

Abstract
Rod‐cone degenerations, for example, retinitis pigmentosa are leading causes of blindness worldwide. Despite slow disease progression in humans, vision loss is inevitable; therefore, development of vision restoration strategies is crucial. Among others, promising approaches include optogenetics and prosthetic implants, which aim to bypass lost photoreceptors (PRs). Naturally, the efficacy of these therapeutic strategies will depend on inner retinal structural and functional preservation. The present study shows that in photoinducible I307N rhodopsin mice (Translational Vision Research Model 4 [Tvrm4]), a 12k lux light exposure eliminates PRs in the central retina in 1 week, but interneurons and their synapses are maintained for as long as 9 weeks postinduction. Despite bipolar cell dendritic retraction and moderate loss of horizontal cells, the survival rate of various cell types is very high. Significant preservation of conventional synapses and gap junctions in the inner plexiform layer is also observed. We found the number of synaptic ribbons to gradually decline and their ultrastructure to become transiently abnormal, although based on our findings intrinsic retinal architecture is maintained despite complete loss of PRs. Unlike common rodent models of PR degeneration, where the disease phenotype often interferes with retinal development, in Tvrm4 mice, the degenerative process can be induced after retinal development is complete. This time course more closely mimics the timing of disease onset in affected patients. Stability of the inner retina found in these mutants 2 months after PR degeneration suggests moderate, stereotyped remodeling in the early stages of the human disease and represents a promising finding for prompt approaches of vision restoration.
Keywords: bipolar cell, dendrites, retinitis pigmentosa, rhodopsin, synapse
Tvrm4 rhodopsin mutant mice undergo rapid photoreceptor loss upon bright light exposure. Early consequences on the inner retina involve mainly second‐order neurons and include quick dendritic retraction and axonal shrinkage in rod bipolar cells (RBCs) and, later, in cone BCs and the presence of anomalous globular synaptic ribbons (GR). Yet, BC survival is nearly 100% and retinal architecture is overall well preserved. Hence, early remodeling in this adult mutant is moderate and similar to what observed in developmental paradigms of retinitis pigmentosa.
1. INTRODUCTION
Degeneration of retinal rods and loss of scotopic vision followed by secondary degeneration of cones progressing to severe, blinding visual impairment are the main clinical manifestations of retinitis pigmentosa (RP), a family of inherited diseases with an estimated incidence of 1:4,000 (Hamel, 2006). RP can be caused by mutations in any of up to 65 different genes identified so far (RetNet: https://sph.uth.edu/retnet/sum-dis.htm). The first symptoms typically manifest during young adulthood, after the retina is fully developed. However, numerous experimental studies on RP have been conducted using naturally occurring or transgenic rodent models (i.e., rd1, rd10, P23H rhodopsin mutant mice), in which photoreceptor (PR) death initiates concomitantly with the late phases of retinal differentiation and refinement of the synaptic circuitry, complicating comparisons to the human pathology. Developing retinal neurons may be intrinsically more vulnerable to the effects of PR death or, conversely, show increased adaptation to the consequences of the PR degeneration.
Translational Vision Research Model 4 (Tvrm4) mice, isolated at the Jackson Laboratory in 2010, bear a single nucleotide polymorphism (SNP) in the rhodopsin gene (rho), resulting in an I307N mutation (substitution of isoleucine to asparagine at Position 307) in the seventh transmembrane domain of the rhodopsin protein (RHO) (Budzynski et al., 2010). The mutation does not affect the structure or function of RHO in the retina if mice are maintained in normal, ambient lighting levels. The mutant RHO is folded normally and transported to rod outer segments. However, if the retina is briefly exposed to a very strong light (i.e., 1,000 lx), the phototransduction cascade leads to a lower binding rate of the mutant opsins to 11‐cis‐retinal and a higher than normal activation of transducin by unbound opsins (Budzynski et al., 2010). Free opsins are known to have a toxic effect on PRs and light induction causes a massive degeneration of rods, particularly in the central retina where light reaches the highest intensity. Cone death accompanies rod degeneration as a secondary phenomenon (Budzynski et al., 2010) as observed in RP (Léveillard et al., 2004). The observed phenotype of Tvrm4 mutants is similar to human class B1 RP due to mutations of RHO with a dominant inheritance pattern. Patients also exhibit focal PR degeneration which can be triggered or accelerated by exposure to bright light flashes and a moderate disease progression (Cideciyan et al., 1998; Gal, Apfelstedt‐Sylla, Janecke, & Zrenner, 1997; Mendes, Van Der Spuy, Chapple, & Cheetham, 2005; Rakoczy et al., 2011).
Previous studies in our laboratory confirmed normal retinal morphology in heterozygous (HT) Tvrm4 mice kept in normal light conditions (animal house standards) and report that 2 days after photoinduction at 12,000 lx for 2′, a dramatic decrease in the a and b waves of the electroretinogram (ERG) is seen. By ~1 week, PRs are completely lost from the central retina and condensed, DNA‐filled vacuoles remain. PR death is accompanied by Müller glial activation, also confined to the central, degenerating retina. Similarly, Iba1‐positive microglial cells with a highly activated, amoeboid morphology, invade the outermost layers of central retina, whereas resident microglia retain a normal morphology in peripheral areas (Gargini, Novelli, Piano, Biagioni, & Strettoi, 2017). The first sign of morphological remodeling of the inner retina is retraction of dendritic arbors from rod bipolar cells (RBCs), decreased metabotropic glutamate receptor 6 (mGluR6) immunoreactivity in the outer plexiform layer (OPL), and displacement of mGluR6 puncta in the inner nuclear layer (INL). It is reported that the degenerating central area does not spread over time; but becomes progressively smaller 1 month after the initial photoinduction. Consequently, the amplitude of the scotopic ERG recovers partially, likely because PR cells at the mid‐periphery of the retina can regenerate damaged outer segments (Gargini et al., 2017).
Outer retinal degeneration is well described in various rodent models of RP, while our knowledge about inner retinal response to PR death, especially in mature animals, is significantly more limited, particularly regarding the maintenance of synaptic connections in the inner plexiform layer (IPL) and functional consequences of synaptic rearrangements (Euler & Schubert, 2015; Greferath et al., 2015; Soto & Kerschensteiner, 2015; Strettoi, 2015). Improvements in restoration of light sensitivity in degenerated retinas (e.g., by prosthetic implants or optogenetic expression of photosensitive molecules) will require a better understanding on how PR loss affects inner retinal neurons and their fundamental circuitry. It is particularly important to document remodeling and fate of neurons of the retinal vertical pathway, as bipolar cells (BCs) are a primary target of vision restoration strategies. BCs are very numerous and play a crucial role in parallel processing of visual signals and proper transfer of information to ganglion cells (GCs). Clearly for this strategy to succeed, the GC cells need to survive as they are the singular output of electrical signals from the retina.
In the present study, we used the I307N Tvrm4 mice to study inner retinal remodeling in an adult model of RP, at early stages following complete PR degeneration.
2. METHODS
2.1. Animals and experimental design
Animal experimental protocols were approved by the Italian Ministry of Health (Protocol #14/D‐2014, CNR Neuroscience Institute) and by the Ethical Committee of the CNR Institution. Animals were treated in accordance with the Association for Research in Vision and Ophthalmology statement for the use of animals in research. Experiments were conducted on HT Tvrm4 mice (RhoTvrm4/Rho+) carrying the I307N mutation on the rho gene and on C57Bl6/J (WT) littermates, both strains originally from Jackson (Bar Harbor, ME). Genotyping was carried out according to Budzynski et al. (2010). Before photoinduction, both mouse pupils were dilated with one drop/eye atropine if the eyes were used for immunohistochemistry, and WT animals were used as controls. For electron microscopy analysis, the right pupils were dilated, while the left eyes served as internal controls (CTRL). Nonilluminated areas at the retinal periphery were also used as internal controls as their morphology was also unchanged. A total of 30 (15 mutants and 15 WT) adult mice (aged 3–5 months) were used for immunohistochemistry; six additional HT mice were used for electron microscopy. Retinal structure was examined at 3, 6, and 9 weeks after photoinduction. A minimum of n = 3 animals were used per experimental group.
2.2. Photoinduction protocol
Following 4 hr of dark adaptation in a black cage, Tvrm4 mice (aged 3–5 months) received bi‐ or unilateral (right eye‐only) eye drops of 1 μl, 0.5% atropine (Allergan). After 10 min, mice were placed in the photoinduction chamber: a black box (140 × 30 cm2 size; 30 cm height) lined with mirrors on the inner sides and covered with a lid holding four fluorescent bulbs (Philips Master TL5 HE 28 W/840, length 115 cm; 16 cm in diameter; Correlated Color Temperature (Nom) 4,000 K Luminous Efficacy (@Max Lumen, Rated) 104 lm/W cool White mercury lamps). Intensity of light was monitored by an inbox light meter sensor (mod. LX103, Lutron, Digital Instruments), at 30 s intervals. Photoinduction duration was controlled by a digital clock; during the procedure animals were fully awake and freely moving. Temperature did not change significantly during light exposure (Gargini et al., 2017). Animals were exposed to 12.000 lx light for 2 min, after which the animals were kept in the chamber in the dark for one additional minute (“cooldown” time) postinduction (PI), then returned to the local animal facility where they were kept in ambient light conditions (below 60 lx) on a 12/12‐hr light/dark cycle, with water and food ad libitum until the time of retinal examination.
2.3. Tissue preparation and immunohistochemistry
Retinal tissue was harvested at 3, 6, or 9 weeks PI. Mice were anesthetized with intraperitoneal Avertin (3‐bromo‐ethanol in 1% tert‐amyl alcohol; 0.1 ml/5 g body weight) and euthanized by quick cervical dislocation following enucleation of eyes, previously labeled on the dorsal pole with a surgical skin marker (Secureline Surgical Skin Marker, Aspen Surgical). Eyecup preparation was performed in 0.1 M phosphate buffer (PB) by removing the anterior segments (cornea, iris, lens) followed by immediate fixation in 4% paraformaldehyde (PFA) in 0.1 M PB, pH 7.4, for 30 min or 1 hr at room temperature. Fixation was followed by 3 × 5 min washes in 0.1 M PB, infiltration in 30% wt/vol sucrose dissolved in 0.1 M PB and infiltration in Tissue‐Tek O.C.T. compound (4583, Sakura, Olympus, Italy). Sample freezing in tissue molds was achieved with isopentane in dry ice; samples were stored at −25 °C until use. Frozen eyecups were later processed for either whole mount preparation or cryosectioning.
Frozen, vertical sections were cut at a cryostat at 12 μm thickness and collected on Super Frost slides for immunohistochemistry (IHCH). For whole mount preparations, defrosted eyecups were extensively washed in phosphate buffered saline (PBS); the retina was gently detached from the sclera and flattened by making four radial cuts toward the optic nerve head maintaining a reference on the dorsal pole.
For IHCH, both retinal whole mounts and sections were incubated in: blocking solution (0.03% Triton‐X 100 + 5% serum from the species in which the secondary antibody was generated, in 0.01 M PBS), for 2 hr at room temperature if sections and overnight at 4°C if whole mounts; primary antibody (Ab) solution (diluted in PBS + 0.01% Triton‐X 100 + 1% serum) overnight at 4°C if sections and 3 days at 4°C if whole mounts; in fluorescent secondary Ab solution—diluted as the primary Ab—for 2 hr at room temperature if sections and 2 days at 4°C if whole mounts (Barone, Novelli, Piano, Gargini, & Strettoi, 2012). Secondary antibodies were conjugated with Alexa Fluor 488 or Rhodamine Red X (Table 1).
Table 1.
Primary and secondary antibodies used in this study
| Mouse monoclonal primary antibodies | Antibody manufacturer, catalog #, RRID | Dilution rate |
|---|---|---|
| Rhodopsin | Sigma‐Aldrich, Italy; O4886 | 1:1000 |
| Protein kinase Cα (PKCα) | Sigma‐Aldrich; P5704 RRID:AB_477375 | 1:800 |
| CD68 (KP1) | Santa Cruz Biotechnology; sc‐20060 RRID:AB_627158 | 1:250 |
| C‐terminal binding protein‐2 (CtBP2/RIBEYE) | BD biosciences; 612044 | 1:800 |
| Glutamine synthetase (GS) | Merck‐Millipore, Italy; MAB302 | 1:1000 |
| Nonphosphorylated neurofilaments (SMI32) | Covance, CA; SMI‐32R RRID:AB_10719742 | 1:800 |
| Glutamic acid decarboxylase‐67 (GAD67) | Merck‐Millipore; MAB5406 | 1:500 |
| Rabbit polyclonal primary antibodies | ||
| Cone Arrestin | Merck‐Millipore; AB15282 | 1:5000 |
| Calbindin D | Swant Ltd., Switzerland; CB38a | 1:500 |
| Connexin36 (Cx36) | Invitrogen, Carlsbad, CA; 51‐6200 | 1:500 |
| Protein kinase Cα (PKCα) | Sigma‐Aldrich; P4334 | 1:800 |
| Glial fibrillary acidic protein (GFAP) | Sigma‐Aldrich; G9269 RRID:AB_477035 | 1:1000 |
| Ionized calcium binding adapter molecule 1 (IBA1) | Wako Chemicals GmbH, Germany; 019‐19741 | 1:500 |
| Disabled‐1 (DAB1) | Kind gift of Prof. Botond Roska, Basel, Switzerland | 1:200 |
| RBPMS | PhosphoSolutions, Aurora, CO; 1830‐RBPMS RRID:AB_2492225 | 1:500 |
| Tyrosine hydroxylase (TH) | Merck‐Millipore; AB152 | 1:1000 |
| Sheep polyclonal primary antibody | ||
| Secretagogin (SCGN) | BioVendor GmbH, Germany; RD184120100 | 1:2000 |
| Goat polyclonal primary antibody | ||
| Choline acetyltransferase (ChAT) | Merck‐Millipore; AB144P RRID:AB_11212843 | 1:500 |
| Secondary antibodies | All from Jackson ImmunoResearch Laboratories, Inc. | |
| Donkey anti‐mouse Alexa Fluor 488 | 715‐545‐151 RRID:AB_2341099 | 1:800 |
| Donkey anti‐rabbit Alexa Fluor 488 | 711‐546‐152 | 1:800 |
| Donkey anti‐goat Alexa Fluor 488 | 705‐546‐147 RRID:AB_2340430 | 1:800 |
| Donkey anti‐sheep Alexa Fluor 488 | 713‐546‐147 | 1:800 |
| Donkey anti‐mouse Rhodamine Red X | 715‐295‐151 | 1:800 |
| Donkey anti‐rabbit Rhodamine Red X | 711‐295‐152 | 1:800 |
2.4. Antibody characterization
Technical information regarding the antibodies used in this study are summarized in Table 1.
