

Abstract
The scientific interest in catalytic hydroaminoalkylation reactions of alkenes has vastly increased over the past decade, but these reactions have struggled to become a viable option for general laboratory or industrial use because of reaction times of several days. The titanium‐based catalytic system introduced in this work not only reduces the reaction time by several orders of magnitude, into the range of minutes, but the catalyst is also demonstrated to be easily available from common starting materials, at a cost of approximately 1 € per millimole of catalyst. We were also able to formally perform C−H activation of methylamine and achieve coupling to a broad variety of alkenes, through silyl protection of the amine and simple deprotection by water.
Keywords: amines, hydroaminoalkylation, methylamine, phenethylamine, titanium
Methylamine can be coupled to alkenes by using a titanium hydroaminoalkylation catalyst that is inexpensive (1 €/g) and easily accessible. Compatibility of methylamine with the catalyst is achieved by silylation; the products can be isolated with the protective group intact or, through simple deprotection with water, as the free amine.

Introduction
The catalytic hydroaminoalkylation of alkenes with N‐methylamines is of high interest, especially for industry, because it uses the same alkene substrates and produces the same products as the already industrially applied hydroformylation with subsequent reductive amination, and avoids multiple reaction steps.1 This direct approach has the potential of severely decreasing the cost, energy, and amount of workup associated with the synthesis of sophisticated amines from alkenes. A lot of research began in 2007 when Herzon and Hartwig reported the first use of N‐arylalkylamines;2 the following decade afforded a vastly increased scope of substrates and several different catalytic systems, most notably based on group 4 and group 5 metals.3 However, long reaction times have been a persistent problem throughout, with common durations ranging from 24 to 96 h.3 Very recently, a remarkable advance was made by DiPucchio, Roşca, and Schafer by using a tantalum ureate complex, putting the shortest reported reaction time for a catalytic hydroaminoalkylation of an alkene at only 2 h (4‐vinylcyclohexene with N‐methylaniline) while maintaining an excellent yield of isolated product.4 This report, unfortunately, has so far been an exception in this field of chemistry.
Results and Discussion
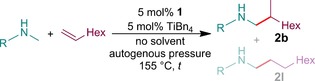
The new catalyst introduced in this work, based on a titanium center and a formamidinato ligand, is now able to convert terminal alkenes with N‐methylanilines within minutes. Formamidinato–titanium complexes have already been used as catalysts for hydroaminoalkylation reactions, but were based on a Ti(NMe2)4 precursor.5, 6 We found in recent work that dimethylamine is a very difficult substrate for hydroaminoalkylation reactions of alkenes7 and as such, it acts as a very potent inhibitor in catalytic systems that employ Ti(NMe2)4 as the titanium source. We wanted to solve this problem by using TiBn4, which is known to potentially be a better precursor,8 but could not isolate a well‐defined complex of this precursor with the formamidine 1. Instead, generation of the catalyst in situ by quenching TiBn4 with the substrate amine and subsequent addition of the ligand (see Scheme 1) proved to be a very simple, quick, and reliable method to access a highly active catalyst.4, 9 We specifically chose TiBn4 as the desired precursor because it can be easily prepared on a multigram scale from very inexpensive starting materials (TiCl4 and BnCl) within only a few hours, making it available to any laboratory that is equipped with Schlenk glassware. We developed a very cost‐ and time‐effective protocol for the synthesis of this catalyst (see the Supporting Information) and likewise, we have provided a synthesis for ligand precursor 1 that is focused on general availability; this precursor can be reliably synthesized from p‐anisidine and cyclopentadiene without any need for Schlenk techniques. We calculated the total material cost to be 1.12 € per millimole of catalyst (see the Supporting Information).
Scheme 1.

Generation of the catalyst in situ and catalytic cycle.
