

Abstract
Background
Runs of homozygosity (ROHs) are homozygous segments of the genome where the two haplotypes inherited from the parents are identical. The current availability of genotypes for a very large number of single nucleotide polymorphisms (SNPs) is leading to more accurate characterization of ROHs in the whole genome. Here, we investigated the occurrence and distribution of ROHs in 3,692 Large White pigs and compared estimates of inbreeding coefficients calculated based on ROHs (FROH), homozygosity (FHOM), genomic relationship matrix (FGRM) and pedigree (FPED). Furthermore, we identified genomic regions with high ROH frequencies and annotated their candidate genes.
Results
In total, 176,182 ROHs were identified from 3,569 animals, and all individuals displayed at least one ROH longer than 1 Mb. The ROHs identified were unevenly distributed on the autosomes. The highest and lowest coverages of Sus scrofa chromosomes (SSC) by ROH were on SSC14 and SSC13, respectively. The highest pairwise correlation among the different inbreeding coefficient estimates was 0.95 between FROH_total and FHOM, while the lowest was − 0.083 between FGRM and FPED. The correlations between FPED and FROH using four classes of ROH lengths ranged from 0.18 to 0.37 and increased with increasing ROH length, except for ROH > 10 Mb. Twelve ROH islands were located on four chromosomes (SSC1, 4, 6 and 14). These ROH islands harboured genes associated with reproduction, muscular development, fat deposition and adaptation, such as SIRT1, MYPN, SETDB1 and PSMD4.
Conclusion
FROH can be used to accurately assess individual inbreeding levels compared to other inbreeding coefficient estimators. In the absence of pedigree records, FROH can provide an alternative to inbreeding estimates. Our findings can be used not only to effectively increase the response to selection by appropriately managing the rate of inbreeding and minimizing the negative effects of inbreeding depression but also to help detect genomic regions with an effect on traits under selection.
Keywords: Candidate genes, Inbreeding coefficients, Runs of homozygosity, Sus scrofa
Background
The inheritance of identical haplotypes from a common ancestor creates long tracts of homozygous genotypes known as runs of homozygosity (ROHs) [1, 2]. Longer haplotypes are inherited from recent common ancestors, and shorter haplotypes are inherited from distant ancestors [2]. Hence, ROHs can provide accurate predictions of alleles at loci that are identical by descent (IBD) and has been widely applied to accurately estimate levels of autozygosity, which is the homozygous state of IBD alleles in human and livestock populations [2, 3]. There are several factors that increase ROH in a population, such as genetic drift, population bottlenecks, mating of close relatives, and natural and artificial selection [2, 3]. Therefore, we can assess the ROH patterns in one population to understand its demographic history and decipher the genetic architecture of economically important traits.
On the basis of levels of autozygosity, ROHs can be used to estimate the inbreeding coefficient of an individual (F), which is defined as the probability that both alleles at any locus within an individual are IBD [4]. Inbreeding leads to harmful effects, such as a reduction in genetic variance and higher frequencies of homozygosity for recessive detrimental mutations, reduction in individual performance (inbreeding depression) and lower population viability [5]. Since inbreeding has been implicated in reduced fitness, there is growing interest in characterizing and monitoring autozygosity to allow accurate estimation of F. Traditionally, F has been estimated based on pedigree information (FPED). Hence, the estimation of FPED relied strongly on the accuracy and amount of pedigree data available [6]. Currently, genotyping technology is no longer a limiting factor in marker-based studies of inbreeding [7]. The inbreeding estimates can be directly derived from the genomic relationship matrix (FGRM) using genome-wide single nucleotide polymorphism (SNP) data [8]. Genomic F can also measure homozygosity (FHOM) directly and, thus, can accurately reflect the actual percentage of the genome that is homozygous. Furthermore, genomic F allows us to estimate inbreeding and inbreeding depression for specific genomic regions, which is not possible with FPED. In addition, genomic F can be estimated in populations without pedigree information [9].
When computing the more accurate genomic F using genotypic data, there is a need to distinguish between alleles that are IBD or identical by state (IBS). This distinction is not easy to make when single markers are analysed, but the use of ROH allows it. Hence, FGRM and FHOM always overestimate inbreeding levels compared to FPED [9], and an alternate approach is to use estimates that are obtained from observed ROHs.
The objective of this study was to identify and characterize ROH patterns in a Large White pig population. We further computed the inbreeding coefficients based on the ROHs identified (FROH) and estimated their correlations with those from other methods, including genomic relationship matrix (FGRM), homozygous coefficients (FHOM) and pedigree-based coefficients (FPED). Moreover, ROH islands may have occurred due to selection for functionally important traits.
Methods
Ethics statement
All animals were treated following the guidelines established by the Council of China for Animal Welfare. The experimental protocols were approved by the Science Research Department of the Institute of Animal Sciences, Chinese Academy of Agricultural Sciences (CAAS) (Beijing, China).
