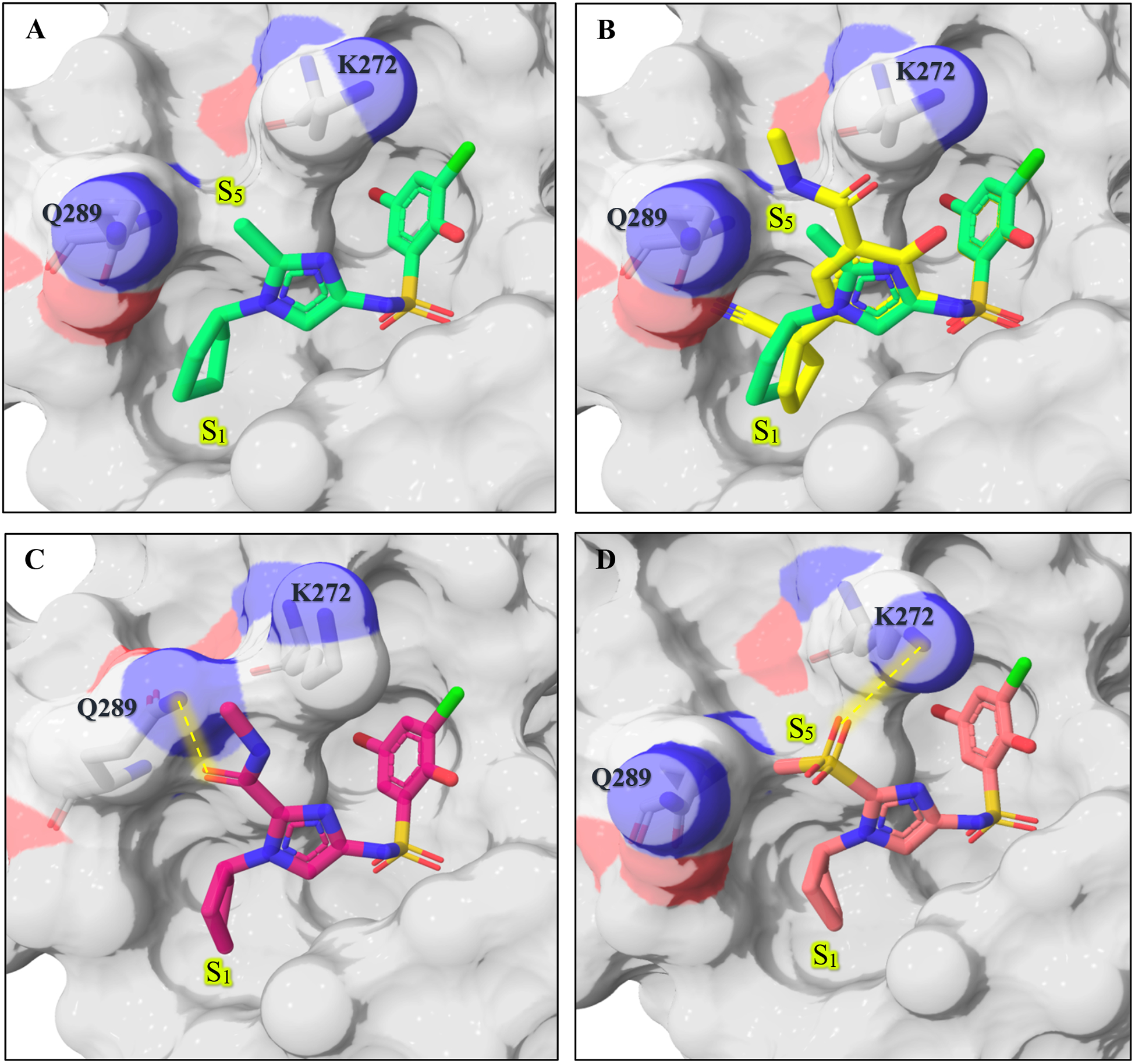
(A) X-ray co-crystal structure of 4 (green, PDB: 6UJ4) bound to WDR5. (B) Overlay of compound 4 and 30 (yellow, PDB: 6U5Y). (C) X-ray co-crystal structure of 8 (magenta, PDB: 6UIF) bound to WDR5. (D) X-ray co-crystal structure of 12 (white, PDB: 6UOZ).
Finally, we decided to evaluate methyl sulfone 12 based on the successful incorporation of this group in the previous series. This led to one of the strongest binders observed in this series so far. The X-ray co-crystal structure of 12 with WDR5 shows one of the sulfone’s oxygens engaged in a hydrogen bond with Lys272. The position of the sulfone in 12 mimics the position of the amide in the biaryl sulfonamide 30 (Figure 7b, see structure in Supplemental Figure 1) and keeps the S5 shelf accessible.
The fragment screen allowed the identification of new chemical matter that improved the WDR5-MYC inhibitors. The compounds shown here are weaker binders relative to the best biaryl sulfonamide analogs; however, they have the potential to display improved drug-like characteristics (Supplemental Table 2). While we did not succeed in significantly improving the Fu, we were able to accomplish one of the other goals of the effort: the deletion of one of the phenols. The most potent compounds reported earlier contain two phenols.16 Likewise, the substitution of the left-hand side phenyl ring for the imidazole furnished compounds with reduced polar surface area relative to the biaryl sulfonamides without raising the lipophilicity (Supplemental Table 3). In addition, most of the compounds shown here were able to maintain good Ligand Efficiencies (LE), which ranged between 0.27 and 0.4, suggesting they remain promising tools for further optimization.
Determination of target engagement.
A collection of eight compounds were assayed in cell lysates of HEK293 (engineered to overexpress HA-tagged c-MYC and Flag-tagged WDR5)16 to determine whether they could disrupt the interaction between WDR5 and MYC at the protein level. Cell lysates were treated with 50 μM of compound and WDR5 was immunoprecipitated (Figure 8A). Pleasingly, most of the tested compounds disrupt this interaction (12, 6, 8, 5, 2i, and 3d), the thiazole 3e shows no demonstrable effect. The limited disruption shown by the amide was an unexpected result, as its binding affinity is superior (Kd = 0.17 μM) to other compounds that did show disruption of the interaction, such as 2i (Kd = 0.4 μM) or 3d (Kd = 0.5 μM). Finally, a structurally related methoxy-containing negative control compound (N.C., Kd > 40 μM, Supplemental Figure 1) was tested and, as expected, it did not disrupt the WDR5-MYC complex, confirming the on-target nature of this disruption.
Figure 8.
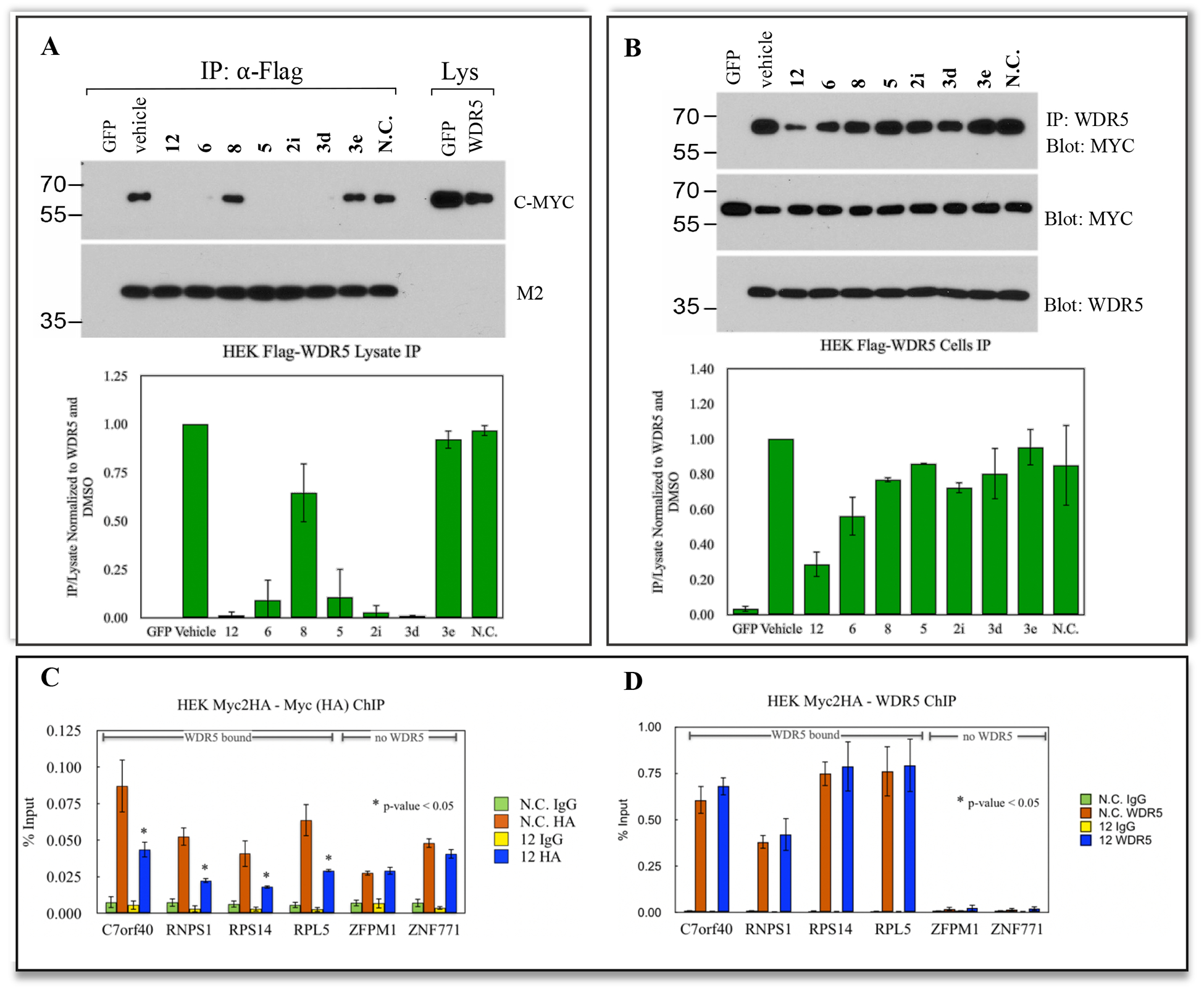
(A) Compound target engagement in cell lysates. Levels of c-MYC proteins co-precipitating with WDR5 were assessed in cell lysates from WDR5-FLAG and c-MYC-HA co-overexpressing HEK293 cells. A fraction of the lysate before addition of compound is shown as a control lane. Immunoprecipitation with α-FLAG was used to isolate WDR5, which is not detectable in the input due to the excess of WDR5 in the immunoprecipitation (IP) fraction. Quantification was performed using ImageJ with the Fiji plugin and normalized to the vehicle control. Data are representative of minimum two independent biological repeats. (B) Compound target engagement in living cells. Transformed HEK293 cells were treated for 24 h with 5 μM compound and lysates were prepared and analyzed as in (A). A fraction of the input lysate was blotted to monitor MYC levels in treated cells. Data are representative of minimum two independent biological repeats (C) Binding of MYC to target genes. Chromatin Immunoprecipitation (ChIP) in c-MYC-HA overexpressing HEK293 cells treated with 20 μM compound for 10 h was performed using an α-HA antibody. Percent recovery was monitored by qPCR at genes dependent on WDR5 for MYC recruitment or two genes independent of WDR5 (ZFPM1 and ZNF771). (D) WDR5 binding to chromatin at MYC target genes. The p values were calculated using a one-tailed student t-test. Data are representative of minimum three independent biological repeats.
In light of the positive data, we conducted Co-IP experiments in whole cells treated with the same eight compounds, Figure 8B. Compounds 12, 6, 8, 5, and 2i decreased complex formation (WDR5-MYC) relative to DMSO. We speculate that the poor performance of 3d and 3e is not necessarily due to their physicochemical properties, as the data available for this class of compounds suggest that they are all similar, with high solubility and permeability and high binding to serum albumin (Supplemental Table 2). However, the lower binding affinity, in combination with other factors such as other non-specific binding to cellular membrane or proteins, efflux, and/or binding kinetics might decrease their activity in cells. We determined that the results shown here were not due to altered protein expression after treatment with compound, as the levels of MYC and WDR5 protein were comparable to DMSO treated cells in the lysates used for immunoprecipitation, confirming an on-target effect.
Since 12 showed the best results at disrupting the MYC-WDR5 interaction in cells, we postulated that it should also break the tether of MYC to chromatin at genomic loci where recruitment of MYC relies on interaction with WDR5. We performed chromatin immunoprecipitation (ChIP) assays and measured MYC binding at known target genes using quantitative PCR. We observed a reduction in the amount of MYC at loci where MYC recruitment is dependent on WDR5 (C7orf40, RNPS1, RPS14, and RPL5)14 when cells were treated with 20 μM of 12 for 10 h. Importantly, the amount of MYC remained nearly unaffected at ZFPM1 and ZMF771, two sites where MYC binding is independent of WDR5 (Figure 8C). Finally, this result was not due to altered expression or binding of WDR5 to these loci because ChIP experiments for WDR5 demonstrated comparable levels between cells treated with 12 and N.C. (Figure 8D).