We tested whether 1 or TiBn4 alone would catalyze hydroaminoalkylation and found that TiBn4 was able to produce slight traces of product, yet not to a usable degree. We also tested the catalytic activity of 1 with Ti(NMe2)4 as the titanium source and only isolated traces of product.10 We found that the catalysis works better when the substrate concentrations are high, with solvent‐free execution yielding the best results. An excess of alkene not only decreases the reaction time further, but also ensures that the solution can still be stirred at high conversions without adding solvent, as the pure reaction products can be quite viscous. Conducting the reactions in ampoules under autogenous pressure resulted in high reliability and good comparability of the results, especially with low‐boiling substrates (see Table 1, entry 12 and Table 2, entries 4, 5, 6) that could otherwise not be heated to 155 °C. It should be noted that we had no trouble reproducing the results in regular glass flasks with clamped glass plug for substrates with a boiling point above 120 °C, as long as the heating time of the respective vessel was accounted for (we increased the heating time by one minute for regular 10 mL flasks and found no significant difference in yield). For the ampoules used in these experiments (Table 1 and Table 2), the reaction time over which the reaction mixture is above 150 °C (which is necessary for this catalyst to exhibit good performance)11 is shorter than the heating time by about one minute. For consistency and simplification of our experiments, the heating times depicted in both tables are the total times over which the ampoules are in contact with an oil bath at 155 °C.12 This means that the actual reaction time of the hydroaminoalkylation of 1‐octene with N‐methylaniline (Table 1, entry 1) amounts to only 3–4 s per catalytic turnover. To enhance the comparability of the heating times of different substrates, we tried to stop the reactions at a yield of isolated product of between 90–95 %; it should be noted that the time could be increased slightly to achieve a quantitative yield. For Table 1, entries 2, 8, 11, 12 and Table 2, entries 7, 8, 9, 11, we noticed that a simple increase of heating time did not increase the yield much further. Unwanted side reactions could be responsible for the incomplete conversion of 4‐vinylcyclohexene (Table 2, entry 7), whereas most other incomplete conversions are likely caused by product inhibition of the catalyst. For o‐methyl‐N‐methylaniline, as well as o‐methylstyrene (Table 1, entry 2 and Table 2, entry 11), it is shown that steric bulk of the substrates too close to the titanium center can inhibit the reaction almost entirely. In these cases, it should be expected that a full conversion of the substrates cannot be achieved by a longer reaction time alone, since the catalyst will likely deteriorate at 155 °C over 12 h. We also encountered this effect for other substrates with steric bulk near the functional group (2‐octene, α‐methylstyrene, 3,3‐dimethylbut‐1‐ene, and N‐methyl‐tert‐butylamine), where a reaction time of several hours would only deliver traces of product and appreciable yields could not be achieved. We were however delighted to see that this catalyst is active enough to give good yields with notoriously difficult substrates such as dialkyamines (Table 1, entries 11 and 12) and cycloalkenes (Table 2, entry 5) within reasonable reaction times. All substrates, except styrenes and trimethylvinylsilane (Table 2, entries 4, 10–13), exhibit excellent regioselectivity, which is consistent with the previously reported uses of formamidinato–titanium catalysts.6, 7 The overall dramatic increase in activity compared to the already reported uses of a formamidinato–titanium complex (several minutes instead of 96 h)5, 6 stems largely from the precursor and reaction conditions; the use of 1 doubles the catalytic activity compared to the ligand reported in 2015.6, 13
Table 1.
Scope of the hydroaminoalkylation of 1‐octene with various secondary amines.
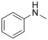
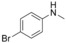
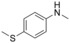
|
Entry |
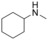
Amine substrate[a] |
Heating time[b] [min] |
Yield of isolated 2 b [%] |
S regio [c] 2 b/2 l |
|---|---|---|---|---|
|
1 |
|
2 |
94 |
97:3 |
|
2 |
|
60 |
8 |
99:1 |
|
3 |
|
7 |
94 |
98:2 |
|
4 |
|
10 |
95 |
97:3 |
|
5 |
|
15 |
91 |
97:3 |
|
6 |
|
6 |
90 |
96:4 |
|
7 |
|
15 |
92 |
96:4 |
|
8 |
|
15 |
82 |
97:3 |
|
9 |
|
2 |
91 |
96:4 |
|
10 |
|
10 |
90 |
96:4 |
|
11 |
|
300 |
49 |
98:2 |
|
12 |
|
120 |
71 |
97:3 |








[a] Reaction conditions: amine (2 mmol), TiBn4 (42 mg, 0.10 mmol, 5 mol %), 1 (53 mg, 0.10 mmol, 5 mol %), 1‐octene (561 mg, 5 mmol) in a 5 mL ampoule, 155 °C, t. [b] Heating time refers to the total time over which the ampoule is held in an oil bath at 155 °C. [c] Selectivity was determined by GC analysis prior to chromatography.
Table 2.
Scope of the hydroaminoalkylation of various alkenes with N‐methylaniline.