Animals and genotyping data
In this study, ear tissue samples were collected from 3,692 Large White pigs from two commercial companies. In these two companies, there were 1,466 and 2,226 pigs, respectively. All samples were genotyped with the GeneSeek GGP Porcine HD array. The SNP chip comprises 50,915 probes that are distributed across 18 autosomes and two sex chromosomes according to the Sus scrofa 10.2 genome version. To update the SNP positions, we reordered the SNPs according to the newest version of the pig genome, Sus scrofa 11.1. Following this, 34,689 autosomal SNPs remained, with a mean distance of 67.052 kb between adjacent SNPs.
Quality filtering
Quality control was performed using PLINK v1.90 software [10] according to the following criteria: (1) the call rate was higher than 0.9; (2) the minor allele frequency (MAF) was higher than 0.01; and (3) SNPs were filtered to exclude loci assigned to unmapped contigs and to sex chromosomes. After quality control, 3,569 pigs and 33,723 variants were retained.
Effective population size
The effective population size (Ne) was calculated from linkage disequilibrium (LD) [11] using the following equation:
where NT(t) is the effective population size estimated t generations in the past, ct is the recombination rate defined for a specific physical distance between SNP markers t generations in the past, f(ct) is the Haldane mapping function of the genetic distance in Morgans between SNPs, is the linkage disequilibrium estimation after adjusting for sampling bias, and α = {1, 2, 2.2} is a correction for the occurrence of mutations [12].
In this study, Ne was computed by implementing SNeP software [12]. Considering the relatively small number of SNPs per chromosome [13], we used an α = 2 and estimated the recombination rate between a pair of genetic markers according to Sved and Feldman [14]. The minimum and maximum distances used between SNPs for Ne estimation were 0.05 and 5 Mb, respectively. The other parameters were set by default [12].
Runs of homozygosity detection and classification
ROHs were identified for each individual using PLINK v1.90 software [10], which uses a sliding window approach to scan each individual’s genotype at each marker position to detect homozygous segments [15]. To define a ROH, the following criteria must be fulfilled: (1) a minimum ROH length of 1 Mb; (2) a minimum of 50 consecutive SNPs included in a ROH, which was calculated using the equation proposed by Lencz et al. [16]:
where α is the percentage of false positive ROHs (set to 0.05 in the present study), ns is the number of SNPs per individual, ni is the number of individuals, and is the heterozygosity across all SNPs; (3) a maximum gap between consecutive SNPs of 1 Mb; (4) a minimum density of one SNP in 100 kb; (5) a sliding window of 50 SNPs across the genome that moves one SNP at a time; (6) a maximum of five missing genotypes and one heterozygous genotype in a ROH to avoid underestimation of long ROHs; and (7) a window threshold of 0.01.
In this study, the ROHs identified were further divided into three classes: 1~5, 5~10 and > 10 Mb.
Inbreeding coefficient estimation
Different estimates of inbreeding coefficients (F) were used for all animals:
The genealogical inbreeding coefficients (FPED) were computed for 3,569 pigs using pedigree information recorded between 2011 and 2019. For all individuals who passed quality control, complete pedigree records were available for 3 to 10 generations with an average depth of 6.86. Pedigree information on a total of 7,572 animals between 2011 and 2019 was available. The FPED was estimated for datasets (n = 3,569) according to Wright’s coefficient [17] with the R package pedigreemm [18].
Genomic inbreeding based on homozygous SNPs was determined using PLINK v1.90 software [10]. The inbreeding coefficient for an individual (FHOM) was computed as FHOM = (O − E)/(L − E), where O is the number of observed homozygotes, E is the number expected by chance, and L is the number of genotyped autosomal SNPs.
Genomic SNP-by-SNP inbreeding coefficient (FGRM) estimates were calculated by GCTA software [19]. The FGRM was calculated as , where xi is the number of copies of the reference allele for the ith SNP, m is the number of SNPs, and pi is the frequency of the reference allele.
Genomic inbreeding coefficients were also estimated based on ROHs (FROH). The FROH for each animal was calculated as , where is the length of ROHi of individual i, and Lauto is the autosomal genome length covered by the SNPs included in the chip.
The inbreeding coefficients estimated by these four methods were compared using Pearson’s correlation.
Detection of common runs of homozygosity and gene annotation
To identify the genomic regions that were most commonly associated with ROHs, the percentage of occurrences of SNPs in ROHs was calculated by counting the number of times a SNP was detected in those ROHs across individuals. The genomic regions most commonly associated with ROHs were identified by selecting the top 1% of SNPs observed in ROHs. Adjacent SNPs over this threshold were merged into genomic regions called ROH islands [20, 21].
The gene content of the ROH islands was annotated using the annotation database provided by NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/vertebrate_mammalian/Sus_scrofa/latest_assembly_versions/GCF_000003025.6_Sscrofa11.1). The biological function of each annotated gene within the ROH islands was determined through an extensive accurate literature search.
Results
Effective population size
The tendency of effective population sizes (Ne) estimated based on linkage disequilibrium (LD) is illustrated in Fig. 1. The historical Ne from 197 generations to 10 generations ago of each breed was estimated based on the LD decay. An increasing Ne as a function of the number of generations was observed, with a Ne of 99 estimated at 10 generations ago and 288 estimated at 197 generations ago.
Fig. 1.