Rhodopsin antibody recognizes an epitope at the N‐terminus of the rhodopsin molecule (Hargrave et al., 1986; Hicks & Barnstable, 1987; Silver et al., 1988). In rat retinal protein extract immunoblotting, two closely spaced 39 kDa bands as well as bands at 78 and 115 kDa with lower intensity representing the rhodopsin monomer and its aggregates are marked (Fekete & Barnstable, 1983). The antibody specifically labels rod PR cells (but not cones) in the rat retina via immunohistochemistry (Barnstable, 1980). In the mouse retina the product labels rod outer segments exclusively (Roof, Adamian, & Hayes, 1994) and the staining observed in our study was consistent with these reports.
Protein kinase Cα (PKCα) antibody recognizes an 80 kDa polypeptide of PKC in bovine brain lysates (manufacturer's data sheet). PKC is a well‐established marker of RBCs in the rat (Greferath, Grünert, & Wässle, 1990) and mouse retina (Barone et al., 2012; Haverkamp, Ghosh, Hirano, & Wässle, 2003) and the same staining consistency was observed in our study.
CD68 is homologous to the mouse antigen macrosialin. Apart from nonhematopoietic tissues like liver and kidney, where CD68 expression is the highest, the antigen can be found on the outer surface of macrophages, monocytes, neutrophils, basophils, and large lymphocytes. The CD68 (KP1) mouse monoclonal antibody was raised against human alveolar macrophages and it stains a 101 kDa and a 70 kDa band on immunoblots of whole cell lysates (manufacturer's data sheet). In the mouse retina CD68 is a marker of microglia and is often used in conjunction with Iba‐1 (Santos et al., 2008), resulting in a similar staining pattern to the one reported here.
C‐terminal binding proteins (CtBP1 and CtBP2) are a class of corepressors capable of interacting with certain transcription factors via a PLDLSL sequence motif. Immunoblotting of mouse brain and retinal tissue lysates revealed that the B‐domain of RIBEYE also labels the 48–50 kDa CtBP2 band besides being present both in PR and BC ribbon synapses in the mouse retina based on IHCH (Dieck et al., 2005). Our observations confirm similar immunoreactivity.
Glutamine synthetase (GS) is an enzyme responsible for catalyzing the amination of glutamic acid and plays a pivotal role in glutamate neurotransmitter recycling. GS immunoblotting of sheep or rat brain lysates shows a strongly labeled 45 kDa band (manufacturer's data sheet), reflecting the fact that mammalian GS is composed of eight 45 kDa subunits. Müller glia is reported to show intense GS immunoreactivity (Riepe & Norenburg, 1977) identical to what observed here.
SMI32 reacts with a nonphosphorylated epitope in neurofilament H of most mammals. In western Blots, SMI32 antibodies show two bands at 180 and 200 kDa, which tend to merge into a single band on 2D blots (manufacturer's data sheet). In the retina of most mammalian species, SMI32 antibodies label the soma and axons of GCs (Lewis & Nixon, 1988).
Glutamic acid decarboxylase (GAD) catalyzes the synthesis of γ‐aminobutyric acid (GABA), a major inhibitory neurotransmitter. GAD is present in the mammalian brain in two forms: the 65 kDa GAD65 and the 67 kDa GAD67, respectively, more abundant in axon terminals and generally distributed throughout the neuron (Kaufman, Houser, & Tobin, 1991). In the mouse retina, GAD67 labels GABA‐ergic amacrine cells (ACs) in the INL and IPL with intense immunofluorescence (Müller, Azar, de los Santos, & Brecha, 2017). Patterns of staining found here coincide with previous reports.
Cone Arrestin or Arrestin‐C is encoded by the Arr3 gene in murine species. Arrestin‐C is predominantly found in inner and outer segments of cone PRs and the IPL of the retina (Gurevich & Benovic, 1995). Cone Arrestin antibody evaluated by the manufacturer by immunoblotting of mouse retinal tissue lysate labeled a 50 kDa band. Immunohistochemical characterization of Cone Arrestin expression pattern performed by Zhu et al. (2002a) and (2002b) reported similar staining intensities and pattern observed in the present study.
Calbindin D is a member of the intracellular calcium‐binding proteins family characterized by EF‐hand motifs. Calbindin D antibodies recognize a single band of approximately 27–28 kDa in immunoblots (Airaksinen et al., 1997). In the mouse and rat retina, Calbindin D antibody labels horizontal cells and a subpopulation of ACs (Hamano et al., 1990). Staining pattern shown in this study confirms previous reports.
Connexin 36 (Cx36), a prime component of neuronal gap junction connexins is known to play a pivotal role in the rod pathway; it was first identified in the rat inferior olive and confirmed to be strongly expressed in neural tissue, including the retina (Condorelli et al., 1998; Demb & Pugh, 2002; Söhl, Degen, Teubner, & Willecke, 1998). Western blots detect monomeric Cx36 migrating at 36 kDa in homogenates of retina, olfactory bulb, and inferior olive (Rash et al., 2000). In the mouse retina Cx36 immunolabeling is concentrated in the IPL. Staining patterns found in this study match previous reports (Ghinia, Novelli, Sajgo, Badea, & Strettoi, 2019).
The glial fibrillary acidic protein (GFAP) is a fundamental protein component of intermediate filaments of CNS macroglia (Eng, 1985). The GFAP positive band on immunoblots of mouse, rat, and human brain lysates appears at approximately 50 kDa (manufacturer's data sheet). In the healthy mouse retina, GFAP is a reliable marker of astrocytes, while in the diseased tissue, it is widely used as a marker of Müller glial hypertrophy (Eisenfeld, Bunr‐milam, & Sarrhy, 1984). Our observations confirm similar immunoreactivity.
Iba1 is a calcium‐binding protein with a molecular weight of 17 kDa specifically expressed in macrophage/microglia (manufacturer's data sheet). A widely used microglial marker in mouse retina (Gargini et al., 2017; Guadagni et al., 2019) produces a staining pattern similar to what observed in our results.
Mouse disabled‐1 (Dab‐1) cytoplasmic adaptor protein is the product of the disabeled‐1 gene and is expressed throughout the CNS during embryonic development. Cell lysates of differentiated, P19 brain cells were found to express 60, 80, and 120 kDa mouse Dab‐1 isoforms by immunoblot analysis (Howell, Gertler, & Cooper, 1997). In the mature mouse retina, Dab‐1 was confirmed to be expressed in AII ACs by IHCH (Rice & Curran, 2000) with staining patterns similar to what we report here.
RBPMS or RNA binding protein with multiple splicing also known as HERMES belongs to the family of RNA binding proteins possibly playing a role in transcription and translation through binding to RNA. RBPMS antibody recognizes a 24 kDa protein band on immunoblots (manufacturer's data sheet). RBPMS is specifically expressed in retinal GCs (RGCs) in multiple mammalian species (Kwong, Caprioli, & Piri, 2010; Rodriguez, de Sevilla Müller, & Brecha, 2014). Patterns of staining found here coincide with previous reports.
Tyrosine hydroxylase (TH) is the enzyme responsible for the biosynthesis of the catecholamines dopamine and norepinephrine from tyrosine and is a useful marker for dopaminergic and noradrenergic neurons. By western blot, the TH antibody selectively labels a single band of about 62 kDa (reduced), corresponding to the protein itself (manufacturer's data sheet). Anti‐TH is a robust marker for dopaminergic ACs (DACs) of multiple mammalian species including mice (Wulle & Schnitzer, 1989). We report a similar staining pattern here.
Secretagogin (SCGN) belongs to the EF‐hand (E‐helix‐loop–F‐helix‐hand) superfamily of calcium‐binding proteins. The sheep anti‐SCGN antibody recognizes a 32 kDa band on western blots of mouse cerebellum and retina extracts. It is a marker for Type 2, 3, 4, 5, 6, and possibly 8 cone bipolar cells (CBCs) (Puthussery, Gayet‐Primo, & Taylor, 2010). We observed staining results similar to what was found in previous publications.
The choline acetyltransferase (ChAT) enzyme is responsible for the synthesis of acetylcholine (Ach). The goat‐anti‐ChAT antibody recognizes a 70/74 kDa protein band on immunoblots (manufacturer's data sheet). In the mouse retina, ChAT antibodies specifically label cholinergic or starburst ACs (Jeon, Strettoi, & Masland, 1998). Our observations confirm a similar staining pattern.
2.5. Transmission electron microscopy
To achieve optimal preservation of mouse retina, we implemented a protocol combining methods from several laboratories (Ghinia et al., 2019; Morrow, Furukawa, Raviola, & Cepko, 2005).
Animals were deeply anesthetized with intraperitoneal (0.1 ml/5 g body weight) Avertin injections, eyes quickly explanted, and mice euthanized by cervical dislocation. Enucleated eyes—with a blue mark on the dorsal pole—were immediately transferred in oxygenated artificial cerebrospinal fluid where the cornea, iris, and lens were removed, and the eyecups were immersed in primary fixative (1% vol/vol glutaraldehyde +2% vol/vol PFA in Sorenson's buffer M/30, pH 7.4 + 5% wt/vol sucrose) for 2 hr at 4°C. Following primary fixation, the eyecups were cut into four equal pieces corresponding to the retinal quadrants (dorsal, nasal, ventral, and temporal) without detaching the retina from the sclera and transferred into secondary fixative (3% vol/vol glutaraldehyde in Sorenson's buffer M/30, pH 7.4 + 5% wt/vol) sucrose) overnight at 4°C. Afterward, samples were extensively washed in buffer at room temperature (Sorenson's buffer M/30, pH 7.4 + 5% wt/vol sucrose—10 × 5 min) and immersed in 3% wt/vol potassium ferrocyanide +2% (wt/vol) OsO4 in H2O, at 4°C for 2 hr. Osmication was followed by washing steps (0.05 M maleate buffer, pH 5.15—10 × 5 min) and by staining en bloc with 1% (wt/vol) uranyl acetate (in 0.05 M maleate buffer, pH 6) for 1 hr at 4°C. Blocks were then washed at room temperature (0.05 M maleate buffer, pH 5.15—5 × 5 min), dehydrated with a graded series of ethanol and infiltrated first with propylene oxide (2 × 15 min), then with a mixture of propylene oxide and epon–araldite resin 50% (vol/vol) catalyzed with 2% DMP30 overnight at room temperature. Final epon–araldite embedding was performed in flat molds at 65°C. Sectioning was performed using a Leica Ultracut ultramicrotome and a diamond knife. Ultrathin sections, 90 nm thick, were collected on single‐hole copper grids (Formvar Support Film Slots, 2 × 1 mm2 Cu grids; FF2010‐CU; Electron Microscopy Science, Inc.). No grid staining was necessary given the high contrast of the specimens and the sensitivity of the EM camera.
2.6. Imaging and analysis
2.6.1. Cell counts in retinal whole mounts
Retinal whole mounts were imaged with a Zeiss Axioplan light microscope equipped with a Zeiss Axiocam color camera and a Plan Neofluar 1.25x/0.035 objective to measure retinal surface prior and post confocal microscopy. Image acquisition for cell number quantification was performed with a Leica TCS‐SL confocal microscope equipped with 488 and 453 lasers, using a Plan Apochromat 40x/1.40 oil objective, employing identical laser and imaging settings for all samples. Four retinal fields (250 × 250 μm2 each) were acquired at each eccentricity of the tissue (dorsal, nasal, ventral, and temporal), covering ~1/15 of the whole area of the retina and spanning the entire INL thickness on the z axis. Confocal stacks were saved as TIFF files and transferred to a MetaMorph image analyzer, where cell counts were performed. Cells were identified on single focal planes, labeled and followed across the entire z stacks. Average numbers of cells/mm2 were multiplied for the corresponding retinal surface, taking a value averaged between the surface before and after confocal imaging. Statistical comparisons of experimental groups were executed in SigmaPlot applying one‐way analysis of variance (ANOVA) test with the Holm–Sidak post hoc method.
2.6.2. Image thresholding analysis
To estimate the area occupied by axonal endings of BCs in retinal whole mounts and vertical sections, or the area covered by immunolabeled synaptic contacts in the IPL, images were obtained with a Leica TCS‐SL confocal microscope using identical laser and imaging settings for all samples, using Plan Apochromat 40x/1.40 or 100x/1.30 oil objectives. High definition projection images were acquired (typically spanning 4 μm across the z axis); brightness and contrast were adjusted with the Zeiss software ZENPRO 2012 or with Adobe Photoshop using consistent settings. An identical region of interest (ROI) was applied to each image for quantification using an image thresholding analysis tool of MetaMorph software. First, each ROI was processed by subtracting the background; then, pixels exceeding the threshold were measured automatically and corresponding areas normalized to mm2. Data from different ROI were averaged for each experimental group. Statistical evaluation was done in SigmaPlot using one‐way ANOVA tests with the Holm–Sidak post hoc method.