|
Entry |
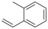
Alkene substrate[a] |
Heating time[b] [min] |
Yield of isolated 2 b [%] |
S regio [c] 2 b/2 l |
|---|---|---|---|---|
|
1 |
|
10 |
97 |
98:2 |
|
2 |
|
7 |
91 |
97:3 |
|
3 |
|
5 |
94 |
97:3 |
|
4 |
|
25 |
75[d] |
82:18 |
|
5 |
|
60 |
96 |
– |
|
6 |
|
120 |
90 |
100:0 |
|
7 |
|
20 |
79[e] |
97:3 |
|
8 |
|
20 |
82 |
97:3 |
|
9 |
|
6 |
85 |
94:6 |
|
10 |
|
4 |
91[f] |
73:27 |
|
11 |
|
60 |
9[f] |
78:22 |
|
12 |
|
13 |
92[f] |
68:32 |
|
13 |
|
2 |
90[f] |
77:23 |












[a] Reaction conditions: N‐methylaniline (214 mg, 2 mmol), TiBn4 (42 mg, 0.10 mmol, 5 mol %), 1 (53 mg, 0.10 mmol, 5 mol %), alkene (5 mmol) in a 5 mL ampoule, 155 °C, t. [b] Heating time refers to the total time over which the ampoule is held in an oil bath at 155 °C. [c] Selectivity was determined by GC analysis prior to chromatography. [d] The regioisomers could not be separated; the isolated yield refers to the fraction of branched product according to GC analysis. [e] The reaction was observed at the terminal alkene exclusively. [f] For styrenes, the yield of isolated 2 b+2 l is given. Both regioisomers were isolated separately.
Contrary to the catalyst itself, the ligand precursor 1 possesses an extremely low solubility in most solvents, and we were only able to dissolve it in chloroform and boiling toluene. We were able to account for this by grinding 1 into a very fine powder with a ball mill, which proved to be essential for the effective use in all cases with short reaction times. After catalysis however, the reaction mixture can be quenched with moist solvents (aliphatic or aromatic hydrocarbons, alcohols, ethers, dichloromethane) and the ligand precursor (as well as TiO2) precipitates. Precursor 1 can then be reisolated by simple extraction of the solid residue with chloroform. Although recycling may not be of high interest for laboratory use because of the inexpensive synthesis of the ligand, the possibility of recycling could be desirable in industrial applications.
We also discovered that this catalytic system is active enough to even convert silyl‐protected primary amines such as the TBDMS‐protected methylamine 3 (Scheme 2) giving direct access to silyl‐protected primary amines by hydroaminomethylation. Although the conversion of primary aminoalkenes in an intramolecular hydroaminoalkylation has been achieved in the past,3, 8 the conversion of primary amine substrates (and consequently the synthesis of primary amine products) in an intermolecular hydroaminoalkylation has unfortunately been almost impossible, most notably because of the formation of metal–imido species or bridging towards multinuclear metal complexes, which deactivates the catalyst.14 This limited scope poses a very severe restriction to the use of hydroaminoalkylation reactions, especially because it prohibits the use of the most important primary amine, methylamine. During the writing of this manuscript, a method for the conversion of silyl‐protected benzylamine was published, but was limited exclusively to the activated amine and a narrow scope of alkenes, with reaction times of 24–240 h.15
Scheme 2.

Hydroaminoalkylation of alkenes with TBDMS‐protected methylamine.
By using the already described method with the in situ generated catalyst and TBDMS‐protected methylamine 3 as the amine substrate (see Scheme 2), we were able to generate primary amine products in good yield after reaction times of 12 h. After completion of the catalysis, the free primary amine can be obtained by hydrolysis. The simplest method we found was to simply treat the diluted crude reaction mixture with wet solvent (CH2Cl2/MeOH/H2O=75:25:1) overnight at room temperature. Alternatively, if a faster workup is required, the deprotection can also be achieved by stirring the crude reaction mixture in boiling EtOH/H2O (4:1) for one minute.