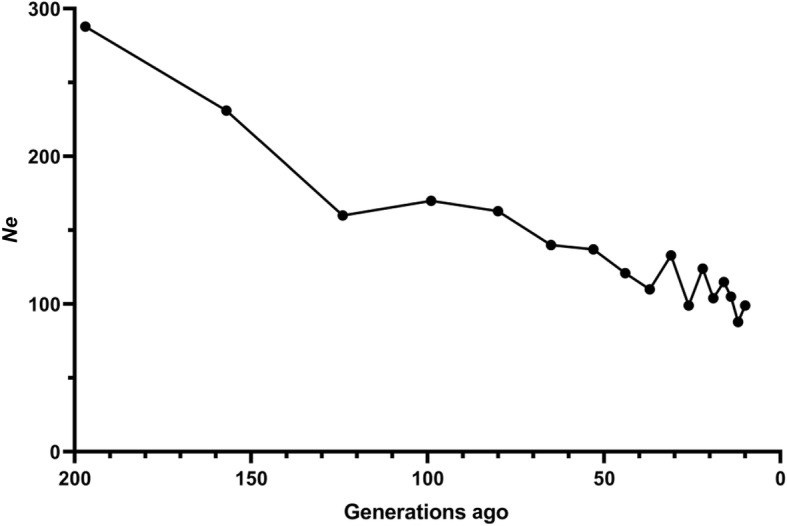
Effective population size (Ne) of the Large White pig population
Summary of runs of homozygosity
In total, 176,182 ROHs were identified from 3,569 animals. All of the Large White individuals exhibited at least one ROH longer than 1 Mb. Table 1 summarizes the basic statistics of the three classes of ROH. As seen in Table 1, the different classes of ROH length for the genotyped animals revealed that ROHs shorter than 10 Mb predominated. The segments shorter than 10 Mb account for approximately 86.27% of ROHs. Although the number of ROHs 1 ~ 5 Mb was the largest, the proportion of the genome covered by them was relatively small compared with that of ROH segments longer than 10 Mb.
Table 1.
Descriptive statistics of three classes of runs of homozygosity
| Type of ROH | n | Number percentage, % | Mean ± SD, Mb |
Total length, Mb | Length percentage, % |
|---|---|---|---|---|---|
| ROH 1~5 Mb | 102,341 | 58.09 | 3.24 ± 0.93 | 331,612.8 | 29.34 |
| ROH 5~10 Mb | 49,645 | 28.18 | 6.85 ± 1.35 | 340,110.2 | 30.09 |
| ROH > 10 Mb | 24,196 | 13.73 | 18.95 ± 12.00 | 458,605.3 | 40.57 |
As seen in Fig. 2, the numbers of ROHs on autosomes varied, which indicates that the ROHs identified were unevenly distributed on autosomes. The numbers of ROH per chromosome tended to increase with chromosome length. The smallest number of ROHs per chromosome was found on SSC 12, while the largest number of ROHs was on SSC 6. The highest ROH coverage was observed on SSC14, whereas the lowest was on SSC13 (Fig. 2).
Fig. 2.

The frequency distribution of the average number of ROHs per chromosome (bars) and average percentage of each chromosome covered by ROHs (lines)
The relationship between the number of ROHs and the length of the genome covered by ROHs per individual varied considerably among animals (Fig. 3). As the number of ROHs increased, the cumulative length of the ROHs also increased. In this population, one animal with extremely long ROHs had a length of ~ 900 Mb (925.603 Mb), and one animal with extremely short ROHs had a length of ~ 20 Mb (18.868 Mb).
Fig. 3.
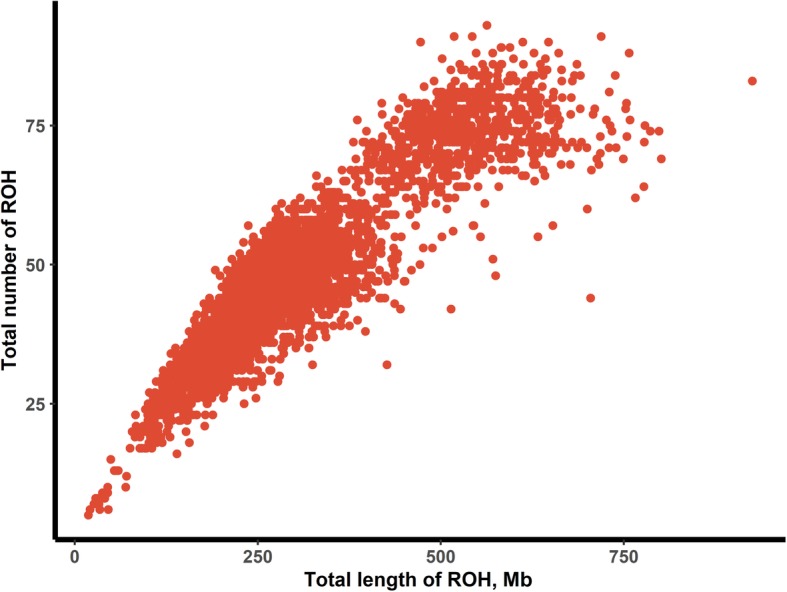
Total number of runs of homozygosity (ROHs) longer than 1 Mb and total length of genome (Mb) covered by ROH segments per individual
Inbreeding coefficient estimates based on pedigree and genomic data
In the present study, seven kinds of inbreeding coefficients were estimated based on the pedigree or genomic data of all individuals. The pedigree-based inbreeding estimates (FPED) obtained using all pedigree data available ranged from 0 to 0.156 with an average of 0.011. The four kinds of FROH were calculated based on three classes of ROH and the total ROH lengths. The average genomic inbreeding based on the total observed ROHs (FROH_total) was 0.140 with a range from 0.008 to 0.409 in this population. The estimated FGRM inbreeding coefficients ranged from −0.168 to 1.359 (mean = 0.099), and the estimated FHOM inbreeding coefficients ranged from −0.177 to 0.431 (mean = 0.103).
Figure 4 depicts the pairwise correlations among seven kinds of inbreeding coefficients. Among all pairwise correlations, the highest correlation was 0.95 between FROH_total and FHOM, while the lowest correlation was − 0.083 between FGRM and FPED. The correlations between the inbreeding coefficients calculated by different classes of ROH with FPED ranged from 0.18 to 0.37, and the highest correlation was found between FROH > 10 Mb and FPED (0.37).
Fig. 4.

Scatterplots (lower panel) and Pearson’s correlations (upper panel) of the genomic inbreeding coefficients FROH (FROH_total, FROH 1~5 Mb, FROH 5~10 Mb, FROH > 10 Mb), FGRM and FHOM, and pedigree-based inbreeding coefficient (FPED)
Functional annotation of genes
The percentage of SNPs in ROHs was plotted against the positions of the SNPs along the chromosomes in Fig. 5. In this study, the threshold used to define a ROH hotspot in the genome was 38.19%, above which the top 1% of SNPs most commonly observed in ROHs could be selected. The SNP with the highest proportion (63.27% of occurrences) was INRA0044866 on SSC14, which was annotated as within the SIRT1 gene. SIRT1 is related to porcine ovarian cell function, suppresses adipogenesis [4, 22], and affects preadipocytes [23]. The region on SSC14 with the second strongest signal harboured one gene: MYPN (63.21% of occurrences). The MYPN gene has been documented to be associated with meat and carcass traits in Italian Large White pigs [24]. Twelve ROH islands located on four chromosomes (SSC1, 4, 6 and 14) ranged in sizes from 3 SNPs on SSC4 and SSC14 to 107 SNPs on SSC4 (Table 2). These ROH islands harboured important candidate genes, which are shown in Table S1.
Fig. 5.

Manhattan plot of occurrences (%) of a SNP in ROHs across individuals
Table 2.
List of genomic regions of extended homozygosity detected in the Large White pig population within each ROH island
| Chr. | Start, bp | End, bp | Length, bp | No. SNPs | No. Genes |
|---|---|---|---|---|---|
| 1 | 45,159,055 | 45,580,780 | 421,725 | 4 | 3 |
| 1 | 46,051,365 | 46,251,692 | 200,327 | 4 | 1 |
| 1 | 47,215,836 | 49,995,943 | 2,780,107 | 50 | 3 |
| 1 | 66,162,636 | 67,278,674 | 1,116,038 | 18 | 9 |
| 4 | 96,116,417 | 102,143,358 | 6,026,941 | 107 | 158 |
| 4 | 102,523,866 | 102,618,589 | 94,723 | 3 | 1 |
| 4 | 104,299,568 | 104,397,592 | 98,024 | 3 | 2 |
| 4 | 106,857,958 | 107,236,227 | 378,269 | 12 | 2 |
| 6 | 26,918,929 | 31,001,440 | 4,082,511 | 71 | 86 |
| 14 | 44,919,089 | 45,412,495 | 493,406 | 13 | 3 |
| 14 | 47,355,668 | 47,467,063 | 111,395 | 3 | 5 |
| 14 | 68,649,520 | 73,203,453 | 4,553,933 | 70 | 35 |
Chr. chromosome
Discussion
Traditionally, the inbreeding coefficient was estimated based on pedigree data. In reality, pedigree information might be erroneous, such as having a missing parent or incorrect parent information. Furthermore, Large White pigs in the base population could be hypothesized to be unrelated. Moreover, the FPED value is the statistical expectation of the probable IBD genomic proportion [25], and FPED does not take into account the stochastic events of recombination during meiosis [26]. Thus, FPED could not completely show the actual relatedness among individuals in the population. With the development of high-throughput genotyping technologies and the reduction in genotyping costs, the inbreeding coefficient can be computed based on molecular information [27–29]. Genetic markers can provide a more accurate relationship estimate than pedigree [30]. Since FHOM cannot distinguish IBD alleles from IBS alleles, it might overestimate inbreeding levels [31]. In addition, FHOM and FGRM values can be negative for some individuals. Therefore, using the above three methods to estimate the inbreeding coefficient is not sufficiently accurate. FROH can alleviate the issues mentioned above, and thus, FROH might be a more effective and accurate alternative for quantifying animal relatedness and inbreeding levels in theory [9, 15, 32].
In the present study, the level of inbreeding based on pedigree was expected to be lower than FROH in the Large White pig population. The correlation between the FPED and FROH estimates was low (0.18–0.37) (Table 1). These results are consistent with previous studies in other pig populations [33, 34] and Nellore cattle [35]. Pearson’s correlation between FROH > 10 Mb and FPED was the highest (0.37) among those between different classes of ROH and FPED in the Large White pig population, while the correlation between FPED and FROH_total was slightly lower (0.35) (Fig. 4). Compared to the other two classes of ROH, the percentage of ROHs with a length > 10 Mb among ROHs of all lengths was the highest at 40.57%. These results suggested that ROH > 10 Mb was the major contribution to FROH_total.
ROHs can reveal the time that inbreeding occurred given the approximate correlation between the length of a ROH and the distance to the common ancestor due to the occurrence of recombination events over time. Fisher [36] reported that the expected length of a DNA segment that is IBD follows an exponential distribution with a mean equal to 1/2 g Morgans, where g is the number of generations since the common ancestor. Recombination events can interrupt long chromosome segments, so long ROHs (~ 10 Mb) arise as a result of recent inbreeding (up to five generations ago), and short ROHs (~ 1 Mb) are produced by IBD genomic regions from old ancestors [15], which are frequently unaccounted for in the recorded pedigree of an individual.
ROHs can be used to improve the accuracies of genomic breeding values (GEBV). EBVs estimated by the traditional BLUP method are based on pedigrees, which are used to construct a numerator relationship matrix (A matrix). If the A matrix is replaced by a relationship matrix based on genotype data (G matrix), GEBV can be obtained with BLUP. Based on the genotype matrix, it is not possible to identify whether IBS marker alleles are IBD or not. ROHs can identify alleles in the same or different individual(s), which indicates IBD [37]. Luan et al. [38] proposed a novel method to predict GEBV based on ROHs. The results showed that the accuracy of GEBV determination was higher with GROH than with GGRM by simulation study.
Here, the ROH islands harboured many candidate genes controlling economically important traits of Large White pigs (Table S1). We identified several candidate genes associated with reproduction. SIRT1 may regulate granulosa cell apoptosis during follicular atresia in porcine ovaries [39] and can reduce ovarian cell viability in rats [40]. SETDB1 plays an essential role in the maintenance of gonocyte survival in pigs, and knockdown of SETDB1 can induce gonocyte apoptosis [41]. PSMD4 effectively inhibits sperm-oocyte binding [42]. GNRHR2 is involved in regulating reproductive behaviour in pigs [43]. CES5A, GAL3ST1 and SPAG17 are essential for spermatogenesis and male fertility [44–46]. Some of the candidate genes have been documented as important candidate genes for muscular development and fat deposition. MYPN showed an association with traits related to muscularity in Pietrain×(Landrace×Large White) and Duroc × Pietrain, particularly association with ham weight and lean content in Duroc × Pietrain [47]. SLC12A4 is differentially expressed between the white and red skeletal muscle of Chinese Meishan pigs [48]. SIRT1 may downregulate pig preadipocyte proliferation and differentiation [23]. FMO5 plays a role in increasing glucose metabolism and insulin sensitivity in brown adipose tissue [49]. SIM1 is involved in the regulation of energy homeostasis [50], and KIF1BP and MCHR2 are involved in the regulation of food intake [51, 52], which in turn affects obesity risk [53]. HORMAD1, TBX15 and WARS2 are also associated with obesity [54, 55]. In addition, ADGRB3 is related to environmental information processing and environmental adaptation in domestic yak [56].
All the candidate genes residing in ROH islands were further analysed using the DAVID v6.8 tool [57] and the Sus scrofa annotation file as background to identify significant (P < 0.05) GO terms and KEGG pathways. Several GO terms (12 biological process, 5 molecular function and 2 cellular component) were significant, and two were significant for KEGG (Additional file: Table S2). The GO term spermatogenesis (0007283) was of particular note, where there were 6 genes. These results reflected that most quantitative phenotypic traits are likely to be influenced by multiple genes. The enrichment results provide novel insights into the genetic architecture of traits under selection. However, the information provided by GO analysis is limited.
Conclusions
In this study, we investigated the occurrence and distribution of ROHs on the autosomes of Large White pigs. The number of ROHs shorter than 10 Mb was the highest, while the genome sequence length covered by ROHs was the longest for ROHs longer than 10 Mb. Among the correlations between the genomic inbreeding coefficients calculated by different methods and the correlation coefficient based on pedigree calculation, the correlation between FPED and FROH > 10 Mb was the highest. FROH might be an effective and accurate alternative for assessing animal relatedness and inbreeding levels. ROH islands harboured many candidate genes controlling reproductive, muscular development, fat deposition and adaptation. Our findings contribute to an understanding of inbreeding effects when assessing ROHs at the genome level and how selection can shape the distribution of ROH islands in the swine genome.
Supplementary information
Additional file 1: Table S1. Gene content inside run-of-homozygosity islands.
Additional file 2: Table S2 GO terms and KEGG pathways enriched (P < 0.05) based on run-of-homozygosity islands.
Acknowledgements
We thank the researchers at our laboratories for their dedication and hard work. We would like to thank everyone who made this thesis possible.
Abbreviations
- F
Inbreeding coefficient
- GEBV
Genomic breeding values
- GRM
Genomic relationship matrix
- HOM
Homozygosity
- IBD
Identical by descent
- IBS
Identical by state
- LD
Linkage disequilibrium
- Ne
Effective population sizes
- PED
Pedigree
- ROH
Runs of homozygosity
- SNPs
Single nucleotide polymorphisms
- SSC
Sus scrofa chromosomes
Authors’ contributions
LYS performed analyses and drafted the manuscript. LGW and LXW contributed to the acquisition of data. LXW and FPZ conceived and designed the experiments. JXL, TYD, HY, LCZ, XL, HMG and XHH gave tremendous help in conducting experiments. LXW and FPZ improved the manuscript. All author(s) read and approved the final manuscript.
Funding
This work was supported by the Natural Science Foundation of China (No. 31572357) to FPZ, and the National Swine Industry Technology System (CARS-35) to LXW.
Availability of data and materials
The data and computing programs used in this manuscript are available from the corresponding authors on request.
Ethics approval and consent to participate
The experiments involving animals were approved by the Science Research Department of the Institute of Animal Science, Chinese Academy of Agricultural Sciences (CAAS) (Beijing, China). There was no use of human participants, data or tissues.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Liangyu Shi and Ligang Wang contributed equally to this work.
Contributor Information
Liangyu Shi, Email: 82101181190@caas.cn.
Ligang Wang, Email: ligwang@126.com.
Jiaxin Liu, Email: 753779828@qq.com.
Tianyu Deng, Email: 970997375@qq.com.
Hua Yan, Email: zcyyh@126.com.
Longchao Zhang, Email: zhlchias@163.com.
Xin Liu, Email: firstliuxin@163.com.
Hongmei Gao, Email: gaohongmei_123@126.com.
Xinhua Hou, Email: 7hxh73@163.com.
Lixian Wang, Email: iaswlx@263.net.
Fuping Zhao, Email: zhaofuping@caas.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40104-020-00447-0.
References
- 1.Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15(5):789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 2.Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF. Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet. 2018;19(4):220–234. doi: 10.1038/nrg.2017.109. [DOI] [PubMed] [Google Scholar]
- 3.Peripolli E, Munari DP, Silva M, Lima ALF, Irgang R, Baldi F. Runs of homozygosity: current knowledge and applications in livestock. Anim Genet. 2017;48(3):255–271. doi: 10.1111/age.12526. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Y, Song T, Peng J, Zhou Z, Wei H, Zhou R, et al. SIRT1 suppresses adipogenesis by activating Wnt/beta-catenin signaling in vivo and in vitro. Oncotarget. 2016;7(47):77707–77720. doi: 10.18632/oncotarget.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma RK, Hedrick PW. Conservation genetics in transition to conservation genomics. Trends Genet. 2010;26(4):177–187. doi: 10.1016/j.tig.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Sorensen P, Janss L, Ostersen T, Edwards D. Genome-wide and local pattern of linkage disequilibrium and persistence of phase for 3 Danish pig breeds. BMC Genet. 2013;14:115. doi: 10.1186/1471-2156-14-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silio L, Rodriguez MC, Fernandez A, Barragan C, Benitez R, Ovilo C, et al. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. J Anim Breed Genet. 2013;130(5):349–360. doi: 10.1111/jbg.12031. [DOI] [PubMed] [Google Scholar]
- 8.Saura M, Fernandez A, Rodriguez MC, Toro MA, Barragan C, Fernandez AI, et al. Genome-wide estimates of coancestry and inbreeding in a closed herd of ancient Iberian pigs. PLoS One. 2013;8(10):e78314. doi: 10.1371/journal.pone.0078314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saura M, Fernandez A, Varona L, Fernandez AI, de Cara MA, Barragan C, et al. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data. Genet Sel Evol. 2015;47:1. doi: 10.1186/s12711-014-0081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corbin LJ, Liu AY, Bishop SC, Woolliams JA. Estimation of historical effective population size using linkage disequilibria with marker data. J Anim Breed Genet. 2012;129(4):257–270. doi: 10.1111/j.1439-0388.2012.01003.x. [DOI] [PubMed] [Google Scholar]
- 12.Barbato M, Orozco-terWengel P, Tapio M, Bruford MW. SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front Genet. 2015;6:109. doi: 10.3389/fgene.2015.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME, et al. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007;17(4):520–526. doi: 10.1101/gr.6023607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sved JA, Feldman MW. Correlation and probability methods for one and two loci. Theor Popul Biol. 1973;4(1):129–132. doi: 10.1016/0040-5809(73)90008-7. [DOI] [PubMed] [Google Scholar]