2.6.3. Whole mount reconstructions
Retinal whole mounts were imaged with an Imager.Z2 Zeiss microscope (Zeiss, Milan, Italy) using EC Plan‐Neofluar 5x/0.16 M27, 10x/0.3 M27, and 20x/0.50 M27 objectives; images were tiled with ZEN module “Tiles & Positions” software to reconstruct the entire retinal surface. TH positive DACs were counted on tiled images of entire retinal surface obtained with a 10x objective.
Additional images of vertical sections were obtained with the same Zeiss Imager.Z2 microscope equipped with an Apotome2 device (Zeiss) using an EC Plan‐Neofluar 20x/0.50 M27 objective.
2.6.4. Transmission electron microscopy analysis
Images were obtained using a JEOL 1200EX II electron microscope (JEOL, Milan, Italy) equipped with a charge‐coupled device Olympus Veleta Megaview camera (Olympus, Münster, Germany) at a magnification of 40.000×, covering systematically the entire width of the IPL represented in each section. A total of 400 transmission electron microscopy (TEM) micrographs (12.96 μm2/micrograph) per retinal sample (corresponding to a sampled area of 0.0052 mm2/sample) were imaged. Image files were saved as TIFF and transferred to a MetaMorph image analysis workstation, where synapses were counted manually. Criteria applied to identify conventional synapses were the presence of synaptic vesicle clusters abutting the presynaptic membrane, the occurrence of synaptic densities and the unequivocal presence of pre‐ and postsynaptic processes. For ribbon synapses, identification was based on the presence of an osmiophilic ribbon, either bisecting the presynaptic wedge or freely floating in the cytoplasm but unequivocally tethering synaptic vesicles; the presence of an arciform density at the active zone; and the obvious presence of presynaptic and postsynaptic processes. Counts were averaged per experimental group and statistically analyzed in SigmaPlot applying one‐way ANOVA tests with Holm–Sidak post hoc method.
3. RESULTS
3.1. PRs of the Tvrm4 mouse retina 3 and 6 weeks PI
By 3 weeks PI by light, PR death in the central retina is largely complete, and no rows of nuclei are visible in the outer nuclear layer (ONL). Surviving, intact rods, and cones can only be found at the far retinal periphery. A similar phenotype is also observed at 6 weeks PI (Figure 1). Large bodies that stain intensely with DNA‐binding molecules are visible in the outer retina. These products of condensation of dying PRs persist in the subretinal space after PR death and remain visible at 9 weeks PI, the latest time point examined. Observations on remodeling were conducted in PR‐free, central retinal areas. The retina of wild type, age matched mice, littermates of Tvrm4 animals, born by crossing HT Tvrm4 mice, were used as controls, as specified in the text.
Figure 1.
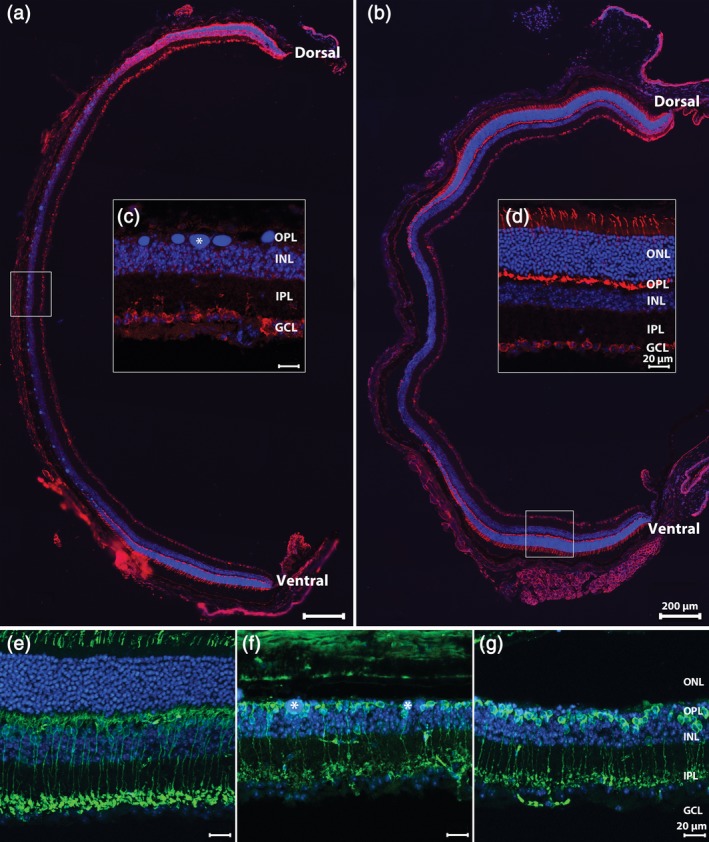
Dorsoventral, longitudinal sections of Tvrm4 retinas 3 (a) and 6 (b) weeks after photoinduction, stained for anti‐Cone Arrestin (red) and Hoechst nuclear marker (blue). (c) During this time interval, photoreceptor (PR) degeneration becomes nearly complete in the central retina where DNA clusters (asterisk) are leftovers from PR nuclear condensation. At both time points examined, intact PRs can be found in more retinal regions, adjacent to the area where the ONL is totally missing, as highlighted in (d). (In this and other pictures: GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer.). Vertical sections of the Tvrm4 retina without photoinduction (e) or 3 (f) and 6 (g) weeks postinduction (PI) stained for anti‐PKCα (green) and Hoechst nuclear marker (blue). In the areas devoid of photoreceptors (f,g) rod bipolar cells retract dendrites and their axonal endings exhibit a visible shrinkage (g) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.2. Rod BCs
Typical atrophy and retraction of dendritic arbors (described by Gargini et al. (2017)) is first observed in RBCS at 7 days PI. By 3 weeks, RBCs in the central, most degenerated part of the retina, are completely devoid of dendrites, their progressive shortening and retraction being accompanied by concurrent shrinkage of RBC axonal endings in the IPL. These changes are still seen at 6 weeks PI (Figure 1). In addition to examining the morphological changes, we determined the number of surviving RBCs, by counting cell bodies in retinal whole mounts. The overall RBC number did not show any change during this time period (one‐way ANOVA, p = .295; n = 3 retinas/experimental group; 16 images/retina: 4/retinal eccentricity) demonstrating a survival rate of nearly 100% (Figure 2) despite regression of dendritic trees (Figures 1 and 2). A significant shrinkage of axonal endings was found at the same time points and quantified in retinal whole mounts by analyzing the surface covered by RBC axonal arborizations in sublamina 5 of the IPL. A decrement larger than 50% was observed in the surface occupied by RBC axonal arbors and lying within the PR devoid area (one‐way ANOVA, p ≥ .0001, n = 3 retinas) compared to the peripheral retina where PRs are preserved (Figure 2). This confirms that the regression of axonal arbors is secondary to the degeneration of PRs.
Figure 2.
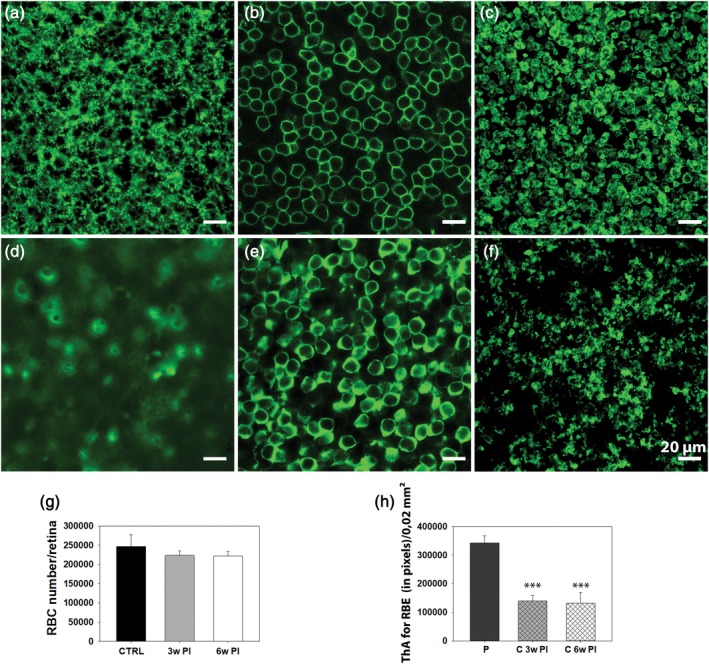
Images of Tvrm4 mouse retina whole mount 3 weeks postinduction (PI), showing, respectively, a peripheral, intact region (a–c) and a central area (d–f). Focal planes are on the (a,d) dendrites, (b,e) cell bodies, and (c,f) axonal endings of RBCs, stained with anti‐PKCα. (a) At all time points examined, dendrites are preserved in the periphery but fully retracted in the central, degenerated retina, where cell bodies look virtually “naked” (d,e) and axonal endings impoverished (f). (g) RBC counting in retinal whole mounts (3 mice/exp. group; 16 images [150 × 150 μm2]/retina: 4 images/retinal eccentricity) shows no statistical difference in the overall number of cell bodies of Tvrm4 retinas compared to WT controls (CTRL) (p = .295). (h) A significant shrinkage of axonal arborizations of RBCs (*** = p ≤ .0001) was evident 3 (3w PI) and 6 weeks PI (6w PI) when comparing central to peripheral retina. (ThA, thresholded area; PKC + RBE, PKC‐positive rod bipolar endings; P, periphery; C, central retina; data are shown as average and SE; one‐way analysis of variance [ANOVA]) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.3. Horizontal cells
Our analysis of remodeling of second‐order neurons also consisted of examining HCs in retinal whole mounts stained with anti‐Calbindin D. Analyses of retinas at 6 weeks PI demonstrated that HCs completely disappeared from islands of variable sizes and shapes in the central retina, creating transition zones with extremely sharp edges between regions of surviving and dead cells. Interestingly, these islands were not precisely overlapping with PR‐lacking areas and HCs were typically missing in the transition zones of PR degeneration (Figure 3a–g); conversely, numerous HCs with hypertrophic processes were preserved in the central retina next to these islands albeit no PRs were present in the corresponding ONL. HC body counts revealed a 37% decrease in cell density (cells/mm2; t test, p ≥ .01; n = 3 retinas/experimental group; 16 images/retina: 4/retinal eccentricity) (Figure 3h), over the central retinal area and in the total number (Figure 3i) of HCs relative to WT controls at 6 weeks PI. Calbindin D and Neurofilament‐200 (NF200) antibody staining of vertical sections confirmed the observation of retained HC bodies and dendrites in central retinal areas devoid of PRs and of decreased HC density in transition zones. Morphological abnormalities (mostly hypertrophy) were first observed in the axonal arbors of HCs (NF200 positive) postsynaptic to rods, while dendrites and cell bodies were preserved for longer times (Figure 3e–g), thus confirming the typical rod‐to‐cone degeneration pattern of this mutant. There is no obvious explanation for the higher severity of HC remodeling in transition zones compared to what is observed in the central retina.
Figure 3.