Terminal alkenes generally work well in this reaction, unless a specific steric hindrance close to the C=C double bond inhibits the reaction. This is most apparent when comparing styrenes, o‐methylstyrene shows almost no conversion, whereas meta‐substitution is tolerated and para‐substitution is tolerated well (Table 3, entries 13–16). 2‐Octene (Table 3, entry 2) shows that an internal alkene can be unreactive, whereas cyclopentene (Table 3, entry 10) exhibits significant reactivity. The same can be observed for 1,1‐disubstituted alkenes, α‐methylstyrene shows almost no reactivity, whereas the conversion of methylenecyclohexane proves that 1,1‐disubstitution can be tolerated and affords good yields (Table 3, entries 11 and 17). As described in Table 1, the catalytic system used in this work (1 with TiBn4) can react good substrate amines such as N‐methylaniline with terminal alkenes within reaction times of only a few minutes and usually tolerates halogenated substrates well. With 3 as a substrate, however, a vastly increased reaction time becomes necessary and side reactions diminish the yield of halogenated phenethylamines, as seen from the reaction of p‐chlorostyrene and p‐bromostyrene (Table 3, entries 20–23). It is also noteworthy that we were unable to successfully convert an alkene containing a tertiary amine (Table 3, entry 9). We suspect the tertiary amine to partially deprotect 3, free methylamine would then immediately disable the catalyst.14 This finding suggests that a successful conversion of a tertiary amine can only be expected when high steric bulk is present near the amino group. Overall, the results show that the tolerance towards steric properties of the substrate varies largely, but a sterically less demanding alkene is generally advantageous. Many alkenes and functional groups are tolerated very well, including dienes, ethers, protected alcohols, and silanes (Table 3, entries 3, 5–8, 12).
Table 3.
Scope and limitations of the hydroaminoalkylation of alkenes with 3.

|
Entry |
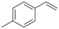
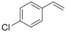
Alkene substrate |
Yield [%][a,b] |
Selectivity 5 b/5 l |
Entry |
Alkene substrate |
Yield [%][a,b] |
Selectivity 5 b/5 l |
|---|---|---|---|---|---|---|---|
|
1 |
|
81 |
>99:1 |
13 |
|
75 |
91:9 |
|
2 |
|
1[c,d] |
29:71 |
14 |
|
1 |
61:39 |
|
3 |
|
57[e] |
>96:4[f] |
15 |
|
39 |
95:5 |
|
4 |
|
19 |
94:6 |
16 |
|
67[g] |
96:4 |
|
5 |
|
57[d] |
84:16 |
17 |
|
3 |
76:24 |
|
6 |
|
81 |
>99:1 |
18 |
|
85 |
>99:1 |
|
7 |
|
44[g] |
96:4 |
19 |
|
76[g] |
>99:1 |
|
8 |
|
77[g] |
>99:1 |
20 |
|
34 |
89:11 |
|
9 |
|
0 |
– |
21 |
|
6 |
76:24 |
|
10 |
|
19 |
– |
22 |
|
14 |
90:10 |
|
11 |
|
63 |
98:2 |
23 |
|
0 |
– |
|
12 |
|
85 |
99:1 |
24 |
|
67[g] |
92:8 |






















[a] Reaction conditions: TiBn4 (42 mg, 0.10 mmol, 5 mol %), 1‐tert‐butyl‐N,1,1‐trimethylsilanamine (3, 291 mg, 2 mmol), 1 (53 mg, 0.10 mmol, 5 mol %), and alkene (5 mmol) in a 5 mL‐ampoule, autogenous pressure, 155 °C, 12 h. Deprotection: crude reaction mixture, MeOH (5 mL), H2O (0.2 mL) in CH2Cl2 (15 mL), rt, overnight. Yield refers to isolated 5 b or the tosylated equivalent, the linear side product 5 l is removed by chromatography. [b] If not otherwise specified, the product was tosylated for isolation owing to volatility: crude deprotected mixture, TosCl (762 mg, 4.0 mmol), NaOH (2 m, 6 mL, 12 mmol) in CH2Cl2 (30 mL), rt, overnight. Regioselectivity was then determined by GC analysis after tosylation and before chromatography. [c] The branched product 5 b in this case refers to 2‐ethylheptan‐1‐amine. [d] Both products were obtained as a mixture that could not be separated. The yield refers to the fraction of major product according to 1H NMR spectroscopy. [e] The product 5 b in this case refers to 2‐methylhex‐5‐en‐1‐amine; one C=C double bond is hydroaminoalkylated under these reaction conditions. [f] Traces of several side products made it impossible to identify the linear product 5 l in GC analyses, we can only say with certainty that the ratio of 5 b:5 l is higher than 96:4. [g] The product was isolated as the free amine without tosylation.