- 15.Howrigan DP, Simonson MA, Keller MC. Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics. 2011;12:460. doi: 10.1186/1471-2164-12-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Todd L, Christophe L, Pamela DR, Burdick KE, Morgan TV, Kane JM, et al. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci U S A. 2007;104(50):19942–19947. doi: 10.1073/pnas.0710021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright S. Evolution in Mendelian genetics. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez AI, Bates DM, Rosa GJ, Gianola D, Weigel KA. Technical note: an R package for fitting generalized linear mixed models in animal breeding. J Anim Sci. 2010;88(2):497–504. doi: 10.2527/jas.2009-1952. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purfield DC, McParland S, Wall E, Berry DP. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS One. 2017;12(5):e0176780. doi: 10.1371/journal.pone.0176780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mastrangelo S, Ciani E, Sardina MT, Sottile G, Pilla F, Portolano B, et al. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim Genet. 2018;49(1):71–81. doi: 10.1111/age.12634. [DOI] [PubMed] [Google Scholar]
- 22.Pavlova S, Klucska K, Vasicek D, Ryban L, Harrath AH, Alwasel SH, et al. The involvement of SIRT1 and transcription factor NF-kappaB (p50/p65) in regulation of porcine ovarian cell function. Anim Reprod Sci. 2013;140(3–4):180–188. doi: 10.1016/j.anireprosci.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 23.Bai L, Pang WJ, Yang YJ, Yang GS. Modulation of Sirt1 by resveratrol and nicotinamide alters proliferation and differentiation of pig preadipocytes. Mol Cell Biochem. 2008;307(1–2):129–140. doi: 10.1007/s11010-007-9592-5. [DOI] [PubMed] [Google Scholar]
- 24.Braglia S, Davoli R, Zappavigna A, Zambonelli P, Buttazzoni L, Gallo M, et al. SNPs of MYPN and TTN genes are associated to meat and carcass traits in Italian large white and Italian Duroc pigs. Mol Biol Rep. 2013;40(12):6927–6933. doi: 10.1007/s11033-013-2812-z. [DOI] [PubMed] [Google Scholar]
- 25.Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2006;2(3):e41. doi: 10.1371/journal.pgen.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curik I, Ferencakovic M, Sölkner J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest Sci. 2014;166:26–34.
- 27.Purfield DC, Berry DP, McParland S, Bradley DG. Runs of homozygosity and population history in cattle. BMC Genet. 2012;13:70. doi: 10.1186/1471-2156-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjelland DW, Weigel KA, Vukasinovic N, Nkrumah JD. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J Dairy Sci. 2013;96(7):4697–4706. doi: 10.3168/jds.2012-6435. [DOI] [PubMed] [Google Scholar]
- 29.Marras G, Gaspa G, Sorbolini S, Dimauro C, Ajmone-Marsan P, Valentini A, et al. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim Genet. 2015;46(2):110–121. doi: 10.1111/age.12259. [DOI] [PubMed] [Google Scholar]
- 30.Goddard M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica. 2009;136(2):245–257. doi: 10.1007/s10709-008-9308-0. [DOI] [PubMed] [Google Scholar]
- 31.Wang J. Marker-based estimates of relatedness and inbreeding coefficients: An assessment of current methods. J Evol Biol. 2014;27:518–30. [DOI] [PubMed]
- 32.Peripolli E, Stafuzza NB, Munari DP, Lima ALF, Irgang R, Machado MA, et al. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genomics. 2018;19(1):34. doi: 10.1186/s12864-017-4365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zanella R, Peixoto JO, Cardoso FF, Cardoso LL, Biegelmeyer P, Cantão ME, et al. Genetic diversity analysis of two commercial breeds of pigs using genomic and pedigree data. Genet Sel Evol. 2016;48:24. doi: 10.1186/s12711-016-0203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes MS, Silva FF, Harlizius B, Duijvesteijn N, Lopes PS, Guimarães SE, et al. Improved estimation of inbreeding and kinship in pigs using optimized SNP panels. BMC Genet. 2013;14:92. doi: 10.1186/1471-2156-14-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peripolli E, Metzger J, de Lemos MVA, Stafuzza NB, Kluska S, Olivieri BF, et al. Autozygosity islands and ROH patterns in Nellore lineages: evidence of selection for functionally important traits. BMC Genomics. 2018;19(1):680. doi: 10.1186/s12864-018-5060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher RA. A fuller theory of “junctions” in inbreeding. Heredity. 1954;8(2):187–197. [Google Scholar]
- 37.Hayes BJ, Visscher PM, McPartlan HC, Goddard ME. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003;13(4):635–643. doi: 10.1101/gr.387103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luan T, Yu X, Dolezal M, Bagnato A, Meuwissen TH. Genomic prediction based on runs of homozygosity. Genet Sel Evol. 2014;46:64. doi: 10.1186/s12711-014-0064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morita Y, Wada-Hiraike O, Yano T, Shirane A, Hirano M, Hiraike H, et al. Resveratrol promotes expression of SIRT1 and StAR in rat ovarian granulosa cells: an implicative role of SIRT1 in the ovary. Reprod Biol Endocrinol. 2012;10:14. doi: 10.1186/1477-7827-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao F, Zhao W, Ren S, Fu Y, Fang X, Wang X, et al. Roles of SIRT1 in granulosa cell apoptosis during the process of follicular atresia in porcine ovary. Anim Reprod Sci. 2014;151(1–2):34–41. doi: 10.1016/j.anireprosci.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Liu T, Zhang P, Li T, Chen X, Zhu Z, Lyu Y, et al. SETDB1 plays an essential role in maintenance of gonocyte survival in pigs. Reproduction. 2017;154(1):23–34. doi: 10.1530/REP-17-0107. [DOI] [PubMed] [Google Scholar]
- 42.Xuan B, Li ZC, Wang QY, Xu M, Chen X, Jin Y. Inhibition of PSMD4 alters ZP1 ubiquitination state and sperm-oocyte-binding ability in pigs. Reprod Domest Anim. 2018;53(3):688–694. doi: 10.1111/rda.13158. [DOI] [PubMed] [Google Scholar]
- 43.Ding H, Liu M, Zhou C, You X, Suo Z, Zhang C, et al. Expression and regulation of GnRHR2 gene and testosterone secretion mediated by GnRH2 and GnRHR2 within porcine testes. J Steroid Biochem Mol Biol. 2019;190:161–172. doi: 10.1016/j.jsbmb.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Ru YF, Xue HM, Ni ZM, Xia D, Zhou YC, Zhang YL. An epididymis-specific carboxyl esterase CES5A is required for sperm capacitation and male fertility in the rat. Asian J Androl. 2015;17(2):292–297. doi: 10.4103/1008-682X.143314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazarian E, Son H, Sapao P, Li W, Zhang Z, Strauss JF et al. SPAG17 Is Required for Male Germ Cell Differentiation and Fertility. Int J Mol Sci. 2018;19(4):1252. [DOI] [PMC free article] [PubMed]
- 46.Suzuki T, Kosaka-Suzuki N, Pack S, Shin DM, Yoon J, Abdullaev Z, et al. Expression of a testis-specific form of Gal3st1 (CST), a gene essential for spermatogenesis, is regulated by the CTCF paralogous gene BORIS. Mol Cell Biol. 2010;30(10):2473–2484. doi: 10.1128/MCB.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wimmers K, Murani E, Te Pas MF, Chang KC, Davoli R, Merks JW, et al. Associations of functional candidate genes derived from gene-expression profiles of prenatal porcine muscle tissue with meat quality and muscle deposition. Anim Genet. 2007;38(5):474–484. doi: 10.1111/j.1365-2052.2007.01639.x. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Xu Z, Li H, Xiong Y, Zuo B. Differential transcriptional analysis between red and white skeletal muscle of Chinese Meishan pigs. Int J Biol Sci. 2010;6(4):350–360. doi: 10.7150/ijbs.6.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pravenec M, Saba LM, Zidek V, Landa V, Mlejnek P, Silhavy J, et al. Systems genetic analysis of brown adipose tissue function. Physiol Genomics. 2018;50(1):52–66. doi: 10.1152/physiolgenomics.00091.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xi D, Roizen J, Lai M, Gandhi N, Kublaoui B. Paraventricular nucleus Sim1 neuron ablation mediated obesity is resistant to high fat diet. PLoS One. 2013;8(11):e81087. doi: 10.1371/journal.pone.0081087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sailer AW, Sano H, Zeng Z, McDonald TP, Pan J, Pong SS, et al. Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proc Natl Acad Sci U S A. 2001;98(13):7564–7569. doi: 10.1073/pnas.121170598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin P, Taussat S, Vinet A, Krauss D, Maupetit D, Renand G. Genetic parameters and genome-wide association study regarding feed efficiency and slaughter traits in Charolais cows. J Anim Sci. 2019;97(9):3684–3698. doi: 10.1093/jas/skz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cirera S, Clop A, Jacobsen MJ, Guerin M, Lesnik P, Jorgensen CB, et al. A targeted genotyping approach enhances identification of variants in taste receptor and appetite/reward genes of potential functional importance for obesity-related porcine traits. Anim Genet. 2018;49(2):110–118. doi: 10.1111/age.12641. [DOI] [PubMed] [Google Scholar]
- 54.Fadason T, Ekblad C, Ingram JR, Schierding WS, O'Sullivan JM. Physical interactions and expression quantitative traits loci identify regulatory connections for obesity and type 2 diabetes associated SNPs. Front Genet. 2017;8:150. doi: 10.3389/fgene.2017.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song QY, Meng XR, Hinney A, Song JY, Huang T, Ma J, et al. Waist-hip ratio related genetic loci are associated with risk of impaired fasting glucose in Chinese children: a case control study. Nutr Metab. 2018;15:34. doi: 10.1186/s12986-018-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.GX E, Basang WD, Zhu YB. Whole-genome analysis identifying candidate genes of altitude adaptive ecological thresholds in yak populations. J Anim Breed Genet. 2019;136(5):371–377. doi: 10.1111/jbg.12403. [DOI] [PubMed] [Google Scholar]
- 57.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Gene content inside run-of-homozygosity islands.
Additional file 2: Table S2 GO terms and KEGG pathways enriched (P < 0.05) based on run-of-homozygosity islands.
Data Availability Statement
The data and computing programs used in this manuscript are available from the corresponding authors on request.