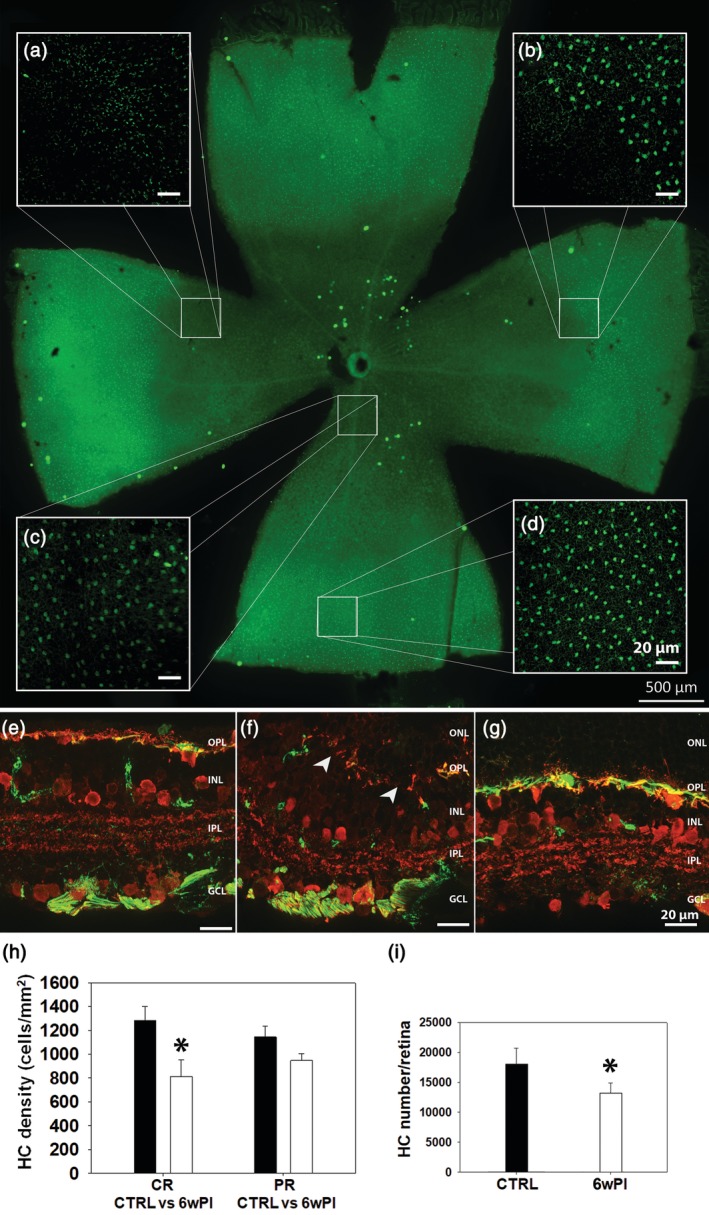
Tvrm4 retinal whole mount, 6 weeks postinduction (PI). Calbindin D antibody staining. Abnormalities in horizontal cells (HCs) are visible even in the low magnification retinal montage, where a central area with reduced immunofluorescence (a–c), corresponding to the light‐induced zone, is evident. This area contains both regions almost devoid of HCs (a) and preserved HCs with a dimmer fluorescence (c). Note the sharp transition zone of HC degeneration (b) and an intact peripheral retina with HCs of normal morphologic features (d). Vertical sections of Translational Vision Research Model 4 (Tvrm4) retina 6 weeks PI stained for anti‐Calbindin D (red) and anti‐NF200 (green) antibodies showing HCs in the central (e), transition zone (f), and peripheral retina (g), respectively. In (e), HC dendrites and cell bodies (red) might be preserved despite degenerating axonal arborizations (green). (f) At the transition zone of PR degeneration, HC dendritic, cellular, and axonal compartments are distorted and irregularly retracted, creating empty areas (arrowheads); (g) in the retinal periphery, all three cellular compartments remain intact. (h) HC body density and total HC body counts (i) (3 mice/exp. group; 16 images [150 × 150 μm2]/retina: 4 images/retinal eccentricity) 6 weeks (6w PI), white columns; compared to WT controls (CTRL), black columns; (CR, central retina; PR, peripheral retina; * = p ≤ .01; data are shown as average and SE; one‐way analysis of variance [ANOVA]) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.4. Cone BCs
CBCs were studied on vertical sections stained for anti‐secretagogin (SCGN), known to label as many as six different types of CBCs (Types 2, 3, 4, 5, 6, and possibly 8) stratifying at various depths in the outer and innermost tiers of the IPL. Similar to other cell types, quantification was done at 3 and 6 weeks PI. In the central retinal areas, lacking PRs, CBC dendritic trees appeared to have lost continuity and complexity, although retraction was not as severe as observed in RBCs and became very evident only at 9 weeks PI (Figure 4a–c). No statistical difference was revealed in the number of cell bodies in any of the studied experimental groups, indicating a survival rate of 100% of CBCs (Figure 4d). However, we found a gradual, significant decrease in the thickness of the three layers revealed by SCGN antibodies in the IPL and corresponding to the axonal arbors of the labeled CBCs (one‐way ANOVA, p ≥ .001; n = 3 retinas). Reduced SCGN staining in sublamina b of the IPL (i.e., SCGN+ Layer 3) increased over time (Figure 4e), despite preservation of the thickness of the whole plexiform layer.
Figure 4.
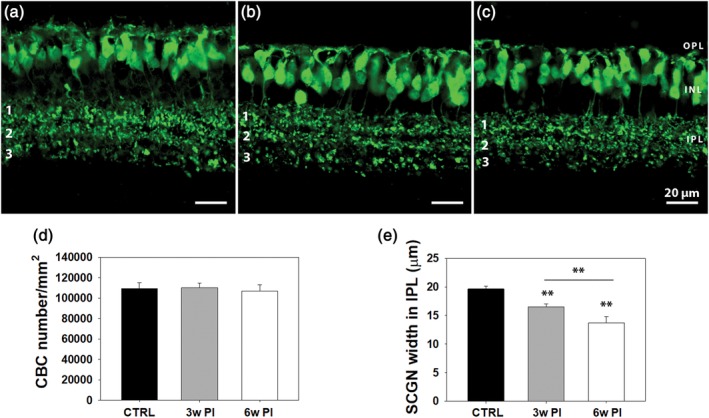
Vertical sections from retinas of WT controls (a) and Tvrm4 mice, 3 (b) and 6 (c) weeks postinduction (PI), with secretagogin (SCGN) staining of cone bipolar cells (CBCs). Three distinct plexa (1, 2, 3) of CBC axonal arbors are visible in the IPL. A gradual, significant decrease in the thickness of SCGN+ axonal arborizations becomes evident over time, mostly due to plexus 3 shrinkage, despite preservation of total IPL width. (d,e) CBC quantitative data. Three vertical sections/animal were analyzed taking 10 micrographs/section, covering five fields, each 150 × 150 μm2. Cell body counting reveals no statistical difference in control and Tvrm4 cases (d). IPL thickness measurements were done on the same micrographs with a MetaMorph longitudinal measuring tool (10 random measurements/micrograph done along the full IPL thickness in the vertical plane (** = p ≤ .001; data are shown as average and SE; one‐way analysis of variance [ANOVA]) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.5. Amacrine cells
Several antibodies were used on vertical retinal sections to label cholinergic (ChAT), GABAergic (GAD67), dopaminergic (TH), AII ACs (DAB1), and Calbindin positive ACs. IHCH confirmed in most cases the preservation of the aforementioned cell types, which comprise large size amacrines (i.e., cholinergic cells) as well as small field, glycinergic amacrines, such as the AIIs. No clear morphological changes were evident in the degenerated retina at 3 and 6 weeks PI (Figure 5). Laminar organization of AC processes also appeared to be normal, as highlighted by the correct positioning of the cholinergic bands at one‐third and two‐third of depth in the IPL and the regular distribution of the three Calbindin positive bands. A noticeable exception was observed in the TH+ DACs, which showed hypertrophic processes in the OPL, exclusively in the central retina and precisely overlapping with the area devoid of PRs. Observations conducted 3 and 6 weeks PI demonstrate that the TH+ hypertrophic processes target and densely entangle some but not all the DNA condensed bodies that accumulate in the outer retina during PR degeneration (Figure 6a–j). Cell counting of TH+ DACs demonstrated no significant change in the total number of these neurons in photo‐induced retinas of Tvrm4 mice 3 and 6 weeks PI (one‐way ANOVA, n = 3 for each group) compared to the published number of WT B6/J mice (Whitney et al., 2009) (Figure 6k). This indicates that hypertrophy of the OPL dopaminergic plexus arises from sprouting of the preexisting dendritic arborizations.
Figure 5.
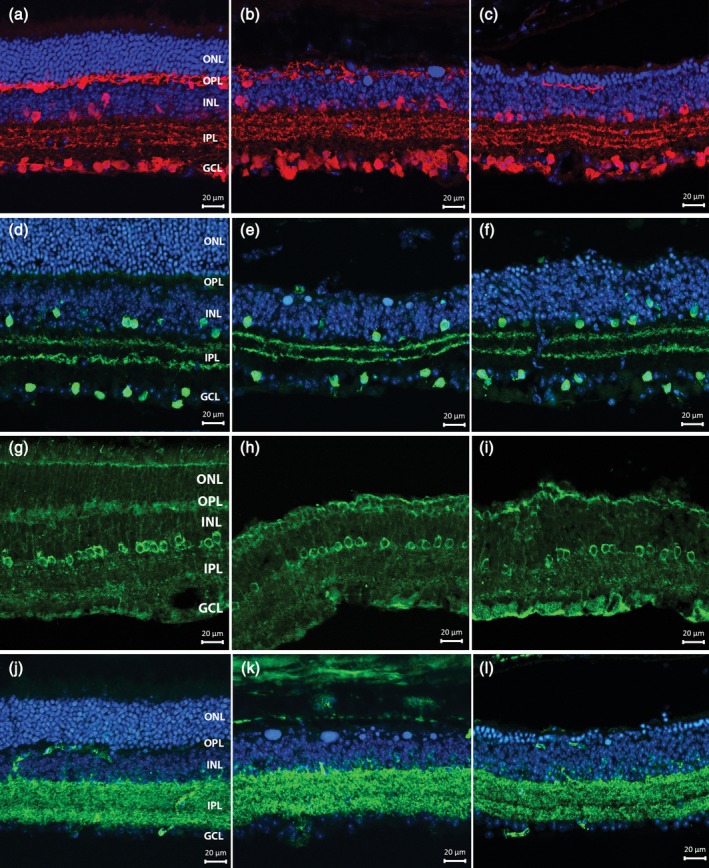
Vertical sections from noninduced control (left column) (a,d,g,j), 3 weeks (central column) (b,e,h,k) and 6 weeks (third column) (c,f,i,l) Tvrm4 retinas. Antibody stainings highlight amacrine cell morphologies and organization: (a–c) Calbindin (red); (d–f) ChAT (green); (g–i) DAB1; and (j–l) GAD67. Blue: Hoechst nuclear counterstaining. The pattern of immunoreactivity in the inner retina is similar to the control at both time points. No major changes are observed in the general morphology and laminar organization of stained bodies and processes [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 6.
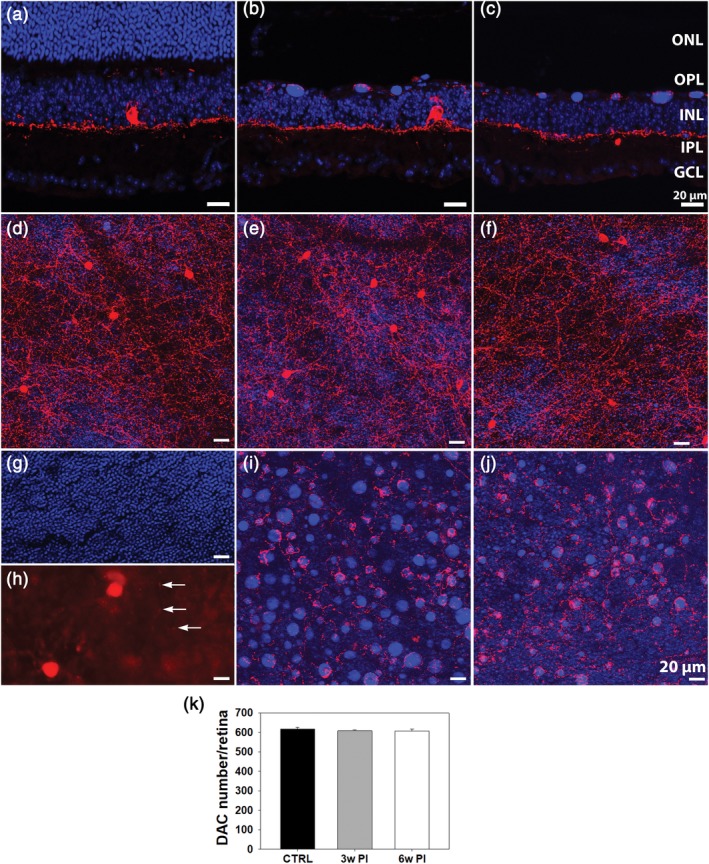
Vertical retinal sections of Tvrm4 mice stained with anti‐TH (red) and Hoechst (blue) in a non‐induced control eye (left column) (a) and 3 weeks (b) or 6 weeks (c) following light exposure. (d–j) Whole mount micrographs of Tvrm4 retinas as above. The focal plane of (d–f) is in the IPL; (g–j) are taken at the ONL/OPL border. (g,h) The inner limit of the ONL and the outer limit of the OPL, respectively, with arrows pointing at TH‐positive processes in the outer retina of a control mouse. In induced retinas (e,f,i,j), the inner plexus (e,f) originating from DACs shows a normal morphology while processes in the OPL are hypertrophic and denser (i,j). Exuberant processes are observed entangling DNA bodies residual from dead PRs. (k) Counts of DAC+ cell bodies (3 retinas/exp. group; counts performed on tiled images of retinal whole mounts) revealed no significant changes 3 and 6 weeks PI. Tvrm4 data are compared to the published number of DACs in WT B6/J (CTRL) mice (Whitney, Raven, Ciobanu, Williams, & Reese, 2009) (data are shown as average and SE; one‐way analysis of variance [ANOVA]) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.6. Ribbon synapses and gap junctions in the IPL
To examine the distribution of connectivity markers in the IPL of Tvrm4 mice, we used RIBEYE/CtBP2 staining for ribbon contacts and examined vertical retinal sections at 3, 6, and 9 weeks PI (n = 3 for each group). Confocal micrographs were acquired and edited with identical settings. Immunoreactivity in the IPL was quantified to estimate the area covered by RIBEYE+ puncta using an image thresholding analysis in an ROI spanning the whole IPL width. A significantly increased area (one‐way ANOVA, p = .010) occupied by RIBEYE+ puncta was revealed 3, but not 6 and 9 weeks after photoinduction with respect to WT controls (Figure 7a–e). A similar method was used to estimate the density of Cx36+ puncta, revealing gap junctions expressing this particular isoform of connexin. Thresholding analysis showed significantly larger areas attributable to Cx36+ signals in the IPL (one‐way ANOVA, p ≥ .001) 3, 6, and 9 weeks after photoinduction with respect to WT controls (Figure 7f–j).
Figure 7.