The TBDMS‐protected methylamine can be replaced by the TMS‐protected equivalent (we present an easy and quick synthesis of both substrates in the Supporting Information). This change drastically decreases the cost of the amine substrate [originating from the price of TMS‐Cl (10 € mol−1) compared to TBDMS‐Cl (245 € mol−1)] and is, therefore, advised for all high‐volume syntheses. The hydroaminomethylation is slower for the TMS‐protected variant,16 but the deprotection of the protected product completes instantly at room temperature when the reaction mixture is in contact with moist solvents. However, we estimate that the TBDMS‐protected methylamine 3 is more suitable for syntheses on a laboratory scale for several reasons: Its relatively high stability means that it can be handled under air and used glassware can be decontaminated in a controlled manner, whereas TMS‐NHMe will react violently with water and alcohols, releasing considerable amounts of MeNH2 gas. The TBDMS‐protected hydroaminomethylation products obtained from 3 can be analyzed by GC and TLC and are mostly stable over 24 h under air, if the solvent (observed for CH2Cl2) is not specifically wet. The stability is unfortunately not high enough to allow for satisfactory purification by column chromatography, we found that a considerable fraction of the product will be deprotected (observed for SiO2 and petroleum ether). Instead, we upscaled the hydroaminomethylation of styrene to multigram scale to demonstrate the possibility of isolating the silyl‐protected product 4 b by distillation (see Scheme 3). Mostly owing to losses at the distillation step, the isolated product was obtained in a reduced yield of 56 % (compared to a yield of 75 % from the reaction shown in Table 3, entry 13). The synthesis of protected amines from alkenes can be a valuable synthetic tool whenever a further functionalization is required.
Scheme 3.

Multigram‐scale isolation of the silyl‐protected phenethylamine.
Another advantage of the bulkier protective group of 3 compared to the TMS‐protected equivalent is the extraordinarily high regioselectivity; TMS‐NHMe produces a mixture of branched and linear product as expected for a formamidinato–titanium catalyst (Table 2, entries 10–13), whereas 3 produces only slight amounts of linear side product for most styrenes (exceptions are shown in Table 3, entries 14, 17, and 21). This finding makes this work especially useful for the direct synthesis of pharmaceutically interesting phenethylamines from styrenes; the hydroaminoalkylation of alkenes with a formamidinato–titanium catalyst would usually be expected to show very poor regioselectivity.6 For most olefins, we were unable to identify a linear product by GC analysis; 2‐octene, vinyltrimethylsilane, o‐methylstyrene, α‐methylstyrene, and pentafluorostyrene (Table 3, entries 2, 5, 14, 17, and 21) were the only olefins yielding poor regioselectivities, which is not unexpected for these substrates.
Conclusion
In summary, we presented a new catalytic system for the hydroaminoalkylation of alkenes with secondary amines, composed of a formamidine and TiBn4. The catalyst is shown to be both easily available and inexpensive; additionally, the ligand can be reisolated after use if needed. A wide spectrum of substrates can be converted with a very short reaction time, excellent selectivity, and quantitative yield. The catalyst can also be used for the hydroaminomethylation of alkenes with silylated methylamine. The presented method allows for the use of either the very inexpensive TMS‐protected or the exceptionally regioselective and easy to handle TBDMS‐protected methylamine, both of which can be synthesized easily.17 The possibility of isolating the protected products is demonstrated and methods for simple deprotection with water are given. A direct catalytic hydroaminomethylation of alkenes with methylamine has not been achieved before and considerable improvement in the reaction times of hydroaminoalkylations with secondary amines has been demonstrated, therefore, we think that this work can be of high value for laboratory work and an important input for potential industrial applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Research Training Group “Chemical Bond Activation” (GRK 2226) funded by the Deutsche Forschungsgemeinschaft for financial support of our research and Jessica Reimer for support of the experimental work. We thank Kirstin Glaser, Karin Grittner, and Frank Fleischer for supplying the ampoules.