Moreover, similar to the effect observed with the previous molecules we demonstrated a functional effect of 3d, 6, 8, and 12 binding to WDR5, as they inhibited the biochemical histone methyltransferase activity of MLL-1 in the full WDR5, RBBP5, ASH2L, and DPY30 (WRAD) complex (Supplemental Table 4). It is known that a functional WRAD MLL complex requires binding of proteins to both the WIN and WBM site of WDR523–25; this data confirms that disrupting the interaction at the WBM site can inactivate the transferase activity of the complex, presumably by disrupting the interaction of WDR5-RBBP5, thus preventing the assembly of the WRAD complex.
Chemistry.
The general strategies developed for compound synthesis are illustrated in Schemes 1–3. The synthesis of fragment-merged compounds with different N-substituents on the imidazole ring started from the commercially available 4-nitro-1H-imidazole, 13 (Scheme 1). Treatment with base and the desired bromoalkene at 135 °C in a microwave reactor gave the compounds 15a, 15d, 15f, and 15h-n. Bulky haloalkenes exhibited very low yields or did not react using these conditions. Therefore, compounds 15c, 15e, and 15g were synthesized utilizing an ANRORC mechanism (Addition of Nucleophile, Ring Opening, and Ring Closure).26 4-Nitro-1H-imidazole was nitrated to form 14, and subsequent addition of the desired amine or aniline (c, e, or g) furnished 15c, 15e, and 15g. An Ullman-type coupling was used to synthesize 15b, and acylation of 13 with cyclobutanecarbonyl chloride or cyclopentanecarbonyl chloride allowed the synthesis of 15o and 15p, respectively. The N-substituted-4-nitroimidazoles were hydrogenated using heterogeneous catalysis (Pd/C); the addition of aqueous HCl was required to stabilize the compounds 16ap. The crude hydrogenation products were reacted with sulfonyl chloride 17, affording the desired sulfonamide analogues 2a-p. Partial hydrogenation of some 4-nitroimidazole intermediates or decomposition of the aminium chlorides contributed to low yields observed within this series.
Scheme 3.
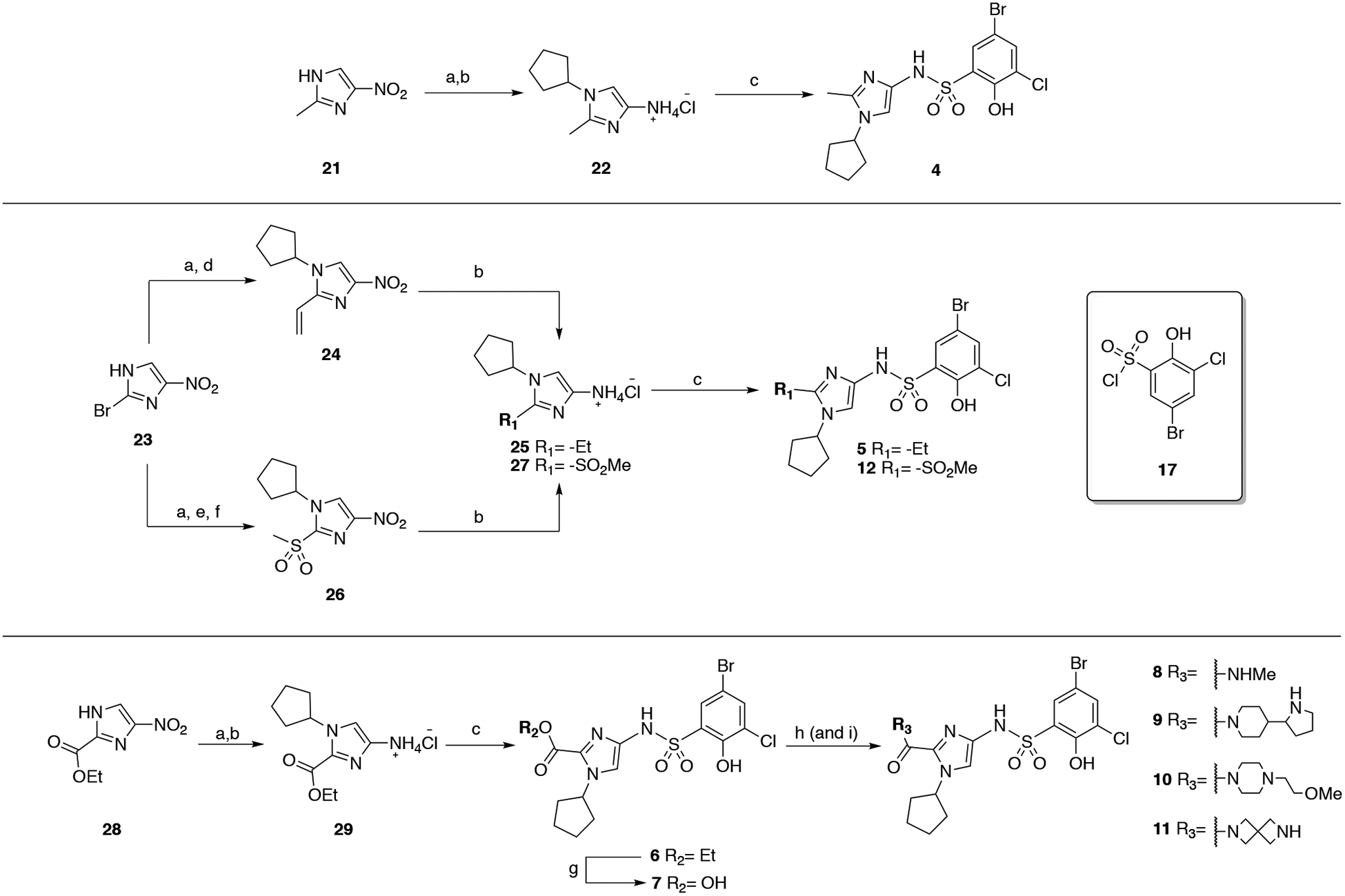
Synthesis of compounds 4, 5, and 6–12. Reagents and conditions: (a) Bromocyclopentane, KI, Cs2CO3, MeCN, MW, 135 °C, 45 min. 24–82%; (b) Pd/C 10%, EtOH, H2, 3 M HCl; (c) 17, pyridine, DCM, 0 °C - r.t., 20–48%; (d) Vinyl boronic pinacol ester, Pd(PPh3)4, Cs2CO3, Dioxane, H2O, 135 °C, 53%; (e) NaSMe, Pd2(dba)3, Xantphos, Et3N, THF, 76 °C, 29%; (f) Oxone, EtOH:H2O (1:1 v/v), 90%; (g) 2 M LiOH, THF, 65 °C, 27%; (h) Amine, PyBOP, DIPEA, CH2Cl2, 49%; (i) HCl 2 M in Dioxane, 40 °C, 40 min, 25%.
The heterocyclic compounds shown in Table 2, 3a-f, were synthesized by routes analogous to those used for the imidazoles. The 1,2,4-triazole 18a was first alkylated with bromocyclopentane and the intermediate was submitted to hydrogenation with addition of 3 M aqueous HCl to produce the aminium chloride 19a. Reaction with 17 allowed the synthesis of the sulfonamide 3a. Compounds 3b-e were synthesized by coupling the commercially available amine or aminium chloride (19b-e) with 17. Analog 3f required a Suzuki coupling between tert-butyl (2-bromothiazol-5-yl)carbamate and cyclopen-1-en-ylboronic acid to produce tert-butyl (2-(cyclopent-1-en-1-yl)thiazol-5-yl)carbamate. This intermediate was then deprotected using 4 M HCl in dioxane. The aminium chloride salt was used to avoid poisoning of the catalyst in the hydrogenation step. Finally, 19f was reacted with the sulfonyl chloride 17 to give the desired product.
The analogs shown in Table 3 were synthesized using multiple approaches. The synthesis of 4 started with 5-methyl-4-nitroimidazole 21, which was N-alkylated with bromocyclopentane and then reduced to the aminium chloride 22. The desired sulfonamide was obtained by reacting 22 with 17. Compound 5 was synthesized by N-alkylating 2-bromo-4-nitro-1H-imidazole (23), and subsequent coupling with vinyl boronic pinacol ester using Suzuki conditions (24). Hydrogenation of the alkene and nitro groups was achieved in the same step (25). Finally, reacting 25 with 17 formed the sulfonamide. Installation of the sulfone group to generate 12 started with N-alkylation of 23, followed by a palladium-catalyzed cross-coupling of 2-bromo-1-cyclopentyl-4-nitro-1H-imidazole with sodium methanethiolate. 1-Cyclopentyl-2-(methylthio)-4-nitro-1H-imidazole was oxidized using Oxone in EtOH:H2O to give 26. The intermediate 27 was accessible by close monitoring of the hydrogenation step; the reaction was stopped after 10–15 min as longer reaction times generated multiple by-products (not-characterized) and disappearance of the desired aminium chloride. The reaction of 17 with 27 allowed the formation of 12. Finally, the synthesis of compounds 6–11 followed a similar route, in which 28 was N-alkylated, reduced (29) and reacted with 17 to produce 6. Hydrolysis using basic conditions allowed the isolation of 7, and PyBOP (or HATU) mediated coupling of the acid with the desired amine furnished 7–11 after Boc-deprotection when required.
CONCLUSIONS
The high frequency of MYC-related alterations in cancers has made it into one of the most studied gene products in oncology. Different strategies to directly target MYC have been pursued. Despite significant effort, these approaches have proved MYC to be a challenging target, and is consider perhaps “undruggable”. Consequently, the examination of alternative and indirect ways to target MYC has become more prominent recently,27–31 and the discovery of WDR5 as a critical co-factor required for the recruitment of MYC to a subset of genomic targets and its later validation through different mutational studies encouraged us to search for small molecules inhibitors of the WDR5-MYC interaction.11,14 We sought to identify small molecules inhibitors with drug-like properties that would enable further studies to examine validation of this approach to treat MYC-driven cancers.
We have previously reported the first set of compounds that bind to the WBM-site of WDR5 with low nanomolar affinity.16 These biaryl sulfonamides proved the feasibility of targeting the challenging and shallow WBM-site and, more importantly, confirmed (by fluorescence polarization assays, differential scanning fluorimetry, histone methyltransferase assays, and CoIP in cell lysates) that disruption of the WDR5-MYC complex with small molecules is possible. While these molecules are some of the most potent reported inhibitors of an external-cleft site of an WD-repeat protein, the undesired physicochemical properties of these compounds (very low free fraction and multiple phenols) discouraged cellular or in vivo studies.
In this report, we used structure-based design and new chemical matter identified in an NMR-based fragment screen to discover a new series of compounds through fragment-merging with our previously established series. Compound 12 was best-in-class of the new hybrid-series, exhibiting high binding affinity (Kd = 0.10 μM) in the FPA and showed robust disruption of the WDR5-MYC interaction in cell lysates. The improvement in cell activity in this series encouraged studies in a whole cell environment. Co-IP studies in transformed HEK293 cells showed a ~4-fold reduction in the amount of the WDR5-MYC complex when cells were treated with 12. Further, the compound was shown by ChIP to reduce the amount of MYC bound to chromatin at loci where recruitment of MYC is dependent on WDR5, but did not affect binding of MYC at loci where it binds independently of WDR5. We have confidence that these data replicate our prior results and that compound 12 demonstrates an on-target effect in whole cells.14 We propose that 12 can be used to further study the cellular implications of disrupting the WDR5-MYC complex and guide further studies towards the development of therapeutic agents to treat MYC-driven cancers.
EXPERIMENTAL SECTION
Protein Expression and Purification
Truncated WDR5 (a.a. 22–334) was cloned into a pET vector with a 6xHis-SUMO tag fused at the N-terminus. The plasmids WDR5 was transformed into E. coli BL21 (DE3) cells. The overnight culture was used to start a 10 L fermentation (BioFlo 415, New Brunswick Scientific) grown at 37 °C. For NMR samples, uniformly 15N-labeled protein was produced in minimal M9 medium, where 15NH4Cl (Cambridge Isotope Laboratories) and D-glucose were used as sole nitrogen and carbon sources. When the cell density reached OD600 = 2.5, the temperature was lowered to 30 °C. The protein was expressed overnight with 1mM isopropyl-β-D-thiogalactoside (IPTG). M peptide (DEEEIDVVSVE) was ordered (Genscript) as HPLC purified synthetic polypeptide. It was dissolved in DMSO for further use.
Cell pellets were dissolved in lysis buffer (1XPBS plus 300 mM NaCl, 20 mM imidazole, 5 mM BME, and 10% glycerol), and broken by homogenization (APV-2000, APV). The lysate was cleared by centrifugation and filtering, and then applied to an affinity column (140 mL, ProBond, Invitrogen). Bound protein was eluted by an imidazole gradient. The His-SUMO-tag was removed by SUMO protease cleavage during dialysis and the subsequent subtractive second nickel-column. WDR5 protein was then purified by size-exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) using NMR or crystallization buffer.
NMR Experiments: Fragment Screening and Kd Determination
NMR samples contained 2 mg/mL (~67 μM) of 15N-labeled WDR5 in 25 mM phosphate buffer, pH = 6.0, 100 mM NaCl, and 1 mM DTT. The fragment library was screened in the format of 12-compound mixtures, and the mixture hits were then deconvoluted.
Nuclear magnetic resonance (NMR) screening was conducted using a Bruker Avance III 600 MHz NMR spectrometer equipped with a 5mm single-axis z-gradient cryoprobe and a Bruker Sample Jet sample changer. Two dimensional, gradient-enhanced 1H-15N heteronuclear multiple-quantum coherence (SOFAST-HMQC)32 spectra were collected at 25 ˚C and used to track chemical shift perturbation upon fragment binding. Spectra were processed and visualized using Topspin (Bruker BioSpin).
NMR titration of WDR5 fragment hits were performed using SOFAST-HMQC to monitor the chemical shift changes (CSC), and the Kd was calculated using the equation
Protein Crystallization, Data Collection, and Structure Refinement.
WDR5 was concentrated to 10 mg/mL (~300 μM) in the buffer of 20 mM HEPES, pH 7.0, 250 mM NaCl, and 5 mM DTT. Apo- and co-crystals were obtained at 18 °C using the hanging drop method. The crystallization condition was 0.1 M Bis-Tris pH 6.0, 0.2 M ammonium acetate, 28% to 32% PEG3350. Soaking was also applied to some compounds using the apo-crystals. Crystals were flash frozen in liquid nitrogen directly.
Diffraction data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-D and G beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. Data were indexed, integrated, and scaled with HKL2000.33 Molecular replacement was achieved with Phaser44 as implemented in CCP4.4534 using a previously determined WDR5 structure (PDB code 3EG6). Refinement of the structural models was conducted with PHENIX35 and included rounds of manual model building in COOT.36 All structure images were prepared with PyMOL.37
Cloning and plasmids
Full length human WDR5 with an N-terminal FLAG tag was cloned into the NgoMIV and SalI sites as a NgoMIV/XhoI fragment. pBabe-GFP was constructed by PCR amplifying the GFP fragment from pEGFPC2 (Clontech) and cloning the product into the EcoRI and BamHI sites of pBabe Puro. Full length human c-MYC with a C-terminal double HA tag was cloned into the BamHI and EcoRI sites of pBabe-IGH.11 HEK293 cells stably expressing MYC2HA were made by retroviral transduction followed by selection in Hygromycin (50 μg/ mL). The mixed population was then infected with pBabe-Puro expressing GFP or WDR5 with selection in puromycin (1 μg/ mL). For retroviral transductions, HEK293T cells were transfected with the appropriate pBabe vector, the pCL10A packaging vector, and pMax-GFP to estimate transfection efficiency. Viral supernatant was collected and used to infect HEK293 class over three days.
Cell culture
HEK293 cells expressing HA-tagged MYC with either Flag-tagged WDR5 or GFP by retroviral transduction have been described.16 Cells were maintained in DMEM supplemented with 10% FBS, Hygromycin B (50 μg/ mL), and puromycin (100 ng/ mL) to maintain transgene expression. Both cell lines were tested and confirmed negative of mycoplasma using the VenorGem PCR test kit (Sigma Aldrich). After thawing from liquid nitrogen, cells were passaged at least twice before use in experiments, and passaged for a maximum of 25 times.
WDR5 Immunoprecipitation
HEK293 cells were harvested and lysates were prepared on ice in lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X100 and supplemented with protease and phosphatase inhibitors). Equal amounts of protein lysate were subject to immunoprecipitation with M2 agarose overnight at 4 °C and immune complexes were recovered, washed in lysis buffer, and resolved by SDS-PAGE. Immunoblotting was performed using the indicated primary antibodies. For experiments in which cells were treated with compound, HEK293 cells were grown to approximately 70% confluence, washed once with PBS then treated for 24 h with compound in OptiMEM media before harvesting.
Antibodies
The following primary antibodies were used for this study: α-C-MYC (#5605), α- WDR5 (#13105), α-HA (#3724) all purchased from Cell Signaling Technology. α-FLAG-HRP (#A8592) and α-FLAG M2 Affinity Gel (#A2220) were from Millipore-Sigma.
Chromatin Immunoprecipitation (ChIP) Experiments
HEK293 cells expressing HA-tagged MYC and GFP were plated in growth media. Upon reaching approximately 70% confluence, cells were washed once with PBS then treated for 10 h with compound in OptiMEM media. Cells were cross linked at room temperature in PBS containing 1% Formaldehyde for 10 minutes, then cells were scraped into 1.5 ml PBS supplemented with 125 mM Glycine. Cells were collected by centrifugation, washed once with PBS + 125 mM Glycine, and pellets stored frozen. Chromatin was prepared by lysing cells at 80×106 per ml in ChIP Lysis Buffer (50 mM Tris pH 8.0, 140 mM NaCl, 1mM EDTA, 1% Triton, 1% SDS and supplemented with protease inhibitor cocktail), Samples were sonicated (BioRuptor) for 30 minutes and cleared by centrifugation. Precipitation was performed essentially as described [Thomas LR 2015 Mol Cell] by diluting chromatin 1:9 in ChIP buffer without SDS. Decrosslinked DNA was diluted to 500 μL with water and 7.5 μL was used for each qPCR reaction. Percent input was calculated by comparison to a 30-fold dilution of decrosslinked chromatin. Primers used for ChIP have been described14.
Fluorescence Polarization Assay
This assay has been previously described.11
Pharmaceutical property determinations
All experiments performed at Q2 Solutions Ltd. (https://www.q2labsolutions.com) using commercially available, standard assay formats. Kinetic solubility determinations: Method QUI-SOL-001. MDCK permeability determinations: Method QUI-PERM-003. Protein binding assessment: Method QUI-PERM-002.
General Chemistry
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Hydrogenation reactions are performed using an atmospheric balloon. Normal phase flash silica gel-based column chromatography is performed using ready-to-connect cartridges from ISCO, on irregular silica gel, particle size 15–40 μM using a Teledyne ISCO Combiflash Rf system. Preparative reversed-phase HPLC was performed on a Gilson instrument equipped with a Phenomenex Kinetex C18 column, using gradients of MeCN in H2O and 0.1% TFA. Compounds that are obtained as a TFA salt after purification were afforded as free base, by dissolving the salt in EtOAc and washing with sat. aq. K2CO3. Proton nuclear magnetic resonance (1H NMR) spectra were recorded at either 400 or 600 MHz on a Bruker spectrometer. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual nondeuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; comp, overlapping multiplets of nonmagnetically equivalent protons; br, broad. All final compounds were of 95% purity or higher, unless otherwise noted, as measured by analytical reversed-phase HPLC. Analytical HPLC was performed on an Agilent 1200 series system with UV detection at 214 and 254 nm, along with evaporative light scattering detection (ELSD). Low-resolution mass spectra were obtained on an Agilent 6140 mass spectrometer with electrospray ionization (ESI). LCMS experiments were performed with the following parameters: Phenomenex Kinetex 2.6 μm XB-C18 100 Å, LC column 50 mm × 2.1 mm; a gradient of 5%−95% MeCN in H2O, and 0.1% TFA, over a 1.2 min or 2.0 min gradient.
General Procedure 1. Chlorosulfonation
Chlorosulfonic acid (7 eq) was cooled to -10 ˚C in a bath of ice/sat. aq. NaCl. To this was added the corresponding aromatic species (1 eq), portion-wise, as required; the mixture was stirred for 1–16 h. The mixture was quenched by careful addition to a slurry of ice/sat. aq. NaCl/CH2Cl2, extracting with CH2Cl2. The crude material was purified by ISCO flash chromatography.
General Procedure 2. N-alkylation
In a microwave (MW) vial, the imidazole (1 eq) was added along with the aryl/alkyl halide (1.2 eq), Cs2CO3 (3 eq) and KI (4 eq). The solids were suspended in MeCN (0.5 M) and the vial was capped and heated in a microwave reactor for 45 min at 135 °C. Upon cooling, the mixture was filtered and the solvent was removed under vacuum. The crude material was purified by ISCO flash chromatography.
General Procedure 3. Hydrogenation for synthesis of compounds
The corresponding intermediate (1 eq) was added to a vial, followed by Pd/C (10% C by wt., 0.05 eq. The solids were suspended in EtOH (at 0.3 M) and the mixture was stirred under a H2 atmosphere for 10 min-2 h. Once the product was observed by LCMS, 10 eq of HCl at 3 M were added. The mixture was filtered through celite, concentrated, and purified by ISCO flash chromatography if required.
General Procedure 4. Sulfonamide Coupling
The corresponding aniline (1 eq) and sulfonyl chloride (1.2–1.5 eq) were combined in CH2Cl2 (at 0.2 M) at r.t., pyridine (3 eq) was added, and the mixture was stirred for 1–16 h. The mixture was concentrated in vacuo and purified by ISCO flash chromatography, unless stated.
General Procedure 5. Nitration
Similar to the procedure previously reported26, a solution of 4-nitroimidazole (1 eq) and HOAc (35 eq) was cooled to 0 °C. HNO3 (14 eq) and Ac2O (14.3 eq) were added and stirring was continued at 0 °C for 10 min. The reaction mixture was then allowed to reach room temperature and stirred for 1 hour. The reaction was quenched by the addition of water and extracted with EtOAc. The organic phase was dried with Na2SO4 and filtered, and the solvent removed under vacuum. The crude material was used without further purification.
General Procedure 6. N-alkylation of 1,4-Dinitro-1H-imidazole
Similar to the procedure previously reported,26 1,4-dinitro-1H-imidazole (1 eq) was dissolved in MeOH:H2O (1:1 at 0.3 M) and cooled to 0 °C. The amine/aniline (1.1 eq) was added dropwise and the mixture was stirred overnight. Water was added and the mixture was extracted with EtOAc. The organic phase was dried with Na2SO4 and filtered, and the solvent removed under vacuum. The crude material was purified using ISCO flash chromatography.
General Procedure 7. Ester Hydrolysis
The ester intermediate (1 eq) was stirred with LiOH (2 M, 5 eq) in THF (at 0.2 M) for 1–16 h at room temperature. The mixture was acidified with hydrochloric acid, extracted with EtOAc, and washed with brine. The crude material was purified by preparative HPLC.
General Procedure 8. PyBOP Mediated Amide Coupling
The corresponding carboxylic acid (1 eq), PyBOP (1.5 eq) and DIPEA (2 eq) were combined in CH2Cl2 (at 0.2 M), followed by the addition of amine (1.5 eq). The solution was stirred at r.t. for 16 h, then diluted with CH2Cl2, washed with sat. aq. NaHCO3, and concentrated. Crude product was purified by ISCO flash chromatography or preparative HPLC.
General Procedure 9. Methyl sulfide Coupling
The aryl/heteroaryl iodide or bromide (1 eq) was combined with sodium methanethiolate (1.05 eq), Pd2(dba)3 (0.025 eq), and Xantphos (0.05 eq). The vial/container was purged with Ar. Then, THF (to 0.34 M) and Et3N (1.25 eq) were added and the mixture was heated to 76 °C for 18 h. Upon cooling the mixture was filtered and the solvent was removed under vacuum. The crude was purified using ISCO Flash chromatography.
General Procedure 10. Sulfide to Sulfone
The sulfide was dissolved in H2O:EtOH (1:1 at 0.32 M) and Oxone (2 eq) was added. The mixture was stirred at r.t. for 4–20 h. Water was added and the mixture was extracted with CH2Cl2. The organic phase was dried using a phase separator and the solvent was removed under vacuum. If required, crude material was purified by ISCO flash chromatography.
General Procedure 12. Suzuki coupling
In a vial, Na2CO3 (3 eq) was dissolved in water (2.0 M). Then, dioxane (0.24 M) was added, followed by the addition of the bromothiazole (1 eq), Pd(PPh3)4 (0.05 eq) and the boronic acid (1.2 eq). The reaction mixture was degassed with Argon and then heated at 100 °C for 18 h. The mixture was diluted with EtOAc and then washed with a saturated solution of NH4Cl. The organic phase was dried over a phase separator and the solvent removed under vacuum. The crude material was purified using ISCO flash chromatography.
General Procedure 13. Boc-deprotection
In a vial, the Boc-protected amine (1 eq) was dissolved in a solution of HCl 4 M in dioxane. The reaction mixture was heated to 50 °C for 1–4 hours. The solvent was removed under vacuum to obtain crude material that was purified by ISCO flash chromatography.
Table 1 Examples
5-Bromo-3-chloro-2-hydroxybenzenesulfonyl chloride (6)
Prepared from 2-chloro-4-bromophenol (2.08 g, 10 mmol) according to General Procedure 1. After purification, 2.19 g (7.15 mmol, 72% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.92 (1H, d, J = 2.4 Hz), 7.86 (1H, d, J = 2.4 Hz); LCMS tR = 1.50 min, m/z = n/a; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclohexyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (7a)
Step A: 1-Cyclohexyl-4-nitro-1H-imidazole (15a)
4-Nitroimidazole (100 mg, 0.88 mmol) was dissolved in 2.2 mL of DMF and K2CO3 (183 mg, 1.33 mmol, 1.5 eq) and TBAB (3 mg, 0.01 mmol, 0.01 eq) were added. The mixture was pre- stirred for ~30 min and bromocyclohexane (180 mg, 1.11 mmol, 1.25 eq) was added. The mixture was stirred at 80 °C for 3 days. Upon cooling of the mixture, water was added and DCM was used to extract the product. Then, the organic phase was dried over a phase separator and the solvent was removed under reduced pressure. The crude was taken forward (175 mg). LCMS (Method B): tR = 1.214 min, m/z = 196.2 [M+H]+.
Step B: 1-Cyclohexyl-1H-imidazol-4-aminium chloride (5a)
1-Cyclohexyl-4-nitro-1H-imidazole (175 mg, crude) was reacted following General Procedure 3. The crude material (24 mg) was taken forward. LCMS tR = 0.565 min, m/z =166.2 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-cyclohexyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclohexyl-1H-imidazol-4-aminium chloride (24 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 4 mg (0.0092 mmol, 1% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.61 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 2.4 Hz, 1H), 7.52 (d, J = 1.5 Hz, 1H), 6.76 (d, J = 1.6 Hz, 1H), 3.93 – 3.81 (m, 1H), 2.12 (d, J = 12.3 Hz, 2H), 1.93 (d, J = 13.5 Hz, 2H), 1.76 (d, J = 13.2 Hz, 1H), 1.68 – 1.53 (m, 2H), 1.49 – 1.35 (m, 2H), 1.32 – 1.21 (m, 1H). LCMS tR = 0.951 min, m/z =434.2, 435.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-phenyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2b)
Step A: 1-Phenyl-4-nitro-1H-imidazole (15b)
4-Nitroimidazole (200 mg, 1.77 mmol) was mixed with iodobenzene (1.77 mmol, 1 eq), CuI (0.27 mmol, 0.15 eq), L-proline (0.27 mmol, 0.15 eq) and K2CO3 (3.54 mmol, 2 eq). The vial was purged with argon and then DMSO (1.4 M) was added. It was capped and stirred overnight at 85 °C. The next day, the reaction mixture was diluted with EtOAc and then filtered. The filtrate was washed with brine and the organic phase was dried over Na2SO4. The solvent was then removed under vacuum and the residue was purified using ISCO flash chromatography 62 mg (0.33 mmol, 19% yield) of the title compound was obtained as a colorless solid. 1H NMR (400 MHz, Chloroform-d) δ 8.11 (s, 1H), 7.84 – 7.75 (m, 1H), 7.62 – 7.54 (m, 2H), 7.54 – 7.49 (m, 1H), 7.45 (dd, J = 7.2, 1.9 Hz, 2H). LCMS tR = 0.127 min, m/z = 190.2 [M+H]+.
Step B: 1-Phenyl-1H-imidazol-4-aminium chloride (5b)
1-Phenyl-4-nitro-1H-imidazole (62 mg, 0.33 mmol) was reacted following General Procedure 3. The crude material (64 mg) was taken forward. LCMS tR = 0.097 min, m/z = 160.2 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-phenyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
Phenyl-1H-imidazol-4-aminium chloride (64 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 50 mg (0.12 mmol, 35% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 9.83 (s, 2H), 8.06 (d, J = 1.7 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.59 – 7.48 (m, 3H), 7.46 – 7.38 (m, 2H), 7.31 (d, J = 1.7 Hz, 1H). LCMS tR = 1.570 min, m/z = 427.8, 428.8 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-2-hydroxy-N-(1-(2-(trifluoromethyl)phenyl)-1H-imidazol-4-yl)benzenesulfonamide (2c).
Step A: 4-Nitro-1-(2-(trifluoromethyl)phenyl)-1H-imidazole (15c)
1,4-Dinitro-1H-imidazole (100 mg, 0.63 mmol) was reacted with 2-(trifluoromethyl)aniline (1.10 mmol) according to General Procedure 6. After purification, 20 mg (0.08 mmol, 12% yield) of the title compound was obtained as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 7.96 – 7.87 (m, 2H), 7.87 – 7.70 (m, 2H), 7.58 (dd, J = 1.6, 0.8 Hz, 1H), 7.52 – 7.45 (m, 1H). LCMS tR = 0.777 min, m/z = 258.2 [M+H]+. Step B: 1-(2-(Trifluoromethyl)phenyl)-1H-imidazole-4-aminium chloride (5c) 4-Nitro-1-(2-(trifluoromethyl)phenyl)-1H-imidazole (20 mg, 0.08 mmol) was reacted following General Procedure 3. The crude material (22 mg) was taken forward. LCMS tR = 0.254 min, m/z = 228.3 [M+H]+.
Step C: 5-Bromo-3-chloro-2-hydroxy-N-(1-(2-(trifluoromethyl)phenyl)-1-imidazol-4-yl)benzenesulfonamide
1-(2-(Trifluoromethyl)phenyl)-1H-imidazole-4-aminium chloride (22 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 18 mg (0.036 mmol, 45% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.86 (d, J = 7.7 Hz, 1H), 7.74 (t, J = 7.7 Hz, 1H), 7.71 – 7.63 (m, 3H), 7.50 (s, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.13 (s, 1H).19F NMR (376 MHz, Chloroform-d) δ -59.42 (3F). LCMS tR = 1.138 min, m/z = 496.2, 497.2 [M+H]+, Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-2-hydroxy-N-(1-isopentyl-1H-imidazol-4-yl)benzenesulfonamide (2d)
Step A: 1-Isopentyl-4-nitro-1H-imidazole (15d)
4-Nitro-1H-imidazole (200 mg, 1.57 mmol) was reacted with 1-bromo-3-methylbutane (2.05 mmol) following General Procedure 2. After purification, 182 mg (0.99 mmol, 63% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (d, J = 1.6 Hz, 1H), 7.41 (d, J = 1.6 Hz, 1H), 4.04 – 3.97 (m, 2H), 1.73 – 1.64 (m, 2H), 1.54 (dt, J = 13.3, 6.7 Hz, 1H), 0.90 (dd, J = 6.7, 0.9 Hz, 6H). LCMS t R = 1.303 min, m/z =184.2 [M+H]+.
Step B: 1-Isopentyl-1H-imidazol-4-aminium chloride (5d)
1-Isopentyl-4-nitro-1H-imidazol (100 mg, 0.54 mmol) was reacted following General Procedure 3. The crude material (103 mg) was taken forward. LCMS tR = 0.192 min, m/z =154.3 [M+H]+.
Step C: 5-Bromo-3-chloro-2-hydroxy-N-(1-isopentyl-1H-imidazol-4-yl)benzenesulfonamide
1-Isopentyl-1H-imidazol-4-aminium chloride (103 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl following General Procedure 4. After purification, 6 mg (0.014 mmol, 3% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 8.07 (s, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 2.4 Hz, 1H), 7.01 (s, 1H), 4.07 (t, J = 7.6 Hz, 2H), 1.75 (dt, J = 8.7, 6.7 Hz, 2H), 1.63 – 1.52 (m, 1H), 0.97 (d, J = 6.7 Hz, 6H). LCMS tR = 0.923 min, m/z = 422.2, 423.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-(dicyclopropylmethyl)-1-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2e)
Step A: 1-(Dicyclopropylmethyl)-4-nitro-1H-imidazole (15e)
1,4-Dinitro-1H-imidazole (100 mg, 0.63 mmol) was reacted with dicyclopropylmethaneamine (1.10 mmol) according to General Procedure 6. After purification, 37 mg (0.18 mmol, 28% yield) of the title compound was obtained as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 7.97 (d, J = 1.6 Hz, 1H), 7.60 (d, J = 1.6 Hz, 1H), 2.85 (t, J = 8.9 Hz, 1H), 1.30 – 1.17 (m, 2H), 0.87 – 0.75 (m, 2H), 0.73 – 0.62 (m, 2H), 0.56 – 0.45 (m, 2H), 0.43 – 0.32 (m, 2H).
Step B: 1-(Dicyclopropylmethyl)-1H-imidazol-4-aminium chloride (5e)
1-(Dicyclopropylmethyl)-4-nitro-1H-imidazole (37 mg, 0.18 mmol) was reacted following General Procedure 3. The crude material (44 mg) was taken forward. LCMS tR = 0.232 min, m/z = 178.3 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-dicyclopropylmethyl-1-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-(Dicyclopropylmethyl)-1H-imidazol-4-aminium chloride (44 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure D. After purification, 8 mg (0.018 mmol, 10% yield) of the title compound was obtained. 1H NMR (400 MHz, Methanol-d4) δ 8.66 (d, J = 1.7 Hz, 1H), 7.82 (d, J = 2.5 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.30 (d, J = 1.7 Hz, 1H), 3.01 (t, J = 9.3 Hz, 1H), 1.39 – 1.26 (m, 2H), 0.82 – 0.72 (m, 2H), 0.62 – 0.48 (m, 4H), 0.37 – 0.25 (m, 2H). LCMS tR = 1.615 min, m/z =445.8, 446.8 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-2-hydroxy-N-(1-isopropyl-1H-imidazol-4-yl)benzenesulfonamide (2f)
Step A: 1-Isopropyl-4-nitro-1H-imidazole (15f)
4-Nitroimidazole (200 mg, 1.77 mmol) was reacted with 2-bromopropane (2.12 mmol) following General Procedure 2. After purification, 225 mg (1.45 mmol, 82% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.82 (d, J = 1.6 Hz, 1H), 7.49 (d, J = 1.6 Hz, 1H), 4.43 (p, J = 6.7 Hz, 1H), 1.55 (d, J = 6.7 Hz, 6H). LCMS tR = 0.366 min, m/z = 156.3 [M+H]+.
Step B: 1-Isopropyl-1H-imidazol-4-aminium chloride (5f)
1-Isopropyl-4-nitro-1H-imidazole (225 mg, 1.45 mmol) was reacted following General Procedure 3. The crude material (295 mg) was taken forward. LCMS tR = 0.086 min, m/z =126.3 [M+H]+.
Step C: 5-Bromo-3-chloro-2-hydroxy-N-(1-isopropyl-1 -imidazol-4-yl)benzenesulfonamide
1-Isopropyl-1H-imidazol-4-aminium chloride (50 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 21 mg (0.053 mmol, 4% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 9.77 (s, 1H), 8.08 (d, J = 1.8 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.05 (d, J = 1.8 Hz, 1H), 4.46 (p, J = 6.7 Hz, 1H), 1.57 (d, J = 6.7 Hz, 6H). LCMS tR = 0.809 min, m/z = 394.1, 396.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopropyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2g)
Step A: 1,4-Dinitro-1H-imidazole (3)
4-Nitroimidazole (500 mg, 4.42 mmol) was reacted following General Procedure 5. Further purification was not required. 538 mg (3.40 mmol, 77% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 8.51 (d, J = 1.7 Hz, 1H), 8.38 (d, J = 1.7 Hz, 1H). LCMS tR = 0.081 min, m/z= 158.3 [M+H]+ ; Purity (AUC) ≥ 95%.
Step B: 1-Cyclopropyl-4-nitro-1H-imidazole (15g)
1,4-Dinitro-1H-imidazole (150 mg, 0.39 mmol) was reacted with cyclopropaneamine (1.04 mmol) according to General Procedure 6. After purification, 105 mg (0.69 mmol, 72% yield) of the title compound was obtained as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 7.78 (d, J = 1.2 Hz, 1H), 7.48 (s, 1H), 3.50 – 3.40 (m, 1H), 1.15 – 1.07 (m, 2H), 1.03 (dt, J = 6.0, 4.1 Hz, 2H). LCMS tR = 0.109 min, m/z = 154.2 [M+H]+.
Step C: 1-(Cyclopropyl)-1H-imidazol-4-aminium chloride (5g)
1-Cyclopropyl-4-nitro-1H-imidazole (50 mg, 0.33 mmol) was reacted following General Procedure 3. The crude material (29 mg) was taken forward. LCMS tR = 0.086 min, m/z =124.2 [M+H]+.
Step D: 5-Bromo-3-chloro-N-(1-(cyclopentanecarbonyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclopropyl-1H-imidazol-4-aminium chloride (29 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 13 mg (0.033 mmol, 10% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.93 (s, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 2.4 Hz, 1H), 7.07 (d, J = 1.5 Hz, 1H), 3.57 – 3.47 (m, 1H), 1.26 – 1.14 (m, 2H), 1.14 – 1.06 (m, 2H). LCMS tR = 0.808 min, m/z = 392.2, 393.4 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclobutyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2h)
Step A: 1-Cyclobutyl-4-nitro-1H-imidazole (15h)
4-Nitroimidazole (100 mg, 0.88 mmol) was reacted with bromocyclobutane (1.11 mmol) following General Procedure 2. After purification, 100 mg (0.59 mmol, 67% yield) of the title compound was obtained. LCMS tR = 0.097 min, m/z = 168.2 [M+H]+.
Step B: 1-Cyclobutyl-1H-imidazol-4-aminium chloride (5h)
1-Cyclobutyl-4-nitro-1H-imidazole (100 mg, 0.59 mmol) was reacted following General Procedure 3. The crude material (50 mg) was taken forward. LCMS tR = 0.090 min, m/z =138.1 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-cyclobutyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclobutyl-1H-imidazol-4-aminium chloride (50 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 14.0 mg (0.033 mmol, 6% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.62 (s, 1H), 7.54 (s, 1H), 7.39 (s, 1H), 6.81 (s, 1H), 4.56 – 4.47 (m, 1H), 2.50 (s, 2H), 2.37 – 2.28 (m, 2H), 1.89 (dt, J = 18.7, 9.5 Hz, 2H). LCMS tR = 1.385 min, m/z = 405.7, 406.8 [M+H]+; ≥ Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2i)
Step A: 1-Cyclopentyl-4-nitro-1H-imidazole (15i)
4-Nitroimidazole (900 mg, 7.96 mmol) was reacted with bromocyclopentane (10.35 mmol) following General Procedure 2. After purification, 1277 mg (7.05 mmol, 71% yield) of the title compound was obtained as a pale-yellow liquid. 1H NMR (400 MHz, Chloroform-d) δ 7.77 (s, 1H), 7.45 (s, 1H), 4.54 – 4.47 (m, 1H), 2.24 (q, J = 8.0 Hz, 2H), 1.89 – 1.68 (m, 6H). LCMS tR = 0.591 min, m/z = 182.3 [M+H]+.
Step B: 1-Cyclopentyl-1H-imidazol-4-aminium chloride (5i)
1-Cyclopentyl-4-nitro-1H-imidazole (50 mg, 0.28 mmol) was reacted according to General Procedure 3. The crude material (51 mg) was taken forward. LCMS tR = 0.114 min, m/z = 152.3 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-cyclopentyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclopentyl-1H-imidazol-4-aminium chloride (51 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 13.8 mg (0.033 mmol, 12% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.61 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 2.4 Hz, 1H), 7.51 (d, J = 1.6 Hz, 1H), 6.75 (d, J = 1.6 Hz, 1H), 4.41 (p, J = 6.7 Hz, 1H), 2.25 – 2.15 (m, 2H), 1.91 – 1.71 (m, 6H). LCMS: tR = 0.874 min, m/z = 420.1, 422.1 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-2-hydroxy-N-(1-(pyrrolidin-3-yl)-1H-imidazol-4-yl)benzenesulfonamide (2j).
Step A: tert-Butyl 3-(4-nitro-1H-imidazole-1-yl)pyrrolidine-1-carboxylate
4-Nitroimidazole (150 mg, 1.33 mmol) was reacted with tert-butyl 3-bromopyrrolidine-1-carboxylate (1.59 mmol) following General Procedure 2. After purification, 185 mg (1.17 mmol, 88% yield) of the title compound was obtained. LCMS tR = 0.800 min, m/z =283.3 [M+H]+.
Step B: 1-(Pyrrolidin-3-yl)-1H-imidazol-4-aminium chloride (15j)
tert-Butyl 3-(4-nitro-1H-imidazole-1-yl)pyrrolidine-1-carboxylate (185 mg, 1.17 mmol) was reacted following General Procedure 3. The Boc-deprotected crude material (183 mg) was taken forward. LCMS t R = 0.084 min, m/z = 153.3 [M+H]+.
Step C: 5-Bromo-3-chloro-2-hydroxy-N-(1-(pyrrolidin-3-yl)-1H-imidazol-4-yl)benzenesulfonamide
1-(Pyrrolidin-3-yl)-1H-imidazol-4-aminium chloride (183 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 8 mg (0.019 mmol, 2% yield) of the title compound was obtained. 13C NMR (151 MHz, Chloroform-d) δ 150.23, 148.22, 138.12, 134.96, 129.75, 125.10, 122.99, 117.44, 111.90, 56.85, 53.37, 45.83, 32.24. 1H NMR (600 MHz, Chloroform-d) δ 7.81 (d, J = 1.6 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 2.4 Hz, 1H), 7.58 (d, J = 1.6 Hz, 1H), 4.93 – 4.87 (m, 1H), 3.80 (dd, J = 11.1, 6.3 Hz, 1H), 3.74 – 3.66 (m, 2H), 3.53 – 3.45 (m, 1H), 2.65 – 2.56 (m, 1H), 2.37 – 2.29 (m, 1H), 1.44 (s, 1H). LCMS tR = 0.918 min, m/z = 453.2, 455.2 [M+CH3OH+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-(cyclopropylmethyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2k).
Step A: 1-(Cyclopropylmethyl)-4-nitro-1H-imidazole (15k)
4-Nitroimidazole (200 mg, 1.77 mmol) was reacted with 1-bromo1-cyclopropylmethane (2.30 mmol) following General Procedure 2. After purification, 226 mg (1.35 mmol, 76% yield) of the title compound was obtained. LCMS tR = 0.485 min, m/z = 168.4 [M+H]+.
Step B: 1-(Cyclopropylmethyl)-1H-imidazol-4-aminium chloride (5k)
1-Cyclopropylmethyl-4-nitro-1H-imidazole (66 mg, 0.39 mmol) was reacted following General Procedure
3. The crude material (60 mg) was taken forward. LCMS tR = 0.089 min, m/z = 138.2 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-(cyclopropylmethyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-(Cyclopropylmethyl)-1H-imidazol-4-aminium chloride (60 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 8 mg (0.020 mmol, 5% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 8.18 (s, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 2.4 Hz, 1H), 7.11 (d, J = 1.7 Hz, 1H), 3.92 (d, J = 7.4 Hz, 2H), 1.30 – 1.19 (m, 1H), 0.87 – 0.81 (m, 2H), 0.46 (q, J = 5.4 Hz, 2H). LCMS tR = 0.758 min, m/z = 406.2 [M+H]+; ≥ 95% (AUC).
5-Bromo-3-chloro-N-(1-(cyclobutylmethyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2l).
Step A: 1-(Cyclobutylmethyl)-4-nitro-1H-imidazole (15l)
4-Nitro-1H-imidazole (200 mg, 1.77 mmol) was reacted with (bromomethyl)cyclobutane (2.30 mmol) following General Procedure 2. After purification, 320 mg (1.77 mmol, quant.) of the title compound was obtained. 1H NMR (400 MHz, Methanol-d4) δ 8.13 (s, 1H), 7.74 (s, 1H), 4.13 (d, J = 7.6 Hz, 2H), 2.79 (p, J = 7.7 Hz, 1H), 2.11 – 2.00 (m, 2H), 1.97 – 1.75 (m, 4H). LCMS (Method B) tR = 0.223 min, m/z = 182.3 [M+H]+.
Step B: 1-(Cyclobutylmethyl-1H-imidazol-4-aminium chloride (5l)
1-(Cyclobutylmethyl)-4-nitro-1H-imidazol (320 mg, 1.77 mmol) was reacted following General Procedure
3. The crude material (500 mg) was taken forward. LCMS tR = 0.090 min, m/z =152.3 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-(cyclobutylmethyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-(Cyclobutylmethyl)-1H-imidazol-4-aminium chloride (100 mg, crude) with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 26 mg (0.062 mmol, approximately 18% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.88 (d, J = 2.4 Hz, 1H), 7.77 (d, J = 2.4 Hz, 1H), 7.32 (d, J = 1.7 Hz, 1H), 6.76 (s, 1H), 6.72 (d, J = 1.7 Hz, 1H), 3.92 (d, J = 7.6 Hz, 2H), 2.70 (p, J = 7.6 Hz, 1H), 2.18 – 2.05 (m, 2H), 2.04 – 1.86 (m, 2H), 1.81 – 1.70 (m, 2H). LCMS tR = 1.111 min, m/z = 421.2, 422.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-(cyclopentylmethyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2m).
Step A: 1-(Cyclopentylmethyl)-4-nitro-1H-imidazole (15m)
4-Nitro-1H-imidazole (200 mg, 1.77 mmol) was reacted with (bromomethyl)cyclopentane (2.30 mmol) following General Procedure 2. After purification, 305 mg (1.56 mmol, 88% yield) of the title compound was obtained. 1H NMR (400 MHz, Methanol-d4) δ 8.18 (d, J = 1.5 Hz, 1H), 7.76 (d, J = 1.5 Hz, 1H), 4.05 (d, J = 7.7 Hz, 2H), 2.37 (p, J = 7.7 Hz, 1H), 1.80 – 1.54 (m, 6H), 1.33 – 1.20 (m, 2H). LCMS tR = 0.722 min, m/z = 196.3 [M+H]+.
Step B: 1-(Cyclopentylmethyl-1H-imidazol-4-aminium chloride (5m)
1-(Cyclopentylmethyl)-4-nitro-1H-imidazole 305 mg (1.56 mmol) was reacted following General Procedure 3. The crude material (204 mg) was taken forward. LCMS tR = 0.095 min, m/z = 166.3 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-(cyclopentylmethyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-(Cyclopentylmethyl)-1H-imidazol-4-aminium chloride (102 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 18 mg (0.041 mmol, 15% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.92 (d, J = 2.4 Hz, 1H), 7.77 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 1.7 Hz, 1H), 6.74 (d, J = 1.7 Hz, 1H), 3.83 (d, J = 7.7 Hz, 2H), 2.26 (p, J = 7.7 Hz, 1H), 1.83 – 1.71 (m, 2H), 1.71 – 1.55 (m, 4H), 1.26 – 1.13 (m, 2H). LCMS tR = 1.151 min, m/z = 435.2, 436.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-2-hydroxy-N-(1-((tetrahydrofuran-2-yl)methyl)-1H-imidazol-4-yl)benzenesulfonamide (2n)
Step A: 4-Nitro-1-((tetrahydrofuran-2-yl)methyl)-1H-imidazole (15n)
4-Nitro-1H-imidazole (200 mg, 1.77 mmol) was reacted with 2-(bromomethyl)tetrahydrofuran (2.30 mmol) following General Procedure 2. After purification, 280 mg (1.093 mmol, 0.62% yield) of the title compound mixed with its regioisomer (3:1, determined by 1H-NMR) was obtained. LCMS tR = 0.098 min, m/z = 198.3 [M+H]+.
Step B: 1-((Tetrahydrofuran-2-yl)methyl)-1H-imidazol-4-aminium chloride (5n)
4-Nitro-1-((tetrahydrofuran-2-yl)methyl)-1H-imidazole (280 mg, 1.09 mmol, regioisomer mixture) was reacted following General Procedure 3. The crude material (205 mg) was taken forward. LCMS tR = 0.088 min, m/z = 168.2 [M+H]+.
Step C: 5-Bromo-3-chloro-2-hydroxy-N-(1-((tetrahydrofuran-2-yl)methyl)-1H-imidazol-4-yl)benzenesulfonamide
1-((Tetrahydrofuran-2-yl)methyl)-1H-imidazol-4-aminium chloride (100 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 13 mg (0.030 mmol, 8% yield) of the title compound was obtained. 1H NMR (400 MHz, Methanol-d4) δ 7.67 (d, J = 2.5 Hz, 1H), 7.64 (d, J = 2.5 Hz, 1H), 7.53 (d, J = 1.6 Hz, 1H), 6.83 (d, J = 1.6 Hz, 1H), 4.12 – 4.05 (m, 2H), 3.98 – 3.90 (m, 1H), 3.78 – 3.66 (m, 2H), 2.01 – 1.92 (m, 1H), 1.87 – 1.76 (m, 1H), 1.66 – 1.55 (m, 1H), 1.49 – 1.39 (m, 1H). LCMS tR = 0.824 min, m/z = 436.2, 437.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-(cyclobutanecarbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2o)
Step A: 1-(Cyclobutanecarbonyl)-4-nitro-1H-imidazole (15o)
Cyclobutanecarbonyl chloride (210 mg, 1.77 mmol, 1 eq) was dissolved in MeCN at 5 M. Then, 4-nitroimidazole (200 mg, 1.77 mmol, 1 eq) and Et3N (250 μL, 1.77 mmol, 1 eq) were added. The reaction mixture was stirred for 16 h at 75 °C. Upon cooling to room temperature, the mixture was filtered and washed with EtOAc. The solvent was dried over a phase separator and the solvent removed under vacuum. The crude was purified using ISCO flash chromatography, 125 mg (0.34 mmol, 19% yield) of the title compound was obtained. LCMS tR = 0.987 min, m/z = does not ionize by ESI.
Step B: 1-(1-(Cyclobutanecarbonyl)-1H-imidazol-4-aminium chloride (5o)
1-Cyclobutanecarbonyl-4-nitro-1H-imidazole (125 mg, 0.34 mmol) was reacted following General Procedure 3. Crude material (70 mg) was taken forward. LCMS tR = 0.347 min, m/z = 166.3 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-(cyclobutanecarbonyl -1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-(Cyclobutanecarbonyl)-1H-imidazol-4-aminium chloride (70 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 3 mg (0.007 mmol, 2% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.94 (d, J = 2.4 Hz, 1H), 7.90 (d, J = 1.5 Hz, 1H), 7.76 (d, J = 2.4 Hz, J = 1.5 Hz, 1H), 7.69 (s, 1H), 7.55 (s, 1H), 3.14 (t, J = 8.4 Hz, 1H), 2.39 – 2.30 (m, 2H), 2.27 – 2.17 (m, 2H), 2.02 (s, 1H), 1.96 – 1.88 (m, 1H). LCMS tR = 1.020 min, m/z = 434.1, 435.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-(cyclopentanecarbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (2p)
Step A: 1-(Cyclopentanecarbonyl)-4-nitro-1H-imidazole (15p)
Cyclopentanecarbonyl chloride (210 mg, 1.77 mmol, 1 eq) was dissolved in MeCN at 5 M. Then, 4-nitroimidazole (200 mg, 1.77 mmol, 1 eq) and Et3N (250 μL, 1.77 mmol, 1 eq) were added. The reaction mixture was stirred for 16 h at 75 °C. Upon cooling to room temperature, the mixture was filtered and washed with EtOAc. The solvent was dried over a phase separator and the solvent removed under vacuum. The crude was purified using ISCO flash chromatography. 41 mg (0.055 mmol, 3% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 8.26 (d, J = 1.7 Hz, 1H), 8.14 (d, J = 1.7 Hz, 1H), 3.46 – 3.34 (m, 1H), 2.13 – 1.98 (m, 4H), 1.86 – 1.71 (m, 5H).
Step B: 1-(1-(Cyclopentanecarbonyl)-1H-imidazol-4-aminium chloride (5p)
1-Cyclopentanecarbonyl-4-nitro-1H-imidazole (41 mg, 0.55 mmol) was reacted following General Procedure 3. The crude material (48 mg) was taken forward. LCMS tR = 0.279 min, m/z =198.4 [M+NH4]+.
Step C: 5-Bromo-3-chloro-N-(1-(cyclopentanecarbonyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-(Cyclopentanecarbonyl)-1H-imidazol-4-aminium chloride (48 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 4 mg (0.009 mmol, 33% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 8.02 (br s, 1H), 7.95 (t, J = 2.4 Hz, 2H), 7.76 (d, J = 2.4 Hz, 1H), 7.69 (d, J = 1.6 Hz, 1H), 2.74 – 2.68 (m, 1H), 1.93 (d, J = 8.2 Hz, 2H), 1.88 – 1.69 (m, 4H), 1.66 – 1.56 (m, 2H). LCMS tR = 1.068 min, m/z = 448.1, 449.3 [M+H]+; Purity (AUC) ≥ 95%.
Table 2 Examples
5-Bromo-3-chloro-N-(1-cyclopentyl-1H-1,2,4-triazol-3-yl)-2-hydroxybenzenesulfonamide (3a)
Step A: 1-Cyclopentyl-3-nitro-1H-1,2,4-triazole
3-Nitro-1,2,4-triazole (100 mg, 0.88 mmol) was reacted with bromocyclopentane (1.14 mmol) following General Procedure 2. After purification, 120 mg (0.66 mmol, 75% yield) of the title compound was obtained. LCMS tR = 0.800 min, m/z = 283.3 [M+H]+.
Step B: 1-Cyclopentyl-1H-1,2,4-triazole-3-aminium chloride (19a)
1-Cyclopentyl-3-nitro-1H-1,2,4-triazole (120 mg, 0.66 mmol) was reacted following General Procedure 3. The crude material (124 mg) was taken forward. LCMS tR = 0.089 min, m/z =153.3 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(1-cyclopentyl-1H-1,2,4-triazol-3-yl)-2-hydroxybenzenesulfonamide
1-Cyclopentyl-1H-1,2,4-triazole-3-aminium chloride (50 mg, 0.27 mmol) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 33 mg (0.030 mmol, 20% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.95 (s, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.68 (d, J = 2.4 Hz, 1H), 4.60 (t, J = 6.2 Hz, 1H), 2.19 – 2.12 (m, 2H), 2.00 – 1.85 (m, 4H), 1.77 – 1.69 (m, 2H). LCMS tR = 1.038 min, m/z = 421.2, 423.1 [M+ H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(5-cyclopentyl-1H-pyrazol-3-yl)-2-hydroxybenzenesulfonamide (3b)
5-Cyclopentyl-1H-pyrazol-3-amine (50 mg, 0.33 mmol) was reacted with bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General procedure 4. After purification, 25 mg (0.059 mmol, 18% yield) of the title compound was obtained. 13C NMR (101 MHz, Chloroform-d) δ 164.24, 151.42, 149.78, 139.14, 129.16, 127.28, 127.03, 110.52, 88.89, 39.16, 32.23, 25.38. 1H NMR (400 MHz, Chloroform-d) δ 7.84 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 5.32 (s, 1H), 2.95 (p, J = 8.0 Hz, 1H), 2.05 – 1.94 (m, 2H), 1.77 – 1.57 (m, 6H). LCMS tR = 1.232 min, m/z = 420.2, 422.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-1H-pyrazol-3-yl)-2-hydroxybenzenesulfonamide (3c)
1-Cyclopentyl-1H-pyrazol-3-amine (30 mg, 0.20 mmol) was reacted with bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General procedure 4. After purification, 71 mg (0.17 mmol, 84% yield) of the title compound was obtained. 13C NMR (101 MHz, Chloroform-d) δ 150.50, 143.65, 137.57, 129.94, 129.36, 125.81, 124.57, 111.23, 99.09, 63.42, 32.93, 24.05. 1H NMR (400 MHz, Chloroform-d) δ 7.61 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 2.4 Hz, 1H), 7.32 (d, J = 2.4 Hz, 1H), 6.15 (d, J = 2.4 Hz, 1H), 4.63 – 4.52 (m, 1H), 2.16 – 2.04 (m, 2H), 1.90 – 1.61 (m, 6H). LCMS tR = 1.118 min, m/z =420.1, 422.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-1H-pyrazol-4-yl)-2-hydroxybenzenesulfonamide (3d)
1-Cyclopentyl-1H-pyrazol-4-aminium chloride (30 mg, 0.15 mmol) was reacted with bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General procedure 4. After purification, 38 mg (0.090 mmol, 59% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.68 (d, J = 2.3 Hz, 1H), 7.60 (d, J = 2.3 Hz, 1H), 7.37 (d, J = 0.8 Hz, 1H), 7.22 (d, J = 0.8 Hz, 1H), 6.45 (s, 1H), 4.65 – 4.54 (m, 1H), 2.19 – 2.08 (m, 2H), 1.97 – 1.86 (m, 2H), 1.86 – 1.65 (m, 4H). LCMS tR = 1.056 min, m/z = 420.2, 422.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(2-cyclopentylthiazol-5-yl)-2-hydroxybenzenesulfonamide (3e)
Step A: tert-Butyl (2-(cyclopent-1-en-1-yl)thiazol-5-yl)carbamate
tert-Butyl (2-bromothiazol-5-yl)carbamate (200 mg, 0.716 mmol) was reacted with cyclopent-1-enylboronic acid (0.860 mmol) following General Procedure 12. After purification, 157 mg (0.589 mmol, 82% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.46 (br s, 1H), 7.25 (s, 1H), 6.33 (t, J = 2.3 Hz, 1H), 2.83 – 2.73 (m, 2H), 2.58 – 2.48 (m, 2H), 2.00 (p, J = 7.5 Hz, 2H). LCMS t R = 0.956 min, m/z = 267.3 [M+ H]+.
Step B: 2-(Cyclopent-1-en-1-yl)thiazol-5-aminium chloride
tert-Butyl (2-(cyclopent-1-en-1-yl)thiazol-5-yl)carbamate (30 mg, 0.11 mmol) was reacted following General Procedure 13. The crude material (75 mg) was taken forward. 1H NMR (400 MHz, Methanol-d4) δ 6.78 (s, 1H), 3.76 – 3.55 (m, 1H), 2.78 (d, J = 6.5 Hz, 2H), 2.69 – 2.61 (m, 2H), 2.11 (p, J = 7.6 Hz, 2H).
Step C: 2-Cyclopentylthiazol-5-aminium chloride (19f)
2-(Cyclopent-1-en-1-yl)thiazol-5-aminium chloride (75 mg, crude) was reacted following General Procedure 3. The crude material (50 mg) was taken forward. LCMS tR = 0.214 min, m/z = 169.3 [M+H]+.
Step D: 5-Bromo-3-chloro-N-(2-cyclopentylthiazol-5-yl)-2-hydroxybenzenesulfonamide
2-Cyclopentylthiazol-5-aminium chloride (50 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 3 mg (0.007 mmol, 1% yield) of the title compound was obtained. 13C NMR (101 MHz, Chloroform-d) δ 176.86, 149.80, 138.00, 137.78, 130.72, 130.46, 124.92, 124.44, 112.06, 44.51, 34.17, 25.50. 1H NMR (400 MHz, Chloroform-d) δ 7.71 (d, J = 2.4 Hz, 1H), 7.68 (d, J = 2.4 Hz, 1H), 7.33 (s, 1H), 3.41 – 3.29 (m, 1H), 2.20 – 2.12 (m, 2H), 1.83 – 1.76 (m, 2H), 1.76 – 1.71 (m, 2H), 1.71 – 1.65 (m, 2H). LCMS tR = 1.088 min, m/z = 437.1, 439.2 [M+H]+; Purity (AUC) ≥ 95%.
N-(Benzo[d]thiazol-2-yl)-5-bromo-3-chloro-2-hydroxybenzenesulfonamide (3f)
Benzothiazol-2-amine (50 mg, 0.33 mmol) was reacted with bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General procedure 4. After purification, 9.0 mg (0.021 mmol, 6% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.82 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.49 – 7.42 (m, 2H), 7.37 – 7.31 (m, 1H). LCMS tR = 1.099 min, m/z = 419.1, 422.2 [M+ H]+; Purity (AUC) ≥ 95%.
Table 3 Examples
5-Bromo-3-chloro-N-(1-cyclopentyl-2-methyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (4)
Step A: 1-Cyclopentyl-2-methyl-4-nitro-1H-imidazole
2-Methyl-4-nitro-1H-imidazole (800 mg, 6.29 mmol) was reacted with bromocyclopentane (8.18 mmol) following General Procedure 2. After purification, 675 mg (3.46 mmol, 55% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.72 (s, 1H), 4.46 (p, J = 7.1 Hz, 1H), 2.46 (s, 3H), 2.30 – 2.18 (m, 2H), 1.99 – 1.86 (m, 2H), 1.85 – 1.73 (m, 4H). LCMS tR = 0.695 min, m/z = 196.3 [M+H]+.
Step B: 1-Cyclopentyl-2-methyl-1H-imidazol-4-aminium chloride (22)
Cyclopentyl-2-methyl-4-nitro-1H-imidazole (21.0 mg, 0.057 mmol) was reacted following General Procedure 3. The crude material (30 mg) was taken forward. LCMS tR = 0.280 min, m/z = 166.4 [M+H]+.
Step C: 5-Bromo-3-chloro-N-(Cyclopentyl-2-methyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclopentyl-2-methyl-1H-imidazol-4-aminium chloride (30 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 6 mg (0.014 mmol, 24%) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.72 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 2.4 Hz, 1H), 6.93 (s, 1H), 4.47 (p, J = 7.2 Hz, 1H), 2.55 (s, 3H), 2.27 – 2.20 (m, 6H), 1.99 – 1.89 (m, 6H), 1.87 – 1.78 (m, 7H). LCMS tR = 0.843 min, m/z = 434.2, 435.2 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-2-ethyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (5)
Step A: 2-Bromo-1-cyclopentyl-4-nitro-1H-imidazole
2-Bromo-4-nitro-1H-imidazole (700 mg, 3.65 mmol) was reacted with bromocyclopentane (4.74 mmol) following General Procedure 2. After purification, 691 mg (2.65 mmol, 72% yield) of the title compound was obtained. 1H NMR (400 MHz, Methanol-d4) δ 8.30 (s, 1H), 4.79 – 4.67 (m, 1H), 2.34 – 2.21 (m, 2H), 1.97 – 1.85 (m, 4H), 1.85 – 1.77 (m, 1H), 1.77 – 1.73 (m, 1H). LCMS tR = 0.837 min, m/z = 260.1, 262.1 [M+H]+.
Step B: 1-Cyclopentyl-4-nitro-2-vinyl-1H-imidazole (24)
In a vial, 100 mg (0.38 mmol, 1 eq) of 2-bromo-1-cyclopentyl-4-nitro-1H-imidazole was added with 3.1:0.700 mL of 1,4-dioxane:H2O. The solution was degassed with Ar for 10 min. Then, 44 mg (0.04 mmol, 0.10 eq) of Pd(PPh3)4 , 65 mg (0.42 mmol, 1.1 eq) of vinyl boronic pinacol ester, and 376 mg (1.15 mmol, 3 eq) of Cs2CO3 were added. The reaction mixture was stirred at 135 °C for 36 hours. Water was added and extracted with DCM. The organic phase was dried over a phase separator and the solvent was removed under vacuum. The crude was purified using ISCO flash chromatography. 60 mg (0.20 mmol, 53% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.74 (s, 1H), 6.59 (dd, J = 17.1, 11.1 Hz, 1H), 6.36 (dd, J = 17.1, 1.4 Hz, 1H), 5.58 (dd, J = 11.0, 1.4 Hz, 1H), 4.65 – 4.54 (m, 1H), 2.24 – 2.17 (m, 2H), 1.89 – 1.73 (m, 6H). LCMS tR = 0.748 min, m/z = 208.3 [M+H]+.
Step C: 1-Cyclopentyl-2-ethyl-1H-imidazol-4-aminium chloride (25)
1-Cyclopentyl-4-nitro-2-vinyl-1H-imidazole (50 mg, 0.21 mmol) was reacted following General Procedure 3. The crude material (52 mg) was taken forward. LCMS tR = 0.092 min, m/z = 180.3 [M+H]+.
Step D: 5-Bromo-3-chloro-N-(1-cyclopentyl-2-ethyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclopentyl-2-methyl-1H-imidazol-4-aminium chloride (52 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 23 mg (0.096 mmol, 48% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.68 (d, J = 2.3 Hz, 1H), 7.54 (d, J = 2.3 Hz, 1H), 6.42 (s, 1H), 4.43 (p, J = 7.2 Hz, 1H), 2.96 (q, J = 7.3 Hz, 2H), 2.21 – 2.10 (m, 2H), 1.91 (s, 2H), 1.84 – 1.70 (m, 4H), 1.30 (t, J = 7.3 Hz, 3H). LCMS tR = 0.901 min, m/z = 448.2, 450.2 [M+H]+; Purity (AUC) ≥ 95%.
Ethyl 4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylate (6)
Step A: Ethyl 1-cyclopentyl-4-nitro-1H-imidazole-2-carboxylate (28)
Ethyl 4-nitro-1H-imidazole-2-carboxylate (300 mg, 1.62 mmol) was reacted with bromocyclopentane (1.94 mmol) following General Procedure 2. After purification, 252 mg (0.995 mmol, 61% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.93 (s, 1H), 5.49 (p, J = 7.1 Hz, 2H), 4.41 – 4.30 (m, 2H), 2.32 – 2.18 (m, 2H), 1.86 – 1.69 (m, 6H), 1.38 – 1.29 (m, 3H). LCMS tR = 0.873 min, m/z = 254.2 [M+H]+.
Step B: Ethyl 1-cyclopentyl-1H-imidazole-4-aminium chloride-2-carboxylate (29)
Ethyl 1-cyclopentyl-4-nitro-1H-imidazole-2-carboxylate (150 mg, 0.592 mmol) was reacted following General Procedure 3. The crude material (159 mg) was taken forward. LCMS tR = 0.625 min, m/z =224.3 [M+H]+.
Step C: Ethyl 4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylate
Ethyl 1-cyclopentyl-1H-imidazole-4-aminium chloride-2-carboxylate (159 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl following General Procedure 4. After purification, 76.5 mg (0.155 mmol, 25% yield) of the title compound was obtained. 13C NMR (101 MHz, Chloroform-d) δ 158.06, 150.81, 137.30, 134.62, 132.77, 130.20, 127.61, 124.45, 112.35, 110.73, 62.02, 59.29, 33.97, 24.15, 13.85. 1H NMR (400 MHz, Chloroform-d) δ 7.70 (d, J = 2.4 Hz, 1H), 7.56 (d, J = 2.4 Hz, 1H), 7.14 (s, 1H), 5.61 – 5.51 (m, 1H), 4.23 (q, J = 7.1 Hz, 2H), 2.30 – 2.19 (m, 2H), 1.90 – 1.71 (m, 6H), 1.15 (t, J = 7.1 Hz, 3H). LCMS tR = 1.128 min, m/z =492.2, 493.3 [M+H]+; Purity (AUC) ≥ 95%.
4-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylic acid (7)
Ethyl 4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylate (27 mg, 0.047 mmol) was reacted following General Procedure 7. After purification, 6 mg (0.013 mmol, 27% yield) of the title compound was obtained. 13C NMR (101 MHz, Chloroform-d) δ 158.06, 150.81, 137.30, 134.62, 132.77, 130.20, 127.61, 124.45, 112.35, 110.73, 62.02, 59.29, 33.97, 24.15, 13.85. 1H NMR (400 MHz, Methanol-d4) δ 7.76 (d, J = 2.4 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.14 (s, 1H), 5.59 (p, J = 6.7 Hz, 1H), 2.25 – 2.15 (m, 2H), 1.87 – 1.80 (m, 2H), 1.80 – 1.69 (m, 4H). LCMS tR = 1.552 min, m/z = 463.8, 464.8 [M+H]+; Purity (AUC) ≥ 95%.
4-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-N-methyl-1H-imidazole-2-carboxamide (8) 4-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylic acid (20 mg, (0.047 mmol) was reacted with methylamine following General Procedure 8. After purification, 11 mg (0.023 mmol, 49% yield) of the title compound was obtained. 13C NMR (151 MHz, Methanol-d4) δ 161.27, 152.28, 137.88, 137.21, 135.76, 131.93, 129.35, 125.49, 112.65, 111.04, 59.69, 34.69, 25.97, 24.98. 1H NMR (600 MHz, Methanol-d4) δ 7.74 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 2.4 Hz, 1H), 6.99 (s, 1H), 5.65 (p, J = 7.3 Hz, 1H), 2.83 (s, 3H), 2.18 – 2.11 (m, 2H), 1.85 – 1.76 (m, 2H), 1.76 – 1.65 (m, 4H). LCMS tR = 1.027 min, m/z = 477.2, 478.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-2-(4-(pyrrolidin-3-yl)piperidine-1-carbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (9)
Step A: tert-Butyl 3-(1-(4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carbonyl)piperidin-4-yl)pyrrolidine-1-carboxylate
4-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylic (33 mg, 0.071 mmol, 1 eq) was mixed with DIPEA (0.21 mmol, 3 eq) and tert-butyl 3-(piperazin-1-yl)pyrrolidine-1-carboxylate (0.14 mmol, 2 eq). The mixture was diluted in DCM (0.29 M) and then HATU (0.082 mmol, 1.15 eq) was added. The reaction mixture was stirred at room temperature for 17 hours, then diluted with water and extracted with EtOAc. The organic phase was dried using a filter separator and the solvent was removed under vacuum. The crude material (97 mg) was taken forward. LCMS (Method B) tR = 0.975 min, m/z = 701.5, 702.5 min [M+H]+.
Step B: 5-Bromo-3-chloro-N-(1-cyclopentyl-2-(4-(pyrrolidin-3-yl)piperidine-1-carbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
tert-Butyl 3-(1-(4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carbonyl)piperidin-4-yl)pyrrolidine-1-carboxylate (50 mg, crude) was reacted following General Procedure 13. After purification, 7.0 mg (0.87 mmol, 15% yield) the title compound was obtained. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (br s, 1H), 7.40 (d, J = 2.7 Hz, 1H), 7.21 (d, J = 2.7 Hz, 1H), 6.98 (s, 1H),
4.78 (p, J = 7.3 Hz, 1H), 3.54 (br s, 2H), 3.43 (br s, 2H), 3.37 – 3.24 (m, 3H), 3.20 – 3.09 (m, 1H), 3.05 – 2.88 (m, 2H), 2.46 – 2.20 (m, 4H), 2.12 – 1.97 (m, 3H), 1.86 – 1.52 (m, 7H). LCMS (Method B) tR = 0.838 min, m/z = 601.3, 603.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-2-(4-(2-methoxyethyl)piperazine-1-carbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (10)
4-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylic acid (40 mg, 0.086 mmol, 1 eq) was mixed with DIPEA (0.26 mmol, 3 eq) and 1-(2-methoxyethyl)piperazine (0.17 mmol, 2 eq). The mixture was diluted in DCM (0.29 M) and then HATU (0.099 mmol, 1.15 eq) was added. The reaction mixture was stirred at room temperature for 17 hours, then diluted with water and extracted with EtOAc. The organic phase was dried using a filter separator and the solvent was removed under vacuum. The crude was purified using preparative HPLC, 16 mg (0.086 mmol, 31% yield) of the title compound was obtained. 1H NMR (400 MHz, Methanol-d4) δ 7.80 (d, J = 2.4 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.14 (s, 1H), 4.98 (p, J = 7.3 Hz, 1H), 3.76 (t, J = 4.9 Hz, 2H), 3.44 (s, 3H), 3.47–3.20 (br s, 10H), 2.24 – 2.15 (m, 2H), 1.91 – 1.80 (m, 2H), 1.73 (s, 4H). LCMS (Method B) tR = 0.879 min, m/z = 590.3, 592.3 [M+H]+; Purity (AUC) ≥ 95%.
5-Bromo-3-chloro-N-(1-cyclopentyl-2-(2,6-diazaspiro[3.3]heptane-2-carbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide (11)
Step A: tert-Butyl 6-(4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carbonyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate
4-((5-Bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carboxylic acid (40 mg, 0.086 mmol, 1 eq) was mixed with DIPEA (0.26 mmol, 3 eq) and tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate (0.17 mmol, 2 eq). The mixture was diluted in DCM (0.29 M) and then HATU (0.099 mmol, 1.15 eq) was added. The reaction mixture was stirred at room temperature for 17 hours, then diluted with water and extracted with EtOAc. The organic phase was dried using a filter separator and the solvent was removed under vacuum. The crude material (98 mg) was taken forward. LCMS (Method B) tR = 1.130 min, m/z = 544.3, 546.3 [M+H]+.
Step B: 5-Bromo-3-chloro-N-(1-cyclopentyl-2-(2,6-diazaspiro[3.3]heptane-2-carbonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
tert-Butyl 6-(4-((5-bromo-3-chloro-2-hydroxyphenyl)sulfonamido)-1-cyclopentyl-1H-imidazole-2-carbonyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (98 mg, crude) was reacted following General Procedure 13. After purification, 7.0 mg (0.87 mmol, 15% yield) of the title compound was obtained. 13C NMR (151 MHz, DMSO-d6) δ 159.2, 136.3, 136.0, 134.6, 128.7, 127.7, 126.5, 110.9, 63.2, 57.9, 52.0, 51.3,
47.8, 46.7, 44.5, 41.8, 40.5, 33.7, 28.1, 24.0. 1H NMR (600 MHz, DMSO-d6) δ 8.88 (s, 2H), 7.42 (s, 1H), 7.23 (s, 1H), 6.99 (s, 1H), 4.78 (p, J = 7.5 Hz, 1H), 3.54 (d, J = 5.2 Hz, 2H), 3.43 (s, 2H), 3.36 – 3.26 (m, 3H), 3.18 – 3.12 (m, 1H), 3.00 (dd, J = 11.4, 7.6 Hz, 1H), 2.97 – 2.89 (m, 1H), 2.47 – 2.37 (m, 2H), 2.37 – 2.30 (m, 1H), 2.26 (dt, J = 11.0, 5.0 Hz, 1H), 2.11 – 1.99 (m, 3H), 1.82 – 1.70 (m, 3H), 1.67 – 1.54 (m, 4H). LCMS (Method B) tR = 0.851 min, m/z = 544.3, 546.3 [M+H]+; Purity (AUC) ≥ 95%.
1-Cyclopentyl-2-(methylsulfonyl)-4-nitro-1H-imidazole (12)
Step A: 1-Cyclopentyl-2-(methylthio)-4-nitro-1H-imidazole
2-Bromo-1-cyclopentyl-4-nitro-1H-imidazole (500 mg, 1.92 mmol) was reacted with sodium methanethiolate following General Procedure 9. After purification, 124 mg (0.55 mmol, 29% yield) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (s, 1H), 4.50 – 4.40 (m, 1H), 2.66 (s, 3H), 2.28 – 2.08 (m, 2H), 1.90 – 1.67 (m, 6H). LCMS tR = 0.881 min, m/z = 228.3 [M+H]+.
Step B: 1-Cyclopentyl-2-(methylsulfonyl)-4-nitro-1H-imidazole (26)
1-Cyclopentyl-2-(methylthio)-4-nitro-1H-imidazole (124 mg, 0.55 mmol) was reacted following General Procedure 10. After purification, 142 mg (0.55 mmol, quant.) of the title compound was obtained. 1H NMR (400 MHz, Chloroform-d) δ 7.92 (s, 1H), 5.33 – 5.22 (m, 1H), 3.47 (s, 3H), 2.41 – 2.30 (m, 2H), 1.93 – 1.74 (m, 6H). LCMS tR = 0.763 min, m/z = 260.2 [M+H]+.
Step C: 1-Cyclopentyl-2-(methylsulfonyl)-1H-imidazole-4-aminium chloride (27)
1-Cyclopentyl-2-(methylsulfonyl)-4-nitro-1H-imidazole (30 mg, 0.13 mmol) was reacted following General Procedure 3. The crude material (25 mg) was taken forward. LCMS tR = 0.186 min, m/z = 230.3 [M+H]+.
Step D: 5-Bromo-3-chloro-N-(1-(cyclobutylmethyl)-2-methyl-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide
1-Cyclopentyl-2-(methylsulfonyl)-1H-imidazole-4-aminium chloride (25 mg, crude) was reacted with 5-bromo-3-chloro-2-hydroxybenzenesulfonyl chloride following General Procedure 4. After purification, 13 mg (0.026 mmol, 20% yield) of the title compound was obtained. 13C NMR (151 MHz, Chloroform-d) δ 149.87, 140.40, 137.65, 133.70, 130.06, 125.71, 124.36, 112.89, 111.50, 59.22, 43.21, 34.21, 24.17. 1H NMR (400 MHz, Chloroform-d) δ 7.88 (d, J = 2.3 Hz, 2H), 7.47 (s, 1H), 5.47 (p, J = 6.8, 6.4 Hz, 1H), 3.44 (s, 3H), 2.56 – 2.45 (m, 2H), 2.10 – 2.04 (m, 2H), 2.04 – 1.92 (m, 4H). LCMS tR = 1.052 min, m/z = 498.2, 500.1 [M+H]+; Purity (AUC) ≥ 95%.
Supplementary Material
Scheme 2.
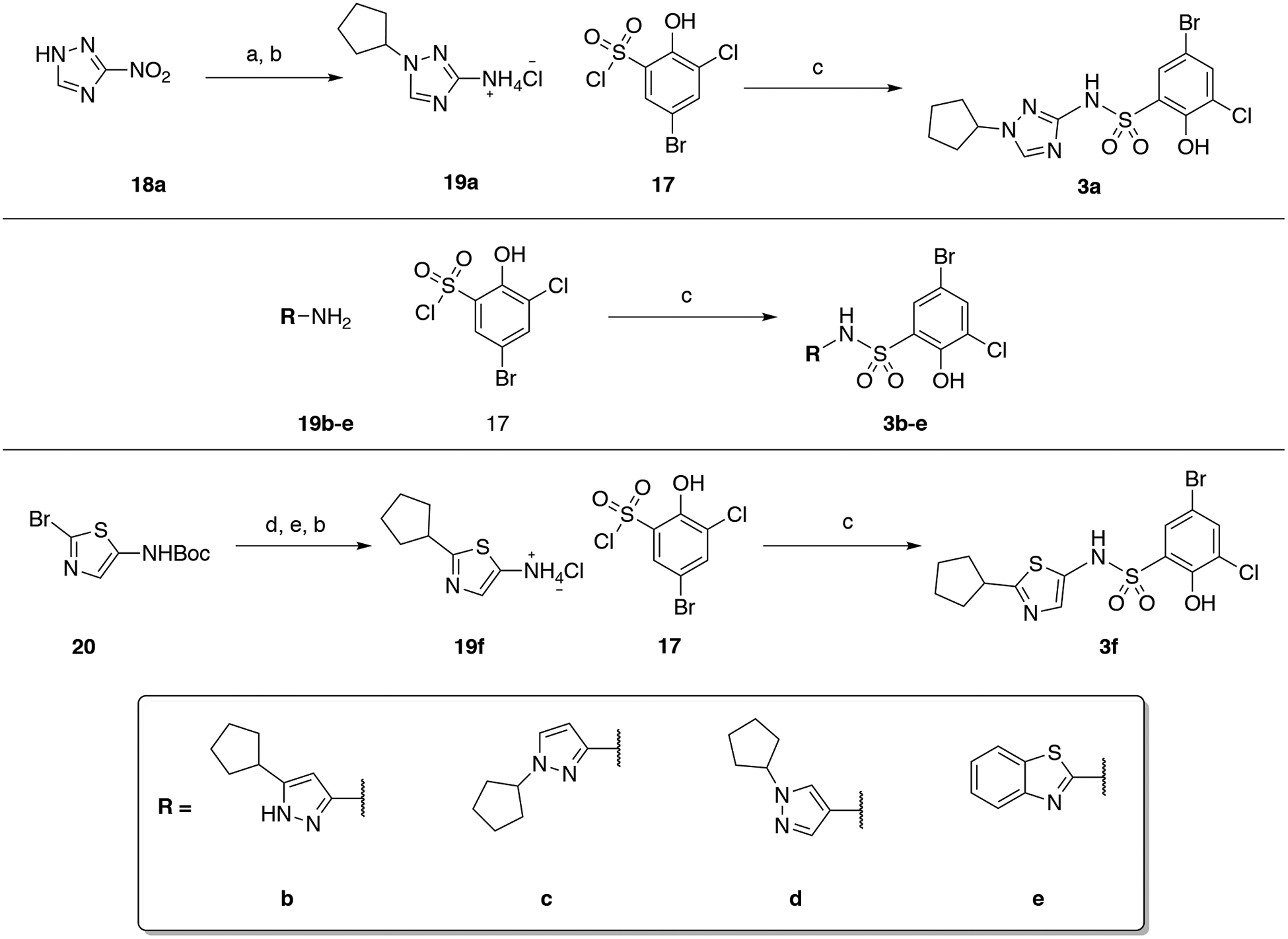
Synthesis of compounds 3a-f. Reagents and conditions: (a) Bromocyclopentane, KI, Cs2CO3, MeCN, MW, 135 °C, 45 min, 75%; (b) Pd/C 10%, EtOH, H2, 3 M HCl; (c) Pyridine, DCM, 0 °C - r.t., 1–84%; (d) Cyclopent-1-en-ylboronic acid, 2 M Na2CO3 (aq), Pd(PPh3)4, H2O, dioxane, 100 °C, 82%; (e) 4 M HCl in dioxane, 50 °C.
Acknowledgements
The authors thank Qi Sun and DeMarco Camper for their contributions to this series of compounds during this project. This work was supported by a grant from the NIH/NCI (CA200709 to W.P.T). This work was also supported by grants from the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation [to W.P.T. and S.W.F.), The TJ Martell Foundation [to W.P.T. and S.W.F.], the Edward P. Evans Foundation [to W.P.T.], Alex’s Lemonade Stand Foundation [to W.P.T.], St. Baldrick’s Foundation [to W.P.T.], CA236733 [to S.W.F. and W.P.T], and CA211305 [to L. R. T.]
Abbreviations Used
- WDR5
WD-repeat protein 5
- WBM
WDR5-binding motif
- WIN
WDR5-interacting site
- RBBP5
retinoblastoma binding protein 5
- KANSL2
KAT8 Regulatory NSL Complex Subunit 2
- MLL
mixed lineage leukemia
- PDAC
pancreatic ductal adenocarcinoma
- HMT
histone methyl transferase
- HTS
high-throughput screen
- FPA
fluorescence polarization assay
- FITC
fluorescein isothiocyanate
- Co-IP
Co-immunoprecipitation
- ChIP
Chromatin immunoprecipitation
Footnotes
- List of Fragment hits; pharmaceutical properties of select compounds; calculated physicochemical properties of selected compounds; histone methyl transferase assay data; X-ray data collection and refinement statistics; chemical structures of N.C and 30; experimental details for the synthesis of negative control.
- Molecular-formula strings
| PDB code | Compound |
|---|---|
| 4Y7R | MbIIIb MYC peptide |
| 6UJJ | F-1 |
| 6UHY | F-2 |
| 6UJH | F-5 |
| 6UHZ | F-6 |
| 6UJL | F-7 |
| 6UJK | 2i |
| 6UJ4 | 4 |
| 6UIF | 8 |
| 6UOZ | 12 |
REFERENCES
- (1).Campaner S; Amati B Two Sides of the Myc-Induced DNA Damage Response: From Tumor Suppression to Tumor Maintenance. Cell Div. 2012, 7 (1), 6 10.1186/1747-1028-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Henriksson M; Luscher B Proteins of the MYC Network: Essential Regulators of Cell Growth and Differentiation. Adv. Cancer Research 1996, 68, 109–182. 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- (3).Dang CV Review MYC on the Path to Cancer. Cell 2012, 149 (1), 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dalla-Favera R; Bregni M; Eriksont J; Pattersonf D; Gallo RC; Crocet CM Human CMyc Onc Gene is Located on the Region of Chromosome 8 that is Translocated in Burkitt Lymphoma Cells. Proc. Natl. Acad. Sci 1982, 79 (December), 7824–7827. 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tansey WP Mammalian MYC Proteins and Cancer. New J. Sci 2014, 1–27. 10.1155/2014/757534. [DOI] [Google Scholar]
- (6).Dang CV C-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol 1999, 19 (1), 1–11. 10.1128/MCB.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chrzan P; Skokowski J; Karmolinski A; Pawelczyk T Amplification of C- Myc Gene and Overexpression of c-Myc Protein in Breast Cancer and Adjacent Non-Neoplastic Tissue. Clin. Biochem 2001, 34, 557–562. 10.1016/S0009-9120(01)00260-0. [DOI] [PubMed] [Google Scholar]
- (8).Prochownik EV; Vogt PK Therapeutic Targeting of Myc. Genes Cancer 2010, 1 (6), 650–659. 10.1177/1947601910377494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Goga A; Yang D; Tward AD; Morgan DO; Bishop JM Inhibition of CDK1 as a Potential Therapy for Tumors Over-Expressing MYC. Nat. Med 2007, 13 (7). 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- (10).Yu C; Niu X; Jin F; Liu Z; Jin C; Lai L Structure-Based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep 2016, 6, 1–11. 10.1038/srep22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Thomas LR; Wang Q; Grieb BC; Phan J; Foshage AM; Sun Q; Olejniczak ET; Clark T; Dey S; Lorey S; Alicie B; Howard GC; Cawthon B; Ess KC; Eischen CM; Zhao Z; Fesik SW; Tansey WP Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58 (3), 440–452. 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stirnimann C; Petsalaki E; Russell R; Muller C WD40 Proteins Propel Cellular Networks. Trends Biochem. Sci 2010, 35 (10), 565–574. 10.1016/j.tibs.2010.04.003. [DOI] [PubMed] [Google Scholar]
- (13).Guarnaccia A; Tansey W Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med 2018, 7 (2), 1–17. 10.3390/jcm7020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Thomas LR; Adams CM; Wang J; Weissmiller AM; Creighton J; Lorey SL; Liu Q; Fesik SW; Eischen CM; Tansey WP Interaction of the Oncoprotein Transcription Factor MYC with its Chromatin Cofactor WDR5 is Essential for Tumor Maintenance. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (50), 1–9. 10.1073/pnas.1910391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Carugo A; Seth S; Heffernan TP In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep 2016, 16, 133–147. 10.1016/j.celrep.2016.05.063. [DOI] [PubMed] [Google Scholar]
- (16).Macdonald JD; Chacón Simon S; Han C; Wang F; Shaw JG; Howes JE; Sai J; Yuh JP; Camper D; Alicie BM; Alvarado J; Nikhar S; Payne W; Aho ER; Bauer JA; Zhao B; Phan J; Thomas LR; Rossanese OW; Tansey WP; Waterson AG; Stauffer SR; Fesik SW Discovery and Optimization of Salicylic Acid-Derived Sulfonamide Inhibitors of the WD Repeat-Containing Protein 5–MYC Protein–Protein Interaction. J. Med. Chem 2019, 62 (24), 11232–11259. 10.1021/acs.jmedchem.9b01411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ionescu C Pathways of Biotransformation - Phase II Reactions In Drug Metabolism: Current Concepts; Caira MR, Eds.; Springer: Dordrecht, 2005; pp 129–170. 10.1007/1-4020-4142-X_3. [DOI] [Google Scholar]
- (18).Rietjens IMCM; Besten C. Den; Hanzlik RP; Van Bladeren PJ Cytochrome P450-Catalyzed Oxidation of Halobenzene Derivatives. Chem. Res. Toxicol 1997, 10 (6), 629–635. 10.1021/tx9601061. [DOI] [PubMed] [Google Scholar]
- (19).Shuker SB; Hajduk PJ; Meadows RP; Fesik SW Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274 (5292), 1531–1534. 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- (20).Scott DE; Coyne AG; Hudson SA; Abell C Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51 (25), 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- (21).Harner MJ; Frank AO; Fesik SW Fragment-Based Drug Discovery Using NMR Spectroscopy. J. Biomol. NMR 2013, 56 (2), 65–75. 10.1007/s10858-013-9740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ichihara O; Barker J; Law RJ; Whittaker M Compound Design by Fragment-Linking. Mol. Inform 2011, 30 (4), 298–306. 10.1002/minf.201000174. [DOI] [PubMed] [Google Scholar]
- (23).Southall SM; Wong PS; Odho Z; Roe SM; Wilson JR Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33 (2), 181–191. 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- (24).Dias J; Van Nguyen N; Georgiev P; Gaub A; Brettschneider J; Cusack S; Kadlec J; Akhtar A Structural Analysis of the KANSL1/WDR5/ KANSL2 Complex Reveals That WDR5 Is Required for Efficient Assembly and Chromatin Targeting of the NSL Complex. Genes Dev 2014, 28 (9), 929–942. 10.1101/gad.240200.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Avdic V; Zhang P; Lanouette S; Groulx A; Tremblay V; Brunzelle J; Couture JF Structural and Biochemical Insights into MLL1 Core Complex Assembly. Structure 2011, 19 (1), 101–108. 10.1016/j.str.2010.09.022. [DOI] [PubMed] [Google Scholar]
- (26).Hao C; Zhao F; Song H; Guo J; Li X; Jiang X; Huan R; Song S; Zhang Q; Wang R; Wang K; Pang Y; Liu T; Lu T; Huang W; Wang J; Lin B; He Z; Li H; Li F; Zhao D; Cheng M Structure-Based Design of 6-Chloro-4-Aminoquinazoline-2-Carboxamide Derivatives as Potent and Selective P21-Activated Kinase 4 (PAK4) Inhibitors. J. Med. Chem 2018, 61 (1), 265–285. 10.1021/acs.jmedchem.7b01342. [DOI] [PubMed] [Google Scholar]
- (27).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective Inhibition of BET Bromodomains. Nature 2010, 468 (7327), 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Delmore JE; Issa GC; Lemieux ME; Rahl PB; Shi J; Jacobs HM; Kastritis E; Gilpatrick T; Paranal RM; Qi J; Chesi M; Schinzel AC; McKeown MR; Heffernan TP; Vakoc CR; Bergsagel PL; Ghobrial IM; Richardson PG; Young RA; Hahn WC; Anderson KC; Kung AL; Bradner JE; Mitsiades CS BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell 2011, 146 (6), 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mertz JA; Conery AR; Bryant BM; Sandy P; Balasubramanian S; Mele DA; Bergeron L; Sims RJ Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc. Natl. Acad. Sci 2011, 108 (40), 16669–16674. 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Andrieu G; Belkina AC; Denis GV Clinical Trials for BET Inhibitors Run Ahead of the Science. Drug Discov. Today Technol 2016, 19, 45–50. 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen H; Liu H; Qing G Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther 2018, 3 (1), 1–7. 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schanda P; Kupce E; Brutscher B SOFAST-HMQC Experiments for Recording 2D Heteronuclear Correlation Spectra of Proteins within Seconds. J. Biomol. NMR 2005, 33, 199–211. 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- (33).Otwinowski Z; Minor W Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- (34).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; Mccoy A; Mcnicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Adams PD; Grosse-Kunstleve RW; Hung LW; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr. Sec. D Biol. Crystallogr 2002; Vol. 58, pp 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- (36).Emsley P; Cowtan K Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sec. D Biol. Crystallogr 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (37).Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.