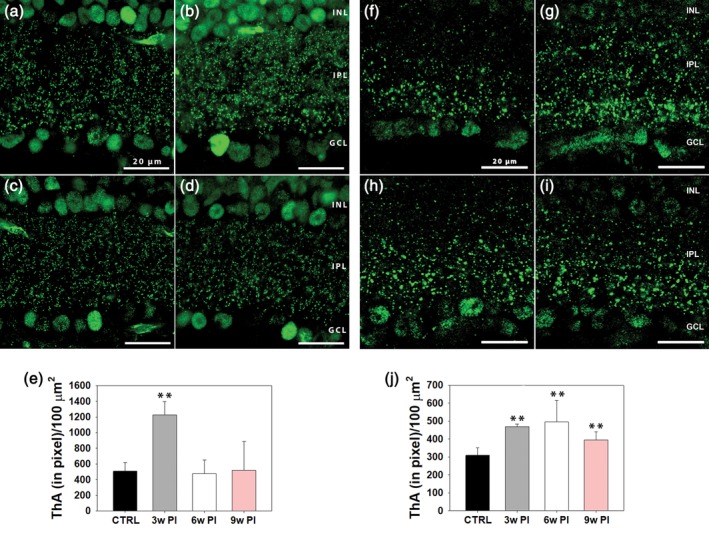
Anti‐CtBP2/RIBEYE staining of ribbon synapses in vertical sections of (a) WT retinas and in Tvrm4 specimens obtained 3 (b), 6 (c), and 9 (d) weeks postinduction (PI), respectively. (e) Quantitative analysis of CtBP2/RIBEYE puncta. A total of three vertical sections/animal and of 10 micrographs/section were acquired. Imaged fields (78 × 78 μm2 acquired with a 100x oil objective) covered both the center (five fields) and the periphery (five fields) of the vertical retinal sections. A significant increase in the density of RIBEYE+ puncta in the IPL was revealed measuring the thresholded area (ThA) (e) 3 weeks PI (** = p ≤ .001; data are shown as average and SE; one‐way analysis of variance [ANOVA]). (f–i) A similar analysis of IPL gap‐junctions labeled with anti‐Cx36 (f–i) shows a significant increase in puncta density 3 (g), 6 (h), and 9 weeks (i) PI compared to (f) WT controls (** = p ≤ .001; data are shown as average and SE; one‐way ANOVA) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.7. Ultrastructural analysis of synaptic contacts in the IPL
To investigate synaptic architecture in the IPL of areas of complete PR degeneration, we performed TEM studies to estimate the density of conventional and ribbon synapses across the whole width of the IPL in retinal samples obtained at 3 and 6 weeks PI (n = 3 for each group) (Figure 8). To limit internal variability, data from each light‐induced retina were compared those obtained from the contralateral (noninduced) retina of the same animals. This was previously shown to exhibit a normal phenotype, identical to that of WT mice (Gargini et al., 2017). Ultrastructural analysis demonstrated general preservation of number and appearance of conventional synaptic contacts in the IPL at both 3 and 6 weeks PI compared to noninduced controls.
Figure 8.
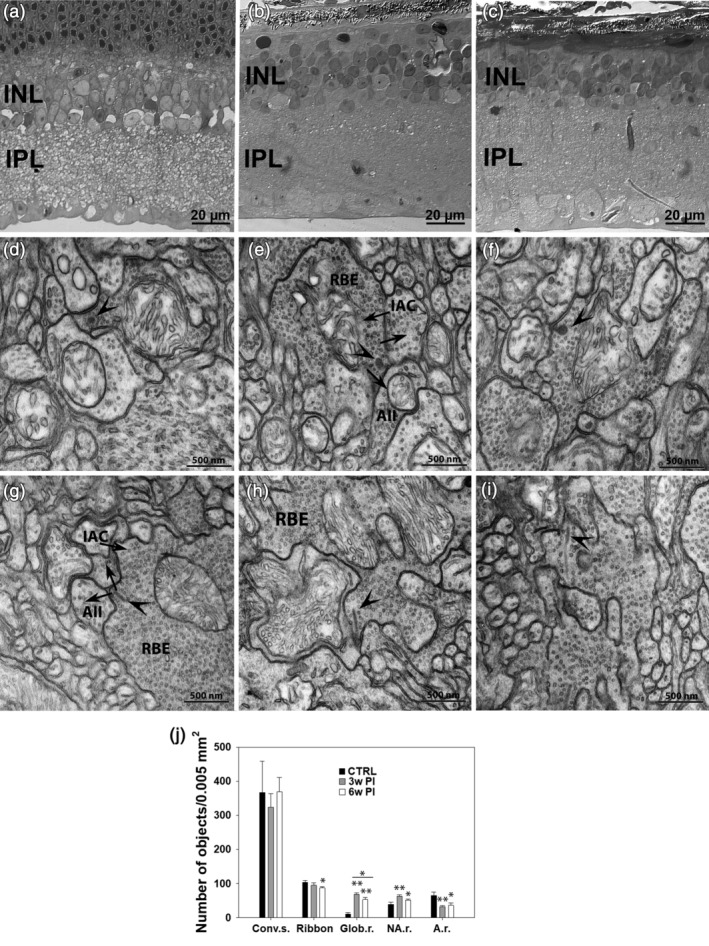
Semithin vertical sections of Translational Vision Research Model 4 (Tvrm4) mouse retina stained with toluidine blue in a noninduced, control eye (a) and 3 (b) and 6 weeks (c) following light exposure. (d–i) TEM micrographs illustrating synaptic ribbons (arrowheads) and conventional chemical synapses (arrows) involving rod bipolar axonal endings (RBE) in a noninduced control retina (d,g) and in Tvrm4 preparations examined 3 (e,h) and 6 (f,i) weeks postinduction (PI). Arrows indicate the direction of neurotransmission. Atypical globular ribbons (d–f, arrowheads) and classical synaptic arrangement involving RBE with elongated ribbons and dendrites of AII and A17‐like amacrine cells (IAC) can be observed in all preparations (e–i). A summary of conventional and ribbon synapse density in the IPL is shown in (j). TEM micrographs (400/sample, 12.96 μm2/micrograph)/sample, corresponding to a sampled area of 0.0052 mm2/retina) were imaged). The number of conventional synapses (Conv. s.) is not affected by photoinduction. The total number of ribbons shows a significant decrement 6 weeks PI (* = p ≤ .01; one‐way analysis of variance [ANOVA]). Also, significantly more ribbons have a globular shape (Glob. r.) (** = p ≤ .001; one‐way ANOVA), 3 and 6 weeks PI, compared to non‐induced controls (CTRL). Globular ribbons represent 72% of all ribbons 3 weeks PI but decrease to 61% 6 weeks PI. The number of nonanchored ribbons (NA. r.) is significantly higher 3 and 6 weeks PI, while the number of anchored ribbons (A.r.) shows the opposite tendency. All data are shown as average and SE
RBC axonal endings could be unequivocally identified by TEM based on their large size, position in sublamina 5 of the IPL and typical cytological features (pale cytoplasm, presence of large number of vesicles, mitochondria, and synaptic ribbons). TEM studies confirmed that RBC axonal endings were visibly smaller with respect to noninduced CTRL specimens.
For ribbon synapses, a decrement in the overall number was observed beginning at 3 weeks PI, although the decrement only became statistically significant at 6 weeks PI. Specifically, we found 9% less ribbons 3 weeks PI and 16% less ribbons 6 weeks PI compared to noninduced CTRLs (Figure 8j) (one‐way ANOVA, p = .034). More noticeable than the change in number, we found in photoinduced specimens the majority of ribbons displayed a globular/spherical shape, very different from the elongated appearance of normal ribbons and more reminiscent of the immature morphology observed during retinal development (Regus‐Leidig, Dieck, Specht, Meyer, & Brandstätter, 2009) (see Figure 8). Quantitative analysis demonstrated that ~72% of all ribbons were globular by 3 weeks PI and 61% of them were similarly rounded at 6 weeks PI, compared to 10% in noninduced controls. In addition, the number of ribbons not anchored to the presynaptic membrane was found to be significantly higher than normal at 3 and 6 weeks PI, while the number of anchored ribbons obviously showed the opposite tendency. Despite these ultrastructural changes, typical dyad synapses established by RBC axonal endings onto two dendritic processes known to represent the AII/A17 AC combination were recognizable and well preserved in all experimental groups (Figure 8).
3.8. Ganglion cells
Previous studies on GCs in retinal degeneration rodent models predict minimal morphological alterations in RGCs at early stages of the retinal disease progression (Damiani, Novelli, Mazzoni, & Strettoi, 2012; Mazzoni, Novelli, & Strettoi, 2008). Indeed, we did not observe any change in the fundamental anatomical features of GCs. Specific staining of cell bodies with RBPMS antibodies showed normal density of somas and appropriate confinement in the homonymous layer. Dendrites and axonal bundles of GCs, labeled by SMI32 (neurofilament) antibodies, also revealing the somas of large‐size GCs, showed absolutely normal distribution in retinal preparations obtained 3 and 6 weeks PI (Figure 9), with no obvious differences with control specimens.
Figure 9.
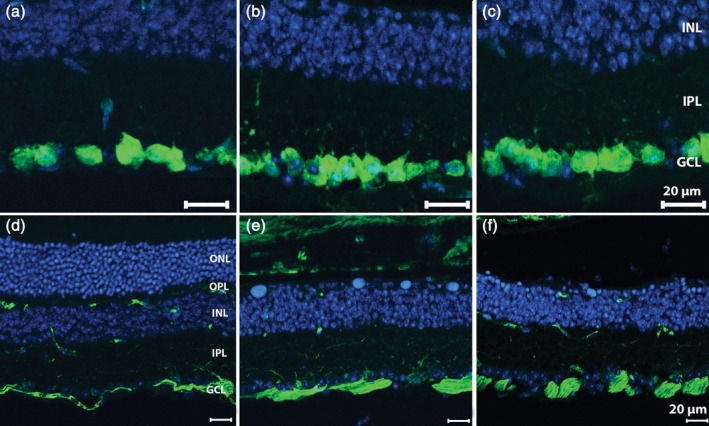
Retinal vertical sections of Tvrm4 mice stained green for anti‐RBPMS (a–c) and anti‐SMI32 (d–f) along with Hoechst nuclear marker (blue). (a,d) Noninduced retinal periphery, with intact ONL. Tvrm4 samples of central retina shown 3 (b,e) and 6 (c,f) weeks postinduction (PI). Cell bodies of ganglion cells, brightly labeled by anti‐RBPMS antibodies, form a continuous layer in both illuminated and control areas (a–c). Dendrites, large‐size cell bodies (arrows) and fascicles of optic nerve fibers revealed by anti‐SMI32 staining show normal morphological features in all experimental groups [Color figure can be viewed at http://wileyonlinelibrary.com]
Altogether, the inner retina of Tvrm4 mice, in which PR degeneration was triggered during adulthood, demonstrated significant cell survival, morphological integrity and preservation of synapses and gap junctions after PR degeneration. Nearly 100% survival rate was found among BCs, in spite of some decrement in HC number. The number of conventional synapses in the IPL was unaffected and the area covered by Cx36‐positive gap junctions showed an increment, albeit we detected a tendency in the total number of ribbon contacts to decrease over time. Quantitative data are summarized in Table 2.
Table 2.
Summary of quantitative data collected at 3 and 6 weeks PI and shown as averages ± SE
| HT3w | HT6w | WT | ||
| RBCs (avg num/retina) | 223,383 ± 12,290 | 221,659 ± 12,000 | 246,416 ± 31,593 | |
| SCGN+ CBCs (avg num/retina) | 110,322 ± 4.538 | 106,822 ± 6,358 | 109,478 ± 5,607 | |
| HCs (avg num/retina) | ND | 13,078 ± 1,835 | 18,104 ± 2,624 | |
| DACs (avg num/retina) | 608 ± 5 | 606 ± 11 | 617 ± 8.3 (Whitney et al., 2009) | |
| HT3w | HT6w | HT9w | WT/CTRL | |
| RIBEYE/CtBP2 (avg ThA [pixels]/100 μm2) | 1,227 ± 169 | 478 ± 175 | 519 ± 366 | 507 ± 109 |
| Connexin36 (avg ThA [pixels]/100 μm2) | 469 ± 15 | 495 ± 210 | 394 ± 45 | 309 ± 43 |
| Conventional synapses (avg num/0.005 mm2) | 324 ± 40 | 369 ± 42 | ND | 367 ± 92 |
| Ribbon synapses (avg num/0.005 mm2) | 95 ± 7 | 87 ± 4 | ND | 104 ± 5 |
| Globular ribbons (avg num/0.005 mm2) | 68 ± 5 | 53 ± 6 | ND | 10 ± 5 |
Abbreviations: avg, average; DAC, dopaminergic amacrine cell; ND, nondetermined; PI, photoinduction; RBC, rod bipolar cell; SCGN, secretagogin; ThA, thresholded area.
4. DISCUSSION
High survival rates of rod and cone BCs demonstrate that the early effects of PR degeneration in the fully developed, adult retina of Tvrm4 mice are no more severe than in the developing retina of other retinal degeneration models (i.e., rd1 and rd10 mice), which are considered representative of human RP, but where there is overlap of PR degeneration and late phases of retinal development. Actually, at 6 weeks PI, the number of rod and cone BCs in Tvrm4 mutants remained close to 100%, unlike that found in rd1 and rd10 mice, where the RBC number dropped significantly by 3 months of age (Gargini, Terzibasi, Mazzoni, & Strettoi, 2007; Strettoi, Porciatti, Falsini, Pignatelli, & Rossi, 2002), corresponding to the stage of retinal degeneration exhibited by Tvrm4 mice at 6 weeks PI.
These results suggest that the mature retina has a higher potential to maintain viability of second‐order neurons than the developing retina, even in retinal degeneration when PR loss is nearly complete.
Dendritic retraction, mostly of RBCs, has been previously shown in rd1 and rd10 mice (Pang et al., 2011; Strettoi et al., 2002; Strettoi & Pignatelli, 2000; Strettoi, Pignatelli, Rossi, Porciatti, & Falsini, 2003) as well as in the P23H transgenic rat (Cuenca et al., 2004); it was speculated that this phenomenon, possibly consequent to synaptic deafferentation, could be worsened by the fact that the dendritic trees of BCs are still incompletely developed when PR degeneration starts, particularly in the Pde mutants (rd1 and rd10). However, RBCs of Tvrm4 mice (light induced in adulthood) fully retract dendrites by 3 weeks PI in the central, PR‐lacking retinal areas similarly to that observed in rd1, rd10 and P23H models of RP. SCGN positive CBCs also show signs of dendritic retraction, with a delay, confirming that CBCs lose dendrites (and glutamate receptors) more slowly than RBCs and following the path of rod‐cone cell loss (Puthussery, Gayet‐Primo, Pandey, Duvoisin, & Taylor, 2009). A previous study in Tvrm4 mice (Gargini et al., 2017) showed that synaptotagmin‐II/ZNP1 positive CBC dendrites attenuate after PRs have died out completely, indicating that different types of CBCs (also belonging to functionally different categories) remodel similarly to RBCs but with some delay, globally reflecting the progression of typical rod‐cone degeneration. These findings demonstrate that dendritic remodeling following PR death neither depends on the age of phenotype onset nor on the underlying genetic mutation or pattern of inheritance: rather, it is a secondary manifestation caused by PR death and represents a true hallmark of RP likely caused by deafferentation.
As observed in the BC axonal terminals, an arrest in growth of RBC axonal arborizations in rd1 mice was previously reported (Strettoi et al., 2002), while the atrophy of RBC axonal endings in rd10 mutants was considered a secondary effect (Barhoum et al., 2008). Recent data also confirm that RBC axon terminals remodel, gradually losing complexity and undergoing progressive reduction as retinal degeneration proceeds (Phillips, Otteson, & Sherry, 2010). Indeed, Tvrm4 mice show these features as early as 3 weeks PI in the central, degenerated retina, where the complexity of RBC axonal endings decreases significantly. SCGN positive axonal endings in the IPL shrink progressively as well, but mainly in the ON sublamina of the IPL, as indicated by a thinning in the width of SCGN positive arborizations located in the innermost plexus (n.3) of the IPL. Altogether, these results confirm that rod‐to‐cone PR degeneration not only affects RBCs but gradually extends to other components of the ON pathway, which can be considered as a highly sensitive indicator of inner retinal remodeling.
Quantitative analysis of HCs at 6 weeks PI revealed a 28% reduction in the number of cells throughout the retina of Tvrm4 mice. This finding is in agreement with reports describing rd10 mutants at ~3 months of age (Gargini et al., 2007). Surprisingly, however, and differently from what was observed in rd10 mice, where HC death overlaps spatially with PR loss, HCs of Tvrm4 mice die in small islands of the mid‐central and mid‐peripheral retina, not necessarily coinciding with the areas devoid of PRs. Remodeling and degeneration of HCs are most severe not in the center of the light exposure zone, but in the “transition” zones between high PR death and survival, respectively, where HC‐free islands are separated by sharp edges from spots of surviving cells, which may persist in the central, most damaged retina, although with decreased intensity of fluorescence staining and hypertrophic processes.
There is no obvious explanation for the higher severity of HC remodeling in transition zones of PR degeneration. An interesting hypothesis is that local signals (i.e. inflammatory molecules such as cytokines and chemokines, released from activating microglia), thought to contribute to cone death and to retinal abnormalities secondary to PR loss (Guadagni et al., 2019) are higher at the boundaries with regions where PR death is not yet complete, as though a wave of adverse signals preceded the actual phase of cell loss. Large‐size neurons, similar to HCs, likely extend across these boundaries and suffer from the adverse effects of diffusible factors. Further investigation is necessary to address the nature of these noncell autonomous events. Yet, one has to remember that the peripheral, non‐induced retina of Tvrm4 mice can be considered entirely normal, unlike the peripheral retina of other mutants, where the phenotype is simply delayed with respect to the central retina. Local signaling is likely to bear intrinsic differences in Tvrm4 and other paradigms of RP.
Morphological analysis of the IPL stained for anti‐CtBP2/RIBEYE revealed excellent preservation of synaptic ribbons for up to 9 weeks PI in adult Tvrm4 mice with respect to WT controls, with a significantly larger area occupied by RIBEYE positive puncta 3 weeks PI. This indicates the possibility of a peak in synaptic remodeling of the IPL at this time point, which might be driven by the peripheral, PR preserving retinal areas in the attempt of saving the flow of remnant visual signals despite extensive PR death in the central retina. Again, it is possible that long‐range, diffusible signals (i.e., acting through intervening glia and blood vessels) may support these attempts of “reconstructive” remodeling, as BCs directly connected to dying PRs are not likely to extend their range of action beyond the light‐induced area. Previous data reported a recovery phase in the Tvrm4 retina at 4 weeks after photoinduction based on ERG recordings and histological results (Gargini et al., 2017). This was speculated to result from only partially damaged PRs at the edge of light‐irradiated area, which could regenerate outer segments in 3–4 weeks provided a connecting cilium was still present. Increased density of synaptic contacts in the IPL could be triggered by this phase of PR recovery in the attempt to preserve the flow of information through the retina or anyway as an effect of a plastic stage of remodeling. The occurrence of migrating, remnant PRs converging in retinal foci has been reported (Busch, Gorgels, & Van Norren, 1999; Lin, Masland, & Strettoi, 2009; Paulus et al., 2008) and could explain the need to accommodate extra synaptic contacts in the IPL.
The IPL of Tvrm4 retinas at 3 and 6 weeks PI was studied with the aid of TEM, confirming that rod bipolar endings have smaller sizes, and yet they are engaged in the typical RBC–AII amacrine–A17 amacrine synaptic arrangements, which could be easily recognized. Quantitative analysis of synaptic contacts revealed no significant changes in the number of conventional (inhibitory) synapses in the overall width of the IPL; however, the total number of ribbon synapses displayed a decreasing tendency over time, with significantly fewer synaptic ribbons at 6 weeks PI compared to non‐induced control retinas. Moreover, approximately 70% of all ribbons exhibited an abnormal globular or amorphous shape at 3 weeks PI while only 10% globular ribbons were found in the controls. Yet, the percentage of anomalous ribbons decreased to 60% by 6 weeks PI. Significantly larger areas covered by RIBEYE positive puncta in the IPL at 3 weeks PI observed by confocal microscopy can be explained by a larger area occupied by the same number of ribbons, implicating an increase of their average size. Most likely, globular ribbons observed at 3 weeks PI result in larger fluorescent profiles in the IPL, causing an increase in the area over the threshold. The decrease in the number of (globular) ribbons at 6 weeks PI agrees with our immunofluorescence observations.
Globular ribbons can be observed during early retinal development (Regus‐Leidig et al., 2009) as precursors preceding full synaptic maturation and anchoring of these organelles at the presynaptic membrane. The globular form found here suggests altered turnover of the ribbons, as previously reported during artificial changes introduced in the dark–light cycle (Adly, Spiwoks–Becker, & Vollrath, 1999; Hull, Studholme, Yazulla, & von Gersdorff, 2006). Spherical (often free‐floating) ribbons reminiscent of the globular organelles we observed have been described in the PR terminals of double‐knockout mice for complexins (Cplx) 3 and 4, two regulatory proteins of vesicle exocytosis (Reim et al., 2009). Cplx3/4 double knockouts display retinal morphological and functional abnormalities altogether indicative of visual defects. Indeed, structural changes in ribbons have been demonstrated to affect coordinated multivesicular release of glutamate at the synapse (Mehta, Snellman, Chen, Li, & Zenisek, 2013). It is possible that RBCs of Tvrm4 retina, deprived from rod input, undergo a downregulation of the dendritic and axonal machinery responsible for sensing and releasing glutamate, respectively. Additional functional studies are required to test the excitability and capability of synaptic transmission of BCs, in view of possible optogenetic therapy targeting these cells. However, our observations suggest overall stability of IPL ribbons, insofar 9 weeks PI the area covered by RIBEYE+ puncta in the IPL did not decrease further based on our IHCH data.
Analysis of CX36 distribution revealed a significantly larger surface occupied by immune positive puncta in the IPL of induced mice at all the time point studied, compared to WT controls, suggestive of an increase of gap junction density following PR degeneration. In degenerating retinal areas, Cx36 positive puncta are visibly distributed more extensively into the OFF layers of the IPL compared to the control samples (see Figure 7). Gap junctions were demonstrated to contribute to the spontaneous electrophysiological oscillations recorded from animal models of RP and human RP patients and pharmacological blockade of gap junctions was proven to decrease aberrant oscillatory activity and restore light responses driven by surviving PRs in the degenerating rd10 retina (Toychiev, Ivanova, Yee, & Sagdullaev, 2013). The increased expression level of Cx36 positive gap junctions observed in induced Tvrm4 mice could represent a maladaptive change, causing oscillatory activity of inner neurons. It would be functionally relevant to prove the existence of oscillatory activity in Tvrm4 mice, by multielectrode array (MEA) recordings from GCs through the time course of degeneration, and to verify the contribution of IPL gap junctions to inner retinal oscillations.
Because cell‐to‐cell coupling and the conductance of gap junctions are highly dependent on and regulated by dopamine levels in the vertebrate retina (Witkovsky, 2004), we studied the DAC network in Tvrm4 mice to correlate the morphology of DACs with changes in Cx36 immunofluorescence. TH labeled DACs showed normal appearance in the inner retina, with normal general architecture, process networks and number of cell bodies 3 and 6 weeks PI. However, processes reaching out to the OPL were found to be markedly hypertrophic in the central retina. Outer DAC dendrites were observed entangling dead PRs, represented by DNA‐filled, membrane‐bound bodies, and constituting typical side products of photoinduction. The area of hypertrophic processes precisely and exclusively colocalized with the area of PR loss. It is known that dopamine is released upon retinal illumination and regulates the gain of retinal neurons to adapt to bright light conditions (Witkovsky, 2004). It seems logical that dopamine release from DA amacrines reached a peak during the acute phase of inducing light exposure. However, there is no obvious explanation of the hypertrophy of the outer dopaminergic plexus observed here and of the obvious contiguity of DA dendrites and DNA‐condensed profiles in the outer retina; moreover, DA dendrite hypertrophy persists well beyond the acute phase of photoinduction.
Calbindin D, ChAT, GAD67, and DAB1 antibodies were used to study GABAergic, cholinergic, and glycinergic (AII) ACs in the retina of Tvrm4 mice 3 and 6 weeks PI. These markers revealed good preservation of all AC types based on anatomical observations. In particular, laminar organization of AC dendrites and cell body layering appeared normal. These results match previous reports of long‐term AC survival rate in rd10 mice, where all AC types were found in normal physiological and morphological organization, except for AII ACs, which were reported to be reduced in number and disorganized in distribution in rd10 animals of 40 days of age (Barhoum et al., 2008). Tvrm4 mice observed 6 weeks PI, a stage corresponding to the phenotype of P40 and older rd10 mice, showed considerable preservation of AII ACs in the central retina, despite complete loss of PRs. Since AII ACs are key members of the rod and cone signaling pathways and are considered a suitable target for optogenetic therapy, their long‐term survival holds promising opportunities for vision restoration in RP. Once again, good preservation of AII amacrine morphology indicates a high capacity of the retina to maintain a normal structural organization even when the disease phenotype manifests in adulthood and plasticity typically associated with a young age has been lost.
Previous studies on GCs in retinal degeneration rodent models predict no morphological alterations in these cells at relatively early stages of the retinal degeneration (Mazzoni et al., 2008; Damiani et al., 2012; (Greferath et al., 2015). We did not observe alterations of the fundamental anatomical features of GCs in Tvrm4, light induced mutants. RBPMS and SMI32 staining showed normal cell density, organization of cell bodies, dendrites, and axonal bundles of these cells in vertical sections both at 3 and 6 weeks PI. Again, we point out to the need of a complete electrophysiological characterization of the Tvrm4 retina, particularly at the GC level, in view of previous reports about the presence of a spontaneous, aberrant, oscillatory activity in the local field potentials of GCs in both rd1 and rd10 retinas recorded with MEA (Goo et al., 2011; Stasheff, Shankar, & Andrews, 2011).
Based on remodeling data obtained from adult Tvrm4 mice in the present study—confirming and expanding previous work in other animal models of RP—we conclude that the process of inherited, rod‐to‐cone degeneration triggers morphological remodeling of inner retinal neurons first acting on components of the rod system and of the ON signaling pathway, as proven by dendritic retraction from RBCs in the OPL and (later) of ON BC axonal ending shrinkage in the ON sublamina of the IPL. Contacts in the IPL are highly preserved, albeit with some ultrastructural abnormalities of synaptic ribbons. Preliminary observations from our TEM analysis in the IPL of Tvrm4 mice 3 and 6 weeks PI show ultrastructural signs typical of autophagy and/or paraptosis (Wei et al., 2015), including the presence of swollen mitochondria with broken or diminished inner membrane system with clear signs of invaginations, empty vacuoles as well as concentric multimembrane compartments. Experimental evidence of autophagy in the IPL would suggest a compensatory response of inner retinal neurons following PR death and loss of input from the outer retina, consequently indicating the possibility of reversing inner retinal remodeling. Quantification of, for example, degenerating mitochondria by TEM analysis and biochemical assessment of LC3‐II (autophagy marker) and related proteins could provide indications about autophagy in the IPL. At cellular level, engineering Tvrm4 mice expressing fluorescent marker proteins like GFP under the promoter of ON BC specific genes would allow single cell transcriptome analysis looking for autophagy‐related expression patterns.
Noticeably, the phenotype of Tvrm4 mutants shares a number of features with non‐neovascular “dry” age‐related macular degeneration (ARMD), including light sensitivity (de Jong, 2006; Marc et al., 2008), focal occurrence of abnormalities in the central retina (de Jong, 2006), rod‐to‐cone PR death (Curcio, Medeiros, & Millican, 1996), and the subsequent dendritic remodeling of second‐order neurons (Jones & Marc, 2005; Sullivan, WoldeMussie, & Pow, 2007). However, clear documentation of inner retinal remodeling in human ARMD is only available for late stages of the disease (Jones et al., 2012), when vision restoration based on remnant retinal tissue is virtually impossible. Therefore, results obtained from photo‐induced Tvrm4 mice may bear important possibilities to translate findings to early and mid‐stages of ARMD and provide evidence of inner retinal ability to resist to PR degeneration, at least in the relatively short run. This encourages timely therapeutic approaches for this devastating disease.
Good preservation of the inner retina in Tvrm4 mice, an excellent model of RHO AD RP Type B1, offers the opportunity to test the efficacy of vision restoration on adult animals by optogenetic tools targeting ON BCs, that is, driving the expression of ChR2 (Lagali et al., 2008; Macé et al., 2015), MAG460 (Gaub et al., 2014), ReaChR (Sengupta et al., 2016), ChrimsonR (Klapoetke et al., 2014), or naturally occurring retinal cone opsins (Berry et al., 2019). This is particularly important in view of the present difficulty to treat dominant (gain‐of function) mutations by gene therapy directed to PRs: maintenance of the inner retina for sometimes after total PR loss offers therapeutic opportunities that exploit second‐order and higher order neurons to restore vision.
ACKNOWLEDGMENTS
The senior author thanks Dr Elena Novelli (IN‐CNR, Pisa) for excellent technical support during the phase of elaboration of experimental protocols necessary for authorized animals experimentation. The authors are grateful to Prof John Flannery (University of California, Berkeley, CA) for carefully editing the manuscript. This work was funded by European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska‐Curie grant agreement No. 674901 (“switchBoard” project).
Stefanov A, Novelli E, Strettoi E. Inner retinal preservation in the photoinducible I307N rhodopsin mutant mouse, a model of autosomal dominant retinitis pigmentosa. J Comp Neurol. 2020;528:1502–1522. 10.1002/cne.24838
Funding information European Union's Horizon 2020 Research and Innovation Programme, Grant/Award Number: 67490
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Adly, M. A. , Spiwoks–Becker, I. , & Vollrath, L. (1999). Ultrastructural changes of photoreceptor synaptic ribbons in relation to time of day and illumination. Investigative Ophthalmology & Visual Science, 40(10), 2165–2172. [PubMed] [Google Scholar]
- Airaksinen, M. S. , Eilers, J. , Garaschuk, O. , Thoenen, H. , Konnerth, A. , & Meyer, M. (1997). Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the Calbindin D28k gene. Proceedings of the National Academy of Sciences of the United States of America, 94(4), 1488–1493. 10.1073/pnas.94.4.1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoum, R. , Martínez‐Navarrete, G. , Corrochano, S. , Germain, F. , Fernandez‐Sanchez, L. , de la Rosa, E. J. , … Cuenca, N. (2008). Functional and structural modifications during retinal degeneration in the rd10 mouse. Neuroscience, 155(3), 698–713. 10.1016/j.neuroscience.2008.06.042 [DOI] [PubMed] [Google Scholar]
- Barnstable, C. J. (1980). Monoclonal antibodies which recognize different cell types in the rat retina. Nature, 286, 231 Retrieved from–235. 10.1038/286231a0 [DOI] [PubMed] [Google Scholar]
- Barone, I. , Novelli, E. , Piano, I. , Gargini, C. , & Strettoi, E. (2012). Environmental enrichment extends photoreceptor survival and visual function in a mouse model of retinitis pigmentosa. PLoS One, 7(11), 1–13. 10.1371/journal.pone.0050726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, M. H. , Holt, A. , Salari, A. , Veit, J. , Visel, M. , Levitz, J. , … Isacoff, E. Y. (2019). Restoration of high‐sensitivity and adapting vision with a cone opsin. Nature Communications, 10(1), 1221 10.1038/s41467-019-09124-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzynski, E. , Gross, A. K. , McAlear, S. D. , Peachey, N. S. , Shukla, M. , He, F. , … Nishina, P. M. (2010). Mutations of the opsin gene (Y102H and I307N) lead to light‐induced degeneration of photoreceptors and constitutive activation of phototransduction in mice. Journal of Biological Chemistry, 285(19), 14521–14533. 10.1074/jbc.M110.112409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch, E.‐M. , Gorgels, T. G. M. F. , & Van Norren, D. (1999). Filling‐in after focal loss of photoreceptors in rat retina. Experimental Eye Research, 68(4), 485–492. 10.1006/EXER.1998.0628 [DOI] [PubMed] [Google Scholar]
- Cideciyan, A. V. , Hood, D. C. , Huang, Y. , Banin, E. , Li, Z. Y. , Stone, E. M. , … Jacobson, S. G. (1998). Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proceedings of the National Academy of Sciences of the United States of America, 95(12), 7103–7108. 10.1073/pnas.95.12.7103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli, D. F. , Parenti, R. , Spinella, F. , Salinaro, A. T. , Belluardo, N. , Cardile, V. , & Cicirata, F. (1998). Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. European Journal of Neuroscience, 10(3), 1202–1208. 10.1046/j.1460-9568.1998.00163.x [DOI] [PubMed] [Google Scholar]
- Cuenca, N. , Pinilla, I. , Sauvé, Y. , Lu, B. , Wang, S. , & Lund, R. D. (2004). Regressive and reactive changes in the connectivity patterns of rod and cone pathways of P23H transgenic rat retina. Neuroscience, 127(2), 301–317. 10.1016/j.neuroscience.2004.04.042 [DOI] [PubMed] [Google Scholar]
- Curcio, C. A. , Medeiros, N. E. , & Millican, C. L. (1996). Photoreceptor loss in age‐related macular degeneration. Investigative Ophthalmology & Visual Science, 37(7), 1236–1249 Retrieved from https://doi.org/ [PubMed] [Google Scholar]
- Damiani, D. , Novelli, E. , Mazzoni, F. , & Strettoi, E. (2012). Undersized dendritic arborizations in retinal ganglion cells of the rd1 mutant mouse: A paradigm of early onset photoreceptor degeneration. Journal of Comparative Neurology, 520(7), 1406–1423. 10.1002/cne.22802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong, P. T. V. M. (2006). Mechanisms of disease: Age‐related macular degeneration. The New England Journal of Medicine, 355(14), 1474–1485. 10.1007/978-3-642-22107-1 [DOI] [PubMed] [Google Scholar]
- Demb, J. B. , & Pugh, E. N. (2002). Connexin36 forms synapses essential for night vision. Neuron, 36(4), 551–553. 10.1016/S0896-6273(02)01062-0 [DOI] [PubMed] [Google Scholar]
- Dieck, S. t. , Altrock, W. D. , Kessels, M. M. , Qualmann, B. , Regus, H. , Brauner, D. , … Brandstätter, J. H. (2005). Molecular dissection of the photoreceptor ribbon synapse. Journal of Cell Biology, 168(5), 825–836. 10.1083/jcb.200408157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenfeld, A. J. , Bunr‐milam, A. H. , & Sarrhy, P. V. (1984). Muller cell expression of gliol fibrillory acidic protein offer genetic and experimental photoreceptor degeneration in the rat retina. Investigative Ophthalmology & Visual Science, 25(11), 1321–1328. [PubMed] [Google Scholar]
- Eng, L. F. (1985). Glial fibrillary acidic protein (GFAP): The major protein of glial intermediate filaments in differentiated astrocytes. Journal of Neuroimmunology, 8(C), 203–214. 10.1016/S0165-5728(85)80063-1 [DOI] [PubMed] [Google Scholar]
- Euler, T. , & Schubert, T. (2015). Multiple independent oscillatory networks in the degenerating retina. Frontiers in Cellular Neuroscience, 9, 444 10.3389/fncel.2015.00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete, D. M. , & Barnstable, C. J. (1983). The subcellular localization of rat photoreceptor‐specific antigens. Journal of Neurocytology, 12(5), 785–803. 10.1007/BF01258151 [DOI] [PubMed] [Google Scholar]
- Gal, A. , Apfelstedt‐Sylla, E. , Janecke, A. R. , & Zrenner, E. (1997). Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Progress in Retinal and Eye Research, 16(1), 51–79. 10.1016/S1350-9462(96)00021-3 [DOI] [Google Scholar]
- Gargini, C. , Novelli, E. , Piano, I. , Biagioni, M. , & Strettoi, E. (2017). Pattern of retinal morphological and functional decay in a light‐inducible, rhodopsin mutant mouse. Scientific Reports, 7(1), 1–14. 10.1038/s41598-017-06045-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargini, C. , Terzibasi, E. , Mazzoni, F. , & Strettoi, E. (2007). Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. Journal of Comparative Neurology, 500(2), 222–238. 10.1002/cne.21144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub, B. M. , Berry, M. H. , Holt, A. E. , Reiner, A. , Kienzler, M. A. , Dolgova, N. , … Isacoff, E. Y. (2014). Restoration of visual function by expression of a light‐gated mammalian ion channel in retinal ganglion cells or ON‐bipolar cells. Proceedings of the National Academy of Sciences of the United States of America, 111(51), E5574–E5583 Retrieved from http://www.pnas.org/content/111/51/E5574.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghinia, M. G. , Novelli, E. , Sajgo, S. , Badea, T. C. , & Strettoi, E. (2019). Brn3a and Brn3b knockout mice display unvaried retinal fine structure despite major morphological and numerical alterations of ganglion cells. The Journal of Comparative Neurology, 527(1), 187–211. 10.1002/cne.24072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo, Y. S. , Ye, J. H. , Lee, S. , Nam, Y. , Ryu, S. B. , & Kim, K. H. (2011). Retinal ganglion cell responses to voltage and current stimulation in wild‐type and rd1 mouse retinas. Journal of Neural Engineering, 8(3), 035003 10.1088/1741-2560/8/3/035003 [DOI] [PubMed] [Google Scholar]
- Greferath, U. , Anderson, E. E. , Jobling, A. I. , Vessey, K. A. , Martinez, G. , de Iongh, R. U. , … Fletcher, E. L. (2015). Inner retinal change in a novel rd1‐FTL mouse model of retinal degeneration. Frontiers in Cellular Neuroscience, 9, 293 10.3389/fncel.2015.00293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greferath, U. , Grünert, U. , & Wässle, H. (1990). Rod bipolar cells in the mammalian retina show protein kinase C‐like immunoreactivity. Journal of Comparative Neurology, 301(3), 433–442. 10.1002/cne.903010308 [DOI] [PubMed] [Google Scholar]
- Guadagni, V. , Biagioni, M. , Novelli, E. , Aretini, P. , Mazzanti, C. M. , & Strettoi, E. (2019). Rescuing cones and daylight vision in retinitis pigmentosa mice. The FASEB Journal, 33(9), 10177–10192. 10.1096/fj.201900414R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich, V. V. , & Benovic, J. L. (1995). Visual Arrestin binding to rhodopsin: Diverse functional roles of positively charged residues within the phosphorylation‐recognition region of Arrestin. Journal of Biological Chemistry, 270, 6010–6016. 10.1074/jbc.270.11.6010 [DOI] [PubMed] [Google Scholar]
- Hamano, K. , Kiyama, H. , Emson, P. C. , Manabe, R. , Nakauchi, M. , & Tohyama, M. (1990). Localization of two calcium binding proteins, Calbindin (28 kD) and Parvalbumin (12 kD), in the vertebrate retina. Journal of Comparative Neurology, 302(2), 417–424. 10.1002/cne.903020217 [DOI] [PubMed] [Google Scholar]
- Hamel, C. (2006). Retinitis pigmentosa. Orphanet Journal of Rare Diseases, 1(1), 1–12. 10.1186/1750-1172-1-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrave, P. A. , Adamus, G. , Arendt, A. , McDowell, J. H. , Wang, J. , Szary, A. , … Jackson, R. W. (1986). Rhodopsin's amino terminus is a principal antigenic site. Experimental Eye Research, 42(4), 363–373. 10.1016/0014-4835(86)90030-8 [DOI] [PubMed] [Google Scholar]
- Haverkamp, S. , Ghosh, K. K. , Hirano, A. A. , & Wässle, H. (2003). Immunocytochemical description of five bipolar cell types of the mouse retina. Journal of Comparative Neurology, 455(4), 463–476. 10.1002/cne.10491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks, D. , & Barnstable, C. J. (1987). Different rhodopsin monoclonal antibodies reveal different binding patterns on developing and adult rat retina. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society, 35(11), 1317–1328. 10.1177/35.11.3655327 [DOI] [PubMed] [Google Scholar]
- Howell, B. W. , Gertler, F. B. , & Cooper, J. A. (1997). Mouse disabled (mDab1): A Src binding protein implicated in neuronal development. EMBO Journal, 16(1), 121–132. 10.1093/emboj/16.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, C. , Studholme, K. , Yazulla, S. , & von Gersdorff, H. (2006). Diurnal changes in exocytosis and the number of synaptic ribbons at active zones of an on‐type bipolar cell terminal. Journal of Neurophysiology, 96(4), 2025–2033. 10.1152/jn.00364.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, C. , Strettoi, E. , & Masland, R. H. (1998). The major cell populations of the mouse retina. Journal of Neuroscience, 18(21), 8936–8946. 10.1523/JNEUROSCI.18-21-08936.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, B. W. , Kondo, M. , Terasaki, H. , Lin, Y. , McCall, M. , & Marc, R. E. (2012). Retinal remodeling. Japanese Journal of Ophthalmology, 56(4), 289–306. 10.1007/s10384-012-0147-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, B. W. , & Marc, R. E. (2005). Retinal remodeling during retinal degeneration. Experimental Eye Research, 81(2), 123–137. 10.1016/j.exer.2005.03.006 [DOI] [PubMed] [Google Scholar]
- Kaufman, D. L. , Houser, C. R. , & Tobin, A. J. (1991). Two forms of the γ‐aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. Journal of Neurochemistry, 56(2), 720–723. 10.1111/j.1471-4159.1991.tb08211.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke, N. C. , Murata, Y. , Kim, S. S. , Pulver, S. R. , Birdsey‐Benson, A. , Cho, Y. K. , … Boyden, E. S. (2014). Independent optical excitation of distinct neural populations. Nature Methods, 11, 338 Retrieved from. 10.1038/nmeth.2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong, J. M. K. , Caprioli, J. , & Piri, N. (2010). RNA binding protein with multiple splicing: A new marker for retinal ganglion cells. Investigative Ophthalmology & Visual Science, 51(2), 1052–1058. 10.1167/iovs.09-4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagali, P. S. , Balya, D. , Awatramani, G. B. , Münch, T. A. , Kim, D. S. , Busskamp, V. , … Roska, B. (2008). Light‐activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nature Neuroscience, 11(6), 667–675. 10.1038/nn.2117 [DOI] [PubMed] [Google Scholar]
- Léveillard, T. , Mohand‐Saïd, S. , Lorentz, O. , Hicks, D. , Fintz, A.‐C. , Clérin, E. , … Sahel, J.‐A. (2004). Identification and characterization of rod‐derived cone viability factor. Nature Genetics, 36(7), 755–759. 10.1038/ng1386 [DOI] [PubMed] [Google Scholar]
- Lewis, S. E. , & Nixon, R. A. (1988). Multiple phosphorylated variants of the high molecular mass subunit of neurofilaments in axons of retinal cell neurons: Characterization and evidence for their differential association with stationary and moving neurofilaments. Journal of Cell Biology, 107(6 II), 2689–2701. 10.1083/jcb.107.6.2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, B. , Masland, R. H. , & Strettoi, E. (2009). Remodeling of cone photoreceptor cells after rod degeneration in rd mice. Experimental Eye Research, 88(3), 589–599. 10.1016/j.exer.2008.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macé, E. , Caplette, R. , Marre, O. , Sengupta, A. , Chaffiol, A. , Barbe, P. , … Dalkara, D. (2015). Targeting Channelrhodopsin‐2 to ON‐bipolar cells with Vitreally administered AAV restores ON and OFF visual responses in blind mice. Molecular Therapy, 23(1), 7–16. 10.1038/MT.2014.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc, R. E. , Jones, B. W. , Watt, C. B. , Vazquez‐Chona, F. , Vaughan, D. K. , & Organisciak, D. T. (2008). Extreme retinal remodeling triggered by light damage: Implications for age related macular degeneration. Molecular Vision, 14(April), 782–806 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18483561%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2375357 [PMC free article] [PubMed] [Google Scholar]
- Mazzoni, F. , Novelli, E. , & Strettoi, E. (2008). Retinal ganglion cells survive and maintain Normal dendritic morphology in a mouse model of inherited photoreceptor degeneration. Journal of Neuroscience, 28(52), 14282–14292. 10.1523/JNEUROSCI.4968-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, B. , Snellman, J. , Chen, S. , Li, W. , & Zenisek, D. (2013). Synaptic ribbons influence the size and frequency of miniature‐like evoked postsynaptic currents. Neuron, 77(3), 516–527. 10.1016/j.neuron.2012.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes, H. F. , Van Der Spuy, J. , Chapple, J. P. , & Cheetham, M. E. (2005). Mechanisms of cell death in rhodopsin retinitis pigmentosa: Implications for therapy. Trends in Molecular Medicine, 11(4), 177–185. 10.1016/j.molmed.2005.02.007 [DOI] [PubMed] [Google Scholar]
- Morrow, E. M. , Furukawa, T. , Raviola, E. , & Cepko, C. L. (2005). Synaptogenesis and outer segment formation are perturbed in the neural retina of Crx mutant mice. BMC Neuroscience, 6(1), 5 10.1186/1471-2202-6-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, L. , Azar, S. S. , de los Santos, J. , & Brecha, N. C. (2017). Prox1 is a marker for AII amacrine cells in the mouse retina. Frontiers in Neuroanatomy, 11, 39 10.3389/fnana.2017.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, J. J. , Dai, X. , Boye, S. E. , Barone, I. , Boye, S. L. , Mao, S. , … Hauswirth, W. W. (2011). Long‐term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Molecular Therapy, 19(2), 234–242. 10.1038/mt.2010.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus, Y. M. , Jain, A. , Gariano, R. F. , Stanzel, B. V. , Marmor, M. , Blumenkranz, M. S. , & Palanker, D. (2008). Healing of retinal photocoagulation lesions. Investigative Ophthalmology and Visual Science, 49(12), 5540–5545. 10.1167/iovs.08-1928 [DOI] [PubMed] [Google Scholar]
- Phillips, M. J. , Otteson, D. C. , & Sherry, D. M. (2010). Progression of neuronal and synaptic remodeling in the rd10 mouse model of retinitis pigmentosa. Journal of Comparative Neurology, 518(11), 2071–2089. 10.1002/cne.22322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthussery, T. , Gayet‐Primo, J. , Pandey, S. , Duvoisin, R. M. , & Taylor, W. R. (2009). Differential loss and preservation of glutamate receptor function in bipolar cells in the rd10 mouse model of retinitis pigmentosa. The European Journal of Neuroscience, 29(8), 1533–1542. 10.1111/j.1460-9568.2009.06728.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthussery, T. , Gayet‐Primo, J. , & Taylor, W. R. (2010). Localization of the calcium‐binding protein secretagogin in cone bipolar cells of the mammalian retina. Journal of Comparative Neurology, 518(4), 513–525. 10.1002/cne.22234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoczy, E. P., Kiel, C., McKeone, R., Stricher, F. & Serrano, L . (2011). Analysis ofdisease‐linked rhodopsin mutations based on structure, function, and proteinstability calculations. J Mol Biol, 405(2), 584–606. 10.1016/j.jmb.2010.11.003 [DOI] [PubMed] [Google Scholar]
- Rash, J. E. , Staines, W. A. , Yasumura, T. , Patel, D. , Furman, C. S. , Stelmack, G. L. , & Nagy, J. I. (2000). Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin‐36 but not connexin‐32 or connexin‐43. Proceedings of the National Academy of Sciences of the United States of America, 97(13), 7573–7578. 10.1073/pnas.97.13.7573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regus‐Leidig, H. , Dieck, S. T. , Specht, D. , Meyer, L. , & Brandstätter, J. H. (2009). Early steps in the assembly of photoreceptor ribbon synapses in the mouse retina: The involvement of precursor spheres. Journal of Comparative Neurology, 512(6), 814–824. 10.1002/cne.21915 [DOI] [PubMed] [Google Scholar]
- Reim, K. , Regus‐Leidig, H. , Ammermüller, J. , El‐Kordi, A. , Radyushkin, K. , Ehrenreich, H. , … Brose, N. (2009). Aberrant function and structure of retinal ribbon synapses in the absence of complexin 3 and complexin 4. Journal of Cell Science, 122(9), 1352–1361. 10.1242/jcs.045401 [DOI] [PubMed] [Google Scholar]
- Rice, D. S. , & Curran, T. (2000). Disabled‐1 is expressed in type AII amacrine cells in the mouse retina. Journal of Comparative Neurology, 424(2), 327–338. [DOI] [PubMed] [Google Scholar]
- Riepe, R. E. , & Norenburg, M. D. (1977). Müller cell localisation of glutamine synthetase in rat retina. Nature, 268, 654 Retrieved from–655. 10.1038/268654a0 [DOI] [PubMed] [Google Scholar]
- Rodriguez, A. R. , de Sevilla Müller, L. P. , & Brecha, N. C. (2014). The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. Journal of Comparative Neurology, 522(6), 1411–1443. 10.1002/cne.23521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof, D. J. , Adamian, M. , & Hayes, A. (1994). Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Investigative Ophthalmology and Visual Science, 35(12), 4049–4062. [PubMed] [Google Scholar]
- Santos, A. M. , Calvente, R. , Tassi, M. , Carrasco, M.‐C. , Martín‐Oliva, D. , Marín‐Teva, J. L. , … Cuadros, M. A. (2008). Embryonic and postnatal development of microglial cells in the mouse retina. Journal of Comparative Neurology, 506(2), 224–239. 10.1002/cne.21538 [DOI] [PubMed] [Google Scholar]
- Sengupta, A. , Chaffiol, A. , Macé, E. , Caplette, R. , Desrosiers, M. , Lampič, M. , … Duebel, J. (2016). Red‐shifted channelrhodopsin stimulation restores light responses in blind mice, macaque retina, and human retina. EMBO Molecular Medicine, 8(11), 1248–1264. 10.15252/emmm.201505699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver, R. , Witkovsky, P. , Horvath, P. , Alones, V. , Barnstable, C. J. , & Lehman, M. N. (1988). Coexpression of opsin‐ and VIP‐like immunoreactivity in CSF‐contacting neurons of the avian brain. Cell and Tissue Research, 253(1), 189–198. 10.1007/BF00221754 [DOI] [PubMed] [Google Scholar]
- Söhl, G. , Degen, J. , Teubner, B. , & Willecke, K. (1998). The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Letters, 428, 27–31. 10.1016/S0014-5793(98)00479-7 [DOI] [PubMed] [Google Scholar]
- Soto, F. , & Kerschensteiner, D. (2015). Synaptic remodeling of neuronal circuits in early retinal degeneration. Frontiers in Cellular Neuroscience, 9, 395 10.3389/fncel.2015.00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasheff, S. F. , Shankar, M. , & Andrews, M. P. (2011). Developmental time course distinguishes changes in spontaneous and light‐evoked retinal ganglion cell activity in rd1 and rd10 mice. Journal of Neurophysiology, 105(6), 3002–3009. 10.1152/jn.00704.2010 [DOI] [PubMed] [Google Scholar]
- Strettoi, E. , & Pignatelli, V. (2000). Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proceedings of the National Academy of Sciences of the United States of America, 97(20), 11020–11025. 10.1073/pnas.190291097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strettoi, E. , Porciatti, V. , Falsini, B. , Pignatelli, V. , & Rossi, C. (2002). Morphological and functional abnormalities in the inner retina of the rd/rd mouse. The Journal of Neuroscience, 22(13), 5492–5504. https://doi.org/20026533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strettoi, E. (2015). A survey of retinal remodeling. Frontiers in Cellular Neuroscience, 9, 494 10.3389/fncel.2015.00494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strettoi, E. , Pignatelli, V. , Rossi, C. , Porciatti, V. , & Falsini, B. (2003). Remodeling of second‐order neurons in the retina of rd/rd mutant mice. Vision Research, 43(8), 867–877. 10.1016/S0042-6989(02)00594-1 [DOI] [PubMed] [Google Scholar]
- Sullivan, R. K. P. , WoldeMussie, E. , & Pow, D. V. (2007). Dendritic and synaptic plasticity of neurons in the human age‐related macular degeneration retina. Investigative Ophthalmology and Visual Science, 48(6), 2782–2791. 10.1167/iovs.06-1283 [DOI] [PubMed] [Google Scholar]
- Toychiev, A. H. , Ivanova, E. , Yee, C. W. , & Sagdullaev, B. T. (2013). Block of gap junctions eliminates aberrant activity and restores light responses during retinal degeneration. The Journal of Neuroscience, 33(35), 13972–13977 Retrieved from http://www.jneurosci.org/content/33/35/13972.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, T. , Kang, Q. , Ma, B. , Gao, S. , Li, X. , & Liu, Y. (2015). Activation of autophagy and paraptosis in retinal ganglion cells after retinal ischemia and reperfusion injury in rats. Experimental and Therapeutic Medicine, 9(2), 476–482. 10.3892/etm.2014.2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney, I. E. , Raven, M. A. , Ciobanu, D. C. , Williams, R. W. , & Reese, B. E. (2009). Multiple genes on chromosome 7 regulate dopaminergic amacrine cell number in the mouse retina. Investigative Ophthalmology & Visual Science, 50(5), 1996–2003. 10.1167/iovs.08-2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkovsky, P. (2004). Dopamine and retinal function. Documenta Ophthalmologica, 108(1), 17–39. 10.1023/B:DOOP.0000019487.88486.0a [DOI] [PubMed] [Google Scholar]
- Wulle, I. , & Schnitzer, J. (1989). Distribution and morphology of tyrosine hydroxylase‐immunoreactive neurons in the developing mouse retina. Developmental Brain Research, 48(1), 59–72. 10.1016/0165-3806(89)90093-X [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Li, A. , Brown, B. , Weiss, E. R. , Osawa, S. , & Craft, C. M. (2002a). Mouse Cone Arrestin expression pattern: Light induced translocation in cone photoreceptors. Molecular Vision, 3(8), 462–471 Retrieved from http://www.molvis.org/molvis/v8/a56 [PubMed] [Google Scholar]
- Zhu, X. , Li, A. , Brown, B. , Weiss, E. R. , Osawa, S. , & Craft, C. M. (2002b). Mouse Cone Arrestin expression pattern: Light induced translocation in cone photoreceptors. Molecular Vision, 3(8), 462–471 Retrieved from http://www.molvis.org/molvis/v8/a56 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.