J. Bielefeld, S. Doye, Angew. Chem. Int. Ed. 2020, 59, 6138.
References
- 1.For a review on hydroformylation reactions, see: Franke R., Selent D., Börner A., Chem. Rev. 2012, 112, 5675–5732. [DOI] [PubMed] [Google Scholar]
- 2. Herzon S. B., Hartwig J. F., J. Am. Chem. Soc. 2007, 129, 6690–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For a review on hydroaminoalkylation reactions, see: Edwards P. M., Schafer L. L., Chem. Commun. 2018, 54, 12543–12560. [DOI] [PubMed] [Google Scholar]
- 4. DiPucchio R. C., Roşca S.-C., Schafer L. L., Angew. Chem. Int. Ed. 2018, 57, 3469–3472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3527–3530. [Google Scholar]
- 5. Elkin T., Kulkarni N. V., Tumanskii B., Botoshansky M., Shimon L. J. W., Eisen M. S., Organometallics 2013, 32, 6337–6352. [Google Scholar]
- 6. Dörfler J., Preuß T., Brahms C., Scheuer D., Doye S., Dalton Trans. 2015, 44, 12149–12168. [DOI] [PubMed] [Google Scholar]
- 7. Bielefeld J., Doye S., Angew. Chem. Int. Ed. 2017, 56, 15155–15158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15352–15355. [Google Scholar]
- 8. Prochnow I., Kubiak R., Frey O. N., Beckhaus R., Doye S., ChemCatChem 2009, 1, 162–172. [Google Scholar]
- 9.We previously found the in situ generation of Ti catalysts to be a successful strategy for hydroaminoalkylation reactions, see: Dörfler J., Doye S., Angew. Chem. Int. Ed. 2013, 52, 1806–1809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1851–1854. The same concept was also shown to be effective for Ta catalysts, see reference [4]. [Google Scholar]
- 10.Reaction conditions equivalent to Table 2, entry 10: N-methylaniline (214 mg, 2 mmol), Ti(NMe2)4 (22 mg, 0.10 mmol, 5 mol %), 1 (53 mg, 0.10 mmol, 5 mol %), styrene (521 mg, 5 mmol) in a 5 mL ampoule, 155 °C, 4 min. The yield of isolated product was 1 %. However, it should be noted that good yields can be obtained if the reaction vessel allows for the escape of gaseous HNMe2 An otherwise identical experiment in a 80 mL Schlenk tube with 500 mL balloon and a reaction time of 20 min gave a 75 % yield of isolated product(2 b+2 l).
- 11.We found that the catalyst will tolerate temperatures at least up to 180 °C with increasing catalytic activity; temperatures below 150 °C will severely decrease the catalytic activity.
- 12.Selected data points for the reaction mixtures core temperature after the ampoule is in contact with the 155 °C oil bath: 120 °C after 35 s, 150 °C (74 s), 155 °C (110 s). Cooldown: 150 °C (14 s), 120 °C (64 s). No significant conversion of substrate is expected below 120 °C. The heating profile (and therefore the exact difference between heating time and reaction time) is expected to be slightly different for substrates with low boiling points. We did not possess the means to determine the heating profile for those substrates under autogenous pressure and thus want to correctly refer to heating times instead.
- 13.Reaction conditions: N-methylaniline (214 mg, 2 mmol), TiBn4 (8.4 mg, 0.02 mmol, 1 mol %), 1 (10.9 mg, 0.02 mmol, 1 mol %), styrene (521 mg, 5 mmol) in a 5 mL ampoule, 155 °C, 20 min. The yield of isolated product was 42 % (2 b+2 l). An otherwise identical experiment with N,N′-bis(2,6-diisopropylphenyl)formamidine instead of 1 as the ligand precursor delivered 18 % of isolated product.
- 14. Stelter L., Teusch T., Bielefeld J., Doye S., Klüner T., Chem. Eur. J. 2018, 24, 12485–12489. [DOI] [PubMed] [Google Scholar]
- 15.During the writing of this manuscript, the following article describing the hydroaminoalkylation of alkenes with TMS-protected benzylamine was published: Koperniku A., Foth P. J., Sammis G. M., Schafer L. L., J. Am. Chem. Soc. 2019, 141, 18944–18948; Selected example: TMS-protected benzylamine (0.5 mmol), 4-phenylbutene (0.9 mmol), Zr(NMe2)4 (10 mol %) in C6D6, 145 °C, 48 h. Combined yield of isolated product 78 %, regioselectivity 75:25 towards the branched product. 4-Phenylbutene was also used as a substrate in our screening (see Table 3, entry 19).31718171 [Google Scholar]
- 16.Reaction conditions with TMS-protected methylamine: TiBn4 (42 mg, 0.10 mmol, 5 mol %), N,1,1,1-tetramethylsilanamine (206 mg, 2 mmol), 1 (53 mg, 0.10 mmol, 5 mol %), and 4-phenylbutene (661 mg, 5 mmol) in a 5 mL-ampoule, autogenous pressure, 155 °C, 24 h. Deprotection: crude reaction mixture, MeOH (5 mL), H2O (0.2 mL) in CH2Cl2 (15 mL), rt. The yield of isolated primary amine product 5 b was 47 %. The equivalent experiment with TBDMS-protected methylamine (Table 3, entry 19) yielded 76 % of 5 b in 12 h.
- 17.For details, see the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary